

Abstract
Spatial organization of G-protein coupled receptors (GPCRs) into dimers and higher order oligomers has been demonstrated in vitro and in vivo. The pharmacological readout was shown to depend on the specific interfaces, but why particular regions of the GPCR structure are involved, and how ligand-determined states change them remains unknown. Here we show why protein-membrane hydrophobic matching is attained upon oligomerization at specific interfaces from an analysis of coarse-grained molecular dynamics simulations of the spontaneous diffusion-interaction of the prototypical beta2-adrenergic (β2AR) receptors in a POPC lipid bilayer. The energy penalty from mismatch is significantly reduced in the spontaneously emerging oligomeric arrays, making the spatial organization of the GPCRs dependent on the pattern of mismatch in the monomer. This mismatch pattern is very different for β2AR compared to the highly homologous and structurally similar β1AR, consonant with experimentally observed oligomerization patterns of β2AR and β1AR. The results provide a mechanistic understanding of the structural context of oligomerization.
The G-protein coupled receptors (GPCRs) comprise a family of more than 800 cell surface receptors characterized by seven transmembrane (TM) segments, and are targeted by a large percentage of therapeutic agents1. The rapid growth of structural information about family A GPCRs from recently determined crystal structures of various members of this protein family2,3,4 has strengthened the basis for understanding their functional mechanisms in a structural context.
A major inference from the crystallographic structures of different family A GPCRs is that they share key motifs for ligand binding and activation2,5,6, and that these are generally consistent with results established from a long record of molecular-level studies employing site-directed mutagenesis and molecular modeling. In particular, the agreement pertains to the identity and putative functional role of the highly conserved sequence motifs (SM) determined from functional studies to constitute functional microdomains (FM) that mediate specific elements of the receptor activation mechanism7, including the “arginine cage” around the conserved E/DRY motif in TM3, the NPxxY motif in TM7, the cluster of aromatic residues in TM6, and the amphiphilic “helix 8”.
The structural specificity of the mechanistic understanding reflected in the identification of these SM/FMs underlying ligand-GPCR function is not yet matched by information regarding the determinant elements for the oligomerization of GPCR proteins in the plane of the lipid membrane, in spite of the abundant evidence that it significantly affects function8,9,10,11,12,13. Crystallographic data have been used to infer on some aspects of dimerization14, and some detailed knowledge of the oligomerization interfaces has emerged8,9,15, including the energetics of particular dimerization interfaces evaluated from computational simulations7,16,17,18. However, the mechanistic basis for GPCR oligomerization in a structural context remains unknown in spite of the ability to predict such interfaces from knowledge-based considerations19. With oligomerization being shown to affect the pharmacological readout of GPCRs8, and its consequences being demonstrated to be physiologically important in vitro and in vivo9,12,13, the absence of structure-based mechanistic insights about oligomerization is increasingly noticeable.
To address these mechanistic aspects, we present here the development of a structure-based understanding of the determinants for oligomerization interfaces in prototypical Class A GPCRs in the context of the membrane surrounding the GPCR monomers and oligomers. A key determinant of the mutual effects of proteins and the lipid environment is the hydrophobic mismatch20, which was also suggested to affect the oligomerization of class-A GPCRs21,22. A major component of the hydrophobic mismatch energy emerges from the inability of membrane deformation around multi-helical membrane proteins, such as GPCRs, to alleviate fully the hydrophobic mismatch23,24. The energy penalty associated with this “residual hydrophobic mismatch” (RHM) is composed of specific local contributions that can be attributed to particular residues23,25. As the RHM can be reduced if the GPCRs oligomerize so as to prevent the exposure of the residues where the mismatch is not alleviated by membrane deformation, the mechanism for GPCR oligomerization in the membrane should depend on the energy cost from the RHM. To calculate the membrane deformation pattern and quantify the energy cost of hydrophobic mismatch, we employed in this study a recently developed approach, Continuum-Molecular Dynamics (CTMD), designed for multi-transmembrane segment (TM) proteins like GPCRs23 (see brief description in Methods).
Here we demonstrate the role of the RHM as a mechanistically important component of GPCR oligomerization in a case study of β2AR oligomerization. From the molecular dynamics simulation of the spontaneous diffusion-reaction of this prototypical GPCR in the standard POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) lipid bilayer, we find that RHM at TMs 1,4, and 5 of the monomeric GPCR is substantially alleviated in the oligomeric arrays that emerge during the simulation. Analysis of these results in contrast to those for the highly homologous and structurally very similar β1AR, explains the role of particular residues in driving GPCR oligomerization in terms of the alleviation of RHM and underscores the mechanistic basis for the agreement with experimental findings for these receptors.
Results
In the analysis of interactions between multi-TM proteins and the membranes surrounding them, the residual hydrophobic mismatch (RHM) at specific TMs, which cannot be alleviated by the remodeling of the membrane, was found to occur in regions in which polar and hydrophobic residues are adjacent on the surface23,25. The energy penalty associated with this RHM was shown to account for a large component of the energy cost of hydrophobic mismatch between such proteins and the membrane23. We reasoned that the demonstrated role of the lipid-protein interactions in GPCR oligomerization21 may involve a reduction of the RHM following the elimination of key residues in such polar/non-polar adjacencies from the interaction with the membrane7,23. The attendant reduction in energy penalty achieved by the oligomerization would thus constitute an important component of the energy drive in the experimentally observed membrane-driven GPCR oligomerization21. Analysis of the simulation trajectories and the energetics of protein-membrane interactions are therefore used here to investigate whether the identification of the specific regions of the GPCR molecule that generate the largest energy drive may broadly define the oligomerization interface.
Oligomerization alleviates the energy cost of residual hydrophobic mismatch in β2AR
A ~18 μs-long simulation trajectory was obtained for the spontaneous diffusion-interaction of nine β2AR molecules in a model POPC lipid bilayer at a lipid/protein ratio of ~110:1. The coarse-grained MD (CGMD) simulation with the MARTINI force field was carried out along the lines of previously described simulations of rhodopsin oligomerization22 (see Methods for further details). The RHM was quantified as described previously23 in the dynamically emerging oligomeric arrays of β2AR, which were seen to remain stably associated within the simulation time scales and to maintain their specific oligomerization interfaces. Indeed, the interfaces identified here as “stable” persisted over the last several microseconds (>3 μs) of the simulation, and Figure 1a illustrates the oligomeric array in the final snapshot of the simulation. To follow how the system evolved to form these oligomeric arrays in the dynamics simulations, Figure 1b quantifies the number (N) of GPCRs that have at least one interaction partner over the course of the simulation. During the first 8–9 μs, N is seen to increase gradually to a constant value, as the nine GPCRs associate in the plane of the membrane. However, this “stable” state was reached after frequent transitions between different values of N, as indicated by the high density of the vertical lines in Figure 1b. Thus, the GPCRs associate and dissociate multiple times locally, en route to forming the oligomeric arrays shown in Figure 1a.
