

Abstract
Delayed post-hypoxic leukoencephalopathy is a rare clinical phenomenon usually observed in a small number of carbon monoxide poisoning survivors. A similar phenomenon is reported here in a patient who successfully recovered from a large overdose of diazepam and methadone, but then abruptly declined 3 weeks after the initial event. Magnetic resnance revealed confluent white matter hyperintensity on fluid-attenuated inversion recovery and T2 weighted sequences, and spectroscopy revealed elevated peaks in choline, creatinine, and lactate. Analysis and review of the literature suggests this phenomenon occurs on average about 19 days after the initial event. Although the pathophysiology remains obscure, it is noted here that the mean lucid interval coincides approximately with the replacement half-life for myelin related lipids and proteins.
Key words: white matter, leukoencephalopathy, myelin, MRI
Introduction
Plum et al.1 recognized as early as 1962 that rarely there may be a delayed deterioration after anoxic injury. Although subsequent autopsy reports by Ginsberg et al.,2 on post-hypoxic leukoencephalopathy noted extensive symmetrical necrotic lesions of the central white matter, with minimal damage to gray matter structures, it was not until later reports using magnetic resonance imaging (MRI) that revealed this could occur as a delayed effect of hypoxic injury, with an intervening lucid interval of many days where the patient is completely normal. Since the initial reports of Plum and colleagues, this phenomenon of delayed neurological deterioration after anoxia has been found in almost 3% of carbon monoxide poisoning cases.3 According to this study of 65 patients by Choi, the lucid interval between the event and delayed deterioration averaged 22.4 days with a mean age of 56.1 years; 75% of patients appeared to have recovered within one year of the event.
Since the publication these early reports, a number of MRI studies have been published which document extensive confluent changes within the white matter as delayed complication to carbon monoxide poisoning,4-8 with the largest series of 15 cases reported by Chang et al.9
Similar changes are reported here for hypoxia in association with a benzodiazepine overdose in a 43 year old who attempted suicide, but was successfully revived and discharged in a fully alert and ambulatory state with a full return to her previously normal mental status, but then suffered a delayed abrupt neurological deterioration with confluent white matter changes seen on both computed tomography (CT) and MRI. The MR spectroscopy changes are also reported here, with a description of the clinical findings to promote greater awareness of this unusual clinical phenomenon.
Case Report
A 43 year old female with chronic back pain developed depression and attempted suicide with an overdose of diazepam and methadone after an argument with her husband in the evening. She intentionally consumed approximately 40 to 50 tablets of diazepam at 5 mg strength as well as an unknown quantity of methadone tablets was found the following day in an unresponsive state with a respiratory state of only 4 breaths per minute but was revived at a local hospital with the use of naloxone. Although she could awaken and answer questions appropriately, this was only transient and required a naloxone drip infusion over 24 hours; no flumazenil had been used. Her potassium was also elevated initially at 7.6 that later normalized and had a leukocytosis secondary to mild aspiration pneumonia that was successfully treated with antibiotics. She was discharged within one week in a much improved state to a psychiatric facility. After two days as in-patient for psychiatric care, she was discharged to home in a markedly improved state with a mental status that was back to her baseline and fully able to be self-sufficient without any cognitive deficits. Interview with family and friends confirmed that she was cognitively normal and fine up until 3 weeks after the event when she abruptly changed midday and seemed confused and forgetful with a rapid worsening the following day. She was first noted not be able to play cards with her family, had developed social withdrawal with a lack of personal hygiene, and became incontinent as well. She appeared very lethargic and not very interactive. After this continued for approximately 5 days without any improvement, she was evaluated at the Emergency Dept. and admitted after a CT head exam disclosed marked and diffuse hypointensity throughout the subcortical white matter (Figure 1A). This was later evaluated by MRI as being a confluent change throughout the entire white matter, and appeared diffusely hyperintense on both FLAIR and T2 weighted sequences (Figure 1B), with hyper-intensity also noted on diffusion weighted imaging. The unusual feature though was that the cerebellar white mater had been spared, and that the change was a strictly supratentorial effect (Figure 2A) without altering signal intensity for the white matter tracts in the internal capsule and brain stem area. Using large voxels placed over the semi-centrum ovale, MR spectroscopic analysis at 1.5 Tesla showed elevated abnormal elevations in the choline and creatinine peaks with a small peak seen for lactic acid as well (Figure 2B). Neurological exam revealed a blunted flat affect with abnormally slow gait and left side neglect, with disorientation and very poor short term recall. As the patient had a detectable benzodiazepine level at approximately 83 ng/mL, 1.0 mg flumazenil was given in 0.2 mg increments every 30 seconds during EEG monitoring. Although background slowing improved slightly, there was a clear and definite transient neurological improvement. Serum benzodiazepine levels continued to decline from 67.9 to 32.8 ng/mL over 4 days, with modest clinical improvement in her mental status also observed. Follow-up evaluation revealed substantial improvement both clinically and radiographically, with a normal mental status and normal MR imaging of the brain within one year of the event, with normal metabolism of both the gray and white matter of the brain, as assessed by 18F-FDG PET imaging (Figure 3).
