

Abstract
Background
Methylocystis sp. strain SC2 can adapt to a wide range of methane concentrations. This is due to the presence of two isozymes of particulate methane monooxygenase exhibiting different methane oxidation kinetics. To gain insight into the underlying genetic information, its genome was sequenced and found to comprise a 3.77 Mb chromosome and two large plasmids.
Principal Findings
We report important features of the strain SC2 genome. Its sequence is compared with those of seven other methanotroph genomes, comprising members of the Alphaproteobacteria, Gammaproteobacteria, and Verrucomicrobia. While the pan-genome of all eight methanotroph genomes totals 19,358 CDS, only 154 CDS are shared. The number of core genes increased with phylogenetic relatedness: 328 CDS for proteobacterial methanotrophs and 1,853 CDS for the three alphaproteobacterial Methylocystaceae members, Methylocystis sp. strain SC2 and strain Rockwell, and Methylosinus trichosporium OB3b. The comparative study was coupled with physiological experiments to verify that strain SC2 has diverse nitrogen metabolism capabilities. In correspondence to a full complement of 34 genes involved in N2 fixation, strain SC2 was found to grow with atmospheric N2 as the sole nitrogen source, preferably at low oxygen concentrations. Denitrification-mediated accumulation of 0.7 nmol 30N2/hr/mg dry weight of cells under anoxic conditions was detected by tracer analysis. N2 production is related to the activities of plasmid-borne nitric oxide and nitrous oxide reductases.
Conclusions/Perspectives
Presence of a complete denitrification pathway in strain SC2, including the plasmid-encoded nosRZDFYX operon, is unique among known methanotrophs. However, the exact ecophysiological role of this pathway still needs to be elucidated. Detoxification of toxic nitrogen compounds and energy conservation under oxygen-limiting conditions are among the possible roles. Relevant features that may stimulate further research are, for example, absence of CRISPR/Cas systems in strain SC2, high number of iron acquisition systems in strain OB3b, and large number of transposases in strain Rockwell.
Introduction
In the global methane cycle, aerobic methanotrophic bacteria are the only biological sink for the greenhouse gas methane. They belong to the Proteobacteria [1] and Verrucomicrobia [2]. The proteobacterial methanotrophs belong to the Alphaproteobacteria and Gammaproteobacteria. Among them, the alphaproteobacterial members of the genus Methylocystis have repeatedly been found to be associated with a wide variety of environments. They have been detected by both cultivation and cultivation-independent molecular techniques in rice paddies [3], [4], different upland and hydromorphic soils [5], [6], [7], landfills [8], [9], peatlands [10], [11], [12], and glacier forefields [13]. These environments are characterized by either oxygen-methane counter-gradients (low-affinity methane oxidation: e.g., rice paddies, peatlands, landfill cover soils) or the consumption of atmospheric methane (high-affinity methane oxidation: e.g., upland soils). Intermediate conditions prevail, for example, in glacier forefields. The ubiquitous distribution of the genus Methylocystis may be due to the fact that its members have greater metabolic flexibility than those of other methanotrophic genera. This is in part related to their facultative nature. For example, Methylocystis sp. strain H2s can utilize acetate [14] and strain SB2 can utilize acetate and ethanol [15], in addition to methane. Methylocystis sp. strain Rockwell has been studied with respect to its ability to utilize different nitrogen sources [16], [17]. Our model organism, Methylocystis sp. strain SC2, contains a novel high-affinity particulate methane monooxygenase (pMMO2), in addition to the conventional pMMO1 [18], [19]. The different methane oxidation kinetics of pMMO1 and pMMO2 allow strain SC2 to adapt to a wide range of methane concentrations and thus to changes in its environment [19]. To understand the total genetic potential of this organism, its genome was sequenced [20].
Another major factor determining methanotrophic activity is the source and availability of nitrogen. Diversity of nitrogen metabolism operating in methanotrophs is well known. N2 fixation is a well-studied feature among methanotrophs. It has been reported for proteobacterial methanotrophs [21], [22], [23] and the distantly related verrucomicrobial member ‘Methylacidiphilum fumariolicum’ SolV [24]. Denitrification is the sequential reduction of nitrate and nitrite to the gaseous compounds nitric oxide (NO), nitrous oxide (N2O), and finally N2. This process is catalyzed by nitrate, nitrite, nitric oxide, and nitrous oxide reductase, respectively [25]. Incomplete denitrification can lead to the emission of N2O, a potent greenhouse gas that contributes to global warming and ozone depletion [26]. Proteobacterial methanotrophs are known to release N2O [27], [28], [29], [30], [31], [32]. Methylococcus capsulatus Bath and Methylosinus trichosporium OB3b have the ability to produce N2O from the oxidation of hydroxylamine [33], [34]. Understanding the release and fate of N2O is of particular importance for the global nitrogen cycle [35]. Thus, in addition to their methane-oxidizing capabilities, knowledge of their nitrogen metabolism is essential for understanding the ecophysiology of methanotrophic bacteria. Based on a genome-inferred inventory, several key enzymes involved in nitrification and denitrification were suggested to be present in methanotrophs [36]. It was proposed that the oxidation of NH3 to nitrite (nitrification) and the production of N-oxides (denitrification) may be interrelated [27], [37], [38]. However, the ability to convert N2O to N2 has not yet been reported for any of the known methanotrophs.
With the advent of next-generation sequencing technologies, the number of sequenced methanotroph genomes has increased considerably. At present, twelve methanotroph genomes are available in public databases and more are being sequenced. The available sequences include those of the alphaproteobacterial methanotrophs Methylosinus trichosporium OB3b [39], Methylocystis parvus OBBP [40], Methylocystis sp. strain Rockwell [41], Methylocystis sp. strain SC2 [20], and the facultative Methylocella silvestris BL2 [42]; and the gammaproteobacterial methanotrophs Methylococcus capsulatus Bath [43], Methylomicrobium album BG8 [44], Methylomicrobium alcaliphilum 20Z [45], Methylomonas methanica MC09 [46], and the psychrotolerant Methylobacter tundripaludum SV96 [47]. In addition, the genome sequences of the acidophilic Verrucomicrobia members Methylacidiphilum infernorum V4 [48] and ‘Ma. fumariolicum’ SolV [49] are available. However, there is no report of any comparative analysis among the methanotroph genomes.
Here, we provide a detailed description of important features of the genome sequence of strain SC2 identified by comparative analysis with the methanotroph genomes available in public databases. In particular, we systematically compared the genome sequence of strain SC2 with those of two other Methylocystaceae members, Methylocystis sp. strain Rockwell and Ms. trichosporium OB3b. Special emphasis was given to genes involved in nitrogen metabolism. Their diverse functional nature in strain SC2 prompted us to perform physiological experiments, in order to verify that this strain is able to fix atmospheric N2, produce N2O and eventually reduce it to N2 by denitrification.
Results and Discussion
Genomic analysis of Methylocystis sp. strain SC2
(a) General features of strain SC2 genome
The genome of strain SC2 totals 4,146,594 bp and consists of three replicons: a circular chromosome of 3,773,444 bp (Figure 1) and two plasmids of 229,614 (pBSC2-1) and 143,536 bp (pBSC2-2), with an average GC content of 63, 61 and 60%, respectively [20], [50].
Figure 1. Genome plot of strain SC2.

The circles represent from outside to inside: circle 1, DNA base position (bp); circle 2, protein-coding regions transcribed on the plus strand (clockwise); circle 3, protein-coding regions transcribed on the minus strand (anticlockwise); circle 4, tRNA genes; circle 5, rRNA genes; circle 6, G+C content plotted using a 10-kb window (sea green and magenta indicate values greater than and less than the average G+C content, respectively); circle 7, GC skew ([G+C]/[G−C]) plotted using a 10-kb window (blue indicates values above average and red indicates values below average). The genome plot was generated using DNAPlotter version 1.4 from Artemis 12.0, Sanger Institute.
The organization of a genome changes through gene rearrangements. The frequency with which rearrangements occur depends on the activity of mobile and repeated elements such as insertion sequences, transposons, prophage sequences, and plasmids [51]. In strain SC2, we manually identified two putative genomic islands, possibly acquired by transduction. These are defined by a 17-kb region (BN69_1471 to BN69_1495) and a 63-kb region (BN69_1579 to BN69_1669). In both genomic islands, CDS with significant BLAST matches encode phage-related proteins including components of phage head protein, tail protein, integrase, recombinase, and lysozyme. However, most of the genomic island CDS had no significant match in the database. When the chromosome of strain SC2 was scanned for prophage sequences using the widely used software Prophinder [52], no such sequences were detected. This might be due to the fact that the identified islands have lost some phage-related features (like the terminal repeats). The large phage-related island also contains a hicAB toxin-antitoxin system (BN69_1608, BN69_1609), which is highly prone to frequent gene rearrangement within a genome and horizontal gene transfer among bacterial and archaeal species [53]. Additional toxin-antitoxin systems encoded on the chromosome include two mazEF systems (BN69_0515, BN69_0516; and BN69_2525, BN69_2526) and one yoeB–yefM system (BN69_3397, BN69_3398). A relBE toxin-antitoxin system was identified in the plasmid pBSC2-1 [50]. All toxin-antitoxin systems encode toxins that target diverse cellular functions like DNA replication, mRNA stability, protein synthesis, cell wall biosynthesis, and ATP synthesis [54]. The toxins (RelE, MazF, and YoeB) predicted to be produced in strain SC2 function as site-specific endoribonucleases that cleave mRNA at specific sites and thereby hamper mRNA stability [55], [56], [57], [58]. The HicA toxin, encoded by the hicAB system, was proposed to function via RNA cleavage [53]. In normally growing cells, these toxins are coexpressed and neutralized by their cognate antitoxins produced from the second gene of the operon [54]. Presence of multiple toxin-antitoxin systems in the chromosome of strain SC2 might help this bacterium to cope with stress or to undergo programmed cell death under stressed conditions [59], [60].
