

Abstract
Network theory applied to protein structures provides insights into numerous problems of biological relevance. The explosion in structural data available from PDB and simulations establishes a need to introduce a standalone-efficient program that assembles network concepts/parameters under one hood in an automated manner. Herein, we discuss the development/application of an exhaustive, user-friendly, standalone program package named PSN-Ensemble, which can handle structural ensembles generated through molecular dynamics (MD) simulation/NMR studies or from multiple X-ray structures. The novelty in network construction lies in the explicit consideration of side-chain interactions among amino acids. The program evaluates network parameters dealing with topological organization and long-range allosteric communication. The introduction of a flexible weighing scheme in terms of residue pairwise cross-correlation/interaction energy in PSN-Ensemble brings in dynamical/chemical knowledge into the network representation. Also, the results are mapped on a graphical display of the structure, allowing an easy access of network analysis to a general biological community. The potential of PSN-Ensemble toward examining structural ensemble is exemplified using MD trajectories of an ubiquitin-conjugating enzyme (UbcH5b). Furthermore, insights derived from network parameters evaluated using PSN-Ensemble for single-static structures of active/inactive states of β2-adrenergic receptor and the ternary tRNA complexes of tyrosyl tRNA synthetases (from organisms across kingdoms) are discussed. PSN-Ensemble is freely available from http://vishgraph.mbu.iisc.ernet.in/PSN-Ensemble/psn_index.html.
Keywords: PSN-Ensemble program, network features, protein structure network, weighted network, MD/NMR ensemble, UbcH5b, beta2-adrenergic receptor, tRNA synthetases
The three-dimensional structural organization in proteins can be easily translated into a network, retaining all critical interaction information. PSN-Ensemble serves as a robust, standalone program, which can easily compute the network features from a protein structure in an automated and user-friendly manner. The major goal of PSN-Ensemble is to bridge the gap between obtaining important biological information from structural/experimental data and complex network-based calculations thereby providing a uniform platform for information exchange across a wide biological community.
Introduction
Network theory has been widely exploited in various fields to identify the emergent features of global connectivity.1 Be it the interactions among different individuals in the social network, the interactions between transactions in the context of economics, transportation in cartography, the interactions among reactions and metabolites in systems biology, or in the mapping of structural topology in the case of macromolecules, network theory has provided important insights into the local topology of interactions from a global perspective.1,2 The expediency of using network theory to study macromolecular structure lies in the fact that it reduces the complex three-dimensional macromolecular organizations to a mathematical representation retaining all the connectivity information in the structure.3 Consequently, such a representation is amenable to analysis using an arsenal of network based mathematical formulations and concepts.1
The choice of interacting units (nodes) for translating a macromolecular structure into a network has been widely experimented in the literature and is largely influenced by the specific questions being addressed.3 One can construct coarse grain protein structure networks by considering the interactions among the C-alpha/C-beta atoms in the amino acid residues, as in a protein backbone network (PBN).4,5 A more detailed network considering the interactions at all atom level on the other hand yields a protein sidechain structure network (PScN).6,7 The interactions in PScN representation can be quantified by considering the residue connections ranging from atom–atom contact to highly packed interactions like aromatic stacking.6 Herein, this representation enables the construction of networks based on weak and strong noncovalent interactions at the sidechain level and this unique approach has been proven to provide valuable biological insights.8–12
Although the network theory is powerful in providing a local view of molecular interactions in a global milieu, a need to understand proteins as macromolecules that undergo constant fluctuations in their conformations has propelled the network analysis of structure ensembles.8–10 Molecular dynamics (MD) simulations capture the dynamics of interactions and the conformational space associated with a given state of macromolecules. Implementation of network theory on conformational ensembles (derived from MD simulations, NMR studies, or multiple X-ray structures) provides the equilibrium (or the average) global properties. Such an approach has found vast applications in the study and analysis of macromolecular structures to obtain valuable insights into several phenomena of biological relevance.8–13 A combination of MD simulation techniques and PScN/PBN to analyze protein structures has led to interesting results in several previous studies, such as ligand induced modulation of rigidity/flexibility/function,8 transition between conformational substates,14 homology modeling and structural refinement,15 understanding of topological features of proteins,7 their folds and functions,11,16 protein structure comparison,17 identification of active site/aromatic clusters, identifying “hotspots” in the structure,11,18 and many more. They have also been conveniently used in understanding allosteric communication pathways or “junction points” responsible for long-range signal transmission in proteins.9,10 Additionally, such network concepts can find newer applications in predicting unprecedented allosteric sites in proteins and binding effect of drugs to such allosteric sites.19–21 Thus, taking a comprehensive view, a judicious usage of network theory in combination with the dynamical information from a structural ensemble provides a generalized tool to address problems with widespread biological implications. The main aim of this study is to present a robust package for such investigations.