Figure 1.
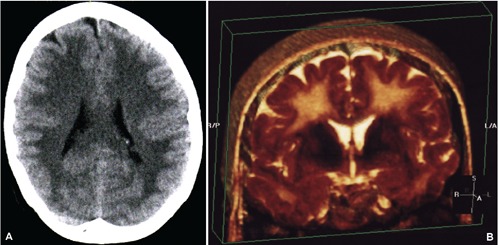
A) Computed tomography appearance at the time of initial development of encephalopathic changes; B) three dimensional reconstruction of coronal T2 sequences obtained with magnetic resonance imaging revealing the extensively confluent subcortical white matter signal hyper-intense change.
Figure 2.
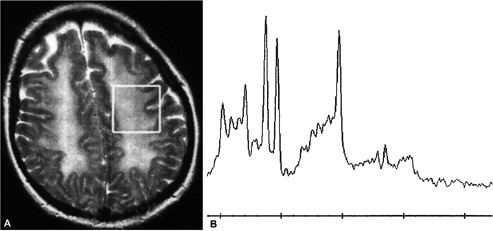
A) Axial T2 weighted image reveals extensive confluent hyper-intense signal abnormality throughout the subcortical white matter with no obvious signal change within the cortex. The white box within the left semi-centrum ovale subcortical white matter represents the voxel used for generating magnetic resonance spectroscopy, shown in figure two; B) magnetic resonance spectroscopy of the subcortical white matter revealing an abnormally high peak for creatinine and choline downstream and to the left of the main central NAA peak with a small yet abnormal presence of a lactic acid peak upstream from the NAA peak.
Figure 3.
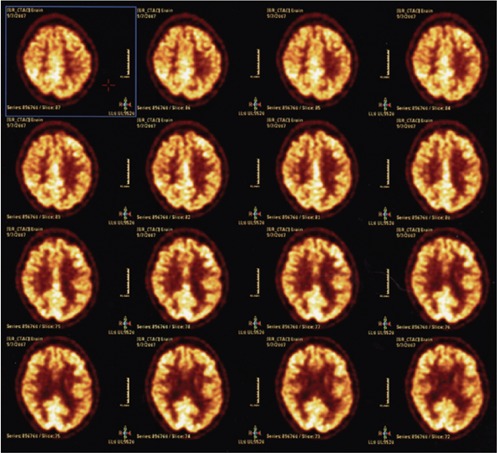
Resolution of symptoms over the next few months with follow-up brain 18FFDG PET scan showing normal uptake patterns for the cerebral cortex and sub-cortical white matter.