Figure 1. Simulation of β2AR oligomerization.

(a) Snapshot from the end of the simulation trajectory, showing the spontaneously evolved higher-order oligomers of β2AR. Note that the simulation cell has periodic boundaries. For the sake of clarity, the membrane is shown as green dots in the simulation cell only, and the water is not shown. The proteins are rendered in VdW representation. To indicate the orientation of the proteins in these arrays, specific parts of proteins are colored as follows: TM1 is rendered in purple, TM4 in red, TM5 in blue, TM6 in yellow and the rest of the protein in silver color. The oligomeric arrays emerging from the simulations are found to involve the typical interfaces such as TM1-TM1, TM5-TM5, and TM4/5 interfaces. (b) Evolution of the number of GPCRs participating in protein-protein interactions over the 18 μs time course of the simulation. A GPCR is designated as interacting if any of its residues is within a cutoff distance of 5 Å from any other GPCR. The plot is densely populated with vertical lines during the first ~8 μs, which shows that the number of interacting GPCRs changes frequently during this time period. This indicates multiple binding-unbinding events (also seen in Supplementary Movie S1). After ~8 μs of such binding-unbinding events, all GPCRs interact continuously. (c) The relative frequency with which the different regions of the protein participate in protein-protein interactions during the last 1.4 μs, shown in a color-coded heatmap projected onto a X-ray structure of β2AR (PDB 2RH16). Light blue, white, and red colors indicate regions involved in protein-protein interactions, but most of protein is colored in the deep blue which identifies regions are not involved in frequent oligomerization contacts during the simulation. The heatmap is presented on three different views of the protein in order to show the entire protein surface. The total number of interactions for each residue is normalized to the maximum frequency of interactions for all residues during this time period.
Even when a protomer maintained a continuous interaction with another GPCR, it was seen to explore locally different interfaces in highly transient interactions (see Supplementary Movie S1) before forming the interface identified as “stable” according to the defined criteria, and shown in Figure 1a. Notably, this organization was achieved from an initial array in which the protomers were positioned equidistantly, and in mutual orientations that had no preference whatsoever for the eventual pattern of oligomerization interfaces (Supplementary Fig. S1). It is noteworthy that the extended oligomeric architectures as seen in Figure 1a have been observed experimentally for GPCRs, e.g., in AFM studies of rhodopsin17,26, and in a recent crystallographic study of ligand-free β1AR27. The β2AR oligomers observed here involve typical interfaces8,9,17,18,26,27,28,29,30, such as TM1-TM1, TM4/5, and TM5-TM5 (Fig. 1a; also see the snapshots in Fig. 2b–d). These interfaces also include residues from neighboring TMs, e.g., the IC end of TM3 in TM4/5 interface (shown in the snapshot in Fig. 2d).
Figure 2. Lipid-protein interactions in the oligomers.

(a) The average energy cost of residual hydrophobic mismatch (RHM) for each TM of β2AR embedded in the POPC membrane bilayer, calculated for a protomer in the oligomeric arrays (in black), and compared to that calculated for the monomeric protein (in red). (b–d) Deformation of the membrane upon β2AR oligomerization. Two profiles are shown in (b), one calculated from the 1.4 μs segment of the trajectory (on the right), and the other from the preceding 1.4 μs segment of the trajectory (on the left); note the similarity which substantiates the robustness of the calculated membrane response observed in all cases. The time-averaged membrane thickness profiles are shown around a protomer that forms a loosely packed TM1-TM1 dimerization interface at one end of an oligomeric array, in (b); around a protomer that forms a tightly packed TM1/H8-TM1/H8 dimerization interface at one end of another oligomeric array, in (c), and around a protomer that interacts with two other GPCRs, forming both a TM1-TM1 interface and a TM5-TM5 interface, in (d). The interacting protomers are taken from simulation snapshots and shown in cartoon representation, with TM1 colored in purple, TM3 in cyan, TM4 in yellow, TM5 in tan, and other TMs in silver. H8 is shown in green color. The membrane deformations were quantified relative to the particular protomer in question, with the trajectory being centered at the protomer. These are shown as time-averaged surfaces composed of phosphate headgroups around the protein calculated on a 2 Å * 2 Å grid, with the color scheme indicating membrane thickening (in red) and thinning (in blue).
A heatmap of the relative frequencies of interaction for the different β2AR residues over the last 1.4 μs of the simulation is shown in Figure 1c superimposed on the molecular structure. Residues in TM1, TM4-TM5, Extracellular (EC) end of TM6, Intracellular (IC) end of TM3, and ICL2 are seen in the heatmap of Figure 1c to appear more frequently in interfacial interactions (red, white, and light blue colors) compared to the other regions of the GPCR (deep blue in the heatmap). This indicates that the early local exploration of dimerization interfaces and neighboring residues is followed by the stabilization of interactions in specific regions of the protein so as to generate the spontaneously evolved, preferred oligomerization interfaces.
While these observations agree with a wealth of prior biophysical data suggesting that oligomerization brings together specific regions of the GPCRs8,9,17,26,27,28,30, it was not possible to establish from the previous data why these specific regions constituted preferred interfaces. Such a quantitative understanding of the mechanism of interface stabilization was obtained from our analysis focusing on the interaction of the residues with the surrounding membrane in the spontaneous aggregation simulations, as described below. In particular, we quantified the extent to which membrane deformations achieve hydrophobic matching in the TM-bundle, and identified the significant reduction in RHM achieved through the formation of the observed oligomerization interfaces.