To identify Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs), the web-based tool “CRISPRFinder” was used [61]. CRISPRs are widespread in prokaryotes. A survey identified them in 83% of 150 archaeal genomes and 46% of 2,356 bacterial genomes analyzed (http://crispr.u-psud.fr/crispr) [62]. CRISPR arrays are composed of highly conserved tandem repeat sequences, varying in size from 23 to 47 base pairs. These repeats are separated by unique ‘spacer’ sequences of similar length, which in most cases have been identified to be of viral origin. CRISPRs are flanked on one side by an AT-rich sequence called the ‘leader’ [62]. CRISPR loci, together with their CRISPR-associated (cas) genes, have recently been shown to constitute a defense system that, in bacteria, restricts propagation of intruding viruses and plasmids. CRISPR systems presumably function as transcriptional regulators or RNA-interference-based immune systems [63], [64], [65]. We could not detect any CRISPR-like sequence in the genome of strain SC2. When a similar search was made with the available genome sequences of methanotrophs, CRISPRs ranging from 2 to 6 per genome were identified by “CRISPRFinder” in all of them, except for the genome of Mce. silvestris BL2 (Table 1). Likewise, cas genes were not found in the genomes of strains SC2 and BL2 but were present in all other methanotroph genomes having CRISPR loci (Table 1). The absence of CRISPRs and cas genes might help strain SC2 to maintain and stabilize its two plasmids, as has also been reported for several strains of multidrug-resistant enterococci [66]. Similar to the situation with Methylocystis sp. strains SC2 and Rockwell, CRISPRs are absent or present among strains of the same species in lactic acid bacteria [67].
Table 1. General features identified in the genomes of the compared methanotrophs.
| Alphaproteobacteria | Gammaproteobacteria | Verrucomicrobia | ||||||
| Features | Methylocystis sp. strain SC2 | Methylocystis sp. strain Rockwell | Ms. trichosporium OB3b | Mce. silvestris BL2 | Mc. capsulatus Bath | Mmo. methanica MC09 | Mm. alcaliphilum 20Z | Ma. infernorum V4 |
| Accession number | HE956757 | AEVM00000000 | ADVE00000000 | CP001280 | AE017282 | CP002738 | FO082060 | CP000975 |
| Status | Complete | 149 contigs | 173 contigs | Complete | Complete | Complete | Complete | Complete |
| Genome size (Mb) | 3.77 | 4.6 | 4.9 | 4.3 | 3.3 | 5.05 | 4.67 | 2.2 |
| G+C content (%) | 63 | 63 | 66 | 63 | 64 | 51 | 49 | 45 |
| Total no. of CDS | 3,666 | 4,637 | 4,4721 | 4,016 | 3,120 | 4,494 | 4,083 | 2,473 |
| rRNA operons | 1 | 1 | 1 | 2 | 2 | 1 | 3 | 1 |
| No. of tRNA genes | All (47) | All (51) | All | All | All (46) | All | All (44) | All (46) |
| tRNA genes in rrn operons 2 | Ile-Ala | Ile-Ala3 | Ile-Ala4 | Ile-Ala (in both) | Ile-Ala (in both) | Ile-Ala | Ile-Ala (in all three) | Ala-Ile |
| pmoCAB1 operon | 2 | 1 | 1 | Absent | 2 | 1 | 1 | 3 |
| pmoCAB2 operon | 1 | Absent | Absent | Absent | Absent | Absent | Absent | Absent |
| Monocistronic pmoC | 3 (1 in plasmid) | 4 | 1 | Absent | 1 | Absent | - | 1 |
| sMMO-encoding operon | Absent | Absent | 1 | 1 | 1 | 1 | - | Absent |
| pxmABC operon | Absent | Absent | Absent | Absent | Absent | Absent | - | Absent |
| Serine pathway genes | Present | Present | Present | Present | Incomplete | Absent | Absent | Present (partial) |
| RuMP pathway genes | Absent | Absent | Absent | Absent | Present | Present | Present | Absent |
| Plasmid(s) | 25 | NR6 | NR | NR | NR | NR | 17 | NR |
| CRISPRs 8 | 0 | 2 | 2 | 0 | 2 | 4 | 3 | 4 |
| No. of cas genes 9 | 0 | 2 | 5 | 0 | 10 | 7 | 18 | 8 |
The number of CDS predicted in the genome announcement is 4,503, while the submitted sequence actually contains 4,472 CDS.
tRNA genes were identified in 16S-23S spacer region of the rRNA operons.
rRNA operon present in contig 219 (AEVM01000005).
rRNA operon present in contig 00159 (NZ_ADVE01000118).
NR – not reported.
Size of plasmid: MEALZ_p (FO082061), 128 kb.
Abbreviation: Clustered Regularly Interspaced Short Palindromic Repeats; identified using the online tool “CRISPR finder”.
CRISPR-associated (cas) genes were predicted using the RAST server.
(b) CDS involved in replication, transcription, and translation
The analysis of GC skewing (Figure 1) did not reveal a clear inversion pattern in the chromosome. Therefore, it was not possible to determine the origin of replication (oriC) by this approach. However, we could identify the putative oriC region using the Ori-Finder program [68], with parameters adjusted to specific DNA boxes of E. coli and one unmatched site permitted. This tool makes predictions based on the following features: (i) compositional strand asymmetry (estimated using the Z-curve program), (ii) distribution of DnaA boxes (either of the Escherichia coli type or species-specific), (iii) location of indicator genes (such as dnaA, hemE, gidA, dnaN, hemB, maf, repC, etc.), and (iv) phylogenetic relationships [68], [69]. The putative oriC was identified within a 1063-bp region (2,008,193 bp to 2,009,255 bp). Its GC content is 53%, which is 10% lower than the GC content of the chromosome as a whole (Figure 2). Three dnaA box motifs could be identified within this region using the E. coli-specific dnaA box sequence as the reference. Two palindromic repeats were also identified in this region (Figure 2A). The predicted oriC is not located in the vicinity of any of the three DnaA-encoding CDS (BN69_0001, BN69_3094, BN69_3291). The Ori-Finder program does not consider dif sites while making oriC predictions [68], [69]. However, we could detect a dif site (276,895 bp to 276,922 bp) located almost halfway of the predicted oriC (Figure 2D). The dif site has been shown to be associated with the termination region of bacterial chromosomes [70] and acts as the recognition site for the XerCD proteins [71]. These are involved in postreplication recombination events. CDS encoding XerCD proteins (BN69_2958 and BN69_2761, respectively) were identified in the chromosome. The sum of these findings provides evidence for the correct prediction of oriC. Nevertheless, experimental validation is needed to unambiguously locate oriC.
Figure 2. Prediction of the oriC region by Ori-Finder.

(A) 1,063-bp sequence (2,008,193 bp to 2,009,255 bp) of the predicted oriC site. Three dnaA box motifs identified using the Escherichia coli specific dnaA boxes are bold-faced and highlighted. Palindromic repeats identified in this region are marked by arrows at the top. (B, C) The Z-curves measuring the disparity between the percent content of AT (red lines), GC (green lines), RY (blue lines) and MK (yellow lines) for the original sequence (B) and the rotated sequence (C). It should be noted that the coordinate origin of the rotated sequence begins and ends in the maximum of the GC disparity curve. Short vertical red lines at the top show the locations of indicator genes, such as dnaA, dnaN, gidA, and hemE. The upward black arrow indicates the position of the predicted oriC. Purple peaks with diamonds indicate DnaA box clusters. (D) Pairwise alignment between the dif sites located in the genomes of E. coli and strain SC2. In strain SC2, the dif-like sequence is located from nucleotide position 276,895 to 276,922 (almost halfway of the deduced oriC) and matches at 20 nucleotide positions with the 28-bp dif sequence of E. coli.
Twenty-four CDS encode proteins whose products are involved in transcription. Among these are the following: Two transcriptional elongation factors (GreA [BN69_0089] and GreB [BN69_2117]), three transcriptional antitermination factors (NusA [BN69_2506], NusB [BN69_0468], and NusG [BN69_1633]), and one transcriptional termination factor rho (BN69_2165). CDS encoding α, β, β′ and ω subunits (BN69_1255, BN69_2895, BN69_2894, and BN69_1074, respectively) of bacterial DNA-directed RNA polymerase were also detected. Nine CDS are devoted to the synthesis and maintenance of the RNA polymerase sigma factor.
Several components of the translation system were identified, including a single copy of the 16S-23S-5S ribosomal RNA operon. The 16S and 23S rRNA genes are interspersed by two transfer RNA (tRNA) genes for isoleucine and alanine. This arrangement is commonly found in proteobacterial rRNA operons [72], [73] and more frequently among members of the Alphaproteobacteria [72]. A similar organization of tRNA genes within the rRNA operon was observed in all the methanotroph genomes examined, except for the Verrucomicrobia member where the arrangement is in the reverse order, Ala-Ile (Table 1). The full complement of 54 ribosomal proteins required for ribosome biosynthesis was identified in the chromosome. This includes 21 and 33 CDS, respectively, encoding components of the small and large ribosomal subunits. In total, 47 tRNA genes covering 20 amino acids were identified. No tRNA for the translation of the amino acid selenocysteine (tRNA-Sec) was found, corroborating that the number of bacteria with tRNA-Sec is much less than previously expected [74]. Other important CDS of the translation system include aminoacyl-tRNA synthetases, responsible for precise attachment of all 20 amino acids to their cognate transfer RNAs. Three bacterial initiation factors (IF-1 [BN69_0875], IF-2 [BN69_2508], and IF-3 [BN69_0601]) and three peptide release factors (RF-1 [BN69_0797], RF-2 [BN69_2418], and RF-3 [BN69_0479]) were identified. The latter are responsible for the recognition of the stop codons UAA, UAG, and UGA to terminate translation.
(c) CDS involved in methanotrophic mode of life
The chromosome of strain SC2 contains all the genes required for a methanotrophic lifestyle, including two copies of the conventional pmoCAB1 operon and a single copy of the novel pmoCAB2 operon (Table S1). In addition, three monocistronic pmoC paralogs were identified, with one present on plasmid pBSC2-2 [50]. As expected for an alphaproteobacterial methanotroph, we could identify the genes involved in the serine pathway of formaldehyde assimilation, but not those involved in the RuMP pathway.
The monocistronic pmoC1Gs (BN69_0852) is identical to the homolog present in the pmoCAB1 operons. Interestingly, the CDS (BN69_0853) present directly upstream of this monocistronic gene encodes an ATP-dependent zinc metalloprotease, FtsH1 protein. No such gene is present in the vicinity of pmoC2Gs. When we searched the genome of strain Rockwell, ftsH genes were found immediately downstream of two of its four monocistronic pmoC genes (ZP_08074599 and ZP_08075129). Characterized in Escherichia coli, FtsH is a membrane-bound ATP-dependent protease that is involved in the degradation of uncomplexed or misfolded integral membrane proteins and short-lived cytoplasmic proteins [75], [76], [77]. FtsH functions as a protein-filtering system and ensures that only correctly folded protein is incorporated into the membrane. Based on the presence of ftsH in close association to monocistronic pmoC, whose exact function is yet to be identified, one may speculate that this monocistronic gene (along with FtsH) might act as a sensor to screen whether properly folded pMMO is incorporated into the membrane of these methanotrophs. However, this needs to be experimentally verified, before claiming an exact function in the two strains, SC2 and Rockwell. No ftsH gene was detected in the vicinity of the monocistronic pmoC in strain OB3b (EFH02634) and Mc. capsulatus Bath (YP_112829). A possible explanation for the absence of this gene might be the additional presence of the soluble form of methane monooxygenase (sMMO) in these bacteria.