Discussion and Conclusions
This case displays some similarities to that described by Gottfired et al.10 who observed delayed post-hypoxic leukoencephalopathy in a 36 year old who overdosed on benzodiazepines, opiates, and amphetamines, and was revived at a local hospital where he was discharged 8 days later in a completely normal state. However, at day 24, he began to act strangely and was became confused and declined thereafter with confluent diffuse white matter changes on MRI and MR Spectroscopy showing elevated choline and lactate. Although the investigators postulated that partial loss of activity for Arylsulfatase A enzyme may have predisposed to the condition, other case reports exist of patients with normal enzyme activities. A case reported by Lee and Lyketsos involved an accidental benzodiazepine overdose in a 71 year old who was discharged in a fully normal condition after a 3 day hospitalization,11 and did fine for 2 weeks until a rapid decline occurred with confluent white matter changes then noted on MRI; arylsufatase levels were normal.
The cause for the delayed post-hypoxic white matter deterioration remains obscure. However, a possible explanation may relate to the half life for myelin related proteins, which in animals has been determined to be in the same approximate time frame as the clinic deterioration seen in these cases of DPHLE (Figure 4; mean time for lucid interval: 19 days). A study by Ando et al.12 found that the half-replacement times as judged by newly synthesized molecule incorporation rate in young adult mice was 20 days for phosphatidylcholine, 25 days for phosphatidylethanolamine. If this process of replacement was to abruptly halt after ischemia, conduction within central myelin could still occur until a critical threshold of loss was achieved where conduction block and failure would occur. Sabri and colleagues noted that half-life of the fast component of myelin basic protein pool was 19-22 days.13 Hayes et al.14 has also found that half-life of the fast turning-over pool of cerebrosides of myelin was 22 days for the adult rat brain, which also is within the same time frame for the lucid interval of 19 days in DPHLE.
Figure 4.
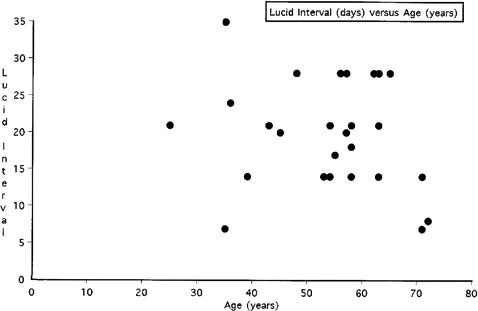
Retrospective review of lucid interval in days (y-axis) in relation age in years (x-axis) for delayed post hypoxic leukoencephalopathy cases from references four through nine, along with the current case.
Of potential theoretical concern is the lack of any use of flumazenil in the acute stage of this patient’s illness; it was not until the patient was symptomatic with DPHLE that flumazenil was first used to successfully improve cognition and improve EEG slowing on a transient basis. Pharmacokinetic studies have shown that the diazepam metabolite, n-desmethyldiazepam, has a elimination half life of about 139 hours, versus 58 hours for the antipyrene related hepatic microsomal enzyme induction rate;15 animal studies show that it accumulates to a high degree within central white matter regions,16 and does not efflux very readily relative to diazepam. It is therefore hypothesized that prolonged retention of diazepam metabolites may play an additional role in the pathogenesis of diazepam related DPHLE, with accumulation of this metabolite likely occurring within central white matter.
In summary, patients with delayed deterioration after benzodiazepine overdose may reflect the above described changes seen in DPHLE, and that further investigation with MRI may be helpful to evaluate for this rare syndrome associated with recent hypoxia. The exact mechanism still remains obscure but further research is clearly warranted. Since flumazenil had a transient beneficial effect here, PET imaging studies using 11C-flumazenil may be of additional value to understand disease pathophysiology.17 DPHLE is relatively rare but fortunately reversible over time with a complete and full recovery documented here for this case and other, and requires extensive support care in the initial phases of the illness;18 however, a comprehensive pathophysiologic explanation for the lucid interval and delayed deterioration phase is not known and largely remains obscure – further research is clearly needed on this unusual and fascinating yet unfortunate neurological problem.