Numerous methods/tools have been developed over the past years by various groups to construct and investigate proteins networks. NetworkAnalyzer and RINalyzer plugins in Cytoscape provide methods for exploring networks derived from protein structures.22,23 NetworkAnalyzer and RINalyzer perform topological analysis of biological network by calculating various network parameters for single structures, which can be visualized using Cytoscape. NetworkView has been reported for the three-dimensional visualization/characterization of networks of protein-RNA complexes24 and Xpyder25 has also been recently proposed as a tool for analysis of dynamic cross-correlation matrices in the context of long-range communication. A webserver GraProStr has been previously developed in our laboratory to calculate various network parameters, like hubs, clusters, cliques/communities for single X-ray structures.26
Although a few programs, as mentioned above, are available for network analysis of protein structures, it is timely to make a consolidated and automated program, including the concept of protein dynamics, easily accessible to the biological community. In this article, we describe the usage and application of a freely available standalone program PSN-Ensemble, which bridges structural dynamics and network analysis, at the detailed level of side-chain interactions, in a highly automated and robust manner. This standalone program operates by providing the coordinates of structure ensembles (from MD simulations, NMR studies, or multiple X-ray structures) as input and yields important network parameters such as hubs, clusters, cliques/communities, paths of allosteric communication between two residues, and important nodes for long-range communication (henceforth denoted as “junction residues”) between domains as outputs. The details of the various steps involved in obtaining these parameters are summarized in the workflow shown in Figure 1. This program is also applicable for computing the above mentioned parameters in case of a single X-ray structure, thus representing an effective standalone version of the GraProStr webserver.26 The overall organization of the program ensures user discretion at every stage in the form of well-defined options and offers an easy, automated visualization of these network parameters on the three-dimensional structure of the protein using Pymol. PSN-Ensemble highlights the following key features.
Figure 1.
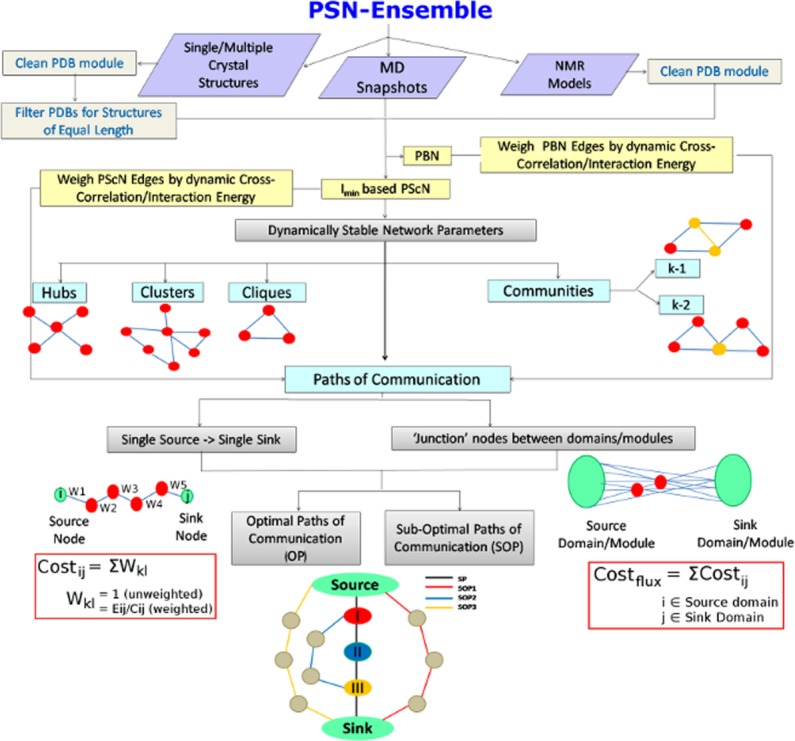
A flowchart representing the workflow in PSN-Ensemble. The package is compatible with crystal structure (single or multiple structures), MD simulation, and NMR ensemble structures. Various network parameters for characterizing the topological organization in proteins [hubs, clusters, cliques, communities (k-1/k-2)] can be obtained at a given Imin (see Methods section) from PScN. The network parameters to characterize long-range communication in terms of paths and cost of communication [OPs and SOPs] can be calculated based on PScN or PBN. A schematic description of each of the network parameters is also included in the flowchart for easy comprehension. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Evaluates the network parameters of Protein structure networks at a desired level of noncovalent, sidechain interaction.
Ensemble network features are derived by taking into account a set of structures from MD simulations, NMR models or multiple X-ray structures.
Variety of weighing schemes, including dynamic stability (frequency of occurrence in the ensemble), dynamic cross-correlation, and energy of interaction, are built into PSN-Ensemble.
Special attention is paid to characterize long-range communication related parameters.
Works effectively with protein multimers and maps back results from network analysis onto the starting structure, retaining the chain information.
Furthermore, we have benchmarked the program using long MD trajectories (150 ns) for UbcH5b, an ubiquitin conjugating enzyme (E2) in its apo form. Statistical coupling analysis (SCA) and experimental studies have previously reported an allosteric communication between the ubiquitin binding site and the ubiquitin ligase (E3) binding domains on UbcH5b.27 It has also been shown that the subtle long-range conformational changes on UbcH5b due to E3 binding enhance its ability to transfer ubiquitin to its substrates.27 Using UbcH5b as a model system, here we describe different network parameters and the context in which they can be used to understand structure-function relationship. Specifically, special attention is paid to the communication paths/important “junction residues” that are responsible for long-range signal transfer in UbcH5b.