The calculation of RHM in the various configurations of the oligomeric array was based on quantification of the exposed surface area SAres,i of the individual residues participating in unfavorable hydrophobic-polar interaction23, as detailed in the Methods. As described before23, the energy cost of the RHM (exposing polar residues to the hydrophobic core of the membrane, and hydrophobic residues to the polar environment) is calculated from the corresponding SAres,i according to equation (1) in Methods. To quantify the change in RHM due to oligomerization, we calculated for each TM the time-averaged RHM per protomer in the trajectory until the first protein-protein interaction occurs, and compared it to the time-averaged RHM per protomer in the stable oligomeric arrays of the last 1.4 μs (see Methods for details). As shown in Figure 2a, the RHM in the group of TMs (TM1, TM4, and TM5) that had the largest RHM in the monomer, is indeed much lower in the oligomeric arrays. The total difference is substantial, ~10 kT (where k is the Boltzmann constant; T is temperature), suggesting that the improved hydrophobic matching to their surrounding membrane environment that is achieved by the β2AR molecules in the oligomeric arrays involving all three TMs (1, 4 and 5), is an important factor in the oligomerization. Note that the alleviation of RHM in the stable oligomeric arrays is robust in the course of the simulation (the RHM differs by <0.2 kT at each TM, and by ~0.3 kT in the entire TM-bundle when calculated from the preceding 1.4 μs of the simulation - see Supplementary Figure S2). While the energetics of oligomeric interactions had been suggested to involve specific protein-protein interactions17, data from FRET experiments and computational studies indicate that hydrophobic mismatch modulates GPCR oligomerization21,22. The present quantitative results show that it is the incomplete hydrophobic matching at specific residues in the monomer, the RHM, that provides an energetically important drive for such spatial organization.
While GPCR oligomerization interfaces can involve many residues (Fig. 1c, see also8), our calculations reveal the key role of specific regions. Thus, the energy gain of ~10 kT from the reduced RHM is due to a relatively small number of residues: D1.28, E1.29, F1.60, N4.40, Q4.62, Q5.63, and E7.33 (generic numbering according to Ballesteros-Weinstein36). It is noteworthy that the reduction in RHM occurs at regions that are diametrically opposite on the molecular circumference. This is essential for the formation of higher-order oligomers (Fig. 1a), which has been shown experimentally to occur for β2AR in both model and cell membranes28,31.
The change in the RHM upon oligomerization depends not only on the removal of key residues from interaction with the membrane, but also on the distinct membrane deformation profile around oligomers, which differs from that around monomers. To understand the impact of these changes we quantified the time-averaged membrane deformations around each protomer in the two oligomeric arrays described above in Figure 1a, over the last 2.8 μs of the trajectory. The stable oligomeric arrays were found to elicit a robust membrane response with a pattern that is sustained in the course of the simulation. Specifically, the pattern of membrane thinning and thickening around each protomer is very similar in two consecutive 1.4 μs segments of the last 2.8 μs simulation (cf. the two panels of Fig. 2b). The change in membrane deformation profile with the formation of tightly packed oligomers is illustrated for the systems in Figure 2b and Figure 2c showing two TM1-TM1 dimeric interfaces, one with much closer packing of the protomers (Fig. 2c) than the other (Fig. 2b). Note that in Figure 2b the β2AR protomer is positioned at one end of the tetrameric array, away from its dimerization partner, so that only a small extracellular part of its TM1 is involved in the dimerization interface. Around this protomer, the membrane is thicker between TM4 and TM5, similar to the red (thick) region around a β2AR monomer on the same 38 Å to 48 Å scale of thickness (see below, in the sub-section comparing β2AR and β1AR). This local thickening is adjacent to regions of local thinning and substantiates the robustness of the membrane deformation pattern which is calculated to be the same for corresponding protomers, or oligomers, when calculated in different circumstances.
At one end of an oligmeric array formed in the simulation, we observe dimerization of the β2AR protomer by means of TM1 to be more closely packed so that the interaction involves both TM1 and H8 (Fig. 2c). Here too, we find local thickening between TM4 and TM5 at the free end of the protomer, but the overall membrane thickness profile around this protomer has considerably less membrane thinning adjacent to the membrane thickening when compared to the case of the loosely dimerizing protomer (Fig. 2b), and to the monomer, as illustrated below.
Cholesterol and the residual hydrophobic mismatch of β2AR monomers
Because RHM was previously found to depend on the lipid composition23, and cholesterol content of the cell membrane is known to be an important modulator of protein function32,33, we evaluated the extent of hydrophobic matching by membrane deformations in β2AR embedded in a POPC bilayer with added 10% Chol. Figure 3 shows the RHM in the TM-bundle of β2AR in a POPC/10% Chol lipid bilayer (blue bars in Fig. 3), and reveals that in the presence of cholesterol, just as in the membranes not containing it, the large RHM occurs at TM1, TM4, and TM5 of the monomer (red bars in Fig. 3). Thus, dimerization in this Chol-containing lipid composition is expected to occur at both the TM1 and the TM4/5 side of the GPCR, just as in the absence of the cholesterol. The total energy cost due to RHM in POPC/10% Chol, ~29–30 kT, is comparable to the corresponding energy cost per protomer in POPC before the formation of oligomeric contacts (Fig. 3).
Figure 3. Residual hydrophobic mismatch: β2AR in POPC vs. POPC/10% Cholesterol, and β2AR vs. β1AR.
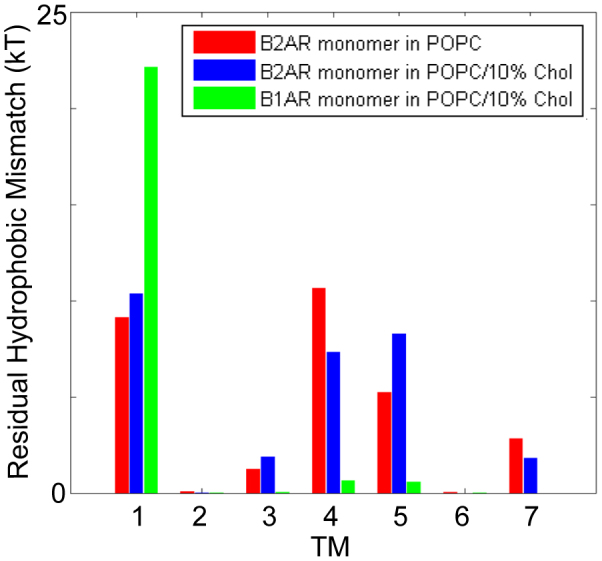
The average energy cost of residual hydrophobic mismatch (RHM) for each TM of a β2AR monomer embedded in POPC/10% Cholesterol bilayer (blue bars), compared to the results for the β2AR monomer in POPC without Cholesterol from Figure 2a (red bars), and the highly homologous β1AR monomer in the same POPC/10% Cholesterol (green bars). The large RHM at the diametrically opposite regions of TM1 and TM4/TM5, observed for β2AR monomer in POPC, occurs as well in POPC/10% Cholesterol. Notably, this pattern is distinct from that in the β1AR monomer, where large RHM occurs only at TM1 but not at TM4/TM5. These differences are consonant with experimentally observed differences in the dimerization patterns of β2AR and β1AR, as described in the text.