(d) Nitrogen metabolism-related genes
Genome sequence analysis revealed the presence of a large number of genes whose products are presumably involved in nitrogen metabolism. This includes N2 fixation, ammonium transport, assimilatory nitrate/nitrite reduction, hydroxylamine detoxification, and denitrification (Table S1) [20]. A full chromosome-encoded complement of N2 fixation-related genes (34 CDS) was identified. The genes mostly clustered together, suggesting that strain SC2 is capable of utilizing N2 as a nitrogen source (see below).
The first step in nitrification is the oxidation of ammonia to hydroxylamine. Ammonia monooxygenase (AMO) performs this step in ammonia-oxidizing bacteria. AMO and pMMO are known to be homologous. They are encoded by three contiguous genes that are organized in the order amoCAB/pmoCAB [78], [79]. Due to their structural homology, pMMO can also oxidize ammonia [80]. Hydroxylamine is highly toxic and bacteria that can oxidize ammonia must have effective mechanisms to detoxify it. All ammonia oxidizers and some methanotrophs are known to use hydroxylamine oxidoreductase (HAO) to oxidize hydroxylamine to nitrite. However, the difference lies in the fact that ammonia oxidizers, but not methanotrophs, use this step for energy production [27]. The haoAB operon, encoding this enzyme, was identified in the chromosome of strain SC2 (BN69_3242, BN69_3241). In addition, the chromosome encodes a hydroxylamine reductase or hybrid cluster protein (BN69_0431) that presumably detoxifies hydroxylamine by reducing it to ammonia. A second copy of hydroxylamine reductase was identified in pBSC2-2 [50]. Thus, strain SC2 apparently possesses two different systems to detoxify hydroxylamine. None of the other genome-sequenced methanotrophs are known to possess both detoxification systems.
Methanotrophs are reported to produce N2O during ammonia oxidation [29], [30], [32]. The chromosome does not encode any enzyme that can contribute to this function in strain SC2. However, a CDS encoding nitric oxide reductase (homolog of norB) is present in each of the two plasmids [50]. And most interestingly, a complete nitrous oxide reductase operon (nosRZDFYX) was identified in pBSC2-2 [50]. The cluster contains the key functional gene nosZ. In addition, it includes nosR and nosX. The two genes are exclusively present in typical nos clusters of denitrifiers as, for example, in Bradyrhizobium japonicum strain USDA 110 [81]. However, the exact origin of this plasmid-borne nos operon could not be predicted as BLAST searches of its individual genes showed homologs from diverse origin (Table S2).
Two genes were predicted to encode ammonium transporters (BN69_0915 and BN69_0931), suggesting that ammonia is an important nitrogen source for strain SC2. We also identified genes encoding the high-affinity ATP-driven potassium transporter (kdpABC). These three genes encoding the potassium transporter ATPase (BN69_2487 to BN69_2489) are located immediately downstream of an osmosensitive signal transduction histidine kinase (kdpD, BN69_2486) and a two-component transcriptional regulator (kdpE, BN69_2485). The potassium transporter has been shown to also transport ammonium ions. This is due to the similarity between ammonium and potassium ions, both in terms of charge and size [82]. Thus, this transporter may facilitate transport of ammonium ions in strain SC2. The chromosome includes a full complement of genes (BN69_2468 to BN69_2473) for transport of nitrate/nitrite across the cytoplasmic membrane and their reduction to ammonia. This is referred as the assimilatory nitrate/nitrite reductase system (Nas). However, genes encoding the Nar or Nap type of nitrate reductase were not found.
Ammonium is the most reduced form of inorganic nitrogen prior to its incorporation into organic nitrogen compounds via glutamate or glutamine, which serve as the key nitrogen donors for biosynthetic processes. The incorporation can occur via the glutamine synthetase/glutamate synthetase (GS) or NADPH-dependent glutamine oxoglutarate amidotransferase (GOGAT) pathway, or the glutamate dehydrogenase (GDH) pathway [83]. In bacteria, GS and GOGAT function as alternative pathways of ammonia assimilation and operate when ammonia is present in the growth medium at low levels [84]. The SC2 chromosome encodes GS (BN69_0652) and both the large (BN69_3582) and small (BN69_3584) subunits of GOGAT. GDH (BN69_0999) was also identified.
In addition to enzymes of the nitrogen metabolism, many potential regulatory components involved in this process are encoded by the chromosome, including sigma factor RpoN (BN69_2202). This factor is essential for the expression of several nitrogen regulons, such as the ntr (nitrogen regulation) and nif (N2 fixation) operons. However, RpoN is not only involved in the nitrogen metabolism, but also controls the regulation of a number of other metabolic processes in bacteria. For example, in Pseudomonas putida, RpoN was found to be involved in processes like motility and expression of plasmid-encoded catabolite operons, and in determining the ability of P. putida to utilize diverse nitrogen and carbon sources [85].
The genes encoding the nitrogen signaling cascade (ntrBC [BN69_0222, BN69_0223] and ntrYX [BN69_0224, BN69_0225]) and a gene for uridyltransferase (glnD [BN69_3100]) were also identified. NtrB and NtrC act as a two-component signal transduction cascade for nitrogen regulation, where NtrB is the bifunctional histidine kinase and NtrC is its cognate response regulator [86]. They are required for maximal GS synthesis. The second transcription regulator, ntrYX, is located immediately upstream to ntrBC. The NtrY and NtrX proteins constitute a two-component regulatory system that is involved in N2 fixation and metabolism [87]. All ntr genes are clustered together in an operon, nifR3-ntrB-ntrC-ntrY-ntrX. The fifth component of this operon, nifR3 (BN69_0221), encodes a tRNA-dihydrouridine.
The identification of a full complement of N2 fixation-related genes and plasmid-borne genes for denitrification prompted us to test strain SC2 for these metabolic capabilities.
Physiological studies on the nitrogen metabolism of strain SC2
(a) N2 fixation
The ability of strain SC2 cells to fix N2 was tested in nitrogen-free mineral salts medium. The serum bottles were flushed with N2 followed by the addition of methane (20%). Different initial concentrations of oxygen were tested. Maximum growth was observed with 10% oxygen in the headspace, while 5% oxygen allowed little growth (Figure 3A). Insignificant increase in OD600 value was observed under lower (1%) and higher (15% and 20%) oxygen concentrations. Most likely, the optimal concentration for growth of strain SC2 under N2-fixing conditions is between 5% and 10% oxygen. In respect to their N2-fixing activities, methanotrophic bacteria are known to vary in oxygen sensitivity. In batch cultivation, the requirement of low oxygen concentration has been demonstrated for Methylobacter luteus (<2%), ‘Ma. fumariolicum’ SolV (<2%), Methylocystis sp. strain T-1 (<6%) and Mc. capsulatus Bath (<10%) [21], [22], [24], [88]. In contrast, some other methanotrophs are able to fix N2 at higher oxygen concentrations including, for example, Ms. trichosporium OB3b (15–17%) and Methylocapsa acidiphila B2T (atmospheric oxygen concentration) [22], [23], [89].
Figure 3. N2 fixation by strain SC2.

(A) Growth dynamics (OD600) of strain SC2 in batch cultures on N-free medium (with atmospheric N2 as sole nitrogen source). Oxygen concentrations of 1% (blue), 5% (green), 10% (red), 15% (brown) and 20% (black) were used to test their effect on N2 fixation-mediated growth. Note that the x-axis is not in scale. (B) Effect of oxygen on the nitrogenase activity (acetylene reduction assay) in strain SC2. Ethylene production was measured after 24 hours of incubation under different concentrations of oxygen in the headspace. Data points are means ±SD of three separate experiments.
The cells growing in N-free medium were tested for nitrogenase activity using the acetylene reduction assay. As methane oxidation or, more precisely, the activity of methane monooxygenase is known to be inhibited by acetylene [90], [91], methanol was used as a source of energy and reducing power in the assay [91]. Ethylene production was detected after 3 hours of incubation with acetylene, and the produced amount increased linearly for more than 24 hours. The 3-hour lag prior to ethylene production was also observed for other methanotrophs, such as Mc. capsulatus Bath and ‘Ma. fumariolicum’ SolV [24], [92]. Ethylene production measured after 24 hours of incubation was found to be affected by oxygen concentration in the headspace, with highest amount produced at 1% oxygen (133 nmol ethylene/mg dry weight of cells). The amount of ethylene produced decreased with increasing concentration of oxygen (Figure 3B). Thus, acetylene reduction activity was affected by the oxygen concentration as also observed in other aerobic diazotrophs including methanotrophs [21]. In principle, both the growth experiments and the nitrogenase activity assays consistently showed the detrimental effect of increasing oxygen concentration to N2 fixation. However, while growth yield was highest at 10% oxygen, nitrogenase activity was greatest at around 1% oxygen in the headspace.