Additionally, the crystal structures of the beta2-adrenergic receptor (β2-AR) in its active28 and inactive29 states are used to exemplify the usage of the program and the valuable information that can be derived, even from a single crystal structure, in terms of various network parameters. The β2-AR is a classical model system to study the GPCR signaling mechanism. Pioneering work by Kobilka and coworkers28,29 provided immense insights into this signaling mechanism through the elucidation of the crystal structures of β2-AR locked in its active (PDB_id: 3SN6) and inactive (PDB_id: 2RH1) states. We have used the network parameters to address the subtle structural differences in terms of topological organization in the active/inactive state of β2-AR as well as probed long-range communication between important residues in this receptor.
A third example is provided to elaborate the application of this program to study proteins which interact with nucleic acids. In this context, the single crystal structure of the ternary complex of tyrosyl tRNA synthetases from three organisms across different kingdoms, namely Thermus thermophilus (bacteria; Pdb_id: 1H3E), Methanococcus janaschii (archea; Pdb_id: 1J1U), and Saccharomyces cerevisiae (yeast; Pdb_id: 2DLC) have been analyzed.30–32 The similarities and differences across the organisms are addressed in terms of long-range communication paths between the aminoacylation site and the anticodon binding site (both at inter and intra subunit level).
In short, PSN-Ensemble offers a rigorous and exhaustive method for obtaining valuable information such as communication paths, regions of rigidity and flexibility, ligand induced conformational variations in terms of the network parameters computed (see individual definitions of the parameters in Methods section). This standalone program is easy to install without the requirement of multiple other packages, with the exception of Matlab (MatlabBGL), CFinder,33 and Pymol (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC). The residues that constitute the results for the network parameters are automatically mapped back onto the original PDB structure, retaining the chain information, if any. The mapping back also allows efficient comparison with available experimental/theoretical data. The features mentioned in the earlier paragraphs make PSN-Ensemble a unique and robust package to evaluate several network properties including those related to long distance communication under one common hood.
Results and Discussions
PSN-Ensemble can be used to investigate proteins (monomers as well as multimers) to characterize them in terms of various network parameters in a highly automated manner. The details of the features of this package are explained in the Methods section. The potential application of PSN-Ensemble to address biologically relevant problems is exemplified here using a long MD simulation trajectory and NMR models for UbcH5b (in apo form), an ubiquitin conjugating enzyme. An Imin of 2% is chosen for this analysis. For UbcH5b, the network analysis is followed by an extensive comparison with experimentally and theoretically available data, which reveals significant agreement. This further substantiates the application of such network concepts in identifying biologically relevant residues, both in terms of topological organization and allosteric communication. Additionally, the usage of the program for a single crystal structure is exemplified using the active and inactive states of β2-AR and the ternary complex of tyrosyl tRNA synthetases from three different organisms, emphasizing the utility of the package to a broader audience of biological community.
Application of PSN-Ensemble to problems in structural biology
MD simulation ensemble of UbcH5b
Proteins undergo ubiquitination, a post translational modification that results in proteosomal degradation of proteins. Ubiquitin activating enzymes (E1), conjugating enzymes (E2), and ligases (E3) are involved in catalyzing this process.34 Ubiquitin forms a thioester linkage with the active site Cys of E1 and is then transferred to E2 as a thioester. Subsequently E3 facilitates the transfer of ubiquitin to the substrate protein resulting in ubiquitination. It has been reported that a long-range signal transfer between the E3 and ubiquitin binding sites regulates the transfer of ubiquitin from E2 to substrates.
In this article, we have chosen UbcH5b, an ubiquitin conjugating enzyme from the yeast Ubc4 family as a model system. SCA and experimental techniques have been used in the past to probe UbcH5b by Özkan et al.27 SCA has been used to identify clusters of co-evolving residues mediating allosteric crosstalk between E3 binding domain and active site of UbcH5b (residuewise summary of the identified important residues in Supporting Information Fig. S3). The role of some of these residues in signal transfer has been also verified through mutagenesis and activity experiments. Herein, we have performed long MD simulations (150 ns) on the apo form of UbcH5b and the equilibrated part of the trajectories (100–150 ns) are used to demonstrate the usage of the PSN-Ensemble package and elaborate on the different biological questions that can be addressed. This is followed by an extensive comparison of our results with available experimental data on UbcH5b.27
Network analysis of UbcH5b MD ensemble using PSN-Ensemble
Topological organization
UbcH5b is analyzed using PSN-Ensemble to examine the topological features of its three dimensional organization in terms of various network parameters like hubs, clusters, cliques, and communities (k-1/k-2). Hubs capture highly connected residues or “hot spots” in the protein, whereas cliques and communities are excellent metrics to investigate higher order connectivity (i.e., regions of rigidity/flexibility in the structure) and percolation of such connectivity through the protein. These parameters can be used to comment on the subtle changes in the conformational organization in a protein on external perturbations, such as ligand binding or even mutations and change in environmental conditions. On the other hand, clusters, especially at the interface of a multimeric protein can illuminate on the features related to protein-protein association and has been shown to be of relevance in previous studies.