The residual hydrophobic mismatch is significantly different for β2AR and β1AR in spite of their similarity in sequence and structure
β2AR shares 67% sequence identity in the TM-bundle with β1AR, and the crystal structures of the two GPCRs are very similar3,5. Yet in spite of the similarity in sequence and structure, β2AR has been shown to differ from β1AR in terms of oligomerization31,34, and localization in the cell membrane35. In particular, β2AR has been shown to form more stable/extensive oligomers than β1AR34, and to form higher order oligomers28,31, whereas β1AR organization was suggested to comprise mainly dimers31. As RHM was demonstrated to be a mechanistically important component of lipid-protein interactions (Figs. 1 and 2), we reasoned that the different spatial organization patterns of these two very similar GPCRs in the membrane may reflect differences in RHM due to small, localized differences in the residues at interfaces with the membrane.
Following the same protocol used for β2AR in the Chol-containing membrane, the RHM was calculated in CGMD simulations of β1AR monomers in the POPC/10% Chol membrane. Figure 3 presents the results for β1AR and compares them with the RHM for β2AR in the identical lipid composition. The remarkable difference is that almost no RHM occurs at TM4 and TM5 of β1AR (green bars, Fig. 3), and the RHM in β1AR monomer is localized to TM1.
The prediction from this pattern of RHM in the β1AR monomer is that dimerization with a TM1-TM1 interface will achieve a significant reduction of RHM energy penalty, but that unlike the case for β2AR, TM5 and TM4 interfaces would not produce such energy drive. This was probed by simulating β1AR dimers with a TM1-TM1 interface, and comparing the resulting RHM to that in a control simulation of a dimer with a typical interface that is distant to TM1, viz. a TM5-TM5 interface. To assess the sensitivity of the calculations to the difference we highlighted between β1AR and β2AR, these simulations were started with a β1AR dimer configuration obtained by aligning a pair of β1AR protomers to a pair of β2AR protomers forming either a TM1-TM1 interface, or a TM5-TM5 interface, in the simulated system shown in Fig. 2 above. All β1AR dimers were embedded in the same POPC membrane model used for the β2AR oligomerization simulation. Each simulation was performed for 2 microseconds, and time-averaged RHM were calculated over the last 1 μs. The results show that with the formation of the TM1-TM1 interface, the average RHM per protomer is reduced to ~7 kT from the RHM calculated to be ~22 kT at TM1 for the β1AR dimer with the TM5-TM5 interface, or the β1AR monomer. Thus, the reduction of RHM in β1AR occurs for the TM1-TM1 interface, but not for the TM5-TM5 interface.
To allow additional freedom in forming the dimerization interface on the timescale of these simulations, two additional simulation were conducted by starting out with the protomers facing each other either in the TM1 region, or the TM4/5 region respectively, but allowing for the presence of lipids between the protomers (see Fig. S3). The resulting RHM at TM1 is ~3 kT per monomer for the TM1-TM1 interface thus formed, which is even smaller than the ~6 kT for the preformed dimer with the TM1-TM1 interface. On the other hand, the RHM at TM1 remains close to ~22 kT for protomers apposed at the TM 4/5 region. The RHM at all other TMs is ~0 kT in all cases (see Table S2 in Supplementary Information), showing that the dimerization drive is as predicted from the RHM and is not complicated by changes in RHM at other TMs. Although a complete spontaneous sampling of dimerization interfaces is well beyond the scope of even such coarse-grained simulations17, the results show that the pattern of RHM in the β1AR monomer contributes an energetically significant component to dimerization only at TM1, and the CGMD simulations reflect the differences between the β1AR and β2AR indicated by the RHM calculations.
Taken together, the above results agree well with the observation that β1AR forms mainly dimers31, which are predicted from the RHM to involve a TM1-TM1 interface. For β2AR, however, both the RHM and the simulation of diffusion-interaction in the POPC membrane bilayers suggest the formation of oligomers. The oligomerization pattern observed in the time scale of the simulation (Fig. 1a), is thus consistent with the evidence that the β2AR forms ordered oligomers with different interfaces28,31. This agreement between the calculated and experimentally determined differences in the oligomerization patterns of the two highly similar GPCRs not only substantiates the importance of the RHM in the oligomerization mechanism, but indicates as well the high sensitivity of this quantity as a predictor of spatial organization in the membrane. It is also noteworthy that for the β2AR monomer, the total energy cost we calculate due to RHM is larger by ~5 kT than for the β1AR monomer, which is further consistent with experimental observations of the more extensive/stable oligomerization of β2AR, compared to β1AR34.
Given the importance of the RHM in the oligomerization, we focused on the large difference in the RHM observed for TM4 and TM5 of β2AR, but not β1AR, to identify the structure-based mechanism for the difference in the ability of membrane deformations to alleviate hydrophobic mismatch for these GPCRs. The reason for this difference is suggested by the observation that the RHM is significantly large when polar membrane-facing residues are immediately adjacent to hydrophobic residues in the TM-bundle of multi-TM proteins, so that the membrane is unable to match the properties of both residues with the same rearrangement25. Indeed, for β2AR in POPC/10% Chol we find such a structural context for the residues that remain exposed to the mismatched environment in TMs 4 and 5 (Fig. 4). The polar residues Q1704.62 and N1484.40 in TM4, and Q1975.36 and Q2245.63 in TM5 are all parts of polar-hydrophobic adjacency. For example, the polar residue Q1704.62 of β2AR is sandwiched, at the same level in the membrane, between two hydrophobic residues, F1043.22 and M171. Figure 4b shows the situation of these four residues in detail.
Figure 4. The structural context of the incomplete hydrophobic matching observed in β2AR, but not in β1AR.