(b) Denitrification
Under standard growth conditions, strain SC2 was found to accumulate only a negligible amount of N2O, both under aerobic as well as anaerobic conditions (Figure 4). One explanation for this result may be the presence of a non-functional (less-active) nitric oxide reductase. Another possibility may be the presence of an active/functional nitrous oxide reductase produced from the plasmid-borne nos operon, thereby resulting in the rapid conversion of N2O to N2. To examine the second possibility, we checked N2O production after blocking the nos activity with purified acetylene [93], [94], [95]. As acetylene is also a potent inhibitor of methane monooxygenase [90], we performed this experiment either by adding methanol or without a carbon source. Under aerobic conditions, N2O production was negligible even after acetylene addition. This was expected as denitrification is an anaerobic process. However, under anaerobic conditions, acetylene inhibition was pronounced and N2O accumulated in the headspace. After 48 hours of incubation, the methanol-fed cells produced 33 nM N2O per mg dry weight of cells (Figure 4). Nearly equal amount of N2O (28 nM per mg dry weight of cells) was produced when cells were incubated under starved condition (data not shown). In the methanol-fed cultures, methanol could act as an alternative electron donor in the absence of methane. However, the ability of the cells to produce N2O under starved conditions needs further experimental investigation. A possible explanation might be that strain SC2 cells are able to use intracellular poly-beta-hydroxybutyrate (PHB) as a source of carbon during starvation periods. PHB was found to be produced by almost all alphaproteobacterial methanotrophs. Actually, strain SC2 produced the maximum amount of PHB among five different Methylocystis strains and the third highest among all alphaproteobacterial methanotrophs tested [96]. During sequence analysis, genes encoding PHB metabolism-related enzymes were detected in the chromosome of strain SC2. These include two PHB depolymerases (BN69_2992 and BN69_3262), one polyhydroxyalkonate synthesis repressor (PhbR [BN69_3069]), an acetyl-CoA acetyltransferase (PhbA [BN69_3068]), one acetoacetyl-CoA reductase (PhbB [BN69_3067]), and two phasin homologs (BN69_0212, BN69_1107). The phbR, phbA, and phbB genes form a cluster, but in an orientation different from that in the PHB-producing methanotroph Methylocystis parvus OBB (phbABR) [40]. The use of PHB as a reducing power for denitrification has been shown in microbial granules in bioreactors [97]. Moreover, strains of Methylocystis parvus were reported to be able to ferment intracellular PHB and use it as a reserve energy source under anoxic conditions [98], [99]. These overall findings support the possibility that PHB degradation and denitrification are interlinked in strain SC2.
Figure 4. N2O production by strain SC2.

Cells were incubated in NMS, either in the presence (filled symbol) or absence (open symbol) of 10% acetylene. Assays were performed both under anaerobic (orange) and aerobic (green) conditions. Data points are means ±SD of three separate experiments. The inset shows the same graph with a y-axis zoomed in for the range 0 to 0.8.
To ultimately prove the emission of N2 and thus the operation of a plasmid-encoded denitrification process in strain SC2, a tracer experiment was performed using 15N-nitrate (K15NO3) as the sole nitrogen source. Under anoxic conditions, we could detect accumulation of about 0.7 nmol 30N2/hr/mg dry weight of cells (Figure 5). Taking all these findings together, strong evidence is provided that strain SC2 possesses a complete denitrification pathway. However, its exact ecophysiological role still needs to be elucidated. Detoxification of toxic nitrogen compounds and energy conservation under oxygen-limiting conditions are among the possible roles.
Figure 5. Denitrification-mediated N2 production by strain SC2.

30N2 production was measured after fifteen days for cells incubated in NMS containing either K15NO3 (blue) or KNO3 (orange). The assays were performed under both anaerobic and aerobic conditions. Data points are means ±SD of three separate experiments.
Comparative genomics
(a) Comparative analysis of methanotroph genomes
Comparative genomics is commonly used for the study of closely related strains of a single species, species of a particular genus, or species of related genera [100]. However, members of broader taxonomic ranks have also been compared, like those belonging to the same family, such as Pseudonocardiaceae [101] and Methylophilaceae [102], or to different families [103]. Here, we compared the genome sequences of eight methanotrophs belonging to different classes and phyla. These include the genomes of four alphaproteobacterial and three gammaproteobacterial methanotrophs, and one from the recently described methanotroph of the phylum Verrucomicrobia. The remaining four publicly available genome sequences were not included in the comparative analysis. This includes the genome of the second verrucomicrobial methanotroph, ‘Ma. fumariolicum’ SolV, which is available in draft form; and three proteobacterial members, Methylocystis parvus OBBP, Mm. album BG8 and Mb. tundripaludum, for which no genome annotations were available. The main features identified in the compared genomes are summarized in Table 1. The genome sequences of strains Rockwell and OB3b are available in draft form and consist of numerous contigs. As strain SC2 is their closest relative, its finished genome sequence was used as the reference for assembling their contigs. The chromosome and the two plasmids of strain SC2 were concatenated to a single sequence containing 4,049 CDS, collectively referred to as the genome. The chromosome and plasmid sequences of Mm. alcaliphilum 20Z were also concatenated into a single file, while the other genomes had no plasmids. The genome sequences were then subjected to comparative analysis, using the EDGAR platform [100].
The pan-genome or the full complement of genes present in the eight methanotroph genomes sums up to 19,358 CDS. On the contrary, the set of genes shared by all eight methanotrophs was represented by only 154 CDS. This core genome represents the conserved genetic backbone and encodes basic cellular machineries, such as DNA replication, DNA repair, transcription, protein biosynthesis, cell division and a few chaperon and heat-shock proteins (Table S3). None of the genes encoded by the plasmids of strain SC2 are included in the core gene set. The number of core genes is remarkably low, presumably due to the fact that the compared methanotrophs are from phylogenetically very distinct groups. However, in the verrucomicrobial genome, 35% of genes were found to be related to proteobacteria [48]. The set of core genes increased to 328 CDS, when this genome was removed from the calculation.
Although the methanotrophs compared in this study exhibit the same basic metabolic capability of utilizing methane as carbon and energy source, none of the key genes involved in the process were shared by all of them. This is due to the fact that methanotrophs have distinct enzyme systems for metabolizing methane. Some possess either pMMO or sMMO, while others have the ability to produce both key enzymes. They use different pathways for assimilation of formaldehyde into cell biomass. While alphaproteobacterial methanotrophs use the serine pathway, gammaproteobacterial methanotrophs employ the RuMP pathway. Similar to our findings, a very small set of core genes was observed between five genera of the family Methylophilaceae [102]. Most interestingly, although the central metabolism in all compared Methylophilaceae members was methylotrophy, their core genome was devoid of genes encoding some of the bona fide methylotrophy-related functions, such as methanol dehydrogenase, methylamine dehydrogenase, and the H4MPT-linked formaldehyde oxidation [102].
A phylogenetic analysis was performed using the concatenated multiple alignments of all 154 core genes (Table S3) and the neighbor-joining method for tree construction. In the core genome tree, members of the proteobacterial methanotrophs were grouped into two distinct clades, with the distantly related genome of Ma. infernorum V4 forming the outgroup (Figure 6). This clustering agreed well with the known phylogeny of methanotrophs as inferred from the comparative analysis of 16S rRNA and pmoA gene sequences [2]. To identify the core gene content that is specific to the genomes of the alphaproteobacterial or gammaproteobacterial methanotrophs, both groups were analyzed separately. While the four alphaproteobacterial methanotrophs shared 1,306 CDS, the three gammaproteobacterial methanotrophs shared 1,193 CDS among themselves (Figure S1).
Figure 6. Neighbor-joining tree constructed for the methanotrophic core genome.
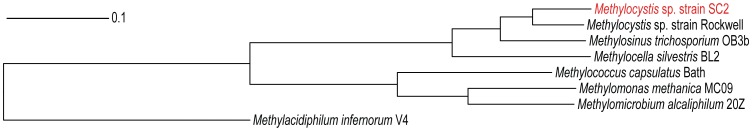
The tree is based on the alignment of 154 CDS that are common to all eight methanotroph genomes used for comparative analysis. Non-matching parts of the alignments were eliminated prior to tree construction. The individual gene alignments were combined into one concatenated alignment. The neighbor-joining tree was constructed using EDGAR. All branches of the phylogenetic tree showed 100% bootstrap support based on 500 replications. See ‘Materials and Methods’ for further details.
(b) Comparative analysis among Methylocystaceae
Three of the eight methanotroph genomes that were comparatively analyzed belong to the family Methylocystaceae in the Alphaproteobacteria. In addition to strain SC2, this includes Methylocystis sp. strain Rockwell and Ms. trichosporium OB3b. Their 16S rRNA gene sequences show a high similarity of respectively 99% and 96% to that of strain SC2. To get an insight into the genomic variation among these three alphaproteobacterial methanotrophs, their genomes were compared in greater detail. Their pan-genome totals 8,374 CDS, while they shared a set of 1,853 CDS among themselves (Figure 7). The predicted products of these common genes are distributed across almost all functional categories of the SEED subsystems (Figure 8). The number of genes assigned to three subsystems, namely, ‘cell wall and capsule’, ‘membrane transport’ and ‘amino acid and derivatives’, showed significant differences between the individual strains and their core genome (Figures 8, S2). Apart from the conserved core genome of all methanotrophs mentioned above, this includes additional genes involved in basic cellular functions. In addition, they also share genes involved in maintaining a methanotrophic lifestyle. These encode, among other proteins, pMMO, methanol dehydrogenase, pyrroloquinoline quinone cofactor biosynthesis proteins, and formate dehydrogenase. Thirty-four nitrogen metabolism-related genes are also shared. These include genes related to N2 fixation, ammonia assimilation, and assimilatory nitrate/nitrite reduction. In addition to the core genes, strains SC2 and Rockwell share, respectively, 228 and 278 genes with strain OB3b, while they share 537 genes among themselves (Figure 7). Genes that need to be specifically mentioned include the different hydroxylamine detoxification systems shared by strain SC2 with either strain Rockwell (haoAB) or strain OB3b (hcp). Eleven percent of the genes shared between strains Rockwell and OB3b are involved in flagella biosynthesis. Although the motility of Ms. trichosporium OB3b is well known [104], all Methylocystis spp. studied so far, including strain SC2, are reported to be non-motile [1], [105], [106], [107], [108], [109]. This is due to the absence of flagella and, as expected, no flagella biosynthesis-related genes were detected in strain SC2 (Figure 8). However, although no published evidence is available for the motility of strain Rockwell, presence of genes responsible for flagella biosynthesis may suggest that this strain is motile. Another interesting finding is that 53 CDS categorized in the subsystem ‘iron acquisition and metabolism’ are present only in the genome of strain OB3b. These include genes involved in iron acquisition and siderophore production (Figure S2). In contrast, strains Rockwell and SC2, respectively, contain only one and two of these genes. The ability of strain OB3b to produce siderophores, albeit in small amounts, was previously shown, using the Fe-chrome azurol S (CAS) plate assay [110].
Figure 7. Venn diagram showing the number of CDS unique to and shared by the Methylocystaceae members.

Data analysis was performed using the genomes of strain SC2 (red), strain Rockwell (blue) and Ms. trichosporium OB3b (green). Numbers in circles indicate the number of unique CDS, while those in intersections represent the number of orthologous CDS common to two or more strains. Orthologs were detected by reciprocal best BLASTP matches with the EDGAR software.
Figure 8. Functional classification of genes identified in members of the Methylocystaceae.