Many residues in UbcH5b that are predicted to be important by Özkan et al. from SCA and mutagenesis studies (F69, H75, F50, L109, W93, W33, and F56) are seen to overlap with those identified from hubs, clusters, cliques, and communities (Fig. 2, Supporting Information Table S1). Additionally, experimentally suggested important residues such as I37, T142, R5, and P25 are also identified by some combination of these four network parameters. PSN-Ensemble identifies a total of 20 hub residues, 90 residues in the largest cluster, 15 cliques, three k-1, and five k-2 communities. About 50% of these residues identified by the various network parameters coincide with important residues identified from previous studies on UbcH5b27 (Supporting Information Table S1).
Figure 2.

Pictorial representation of the various network parameters [hubs, clusters, cliques, communities (k-1/k-2), OPs and SOPs, junction nodes] for an MD ensemble of UbcH5b (PDB_id: 2ESK) using PSN-Ensemble at an Imin of 2%. The protein is represented as a cartoon and the residues identified from various network parameters are shown as van der Waals spheres. The top three communities (blue, red, and green) and the OP (red) and top six SOPs (blue) are plotted. The important residues identified from the previous studies are color coded as explained in the figure. Paths of communication (OPs/SOPs) are obtained by choosing Ser94 (E3 binding site) and Cys85 (active site) as the source and sink, respectively. “Junction nodes” are obtained between the source domain comprising of residues Ser94, Pro95, Phe62, Tyr60, Arg5, Lys8 and sink domain comprising of Cys85 (active site) and Asn77 (catalytic site).
Allosteric communication perspective
The allosteric communication perspective can be examined using the concept of optimal paths (OPs) and suboptimal paths (SOPs) of communication within a network. Whereas identification of linear pathways (both OPs and SOPs) between a single source to sink residue (source and sink are distant residues within which signal transfer takes place) enables recognition of key players in transmission of information, the ‘junction residues’ efficiently capture the critical points/nodes in the complex network of long range communication.
UbcH5b has been shown to exhibit allosteric crosstalk between the E3 binding site and the active site. PSN-Ensemble is used to investigate the key players involved in distal signal transfer between these two sites. For the present calculation of paths of communication, we choose residue S94 from the E3 binding site and the active site residue C85 as the two termini between which the signal transfer is investigated. Both OPs and SOPs are evaluated (residues W93, L109, L89, N77 occur in the majority of the OPs while F69, L52, L103 occur in majority of the SOPs). However, it has been proposed that the communication between the E3 binding and active site is mediated by a complex network of residues rather than a linear pathway. To explore the critical residues comprising this network, the “junction residues” responsible for interdomain signal transfer are further identified using PSN-Ensemble. The E3 binding domain and the catalytic/active site residues are considered as source and sink, respectively, for information flow. The residues that flux maximum number of the OPs as well as SOPs in going from one source to sink are thought to be crucial players in the allosteric communication network (a pictorial summary is presented in Fig. 2). A good agreement between the residues predicted to be involved in allosteric communication and those obtained from previous studies27 assert the robustness of our approach (Supporting Information Table S3 and S4). The cost of communication between the E3 binding site and active site is also computed which gives a quantitative picture of the information transfer (Fig. 3).
Figure 3.
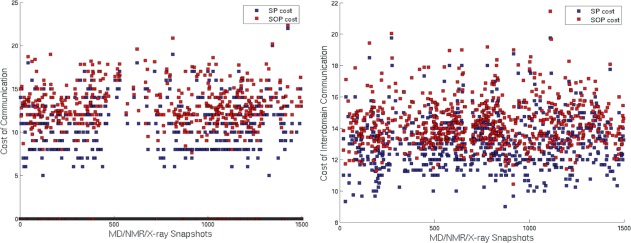
Plots for (a) cost of communication between single source (Ser94)—single sink (Cys85) residues. The SOP cost is averaged over all the SOPs for a given snapshot. (b) Average cost of communication between source domain (Ser94, Pro95, Phe62, Tyr60, Arg5 [E3 binding domain] and sink domain (Cys85 [active site] and Asn77 [catalytic site]). A distinct pattern is evident in the profile for the cost of communication between residues suggesting probable conformational changes at regular intervals. Such a pattern is not evident from the RMSD profile which is relatively flat (data not shown). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Additionally, we evaluated all the above mentioned parameters for the NMR model of UbcH5b, the results of which are pictorially depicted in Supporting Information Figure S4 and a residuewise comparison of the results from MD and NMR ensemble is also presented in Supporting Information Table S2.