(a) Time-averaged membrane deformations around β1AR and β2AR. A snapshot of the protein is shown, with the region of the protein where RHM occurs in β2AR, colored as follows: TM3 is in cyan, TM4 in yellow, and TM5 in tan colors; residues near the ends of the TMs are shown in VdW representation, and are colored according to hydrophobicity properties with hydrophobic residues in orange, and polar residues in purple. The membrane deformations are shown in terms of the time-averaged phosphate surface around the centered protein calculated on a 2 Å * 2 Å grid, with the color scheme indicating membrane thickening (in red) and thinning (in blue). The membrane is seen to thicken (red) near the hydrophobic residues, and to become thinner (blue) in the region immediately adjacent to the polar residues. This shows that in order to alleviate completely the hydrophobic mismatch the membrane would have to become both thicker and thinner in closely neighboring regions, which explains the incomplete hydrophobic matching in β2AR. (b) Neighborhoods of adjacent polar and hydrophobic residues at regions of incomplete hydrophobic matching in β2AR. Hydrophobic residues are shown in orange color and polar residues in purple. Q1704.62 is sandwiched, at the same level in the membrane, between F1043.22 and M171. At the other end of TM4, N1484.40 is in a polar group comprising K1474.39, N1484.40, K1494.41, R1514.43 that is similarly adjacent to the hydrophobic F1323.52 and Y1333.51. In TM5, Q1975.36 is adjacent to hydrophobic residues I2986.60 and V2956.57. Moreover, Q2245.63 is part of a group of polar residues, R2215.60 and K227, which is positioned next to a group of hydrophobic residues F1323.52 and Y1333.51. (c) Structure-based sequence alignment of β1AR5 vs. β2AR for TM4 and TM5, with the loci where the two homologous GPCRs differ in terms of hydrophobic character indicated in bold lettering.
In contrast, in β1AR, the corresponding residues in positions 4.40 and 4.62 of TM4 are non-polar (Ala and Leu), whereas in β2AR they are polar (Asn and Gln) (Fig. 4c). In both β1AR and β2AR, the residues at positions 5.36 and 5.63 of TM5 are polar, but different: in β1AR they are Arg, whereas in β2AR they are Gln. This is an important difference because the longer Arg is known to be able to better accommodate hydrophobic mismatch by means of snorkeling37, and does so as well in our simulations.
Discussion
GPCRs have long been considered to function as monomeric units, but the functional unit is now widely considered to involve dimers or indeed oligomers. Much experimental evidence suggests that family A GPCRs spontaneously undergo association and dissociation in the membrane38,39,40,41, and the analysis here identifies important contributions to the energetics of such spatial organization of the GPCRs.
Although rhodopsin and β2AR can efficiently activate downstream G-protein even as a monomer42, these receptors and several other members of family A GPCRs have been demonstrated with a number of experimental approaches to form dimers or higher-order oligomers in the membrane environment26,34,38,41,43. In particular, the dimerization interface has emerged as critical to the functional properties, by being ligand-sensitive8,28 and by modulating communication between functionally asymmetric protomers in the di-/oligomeric complex9,10. Ligand-GPCR interactions are typically investigated with reference to structural motifs and the SM/FM that have been shown to mediate activation7, but one of the reasons making it more difficult to identify the key structural elements underlying the mechanism of GPCR oligomerization is that the interfaces usually involve a large number of residues, and the total surface area buried in the dimeric interface per se is not indicative of the strength of dimerization17. The role of the protein-membrane interaction and reduction of RHM in the spatial organization of GPCRs, as described here, clarifies in a structure-based quantitative manner some key elements in the mechanism of dimerization and oligomerization. Thus, the quantitative analysis of hydrophobic matching in the spontaneous oligomerization of β2AR in the membrane demonstrates that the large RHM, which occurs at sites where polar and hydrophobic membrane-facing residues are adjacent in the GPCR structure, contributes significantly to the mechanism of GPCR oligomerization. We had previously shown that in multi-TM proteins such adjacencies constrain the ability of membrane deformation to achieve complete hydrophobic matching, thus generating local RHM23,25. In fact, for multi-TM proteins with radially asymmetric hydrophobic surface, it has been demonstrated that RHM can occur even if the average hydrophobic thickness of the protein is well-matched to that of the lipid environment25. The results described here illustrate the underlying mechanism, and its consequences for the formation of oligomers, with the case of the β2AR.
We showed that the structure of monomeric β2AR (but not of the highly homologous β1AR) leaves specific residues of TM4 and TM5 exposed in spite of membrane deformation aimed at reducing hydrophobic mismatch, which produces the corresponding energy penalty. In the β2AR, the unfavorable exposure occurs as well in TM1, and because the RHM is eliminated (or significantly reduced) when monomers come together, oligomeric arrays are formed (from interactions at both the TM4,5 interface and the antipodal TM1 interface), and these remain stable within the timescale of the simulation. The oligomerization involves an energy gain from the alleviation of RHM which we evaluated to be ~10 kT for β2AR in POPC. Notably, the extent and pattern of RHM has been shown to depend on the lipid composition23, and this may explain differences in functional properties of GPCRs in different cells or different membrane environments in the same cell, but for β2AR we showed here that the presence of 10% cholesterol with POPC lipid does not produce significant changes in the oligomerization pattern.
While the relation between reduction of RHM and oligomer formation is demonstrated here for the prototypical GPCR β2AR, this is likely to be a common feature in family A GPCRs. Indeed, a large RHM was found here in the β1AR monomer as well, and previously for rhodopsin and the serotonin 5HT2A receptor7,23. But the RHM depends on the particular GPCR conformation, and different patterns of RHM can occur, as illustrated here even for the highly homologous β1AR and β2AR, when conformational changes modify the adjacency of membrane-facing polar and hydrophobic residues. Indeed, we found that in the structurally very similar β1AR, where only TM1 exhibits a high RHM, the corresponding energy gain is limited to the formation of TM1-TM1 interfaces, suggesting a membrane-driven preference for spatial organization as dimers rather than the oligomers we observed for β2AR.
As noted in the Results, we obtained support for the underlying mechanistic hypotheses concerning the nature of the energy that drives dimerization, from the agreement with available experimental data on (i)-spontaneous oligomerization of family A GPCRs in the membrane28,44,45, (ii)-the identified oligomerization interfaces8,26, and (iii)-the observed differences in oligomerization between β1AR and β2AR31,34. Furthermore, our findings explain the available detailed knowledge regarding the nature and identity of oligomerization interfaces8,9. The insights produced by the present analysis complement the data regarding interaction energies calculated at particular dimerization interfaces from biased MD simulations17,18,29 with a mechanistic explanation for the identity and nature of residues driving the oligomerization.
Together, these insights produce an initial mechanistic understanding of the structural elements underlying GPCR-GPCR interactions in the plane of the membrane, akin to the understanding offered by the SM/FMs7 regarding the mechanisms and outcomes of GPCR-ligand interactions. In particular, the specific identification of residues involved in the RHM offers a practical and informative approach to the validation of conclusions from this type of calculations regarding both dimerization/oligomerization propensities, and the predicted interfaces, by suggesting specific mutations that would affect them. These inferences and predictions offer as well a defined structural basis for mechanistic investigations, e.g., of ligand-induced changes in the extent and/or dimerization interface of GPCR oligomerization7,8,46 by performing mutational studies.