The gene content of strain SC2 (red), strain Rockwell (blue) and Ms. trichosporium OB3b (green) and that of the core genome shared by them (grey) was subjected to functional classification by the RAST server. CDS were classified into 27 functional categories using the SEED subsystem. Numbers in parentheses next to the strain names indicate the number of CDS assigned to the SEED subsystem out of the total number of CDS present in the particular genome. The proportion of CDS (x-axis) assigned to a particular subsystem was calculated by dividing the number of CDS assigned to this category by the total number of CDS assigned to the SEED subsystem database. The functional categories were arranged according to the number of CDS assigned for strain SC2 to each category. The number of CDS classified for the individual strains and their core genome into each SEED subsystem was subjected to statistical analysis using STAMP. A p-value cutoff of 0.05 was used to determine significant differences. Subsystems showing significant differences are marked by an asterisk.
The three Methylocystaceae members shared approximately half of their CDS, while the other half is unique to the respective strain. Presence of a large number of unique genes even among closely related genomes has been frequently observed [100]. Interestingly, the majority of the genes unique to the individual strain are novel or conserved hypothetical. Only a small proportion could be assigned to functional groups in the SEED subsystem, using the RAST server for analysis. This includes 176 (out of 1,441), 100 (out of 1,969) and 302 (out of 2,108) CDS present in the genomes of strains SC2, Rockwell and OB3b, respectively. Among the enzymes encoded by the 1,441 unique genes identified in strain SC2, those that need special mention are the high-affinity pMMO2 and the plasmid-encoded nitric oxide and nitrous oxide reductases. In addition, strain SC2 possesses two pmoCAB1 operons, while strains Rockwell and OB3b harbor a single copy of pmoCAB. Based on manual search, we could identify a large number of unique genes (173) in strain Rockwell to encode different families of transposases. The number of transposase-encoding genes was quite low in the unique gene set of strains SC2 (45) and OB3b (25). Five such genes were found to be shared by strains Rockwell, SC2, and OB3b. In fact, the number of transposases encoded by the genome of strain Rockwell was almost five times more than the average number of such genes (∼38) detected in 2,137 complete genome sequences analyzed [111]. This may suggest that genome rearrangements occur more frequently in strain Rockwell than in the other two Methylocystaceae members. The unique genes present in strain OB3b are those encoding for the soluble methane monooxygenase, iron acquisition systems, urea decomposition system, a large number of membrane transporters and systems imparting resistance to antibiotics and toxic compounds.
Final remarks
Annotation and comparative analysis of the genome sequence of strain SC2 provide detailed insights into the lifestyle and metabolic potential of this bacterium. Genome analysis coupled with physiological experiments confirmed that strain SC2 possesses diverse nitrogen metabolism-related pathways. This includes the capability to fix atmospheric N2 and perform a complete denitrification process, suggesting that strain SC2 is able to thrive under oxygen- and nitrogen-limiting conditions. Its capability to survive in low-methane environments has already previously been shown. The functionality of the plasmid-encoded nitrous oxide reductase is unique to known methanotrophs. Under the tested conditions, the enzyme efficiently converts N2O to N2. The presence of the complete nos operon and a monocistronic pmoC on pBSC2-2 suggests that at least this plasmid confers important metabolic traits to strain SC2. Absence of CRISPR/Cas systems may have allowed strain SC2 to acquire and maintain its two plasmids. Comparative genomics across the major methanotroph groups revealed that, although performing the same key metabolic processes, they have very few genes in common. However, the three Methylocystaceae members share almost half of their genes. These encode (among other) the central metabolic pathways for methane oxidation and nitrogen fixation. On the other hand, they clearly differ in their genetic potential. This includes the presence of the high-affinity pMMO2 and plasmid-encoded nitrous oxide reductase in strain SC2, high number of iron acquisition systems in strain OB3b, and motility-related genes and predicted genome instability in strain Rockwell (the latter derived from the large number of transposase genes).
Materials and Methods
Growth conditions
Strain SC2 was cultivated in nitrogen-containing (1 g KNO3 per litre) mineral salts medium (NMS) without any vitamin supplement [112]. In N-free medium, no nitrogen-containing compound was added. Whenever there was a change in media, cultures were harvested by centrifugation (2,655×g, 15 min, 4°C), washed twice with phosphate buffer (5.4 g Na2HPO4·7H2O and 2.6 g KH2PO4 per litre distilled H2O) and finally resuspended in the desired medium. After each physiological experiment, purity of the culture was confirmed by fluorescence in situ hybridization (FISH) using a strain SC2-specific 16S rRNA-targeted oligonucleotide probe as described earlier [105] (Figure S3).
N2 fixation assay
N2-fixing ability of strain SC2 was tested by batch incubation in N-free medium. Cells used for the assay were initially grown in NMS medium up to early logarithmic phase and washed properly to remove any residual nitrogen source. They were then inoculated in 20 ml N-free medium, resulting in an initial OD600 of 0.05. Incubation was done in 120-ml serum bottles that were sealed with butyl rubber stoppers. The bottles were flushed with N2. Methane (20% [vol/vol]) and the desired amount of oxygen (1, 5, 10, 15 or 20% [vol/vol]) were then injected into the headspace.
The acetylene reduction assay is widely used to test for nitrogenase activity in bacteria and was performed accordingly [21], [24], [90], [113]. To induce enzyme activity, cells were initially grown in N-free medium. Briefly, 5 ml of a suspension of log-phase cells (0.48 mg dry weight) were transferred to 25-ml serum bottles and sealed with butyl rubber stoppers. The bottles were flushed with N2-free helium gas. Oxygen in the headspace was then set to 1, 5, 10, 15, or 20% (vol/vol), as mentioned above. Methanol was added to a final concentration of 0.1% (vol/vol). Acetylene (10% [vol/vol]), which had been purified by successive passage through 2 M sulfuric acid and double-distilled water, was then injected. To measure the ethylene production, 0.5 ml of the gas phase was sampled at fixed time intervals and analyzed using a gas chromatograph. The gas chromatograph (GC 14b; Shimadzu, Griesheim, Germany) was equipped with a stainless steel column filled with Porapak R and a flame ionization detector. N2 was used as the carrier gas. Pure acetylene and ethylene were used for calibration and as standards. All gas chromatography systems were routinely calibrated with certified gas standards (Air Liquide GmbH, Kassel, Germany). In all measurements, signals were processed and chromatograms were integrated using the Peak Simple software (version 2.66, SRI Instruments, Torrence, CA, USA).
Denitrification assay
The acetylene inhibition method [93], [94], [95] was used to verify the production of N2O by strain SC2. Briefly, NMS-grown log-phase cells (3.4 mg dry weight) were resuspended in 5 ml of fresh NMS medium supplemented with methanol (0.1% [vol/vol]) or without a carbon source in 25-ml serum bottles that were capped with butyl rubber stoppers. To make the system anaerobic, the headspace was flushed with N2 for 10 min. If aerobic conditions should be maintained, oxygen (as described above) was injected into the headspace. When needed, purified acetylene was added to inhibit the conversion of N2O to N2. The bottles were then incubated on a rotary shaker at 30°C and periodically analyzed for N2O in the headspace using a gas chromatograph (Carlo Erba Instruments, GC 8000) connected to a 63Ni-electron capture detector (ECD) [95]. Potential rates of N2O production were calculated by linear regression after correcting for N2O dissolved in the liquid phase using the Bunsen coefficient for N2O [114].
Using 15N-nitrate (K15NO3), a tracer experiment was performed to check denitrification-mediated formation of N2. Strain SC2 cells (3.5 mg dry weight) that were pre-grown in NMS up to log phase were washed twice and resuspended in 5 ml of fresh NMS medium containing K15NO3 (isotopic purity of 98% 15N; Sigma-Aldrich) as the only nitrogen source, in 25-ml serum bottles. A control set was also installed where K15NO3 was replaced with KNO3. The serum bottles were sealed tightly with butyl rubber stoppers and, to make the system anaerobic, flushed with N2-free helium for 10 min. Aerobic conditions were maintained as described above. Bottles were incubated at 30°C on a rotary shaker. The production of N2 and the isotopic composition of N2 in the headspace was analyzed with a GC-IRMS system [114]. As K15NO3 was the only nitrogen source, the masses 28 (28N2 [14N14N]) and 29 (29N2 [14N15N]) were ignored and the increase in mass 30 (30N2 [15N15N]) with time was used as a proof of denitrification [114], [115]. 30N2 production was determined after fifteen days of incubation, because production was below the detection limit during the initial days of incubation.
Comparative genome analysis
Annotations of the chromosome and plasmid sequences of strain SC2 were performed using the Silver genome annotation interface (http://www.micro-genomes.mpg.de/). All CDS mentioned in the text have an E-value of 10−10 as cutoff.
Eight methanotroph genomes (Table 1) were used to setup a new comparative genomics project in the EDGAR server of the Center for Biotechnology, Bielefeld University, Bielefeld, Germany (http://edgar.cebitec.uni-bielefeld.de) [100]. For strain SC2 and Mm. alcaliphilum 20Z, concatenated sequences of their chromosome and the plasmid(s) were used. The strain SC2 genome was used as the reference in all comparative analyses. The EDGAR platform calculates so-called BLASTP score ratio values (SRV) and then defines orthologous proteins based on bidirectional best BLAST hits. As the genomes used in this comparative study represent a set of phylogenetically diverse bacteria, a comparably low SRV cutoff of 35 was used. As a consequence, paralogous genes might have been discarded during the analysis.
Construction of a core genome tree
EDGAR was used to construct a phylogenetic tree based on 154 CDS common to all analyzed species (orthology-cutoff 35% SRV) [100]. The genomic sequences that were initially aligned sum up to 1,232 CDS with a total of 473,457 amino acids. Alignments of these core genes were generated using MUSCLE [116], with non-matching parts being masked by GBLOCKS and subsequently removed [117]. The remaining parts of all the individual gene alignments were compiled in one concatenated alignment. Pairwise distances between the concatenated core genome sequences were calculated using Kimura's two-parameter method. The distance matrix was used as input to construct a phylogenetic tree with the neighbor-joining method (implemented in the PHYLIP package). The final tree was created in Newick format and visualized in iTOL, a web server for visualizing phylogenetic trees (http://itol.embl.de/index.shtml).