Active/inactive states of beta2-adrenergic receptor (β2-AR)
PSN-Ensemble can provide a wealth of information for an ensemble of structures from a topological perspective including dynamical information. However, at a simpler level, PSN-Ensemble serves as a powerful tool even for a single crystal structure and is capable of providing answers to biologically important questions. In an effort to demonstrate this, we choose the crystal structures of a G-protein coupled receptor (β2-AR). GPCRs are comprised of three major structural components: the extracellular (EC), the transmembrane (TM), and the intracellular (IC) regions. The TM helix bundle binds ligands and the induced conformational change transmits this signal to the IC, which in turn interacts with cytosolic signaling partners.35 This common structural framework is widely exploited by a large number of G-protein coupled receptors and β2-AR is a popular model system for studying GPCR mediated transmembrane signaling.28
The availability of crystal structures for β2-AR in its inactive (bound to an inverse agonist, carazolol) as well as active (bound to Gs protein, a heterotrimer comprised of Gα and Gβγ subunits and agonist) forms28,29 permit a comparative analysis from the network perspective. Herein, we analyze the active and inactive states of β2-AR in light of the differences in their topological organization and the structural variation that dictates the functional outcome. A special emphasis on the interface between β2-AR and Gs protein (only the Gα subunit contact the β2-AR directly28) and long-range communication between ligand binding pocket and Gs protein coupling interface reveals intriguing structural features. The major questions asked in the context of β2-AR are briefly described in the following subsections and a network perspective is provided for each of them.
Conformational variations: active and inactive states of β2-AR
The active and inactive state crystal structures of β2-AR are analyzed using the PSN-Ensemble package. As detailed in the Methods section, the hubs, cliques, and communities highlight the conformational variations in the β2 adrenergic receptor on binding of the Gs protein [Figs. 4 and 5(a)]. The results from these network parameters show a major overlap with the key residues and motifs identified from previous studies (Supporting Information Table S5). A drastic rewiring of interactions and alteration of rigidity/flexibility is clearly evident from the cliques that involve these important residues [Fig. 5(a)] and community structures [Fig. 4(b,c)], in terms of their location in the active and inactive states. An alteration of rigidity in the region proximal to F139 (F139 is a major determinant of Gs protein recognition) is induced in the active state, which may bear relevance toward recognition [Figs. 4(b,c) and 5(a)]. An enhancement in the overall number of hubs, cliques, and percolating communities is also clear in the active form, indicating an overall tight packing through sidechain interactions [Figs. 4 and 5(a), Supporting Information Table S5].
Figure 4.
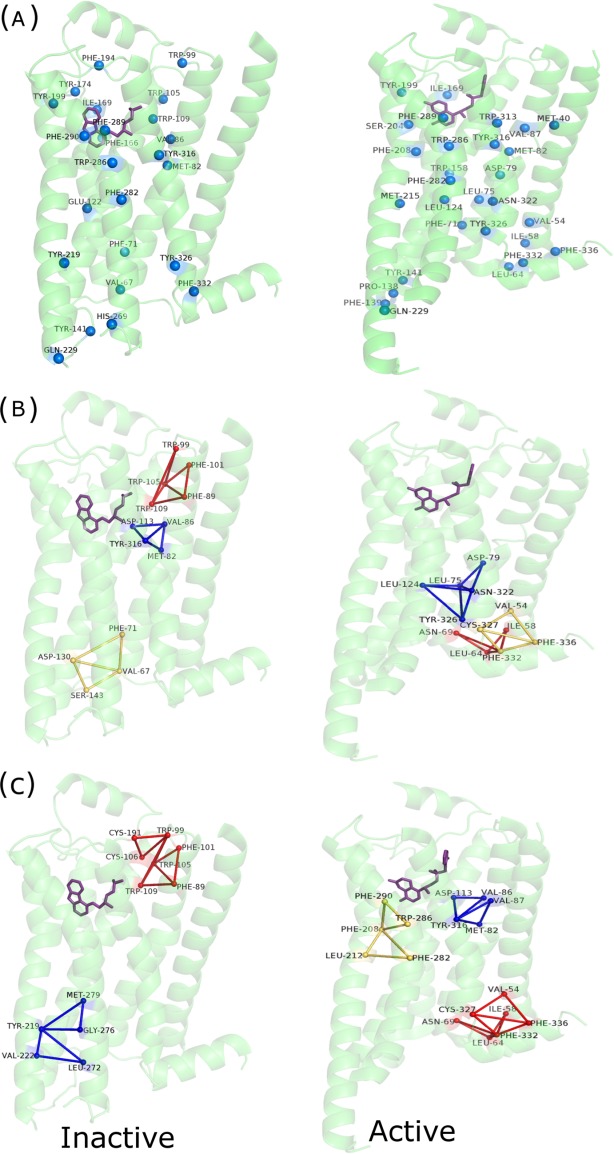
Pictorial depiction of (a) hubs, (b) communities [k-1], and (c) communities [k-2] for the inactive and active forms of β2-AR. The protein backbone is depicted in green cartoon representation. Only the β2-AR is depicted for ease of comparison. The hub residues are depicted as van der Waals spheres and the communities (top three) are depicted using blue, red, and yellow lines, respectively. A residuewise comparison of these results with key residues predicted in earlier studies are summarized in Supporting Information Table S5. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 5.