For the reasons illustrated here, the extent and pattern of RHM in the monomer may change in different lipid environments even for the same receptor23, so that the energy drive from RHM towards oligomerization may depend on the lipid composition. Significant differences in oligomerization were indeed found for rhodopsin in membranes of different hydrophobic thickness21,22. In the cell membrane, the alleviation of RHM may thus conceivably occur by partioning into, or out of, raft-like nanodomains, in additon to homo-oligomerization and also hetero-oligomerization. Therefore, the quantitative understanding of the structural elements underlying the mechanistic role of lipid-GPCR interactions demonstrated here, should allow us to utilize the rapidly increasing structural information about GPCRs from X-ray crystallography to enhance our mechanistic understanding of how GPCRs organize in the cell membrane.
Methods
To quantify the energetics of hydrophobic mismatch between the GPCR and the lipid bilayer we applied throughout the recently described Continuum-Molecular Dynamics (CTMD) hybrid approach23. In this approach, the key issue of the radial asymmetry of the GPCR-membrane interface is treated by using the results of the MD simulation of the system in combination with continuum representations, as described below.
Energetics of membrane-protein interactions
The two components of the energy penalty due to hydrophobic mismatch are the penalties for the RHM (i.e., hydrophobic mismatch unalleviated by the deformed membrane), and for membrane deformation.
Residual hydrophobic mismatch (RHM)
Let SAres,i be the surface area of the ith residue participating in unfavorable hydrophobic-polar interactions (RHM). It is calculated in terms of the residue-specific solvent accessible surface areas (SASA), as follows:
Let SAmem,i = SASA with the solute comprising the protein and the hydrophobic core of lipid bilayer (C2-C2); let SAprot,i = SASA with the solute comprising the protein only.
Then, for hydrophobic residues, the RHM is SAmem,i, i.e., the part of the residue that is exposed outside the hydrophobic core of the lipid bilayer. Interfacial Trp is not penalized as it is favorable for the Trp to be located at the interface47.
For polar residues, the RHM is computed as SAres,i = SAprot,i − SAmem,i, i.e., the part of the residue that is exposed on the surface of the protein, but is not exposed outside of the hydrophobic core of the lipid bilayer. Arg and Lys located close to the membrane headgroups are not penalized as they alleviate hydrophobic mismatch by means of snorkeling37. Ser and Thr are not penalized as their polar parts form H-bonds with the helix backbone of the protein48.
The corresponding energy penalty is taken to be directly proportional to SAres,i, and the energy penalty at the Nth TM (N = 1 to 7) is given by
where the constant of proportionality is taken to be 0.028 kcal/(mol. Å2)49, and NTM is the number of membrane-exposed residues in the TM.
The solvent accessibilities (SASA) are computed from cognate coarse-grained molecular dynamics (CGMD) simulations (described below), with a probe radius of 5.2 Å (as done for MARTINI simulations22), and using the g_sas utility of the GROMACS software50.
The residue-wise comparison of the RHM in β1AR and β2AR is done on the basis of their structure-based sequence comparison (see the report on the β1AR crystal structure5).
Membrane deformation
The membrane deformation is described in terms of the local bilayer thickness d(x,y) and the corresponding membrane deformation 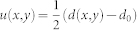 , where d0 is the bulk thickness of the bilayer away from the protein. To calculate d(x,y) around a protein from a MD trajectory, the trajectory is centered at that protein and the time-averaged d(x,y) is computed by fitting a 2 Å * 2 Å rectangular grid to the Phosphate beads over the course of the trajectory, followed by spatial smoothing. Complete details of this procedure are available in the original description of the approach and its application23. In addition, a web accessible stand-alone software, the CTMDapp, is available at memprotein.org/resources/servers-and-software, to quantify with CTMD the u(x,y) variable as well as the energy cost of membrane deformation (described below).
, where d0 is the bulk thickness of the bilayer away from the protein. To calculate d(x,y) around a protein from a MD trajectory, the trajectory is centered at that protein and the time-averaged d(x,y) is computed by fitting a 2 Å * 2 Å rectangular grid to the Phosphate beads over the course of the trajectory, followed by spatial smoothing. Complete details of this procedure are available in the original description of the approach and its application23. In addition, a web accessible stand-alone software, the CTMDapp, is available at memprotein.org/resources/servers-and-software, to quantify with CTMD the u(x,y) variable as well as the energy cost of membrane deformation (described below).
In the continuum framework of the CTMD method, the membrane is treated as an elastic continuum, with the energy cost of deformation taken to be the sum of the compression-extension, splay-distortion, and surface tension components51,52. The corresponding Euler-Lagrange equation is solved without the assumption of radial symmetry to obtain u(x,y) and with specific boundary conditions on the protein-membrane boundary from the cognate MD simulations, as described in complete detail in the original reference23. The elastic constants for POPC/10% Chol were taken to have typical values for such a lipid composition, Ka = 230 mN/m53, Kc = 0.9 * 10−19 J53, α = 3*10−3 N/Å23. The monolayer spontaneous curvature C0 = −0.004 Å−1 was calculated by taking the weighted sum of the spontaneous curvatures of individual components, weighted by their molar fractions32,54. The spontaneous curvature of cholesterol was taken as −0.04 Å−1 from Ref. 54, and that of POPC as 046. The solution to the Euler-Lagrange equation provides the energy cost of the membrane deformation, which we compute here for the simulations of the monomeric GPCRs.
The energy cost of membrane deformation was computed here for the numerically tractable case of monomeric simulations and not for the oligomeric simulation, as it was found to be similarly small for the monomeric B1AR and B2AR (<2 kT), compared to the ~10 kT gain from the alleviation of RHM on oligomerization of B2AR.
Molecular dynamics simulations
The necessary molecular-level information for the monomeric beta-adrenergic receptors was obtained from cognate CGMD simulations29 with the MARTINI force field22,56, using the GROMACS software57 and starting from molecular models based on available X-ray structures. The Martini force field has been used successfully for CGMD simulations of a number of membranes and membrane-protein systems, including the study of rhodopsin oligomerization17,22, and the hydrophobic mismatch between rhodopsin and lipidic cubic phase in the context of GPCR crystallization conditions58. It proved to be particularly well suited for the study of processes involving hydrophobic mismatch, comparing favorably to experiments and all-atom MD in terms of the partitioning of residues between water and hydrophobic media56,59. Of particular relevance to the comparison of β1AR and β2AR here (see Fig. 3 and 4c), such CGMD simulations were shown to be sensitive to the effects of some small mutations in transmembrane helices (e.g., Leu to Ala) on the hydrophobic mismatch related phenotype56, which are relevant to the type of differences in sequence between the βAR we compare. A known limitation of such CGMD simulations is that the assignment of helical conformation to stretches of residues is constrained during the trajectory calculation so that the protein cannot adapt to hydrophobic mismatch by changes in its helical content. However, such adaptation has been reported to occur for GPCRs only under conditions of large mismatch between the GPCR and the bilayer60, much larger than is the case of the systems we studied.