Classification of CDS into functional groups
To classify CDS present in a particular genome or a selected gene set (like the core genome) into functional groups, we used the RAST server (http://rast.nmpdr.org/rast.cgi). To achieve this classification, the gene sets (in GenBank format) were subjected to automated annotation process in the SEED subsystem using RAST, and gene calls were preserved as in the uploaded file [118]. This resulted in an output where the CDS were functionally classified into 27 distinct hierarchical categories. Analysis of significant differences in the number of CDS classified for the individual strains and their core genome into each SEED subsystem was performed using STAMP (Statistical Analysis of Metagenomic Profiles) [119].
Supporting Information
Venn diagrams showing the number of CDS unique to and shared by different methanotrophs. Numbers in circles indicate the total number of CDS unique to each member, while those in intersections represent the number of orthologous CDS common to two or more methanotrophs. (A, B) Comparative genomics was performed between (A) four alphaproteobacterial methanotrophs [(1) Methylocystis sp. strain SC2, (2) Mce. silvestris BL2, (3) Methylocystis sp. strain Rockwell, and (4) Ms. trichosporium OB3b] and (B) three gammaproteobacterial methanotrophs [(1) Mm. alcaliphilum 20Z, (2) Mc. capsulatus Bath, and (3) Mmo. methanica MC09]. Orthologs were detected by reciprocal best BLASTP matches with the EDGAR software.
(TIF)
Strain-specific differences in the number of CDS present in particular SEED subsystems relative to the core genome. The number of CDS classified for the individual strains and their core genome into each SEED subsystem was subjected to statistical analysis using STAMP. A p-value cutoff of 0.05 was used for determining significant differences. Subsystems showing significant differences in strains SC2 (A), Rockwell (B) and OB3b (C) (blue), when compared to their core genome (orange), are shown.
(TIF)
Purity check of strain SC2 by FISH. Representative field of view showing cells of strain SC2: (A) phase contrast microscopy; (B, C) whole-cell hybridization with bacterial probe EUB338 (green) and species-specific probe Mcyst-1256 (red); (D) staining with DAPI (blue). Bar represents 10 µm.
(TIF)
Gene products that are known or likely to be involved in methane oxidation and nitrogen metabolism of Methylocystis sp. strain SC2. Gene homologs identified in the draft genomes of strain Rockwell and Ms. trichosporium OB3b are shown in the last two columns by their respective locus tags.
(DOCX)
BLAST hits of the strain SC2 plasmid-encoded nos genes.
(DOCX)
List of 154 CDS that form the core genome of the eight methanotrophs compared in this study and were used for the construction of the genome tree.
(XLSX)
Acknowledgments
Geshe Braker and Svetlana N. Dedysh are greatly acknowledged for expert advice.
Funding Statement
This work was funded by the LOEWE Research Center for Synthetic Microbiology (SYNMIKRO). BD is grateful to the Alexander von Humboldt Foundation for his fellowship. JB acknowledges funding by the German Federal Ministry of Education and Research (grants 0315599A & 0315599B “GenoMik-Transfer”). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Hanson RS, Hanson TE (1996) Methanotrophic bacteria. Microbiol Rev 60: 439–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Op den Camp HJM, Islam T, Stott MB, Harhangi HR, Hynes A, et al. (2009) Environmental, genomic and taxonomic perspectives on methanotrophic Verrucomicrobia . Environ Microbiol Rep 1: 293–306. [DOI] [PubMed] [Google Scholar]
- 3. Eller G, Frenzel P (2001) Changes in activity and community structure of methane-oxidizing bacteria over the growth period of rice. Appl Environ Microbiol 67: 2395–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Horz HP, Yimga MT, Liesack W (2001) Detection of methanotroph diversity on roots of submerged rice plants by molecular retrieval of pmoA, mmoX, mxaF, and 16S rRNA and ribosomal DNA, including pmoA-based terminal restriction fragment length polymorphism profiling. Appl Environ Microbiol 67: 4177–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Radajewski S, Webster G, Reay DS, Morris SA, Ineson P, et al. (2002) Identification of active methylotroph populations in an acidic forest soil by stable-isotope probing. Microbiology 148: 2331–2342. [DOI] [PubMed] [Google Scholar]
- 6. Knief C, Lipski A, Dunfield PF (2003) Diversity and activity of methanotrophic bacteria in different upland soils. Appl Environ Microbiol 69: 6703–6714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Knief C, Kolb S, Bodelier PL, Lipski A, Dunfield PF (2006) The active methanotrophic community in hydromorphic soils changes in response to changing methane concentration. Environ Microbiol 8: 321–333. [DOI] [PubMed] [Google Scholar]
- 8. Chen Y, Dumont MG, Cebron A, Murrell JC (2007) Identification of active methanotrophs in a landfill cover soil through detection of expression of 16S rRNA and functional genes. Environ Microbiol 9: 2855–2869. [DOI] [PubMed] [Google Scholar]
- 9. Cebron A, Bodrossy L, Chen Y, Singer AC, Thompson IP, et al. (2007) Identity of active methanotrophs in landfill cover soil as revealed by DNA-stable isotope probing. FEMS Microbiol Ecol 62: 12–23. [DOI] [PubMed] [Google Scholar]
- 10. McDonald IR, Murrell JC (1997) The particulate methane monooxygenase gene pmoA and its use as a functional gene probe for methanotrophs. FEMS Microbiol Lett 156: 205–210. [DOI] [PubMed] [Google Scholar]
- 11. Dedysh SN, Dunfield PF, Derakshani M, Stubner S, Heyer J, et al. (2003) Differential detection of type II methanotrophic bacteria in acidic peatlands using newly developed 16S rRNA-targeted fluorescent oligonucleotide probes. FEMS Microbiol Ecol 43: 299–308. [DOI] [PubMed] [Google Scholar]
- 12. Chen Y, Dumont MG, McNamara NP, Chamberlain PM, Bodrossy L, et al. (2008) Diversity of the active methanotrophic community in acidic peatlands as assessed by mRNA and SIP-PLFA analyses. Environ Microbiol 10: 446–459. [DOI] [PubMed] [Google Scholar]
- 13. Nauer PA, Dam B, Liesack W, Zeyer J, Schroth MH (2012) Activity and diversity of methane-oxidizing bacteria in glacier forefields on siliceous and calcareous bedrock. Biogeosciences 9: 2259–2274. [Google Scholar]
- 14. Belova SE, Baani M, Suzina NE, Bodelier PLE, Liesack W, et al. (2011) Acetate utilization as a survival strategy of peat-inhabiting Methylocystis spp. Environ Microbiol Rep 3: 36–46. [DOI] [PubMed] [Google Scholar]
- 15. Im J, Lee S-W, Yoon S, DiSpirito AA, Semrau JD (2011) Characterization of a novel facultative Methylocystis species capable of growth on methane, acetate and ethanol. Environ Microbiol Rep 3: 174–181. [DOI] [PubMed] [Google Scholar]
- 16. Nyerges G, Han SK, Stein LY (2010) Effects of ammonium and nitrite on growth and competitive fitness of cultivated methanotrophic bacteria. Appl Environ Microbiol 76: 5648–5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nyerges G, Stein LY (2009) Ammonia cometabolism and product inhibition vary considerably among species of methanotrophic bacteria. FEMS Microbiol Lett 297: 131–136. [DOI] [PubMed] [Google Scholar]
- 18. Ricke P, Erkel C, Kube M, Reinhardt R, Liesack W (2004) Comparative analysis of the conventional and novel pmo (particulate methane monooxygenase) operons from Methylocystis strain SC2. Appl Environ Microbiol 70: 3055–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baani M, Liesack W (2008) Two isozymes of particulate methane monooxygenase with different methane oxidation kinetics are found in Methylocystis sp. strain SC2. Proc Natl Acad Sci USA 105: 10203–10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dam B, Dam S, Kube M, Reinhardt R, Liesack W (2012) Complete genome sequence of Methylocystis sp. strain SC2, an aerobic methanotroph with high-affinity methane oxidation potential. J Bacteriol 194: 6008–6009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Murrell JC, Dalton H (1983) Nitrogen fixation in obligate methanotrophs. J Gen Microbiol 129: 3481–3486. [Google Scholar]
- 22. Dedysh SN, Ricke P, Liesack W (2004) NifH and NifD phylogenies: an evolutionary basis for understanding nitrogen fixation capabilities of methanotrophic bacteria. Microbiology 150: 1301–1313. [DOI] [PubMed] [Google Scholar]
- 23. Auman AJ, Speake CC, Lidstrom ME (2001) nifH sequences and nitrogen fixation in type I and type II methanotrophs. Appl Environ Microbiol 67: 4009–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Khadem AF, Pol A, Jetten MS, Op den Camp HJ (2010) Nitrogen fixation by the verrucomicrobial methanotroph ‘Methylacidiphilum fumariolicum’ SolV. Microbiology 156: 1052–1059. [DOI] [PubMed] [Google Scholar]
- 25. Zumft WG (1997) Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev 61: 533–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Waibel AE, Peter T, Carslaw KS, Oelhaf H, Wetzel G, et al. (1999) Arctic ozone loss due to denitrification. Science 283: 2064–2069. [DOI] [PubMed] [Google Scholar]
- 27. Klotz MG, Stein LY (2008) Nitrifier genomics and evolution of the nitrogen cycle. FEMS Microbiol Lett 278: 146–156. [DOI] [PubMed] [Google Scholar]
- 28. Wrage N, Velthof GL, van Beusichem ML, Oenema O (2001) Role of nitrifier denitrification in the production of nitrous oxide. Soil Biol Biochem 33: 1723–1732. [Google Scholar]
- 29. Campbell MA, Nyerges G, Kozlowski JA, Poret-Peterson AT, Stein LY, et al. (2011) Model of the molecular basis for hydroxylamine oxidation and nitrous oxide production in methanotrophic bacteria. FEMS Microbiol Lett 322: 82–89. [DOI] [PubMed] [Google Scholar]
- 30. Bergmann DJ, Zahn JA, Hooper AB, DiSpirito AA (1998) Cytochrome P460 genes from the methanotroph Methylococcus capsulatus Bath. J Bacteriol 180: 6440–6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Elmore BO, Bergmann DJ, Klotz MG, Hooper AB (2007) Cytochromes P460 and c′-beta; a new family of high-spin cytochromes c. FEBS Lett 581: 911–916. [DOI] [PubMed] [Google Scholar]