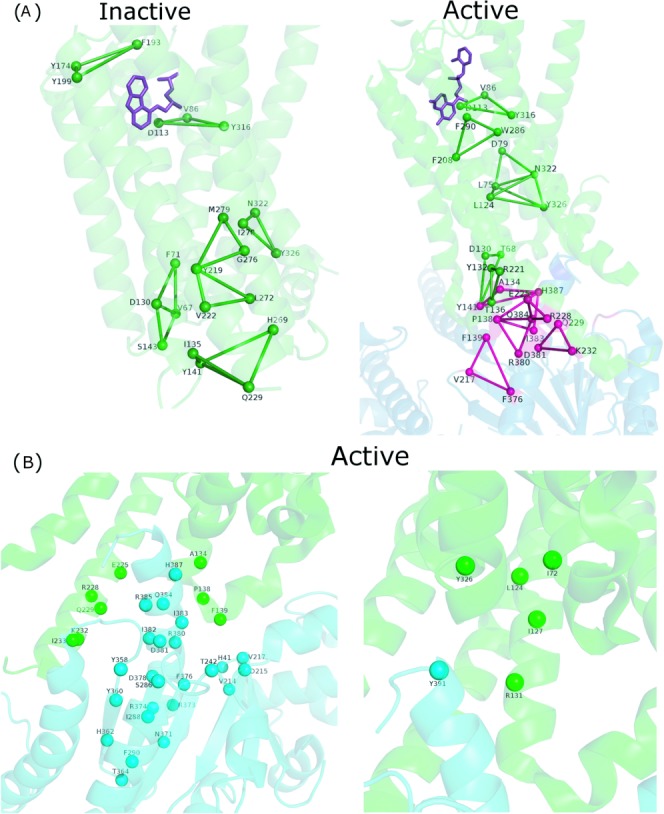
Pictorial depiction of (a) core and interface cliques constituted by key residues (Supporting Information Table S5) for the inactive and active forms and (b) clusters at the interface between the receptor and Gαs for the active form of β2-AR. The β2-AR and Gαs backbones are depicted in green and blue/cyan cartoon representation, respectively. The interface cluster residues are depicted as van der Waals spheres and the cliques are depicted using dark green (core) and dark pink (interface) lines, respectively. The Gs protein coupling interface is beautifully captured by these interface network parameters. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Gs protein coupling interface of β2-AR: Interface clusters
The network parameters like cliques and clusters formed at the interface are excellent metrics to capture the coupling specificity for multimeric organization of different protein subunits. Previous studies in our laboratory have exemplified the application of interface clusters toward the identification and classification of various multimeric organizations in lectins.36 It has been suggested in the literature that the basis for coupling specificity between Gs protein and β2-AR involves subtle structural features. The residues that participate in the interface clusters and cliques for β2-AR (in the active state) reveal interactions among many of the crucial residues predicted from earlier studies (residuewise summary in Fig. 5, Supporting Information Table S5). The interface regions of higher order connectivity, as revealed through interface cliques, capture many important receptor G-protein interactions, for example, those involving the D130 and R132 of the conserved DRY sequence in β2-AR, critical Y141 which stabilizes F139 through interaction with D130, and interactions of F139 with the hydrophobic pocket in Gαs [Fig. 5(a)].28 Additionally, the recognition interface in the active state of β2-AR can also be probed using interface clusters. The interface clusters clearly exhibit the interactions that constitute the interface in the receptor-Gs protein complex, again many of which are in good agreement with the previous experimental data28 [Fig. 5(b)].
Allosteric signaling: active and inactive states of β2-AR
A residue in the ligand binding pocket (D113) and a critical residue (F139) at the Gs protein coupling interface of β2-AR are chosen for probing the paths of allosteric communication across the membrane in both active and inactive states of the receptor. Strikingly, although OP/SOPs are identified between the chosen termini for the active state, such paths till F139 are absent in the inactive form (Fig. 6). When probed further for interdomain communication to identify “junction residues” (Supporting Information Fig. S5), the inactive state offered no such residues between the ligand binding pocket and F139. This implies a blocked communication in the inactive state, which may be re-established on ligand-induced conformational perturbation. We further investigated the structural variations leading to the absence of communication paths in 2RH1 (inactive β2-AR). The paths indeed get blocked (after D130) in the inactive state due the drastically different orientation of F139 and Y141, when compared with their conformation in the active state. It has been proposed earlier that on Gs protein coupling,28 the intracellular loop2 (ICL2) that accommodates the critical F139 is stabilized through an interaction between Y141 and D130, which we believe re-establishes the paths for signal transfer (Fig. 6).
Figure 6.