References
- 1.Plum F, Posner JB, Hain RF. Delayed neurological deterioration after anoxia. Arch Intern Med 1962;110:18-25 [DOI] [PubMed] [Google Scholar]
- 2.Ginsberg MD, Hedley-Whyte ET, Richardson EP., Jr Hypoxic-ischemic leukoencephalopathy in man. Arch Neurol 1976;33:5-14 [DOI] [PubMed] [Google Scholar]
- 3.Choi IS. Delayed neurologic sequelae in carbon monoxide intoxication. Arch Neurol 1983;40:433-5 [DOI] [PubMed] [Google Scholar]
- 4.Kim JH, Chang KH, Song IC, et al. Delayed encephalopathy of acute carbon monoxide intoxication: diffusivity of cerebral white matter lesions. AJNR Am J Neuroradiol 2003;24:1592-7 [PMC free article] [PubMed] [Google Scholar]
- 5.Murata T, Kimura H, Kado H, et al. Neuronal damage in the interval form of CO poisoning determined by serial diffusion weighted magnetic resonance imaging plus 1H-magnetic resonance spectroscopy. J Neurol Neurosurg Psychiatry 2001;71:250-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamada K, Houkin K, Aoki T, et al. Cerebral metabolic changes in delayed carbon monoxide sequelae studied by proton MR spectroscopy. Neuroradiology 1994;36:104-6 [DOI] [PubMed] [Google Scholar]
- 7.Hayashi R, Hayashi K, Inoue K, Yanagisawa N. A serial computerized tomographic study of the interval form of CO poisoning. Eur Neurol 1993;33:27-9 [DOI] [PubMed] [Google Scholar]
- 8.Kinoshita T, Sugihara S, Matsusue E, et al. Pallidoreticular damage in acute carbon monoxide poisoning: diffusion-weighted MR imaging findings. AJNR Am J Neuroradiol 2005;26:1845-8 [PMC free article] [PubMed] [Google Scholar]
- 9.Chang KH, Han MH, Kim HS, et al. Delayed encephalopathy after acute carbon monoxide intoxication: MR imaging features and distribution of cerebral white matter lesions. Radiology 1992;184:117-22 [DOI] [PubMed] [Google Scholar]
- 10.Gottfried JA, Mayer SA, Shungu DC, et al. Delayed posthypoxic demyelination. Association with arylsulfatase A deficiency and lactic acidosis on proton MR spectroscopy. Neurology 1997;49:1400-4 [DOI] [PubMed] [Google Scholar]
- 11.Lee HB, Lyketsos CG.Delayed post-hypoxic leukoencephalopathy. Psychosomatics 2001;42:530-3 [DOI] [PubMed] [Google Scholar]
- 12.Ando S, Tanaka Y, Toyoda Y, Kon K. Turnover of myelin lipids in aging brain. Neurochem Res 2003;28:5-13 [DOI] [PubMed] [Google Scholar]
- 13.Sabri MI, Bone AH, Davison AN. Turnover of myelin and other structural proteins in the developing rat brain. Biochem J 1974;142:499-507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayes LW, Jungalwala FB. Synthesis and turnover of cerebrosides and phosphatidylserine of myelin and microsomal fractions of adult and developing rat brain. Biochem J 1976;160:195-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohnhaus EE, Park BK, Colombo JP, Heizmann P. The effect of enzyme induction on diazepam metabolism in man. Br J Clin Pharmacol 1979;8:557-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Placidi GF, Tognoni G, Pacifici GM, et al. Regional distribution of diazepam and its metabolites in the brain of cat after chronic treatment. Psychopharmacology (Berl) 1976;48:133-7 [DOI] [PubMed] [Google Scholar]
- 17.Meyer M, Koeppe RA, Frey KA, et al. Positron emission tomography measures of benzodiazepine binding in Alzheimer’s disease. Arch Neurol 1995;52:314-7 [DOI] [PubMed] [Google Scholar]
- 18.Shprecher D, Mehta L. The syndrome of delayed post-hypoxic leukoencephalopathy. Neuro Rehabilitation 2010;26:65-72 [PMC free article] [PubMed] [Google Scholar]