For the present work, the human β1AR was modeled with homology to the X-ray structure 2VT4 of turkey β1AR5. A model of the inactive β2AR was constructed based on the PDB entry 2RH16. The missing segments were modeled using Rosetta55. Each of the monomer constructs for β1AR and β2AR, respectively, was embedded in a large patch of POPC/10%Chol lipid bilayer with a lipid/protein ~530:1. After an initial equilibration with an all-atom force field, the systems were simulated for at least 2 microsecond with the MARTINI force field56, at constant volume, with restraints on the BAS particles, and with periodic boundary conditions. Further details on the protein constructs are available in ref. 29.
The MD simulations of the diffusion-interaction of β2AR molecules in the lipid bilayer were performed with a system of 9 β2AR molecules constructed from the starting structure obtained from the CGMD simulation of the monomeric β2AR based on the X-ray structure 2RH1 (see previous subsection). The model lipid bilayer was composed of POPC molecules. The number of proteins chosen corresponds to previously performed structural modeling of GPCR oligomers based on experimentally derived constraints9. A protein/lipid ratio of 1:110 was used, in line with the constructs studied in experiments and computations addressing the role of the membrane in GPCR oligomerization in the membrane21,22. The multi-GPCR system was simulated with constant temperature and pressure, periodic boundary conditions, and a time step of 30 fs.
Author Contributions
S.M. and H.W.* conceived the study. S.M., J.J., G.K., M.F. and H.W.* conceived the experiments. S.M., J.J. and H.W. performed the experiments. S.M. and H.W.* interpreted the data. S.M., G.K., J.J., M.F. and H.W.* wrote the paper.
Supplementary Material
Supplementary Information
Video - Supplementary Movie S1
Acknowledgments
We thank Drs. Olaf S. Andersen at Weill Cornell Medical College, and Davide Provasi at Mount Sinai, for helpful discussions. This work was supported by the National Institutes of Health grants P01DA012923 and U54GM087519 to Harel Weinstein, and DA026434, and DA034049 to M.F. We gratefully acknowledge the allocations of computational resources at (1) the Institute for Computational Biomedicine at Weill Medical College of Cornell University, (2) the New York Blue Gene Computational Science facility housed at Brookhaven National Lab, (3) the Scientific Computing Facility at Icahn School of Medicine at Mount Sinai, (4) the NERSC computational resources allocation m1710 (to H.W. and G.K.), and (5) NSF Teragrid allocations MCB090132 (to G.K.) and MCB080109N (to M.F.).
References
- Kahsai A. W. et al. Multiple ligand-specific conformations of the β2-adrenergic receptor. Nat. Chem. Biol. 7, 692–700 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K. et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289, 739–745 (2000). [DOI] [PubMed] [Google Scholar]
- Rosenbaum D. M., Rasmussen S. G. F. & Kobilka B. K. The structure and function of G-protein-coupled receptors. Nature 459, 356–363 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V., Cherezov V. & Stevens R. C. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 53, 531–556 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T. et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 454, 486–491 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V. et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan J., Khelashvili G., Mondal S., Mehler E. L. & Weinstein H. Ligand-dependent conformations and dynamics of the serotonin 5-HT2A receptor determine its activation and membrane-driven oligomerization properties. PLoS Comput. Biol. 8, e1002473 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Shi L., Filizola M., Weinstein H. & Javitch J. A. Crosstalk in G protein-coupled receptors: changes at the transmembrane homodimer interface determine activation. Proc. Natl. Acad. Sci. USA 102, 17495–17500 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y., Moreira I. S., Urizar E., Weinstein H. & Javitch J. A. Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat. Chem. Biol. 5, 688–695 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jastrzebska B., Orban T., Golczak M., Engel A. & Palczewski K. Asymmetry of the rhodopsin dimer in complex with transducin. FASEB J. 27, 1572–1584 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jastrzebska B., Ringler P., Palczewski K. & Engel A. The rhodopsin-transducin complex houses two distinct rhodopsin molecules. J. Struct. Biol. 182, 164–172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Maeso J. et al. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature 452, 93–97 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivero-Muller A. et al. Rescue of defective G protein-coupled receptor function in vivo by intermolecular cooperation. Sci. Signal. 107, 2319–2324 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V. et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein coupled receptor. Science 318, 1258–1265 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knepp A. M., Periole X., Marrink S. J., Sakmar T. P. & Huber T. Rhodopsin forms a dimer with cytoplasmic helix 8 contacts in native membranes. Biochemistry 51, 1819–1821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provasi D., Johnston J. M. & Filizola M. Lessons from free energy simulations of delta opioid receptor homodimers involving the fourth transmembrane helix. Biochemistry 49, 6771–6776 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periole X., Knepp A. M., Sakmar T. P., Marrink S. J. & Huber T. Structural determinants of the supramolecular organization of G protein-coupled receptors in bilayers. J. Am. Chem. Soc. 134, 10959–10965 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J. M. et al. Making structural sense of dimerization interfaces of delta opioid receptor homodimers. Biochemistry 50, 1682–1690 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filizola M. & Weinstein H. The study of G protein coupled receptor oligomerization with computational modeling and bioinformatics. FEBS J. 272, 2926–2938 (2005). [DOI] [PubMed] [Google Scholar]
- Lundbaek J. A., Collingwood S. A., Ingelfsson H. I., Kapoor R. & Andersen O. S. Lipid bilayer regulation of membrane protein function: gramicidin channels as molecular force probes. J. R. Soc. Interface 7, 373–395 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho A. V. r., Huber T., Sakmar T. P. & Brown M. F. Curvature and hydrophobic forces drive oligomerization and modulate activity of rhodopsin in membranes. Biophys. J. 91, 4464–4477 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periole X., Huber T., Marrink S. J. & Sakmar T. P. G protein-coupled receptors self-assemble in dynamics simulations of model bilayers. J. Am. Chem. Soc 129, 10126–10132 (2007). [DOI] [PubMed] [Google Scholar]