- 32. Poret-Peterson AT, Graham JE, Gulledge J, Klotz MG (2008) Transcription of nitrification genes by the methane-oxidizing bacterium, Methylococcus capsulatus strain Bath. ISME J 2: 1213–1220. [DOI] [PubMed] [Google Scholar]
- 33. Sutka RL, Ostrom NE, Ostrom PH, Gandhi H, Breznak JA (2003) Nitrogen isotopomer site preference of N2O produced by Nitrosomonas europaea and Methylococcus capsulatus Bath. Rapid Commun Mass Spectrom 17: 738–745. [DOI] [PubMed] [Google Scholar]
- 34. Sutka RL, Ostrom NE, Ostrom PH, Breznak JA, Gandhi H, et al. (2006) Distinguishing nitrous oxide production from nitrification and denitrification on the basis of isotopomer abundances. Appl Environ Microbiol 72: 638–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stein LY, Klotz MG (2011) Nitrifying and denitrifying pathways of methanotrophic bacteria. Biochem Soc Trans 39: 1826–1831. [DOI] [PubMed] [Google Scholar]
- 36. Stein LY (2011) Surveying N2O-producing pathways in bacteria. Methods Enzymol 486: 131–152. [DOI] [PubMed] [Google Scholar]
- 37. Arp DJ, Stein LY (2003) Metabolism of inorganic N compounds by ammonia-oxidizing bacteria. Crit Rev Biochem Mol Biol 38: 471–495. [DOI] [PubMed] [Google Scholar]
- 38. Zahn JA, Duncan C, DiSpirito AA (1994) Oxidation of hydroxylamine by cytochrome P-460 of the obligate methylotroph Methylococcus capsulatus Bath. J Bacteriol 176: 5879–5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stein LY, Yoon S, Semrau JD, Dispirito AA, Crombie A, et al. (2010) Genome sequence of the obligate methanotroph Methylosinus trichosporium strain OB3b. J Bacteriol 192: 6497–6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. del Cerro C, Garcia JM, Rojas A, Tortajada M, Ramon D, et al. (2012) Genome sequence of the methanotrophic poly-beta-hydroxybutyrate producer Methylocystis parvus OBBP. J Bacteriol 194: 5709–5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stein LY, Bringel F, DiSpirito AA, Han S, Jetten MS, et al. (2011) Genome sequence of the methanotrophic alphaproteobacterium Methylocystis sp. strain Rockwell (ATCC 49242). J Bacteriol 193: 2668–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen Y, Crombie A, Rahman MT, Dedysh SN, Liesack W, et al. (2010) Complete genome sequence of the aerobic facultative methanotroph Methylocella silvestris BL2. J Bacteriol 192: 3840–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ward N, Larsen Ø, Sakwa J, Bruseth L, Khouri H, et al. (2004) Genomic insights into methanotrophy: the complete genome sequence of Methylococcus capsulatus (Bath). PLoS Biol 2: e303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kits KD, Kalyuzhnaya MG, Klotz MG, Jetten MS, Op den Camp HJ, et al. (2013) Genome sequence of the obligate gammaproteobacterial methanotroph Methylomicrobium album strain BG8. Genome Announc 1: e00170–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vuilleumier S, Khmelenina VN, Bringel F, Reshetnikov AS, Lajus A, et al. (2012) Genome sequence of the haloalkaliphilic methanotrophic bacterium Methylomicrobium alcaliphilum 20Z. J Bacteriol 194: 551–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boden R, Cunliffe M, Scanlan J, Moussard H, Kits KD, et al. (2011) Complete genome sequence of the aerobic marine methanotroph Methylomonas methanica MC09. J Bacteriol 193: 7001–7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Svenning MM, Hestnes AG, Wartiainen I, Stein LY, Klotz MG, et al. (2011) Genome sequence of the Arctic methanotroph Methylobacter tundripaludum SV96. J Bacteriol 193: 6418–6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hou S, Makarova KS, Saw JH, Senin P, Ly BV, et al. (2008) Complete genome sequence of the extremely acidophilic methanotroph isolate V4, Methylacidiphilum infernorum, a representative of the bacterial phylum Verrucomicrobia . Biol Direct 3: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Khadem AF, Wieczorek AS, Pol A, Vuilleumier S, Harhangi HR, et al. (2012) Draft genome sequence of the volcano-inhabiting thermoacidophilic methanotroph Methylacidiphilum fumariolicum strain SolV. J Bacteriol 194: 3729–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dam B, Kube M, Dam S, Reinhardt R, Liesack W (2012) Complete sequence analysis of two methanotroph-specific repABC-containing plasmids from Methylocystis sp. strain SC2. Appl Environ Microbiol 78: 4373–4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kolsto AB (1997) Dynamic bacterial genome organization. Mol Microbiol 24: 241–248. [DOI] [PubMed] [Google Scholar]
- 52. Lima-Mendez G, Van Helden J, Toussaint A, Leplae R (2008) Prophinder: a computational tool for prophage prediction in prokaryotic genomes. Bioinformatics 24: 863–865. [DOI] [PubMed] [Google Scholar]
- 53. Makarova KS, Grishin NV, Koonin EV (2006) The HicAB cassette, a putative novel, RNA-targeting toxin-antitoxin system in archaea and bacteria. Bioinformatics 22: 2581–2584. [DOI] [PubMed] [Google Scholar]
- 54. Yamaguchi Y, Park JH, Inouye M (2011) Toxin-antitoxin systems in bacteria and archaea. Annu Rev Genet 45: 61–79. [DOI] [PubMed] [Google Scholar]
- 55. Pedersen K, Zavialov AV, Pavlov MY, Elf J, Gerdes K, et al. (2003) The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell 112: 131–140. [DOI] [PubMed] [Google Scholar]
- 56. Zhang Y, Zhang J, Hara H, Kato I, Inouye M (2005) Insights into the mRNA cleavage mechanism by MazF, an mRNA interferase. J Biol Chem 280: 3143–3150. [DOI] [PubMed] [Google Scholar]
- 57. Kamada K, Hanaoka F (2005) Conformational change in the catalytic site of the ribonuclease YoeB toxin by YefM antitoxin. Mol Cell 19: 497–509. [DOI] [PubMed] [Google Scholar]
- 58. Maisonneuve E, Shakespeare LJ, Jørgensen MG, Gerdes K (2011) Bacterial persistence by RNA endonucleases. Proc Natl Acad Sci U S A 108: 13206–13211. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59. Kolodkin-Gal I, Engelberg-Kulka H (2006) Induction of Escherichia coli chromosomal mazEF by stressful conditions causes an irreversible loss of viability. J Bacteriol 188: 3420–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hazan R, Sat B, Engelberg-Kulka H (2004) Escherichia coli mazEF-mediated cell death is triggered by various stressful conditions. J Bacteriol 186: 3663–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Grissa I, Vergnaud G, Pourcel C (2007) CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 35: W52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Grissa I, Vergnaud G, Pourcel C (2007) The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8: 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Barrangou R, Horvath P (2012) CRISPR: New horizons in phage resistance and strain identification. Annu Rev Food Sci Technol 3: 143–162. [DOI] [PubMed] [Google Scholar]
- 64. Deveau H, Garneau JE, Moineau S (2010) CRISPR/Cas system and its role in phage-bacteria interactions. Annu Rev Microbiol 64: 475–493. [DOI] [PubMed] [Google Scholar]
- 65. Horvath P, Barrangou R (2010) CRISPR/Cas, the immune system of bacteria and archaea. Science 327: 167–170. [DOI] [PubMed] [Google Scholar]
- 66. Palmer KL, Gilmore MS (2010) Multidrug-resistant enterococci lack CRISPR-cas . MBio 1(4): e00227–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Horvath P, Coute-Monvoisin AC, Romero DA, Boyaval P, Fremaux C, et al. (2009) Comparative analysis of CRISPR loci in lactic acid bacteria genomes. Int J Food Microbiol 131: 62–70. [DOI] [PubMed] [Google Scholar]
- 68. Gao F, Zhang CT (2008) Ori-Finder: a web-based system for finding oriCs in unannotated bacterial genomes. BMC Bioinformatics 9: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sernova NV, Gelfand MS (2008) Identification of replication origins in prokaryotic genomes. Brief Bioinform 9: 376–391. [DOI] [PubMed] [Google Scholar]
- 70. Hendrickson H, Lawrence JG (2007) Mutational bias suggests that replication termination occurs near the dif site, not at Ter sites. Mol Microbiol 64: 42–56. [DOI] [PubMed] [Google Scholar]
- 71. Kono N, Arakawa K, Tomita M (2011) Comprehensive prediction of chromosome dimer resolution sites in bacterial genomes. BMC Genomics 12: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Garcia-Martinez J, Bescos I, Rodriguez-Sala JJ, Rodriguez-Valera F (2001) RISSC: a novel database for ribosomal 16S-23S RNA genes spacer regions. Nucleic Acids Res 29: 178–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nomura M, Morgan EA (1977) Genetics of bacterial ribosomes. Annu Rev Genet 11: 297–347. [DOI] [PubMed] [Google Scholar]
- 74. Matsugi J, Murao K (2004) Genomic investigation of the system for selenocysteine incorporation in the bacterial domain. Biochim Biophys Acta 1676: 23–32. [DOI] [PubMed] [Google Scholar]
- 75. Akiyama Y, Ito K (2003) Reconstitution of membrane proteolysis by FtsH. J Biol Chem 278: 18146–18153. [DOI] [PubMed] [Google Scholar]
- 76. Akiyama Y (2009) Quality control of cytoplasmic membrane proteins in Escherichia coli . J Biochem 146: 449–454. [DOI] [PubMed] [Google Scholar]
- 77. Ito K, Akiyama Y (2005) Cellular functions, mechanism of action, and regulation of FtsH protease. Annu Rev Microbiol 59: 211–231. [DOI] [PubMed] [Google Scholar]
- 78. Holmes AJ, Costello A, Lidstrom ME, Murrell JC (1995) Evidence that particulate methane monooxygenase and ammonia monooxygenase may be evolutionarily related. FEMS Microbiol Lett 132: 203–208. [DOI] [PubMed] [Google Scholar]
- 79. Semrau JD, Chistoserdov A, Lebron J, Costello A, Davagnino J, et al. (1995) Particulate methane monooxygenase genes in methanotrophs. J Bacteriol 177: 3071–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bedard C, Knowles R (1989) Physiology, biochemistry, and specific inhibitors of CH4, NH4 +, and CO oxidation by methanotrophs and nitrifiers. Microbiol Rev 53: 68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sanford RA, Wagner DD, Wu Q, Chee-Sanford JC, Thomas SH, et al. (2012) Unexpected nondenitrifier nitrous oxide reductase gene diversity and abundance in soils. Proc Natl Acad Sci U S A 109: 19709–19714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Buurman ET, Teixeira de Mattos MJ, Neijssel OM (1991) Futile cycling of ammonium ions via the high affinity potassium uptake system (Kdp) of Escherichia coli . Arch Microbiol 155: 391–395. [DOI] [PubMed] [Google Scholar]