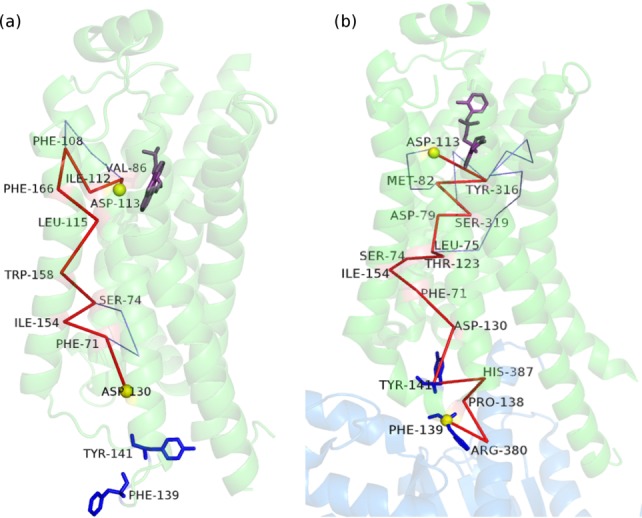
Pictorial depiction of the paths of communication (OP/SOPs) from D133 in the ligand binding pocket to F139 at the Gs protein coupling interface for the (a) inactive and (b) active states of β2-AR. The path is blocked due to lack of appropriate interactions in the inactive state. Binding of Gs protein and agonist in the active state appears to re-establish this path of communication/signaling. The β2-AR and Gαs backbones are depicted in green and blue cartoon representation, respectively. The OP is shown in red and the SOPs are shown in blue lines. The source and the sink residues are highlighted as yellow van der Waals' spheres. The key F139 and Y141 residues and the ligand (inverse agonist/agonist) are shown in blue and violet stick representation, respectively. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
A close and thorough inspection of the results from various network parameters, addressing the topological and long-range communication differences in the active and inactive conformations of β2-AR (residuewise summary in Supporting Information Table S5) reveal the strength of PSN-Ensemble. The key residues predicted from network analysis exhibits a high concordance with those highlighted in complementary biochemical and biophysical studies. Of particular interest are the results from interface cluster analysis which impart insight into the crucial residues that constitute the receptor-G-protein coupling interface. Additionally, it is intriguing to observe the G-protein induced restoration of long-range signaling between ligand binding pocket and G-protein coupling interface in the active state of β2-AR. This highlights the importance of subtle structural changes leading to re-orchestration of interactions (i.e., residue contacts) which may act as a switch to reinstate flow of information across distant sites in going from an inactive to an active state. This also manifests that a small change at the residue level is capable of controlling the global communication and such functional insights are often elusive from conventional techniques of structure comparison.
Ternary complexes of tyrosyl tRNA synthetase from bacteria, archea, and yeast
To illustrate the usage of PSN-Ensemble to analyze proteins in complex with nucleic acids, we have focussed on single crystal structures of tyrosyl tRNA synthetase (TyrRS) obtained from three organisms across different kingdoms. Aminoacyl tRNA synthetases (aaRS) play a crucial role in maintaining the fidelity of protein biosynthesis by ensuring correct translation of the genetic code.37 TyrRS is a member of Class Ic aaRSs and acts as a functional dimer. TyrRS is composed of a N-terminal Rossman fold catalytic domain, two consensus motifs HIGH and KMSKS, central α helical domain, and a C-terminal anticodon binding domain.38 Archeal and eukaryotic TyrRS share greater sequence similarity, when compared with bacterial systems and are not orthogonal to their bacterial counterparts. Ternary complexes of TyrRS [Tyrosine+tRNATyr+TyrRS] from Thermus thermophilus (bacteria; Pdb_id: 1H3E), Methanococcus janaschii (archea; Pdb_id: 1J1U), and Saccharomyces cerevisiae (yeast; PDB_id: 2DLC)30–32 are probed using PSN-Ensemble to evaluate intra and inter subunit communication in these complexes.
Intra and long range communication between the active site and the anticodon binding domain in TyrRS
The ternary complex of TyrRS from Thermus thermophilus (System 1), Methanococcus janaschii (System 2), and Saccharomyces cerevisiae (System 3) consists of TyrRS + tRNATyr +ATP+Tyrosinol, TyrRS + tRNATyr +Tyrosine, and TyrRS + tRNATyr +YMP, respectively. (Sequence and structural similarity details between the three systems obtained using ClustalW39 and TM-Align40 are given in Supporting Information Table 7). They are homodimers and the two subunits are related by crystallographic two-fold symmetry axis. Two tRNA molecules bind to the dimer such that the acceptor stem is bound to one subunit while the anti-codon loop of the same tRNA is bound to the other subunit. Herein, we probe the long-range communication within and across the subunits by considering the aminoacylation site/active site as the source and the anti-codon recognition region as the sink. Many source and sink residues in Methanococcus janaschii and Saccharomyces cerevisiae are conserved, while only a few residues in the source domain of Thermus thermophilus are conserved.30–32 The details of source and sink residues in the three systems are given in Supporting Information Table 6. The intra and inter-subunit paths of communication between the active site and anticodon recognition domain of the two subunits are probed in all three systems.