- Mondal S., Khelashvili G., Shan J., Andersen O. S. & Weinstein H. Quantitative modeling of membrane deformations by multihelical membrane proteins: application to G-protein coupled receptors. Biophys. J. 101, 2092–2101 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh D. Energetics of hydrophobic matching in lipid-protein interactions. Biophys. J. 94, 3996–4013 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S., Khelashvili G., Shi L. & Weinstein H. The cost of living in the membrane: a case study of hydrophobic mismatch for the multi-segment protein LeuT. Chem. Phys. Lipids 169, 27–38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotiadis D. et al. Atomic-force microscopy: rhodopsin dimers in native disc membranes. Nature 421, 127–128 (2003). [DOI] [PubMed] [Google Scholar]
- Huang J., Chen S., Zhang J. J. & Huang X.-Y. Crystal structure of oligomeric beta1-adrenergic G protein coupled receptors in ligand-free basal state. Nat. Struct. Mol. Biol. 20, 419–425 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung J. J. et al. Ligand-regulated oligomerization of β2-adrenoceptors in a model lipid bilayer. EMBO J. 28, 3315–3328 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J. M., Wang H., Provasi D. & Filizola M. Assessing the relative stability of dimer interfaces in G protein-coupled receptors. PLoS Comput. Biol. 8, e1002649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y. et al. Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. J. Biol. Chem. 278, 21655–21662 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calebiro D. et al. Single-molecule analysis of fluorescently labeled G-protein coupled receptors reveals complexes with distinct dynamics and organization. Proc. Natl. Acad. Sci. USA 110, 743–748 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelashvili G., Mondal S., Andersen O. S. & Weinstein H. Cholesterol modulates the membrane effects and spatial organization of membrane-penetrating ligands for G-protein coupled receptors. J. Phys. Chem. B 114, 12046–12057 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson M. A. et al. A specific cholesterol binding site is established by the 2.8 Å Structure of the human β2-adrenergic receptor. Structure 16, 897–905 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsch S., Klotz K. N., Engelhardt S., Lohse M. J. & Bunemann M. Analysis of receptor oligomerization by FRAP microscopy. Nat. Methods 6, 225–230 (2009). [DOI] [PubMed] [Google Scholar]
- Rybin V. O., Xu X., Lisanti M. P. & Steinberg S. F. Differential targeting of β2-adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. J. Biol. Chem. 275, 41447–41457 (2000). [DOI] [PubMed] [Google Scholar]
- Ballesteros J. A. & Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428 (1995). [Google Scholar]
- Sankararamakrishnan R. & Weinstein H. Positioning and stabilization of dynorphin peptides in membrane bilayers: the mechanistic role of aromatic and basic residues revealed from comparative MD simulations. J. Phys. Chem. B 106, 209–218 (2002). [Google Scholar]
- Hern J. A. et al. Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proc. Natl. Acad. Sci. USA 107, 2693–2698 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert N. A. GPCR dimers fall apart. Sci. Signal. 3, pe12 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca J. M. & Lambert N. A. Instability of a class A G protein-coupled receptor oligomer interface. Mol. Pharmacol. 75, 1296–1299 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai R. S. et al. Full characterization of GPCR monomer–dimer dynamic equilibrium by single molecule imaging. J. Cell Biol. 192, 463 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton M. R. et al. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc. Natl. Acad. Sci. USA 104, 7682–7687 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G. & Bouvier M. Methods to monitor the quaternary structure of G protein-coupled receptors. FEBS J. 272, 2914–2925 (2005). [DOI] [PubMed] [Google Scholar]
- Mansoor S. E., Palczewski K. & Farrens D. L. Rhodopsin self-associates in asolectin liposomes. Proc. Natl. Acad. Sci. USA 103, 3060 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding P. J. et al. Constitutive dimerization of the G-protein coupled receptor, neurotensin receptor 1, reconstituted into phospholipid bilayers. Biophys. J. 96, 964–973 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubias O., Teague W. E. Jr, Hines K. G., Mitchell D. C. & Gawrisch K. Contribution of Membrane Elastic Energy to Rhodopsin Function. Biophys. J. 99, 817–824 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau W. M., Wimley W. C., Gawrisch K. & White S. H. The preference of tryptophan for membrane interfaces. Biochemistry 37, 14713–14718 (1998). [DOI] [PubMed] [Google Scholar]
- Gray T. M. & Matthews B. W. Intrahelical hydrogen bonding of serine, threonine and cysteine residues within alpha-helices and its relevance to membrane-bound proteins. J. Mol. Biol. 175, 75–81 (1984). [DOI] [PubMed] [Google Scholar]
- Ben-Tal N., Ben-Shaul A., Nicholls A. & Honig B. Free-energy determinants of alpha-helix insertion into lipid bilayers. Biophys. J. 70, 1803–1812 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhaber F., Lijnzaad P., Argos P., Sander C. & Scharf M. The double cubic lattice method: efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. J. Comput. Chem. 16, 273–284 (1995). [Google Scholar]
- Huang H. W. Deformation free energy of bilayer membrane and its effect on gramicidin channel lifetime. Biophys. J. 50, 1061–1070 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen C., Goulian M. & Andersen O. S. Energetics of inclusion-induced bilayer deformations. Biophys. J. 74, 1966–1983 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawicz W., Olbrich K. C., McIntosh T., Needham D. & Evans E. Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophys. J. 79, 328–339 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. & Rand R. P. The influence of cholesterol on phospholipid membrane curvature and bending elasticity. Biophys. J. 73, 267–276 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohl C. A., Strauss C. E. M., Chivian D. & Baker D. Modeling structurally variable regions in homologous proteins with rosetta. Proteins 55, 656–677 (2004). [DOI] [PubMed] [Google Scholar]
- Monticelli L. et al. The MARTINI coarse-grained force field: extension to proteins. J Chem. Theory Comput. 4, 819–834 (2008). [DOI] [PubMed] [Google Scholar]
- Van Der Spoel D. et al. GROMACS: Fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 (2005). [DOI] [PubMed] [Google Scholar]
- Khelashvili G. et al. Why GPCRs behave differently in cubic and lamellar lipidic mesophases. J. Am. Chem. Soc. 134, 15858–15868 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong D. H. et al. Improved parameters for the Martini coarse-grained protein force field. J Chem. Theory Comput. 9, 687–697 (2012). [DOI] [PubMed] [Google Scholar]
- Soubias O., Niu S. L., Mitchell D. C. & Gawrisch K. Lipid-rhodopsin hydrophobic mismatch alters rhodopsin helical content. J. Am. Chem. Soc. 130, 12465–12471 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Video - Supplementary Movie S1