- 83. Merrick MJ, Edwards RA (1995) Nitrogen control in bacteria. Microbiol Rev 59: 604–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Helling RB (1998) Pathway choice in glutamate synthesis in Escherichia coli . J Bacteriol 180: 4571–4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kohler T, Harayama S, Ramos JL, Timmis KN (1989) Involvement of Pseudomonas putida RpoN sigma factor in regulation of various metabolic functions. J Bacteriol 171: 4326–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Weiss V, Kramer G, Dunnebier T, Flotho A (2002) Mechanism of regulation of the bifunctional histidine kinase NtrB in Escherichia coli . J Mol Microbiol Biotechnol 4: 229–233. [PubMed] [Google Scholar]
- 87. Pawlowski K, Klosse U, de Bruijn FJ (1991) Characterization of a novel Azorhizobium caulinodans ORS571 two-component regulatory system, NtrY/NtrX, involved in nitrogen fixation and metabolism. Mol Gen Genet 231: 124–138. [DOI] [PubMed] [Google Scholar]
- 88. Takeda K (1988) Characteristics of a nitrogen-fixing methanotroph, Methylocystis T-1. Antonie van Leeuwenhoek 54: 521–534. [DOI] [PubMed] [Google Scholar]
- 89. Dedysh SN, Khmelenina VN, Suzina NE, Trotsenko YA, Semrau JD, et al. (2002) Methylocapsa acidiphila gen. nov., sp. nov., a novel methane-oxidizing and dinitrogen-fixing acidophilic bacterium from Sphagnum bog. Int J Syst Evol Microbiol 52: 251–261. [DOI] [PubMed] [Google Scholar]
- 90. Dalton H, Whittenbury R (1976) The acetylene reduction technique as an assay for nitrogenase activity in the methane oxidizing bacterium Methylococcus capsulatus strain Bath. Arch Microbiol 109: 147–151. [Google Scholar]
- 91. De Bont JAM, Mulder EG (1974) Nitrogen fixation and co-oxidation of ethylene by a methane-utilizing bacterium. J Gen Microbiol 83: 113–121. [Google Scholar]
- 92. Zhivotchenko AG, Nikonova ES, Jørgensen MH (1995) Effect of fermentation conditions on N2 fixation by Methylococcus capsulatus . Bioprocess Biosyst Eng 14: 9–15. [Google Scholar]
- 93. Ren T, Roy R, Knowles R (2000) Production and consumption of nitric oxide by three methanotrophic bacteria. Appl Environ Microbiol 66: 3891–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yoshinari T, Knowles R (1976) Acetylene inhibition of nitrous oxide reduction by denitrifying bacteria. Biochem Biophys Res Commun 69: 705–710. [DOI] [PubMed] [Google Scholar]
- 95. Braker G, Schwarz J, Conrad R (2010) Influence of temperature on the composition and activity of denitrifying soil communities. FEMS Microbiol Ecol 73: 134–148. [DOI] [PubMed] [Google Scholar]
- 96. Pieja AJ, Rostkowski KH, Criddle CS (2011) Distribution and selection of poly-3-hydroxybutyrate production capacity in methanotrophic proteobacteria. Microb Ecol 62: 564–573. [DOI] [PubMed] [Google Scholar]
- 97. Qin L, Liu Y, Tay JH (2005) Denitrification on poly-beta-hydroxybutyrate in microbial granular sludge sequencing batch reactor. Water Res 39: 1503–1510. [DOI] [PubMed] [Google Scholar]
- 98. Vecherskaya M, Dijkema C, Saad HR, Stams AJM (2009) Microaerobic and anaerobic metabolism of a Methylocystis parvus strain isolated from a denitrifying bioreactor. Environ Microbiol Rep 1: 442–449. [DOI] [PubMed] [Google Scholar]
- 99. Pieja AJ, Sundstrom ER, Criddle CS (2011) Poly-3-hydroxybutyrate metabolism in the type II methanotroph Methylocystis parvus OBBP. Appl Environ Microbiol 77: 6012–6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Blom J, Albaum SP, Doppmeier D, Pühler A, Vorhölter FJ, et al. (2009) EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinformatics 10: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Strobel T, Al-Dilaimi A, Blom J, Gessner A, Kalinowski J, et al. (2012) Complete genome sequence of Saccharothrix espanaensis DSM 44229T and comparison to the other completely sequenced Pseudonocardiaceae . BMC Genomics 13: 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lapidus A, Clum A, LaButti K, Kaluzhnaya MG, Lim S, et al. (2011) Genomes of three methylotrophs from a single niche reveal the genetic and metabolic divergence of the Methylophilaceae . J Bacteriol 193: 3757–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lukjancenko O, Ussery DW, Wassenaar TM (2012) Comparative genomics of Bifidobacterium, Lactobacillus and related probiotic genera. Microb Ecol 63: 651–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Shonnard DR, Taylor RT, Tompson A, Knapp RB (1992) Hydrodynamic effects on microcapillary motility and chemotaxis assays of Methylosinus trichosporium OB3b. Appl Environ Microbiol 58: 2737–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Dunfield PF, Yimga MT, Dedysh SN, Berger U, Liesack W, et al. (2002) Isolation of a Methylocystis strain containing a novel pmoA-like gene. FEMS Microbiol Ecol 41: 17–26. [DOI] [PubMed] [Google Scholar]
- 106. Dedysh SN, Belova SE, Bodelier PLE, Smirnova KV, Khmelenina VN, et al. (2007) Methylocystis heyeri sp. nov., a novel type II methanotrophic bacterium possessing ‘signature’ fatty acids of type I methanotrophs. Int J Syst Evol Microbiol 57: 472–479. [DOI] [PubMed] [Google Scholar]
- 107. Bowman JP, Sly LI, Nichols PD, Hayward AC (1993) Revised taxonomy of the methanotrophs: Description of Methylobacter gen. nov., emendation of Methylococcus, validation of Methylosinus and Methylocystis species, and a proposal that the family Methylococcaceae includes only the group I methanotrophs. Int J Syst Bacteriol 43: 735–753. [Google Scholar]
- 108. Wartiainen I, Hestnes AG, McDonald IR, Svenning MM (2006) Methylocystis rosea sp. nov., a novel methanotrophic bacterium from Arctic wetland soil, Svalbard, Norway (78° N). Int J Syst Evol Microbiol 56: 541–547. [DOI] [PubMed] [Google Scholar]
- 109. Lindner AS, Pacheco A, Aldrich HC, Costello Staniec A, Uz I, et al. (2007) Methylocystis hirsuta sp. nov., a novel methanotroph isolated from a groundwater aquifer. Int J Syst Evol Microbiol 57: 1891–1900. [DOI] [PubMed] [Google Scholar]
- 110. Yoon S, Dispirito AA, Kraemer SM, Semrau JD (2011) A simple assay for screening microorganisms for chalkophore production. Methods Enzymol 495: 247–258. [DOI] [PubMed] [Google Scholar]
- 111. Aziz RK, Breitbart M, Edwards RA (2010) Transposases are the most abundant, most ubiquitous genes in nature. Nucleic Acids Res 38: 4207–4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Heyer J, Galchenko VF, Dunfield PF (2002) Molecular phylogeny of type II methane-oxidizing bacteria isolated from various environments. Microbiology 148: 2831–2846. [DOI] [PubMed] [Google Scholar]
- 113. Toukdarian AE, Lidstrom ME (1984) Nitrogen metabolism in a new obligate methanotroph, ‘Methylosinus’ strain 6. J Gen Microbiol 130: 1827–1837. [DOI] [PubMed] [Google Scholar]
- 114. Ngugi DK, Brune A (2012) Nitrate reduction, nitrous oxide formation, and anaerobic ammonia oxidation to nitrite in the gut of soil-feeding termites (Cubitermes and Ophiotermes spp.). Environ Microbiol 14: 860–871. [DOI] [PubMed] [Google Scholar]
- 115. Steingruber SM, Friedrich J, Gachter R, Wehrli B (2001) Measurement of denitrification in sediments with the 15N isotope pairing technique. Appl Environ Microbiol 67: 3771–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Talavera G, Castresana J (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56: 564–577. [DOI] [PubMed] [Google Scholar]
- 118. Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, et al. (2008) The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Parks DH, Beiko RG (2010) Identifying biologically relevant differences between metagenomic communities. Bioinformatics 26: 715–721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Venn diagrams showing the number of CDS unique to and shared by different methanotrophs. Numbers in circles indicate the total number of CDS unique to each member, while those in intersections represent the number of orthologous CDS common to two or more methanotrophs. (A, B) Comparative genomics was performed between (A) four alphaproteobacterial methanotrophs [(1) Methylocystis sp. strain SC2, (2) Mce. silvestris BL2, (3) Methylocystis sp. strain Rockwell, and (4) Ms. trichosporium OB3b] and (B) three gammaproteobacterial methanotrophs [(1) Mm. alcaliphilum 20Z, (2) Mc. capsulatus Bath, and (3) Mmo. methanica MC09]. Orthologs were detected by reciprocal best BLASTP matches with the EDGAR software.
(TIF)
Strain-specific differences in the number of CDS present in particular SEED subsystems relative to the core genome. The number of CDS classified for the individual strains and their core genome into each SEED subsystem was subjected to statistical analysis using STAMP. A p-value cutoff of 0.05 was used for determining significant differences. Subsystems showing significant differences in strains SC2 (A), Rockwell (B) and OB3b (C) (blue), when compared to their core genome (orange), are shown.
(TIF)
Purity check of strain SC2 by FISH. Representative field of view showing cells of strain SC2: (A) phase contrast microscopy; (B, C) whole-cell hybridization with bacterial probe EUB338 (green) and species-specific probe Mcyst-1256 (red); (D) staining with DAPI (blue). Bar represents 10 µm.
(TIF)
Gene products that are known or likely to be involved in methane oxidation and nitrogen metabolism of Methylocystis sp. strain SC2. Gene homologs identified in the draft genomes of strain Rockwell and Ms. trichosporium OB3b are shown in the last two columns by their respective locus tags.
(DOCX)
BLAST hits of the strain SC2 plasmid-encoded nos genes.
(DOCX)
List of 154 CDS that form the core genome of the eight methanotrophs compared in this study and were used for the construction of the genome tree.
(XLSX)