System 1
The residues of TyrRS from Thermus thermophilus, interacting with activated tyrosine [Y41, D182, Q179, Q197, G194, Y175, L224] and those interacting with the anti-codon bases of tRNA [D423, Y342, D259, R256, P285] are considered as source and sink domain, respectively. Probing intra and inter subunit paths of communication in the ternary complex reveals the existence of paths of communication only within the same subunit and not across subunits. The absence of any long-range inter subunit communication implies that both tRNAs are required at the same time for charging the tRNAs with tyrosine. This may also suggests that the recognition of the anticodon stem of tRNA1 by subunit A of TyrRS may facilitate the charging of the tRNA2 bound to subunit B and vice versa. The paths of communication within the two subunits are almost identical and the shortest path is from Leu 244 to Asp 259, which consists of only six residues [Fig. 7(a)]. The paths between different residues of the two domains have a considerable overlap and pass through the source residue Leu 224 and residue Leu 262 near the sink region.
Figure 7.

Pictorial depiction of paths of communication between source and sink residues of TyrRS from (a) Thermus thermophilus (bacteria), (b) Methanococcus janachsii (archea), and (c) Saccharomyces cerevisiae (yeast). TyrRS backbone is shown in cartoon representation in green (subunit A) and cyan (subunit B) and tRNA molecules are depicted as cartoons in wheat color. Tyrosine and ATP molecules are shown in stick representation in pink and steel green, respectively. The source/sink residues and the residues common to all paths are depicted as van der Waals spheres in orange/yellow and blue, respectively. Intra subunit, inter subunit and those common to both intra and inter subunit pathways are depicted as lines in blue, red, and green, respectively. As evident, inter subunit communication exists in archeal and yeast TyrRS, while it is absent in its bacterial counterpart. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
System 2
A similar analysis on TyrRS from Methanococcus janaschii reveals the existence of long-range communication both within and across subunits. The residues [Y32, D158, Q155, Q173, Y151, H177, H70] and [F261, H283, D286, C231] are considered from of source and sink domains, respectively. The shortest path within the subunit is from His 177 to Ile 231 and consists of 11 residues while that across subunits is from His70 in one subunit and Cys 231 in the other subunit and is 22 residues long [Fig. 7(b)]. Strikingly, all intra subunit paths pass through source residue His 177 and residue Ile 244, and all paths across subunits also pass through the same two residues in the subunit housing the sink domain.
System 3
TyrRS from Saccharomyces cerevisiae also exhibit patterns of communication similar to System 2. The residues [Y43, D177, Q174, Q192, Y170, D191, G189, Y56, Y101, V219] and [F296, P320, D321, C255, F254, P319, D259, D423, P257] are considered for communication between the source and the sink domains, respectively. Paths exist both within and across subunits [Fig. 7(c)]. For intra subunit communication, the shortest path is 16 residues long. There are two such shortest paths, one between Tyr 101 and Cys 255. Across subunits, the shortest path is 39 residues long. And, two such paths exist: one between 177 in one subunit and 255 in other subunit and other between 170 in one subunit and 255 in other subunit. Again, all intra subunit paths pass through residue Pro 52 and sink residue Cys 255 and all inter subunit paths pass through residues Pro 52 and Cys 255 in the subunit containing the sink domains. It is remarkable that TyrRS from both archeal (Pdb_id: 1J1U) and yeast (Pdb_id: 2DLC) systems exhibit similar patterns of communication between and across the two subunits. The inter subunit paths from the active site of subunit A pass through the active site of subunit B to reach its anticodon binding region. The inter subunit communication paths are symmetric since the starting structure is symmetric. Due to their ability to exhibit both intra and inter subunit communication, both archeal and yeast TyrRS would exhibit higher flexibility in charging their corresponding tRNAs in comparison to their bacterial counterpart. As a result, tRNA1 bound to subunit A can charge itself and also the tRNA 2 bound to subunit B and vice versa. Although significant results are obtained here based on the analysis on single static-crystal structures, it should be noted that an ensemble of simulation snapshots can provide more insights. For example, ensemble from the simulation of human TrpRS (an aaRS belonging to the same class as TyrRS) showed41 asymmetric behavior of the two subunits and also that the frequency of inter-subunit communication was extremely low, suggesting the possibility of intra-subunit communication.
Analysis of interface clusters: structural support for the absence of inter subunit communication in TyrRS from Thermus thermophilus
We further explored the three systems using PSN-Ensemble to establish a rationale for the existence of long-range inter-subunit communication only in archeal and yeast TyrRS and not in bacterial TyrRS. As intra subunit communication pathways exists in all three systems, the possible break in the inter subunit pathway in bacterial TyrRS should occur in the region connecting the two subunits, that is, at the interface. On comparing the largest interface clusters in the three systems at Imin 4 and 6% (see Fig. 8), it is evident that the interface connectivity is greater in the archeal/yeast system when compared with bacterial TyrRS. At an Imin of 4%, the archeal/yeast TyrRS exhibit a single highly connected cluster spanning both subunits. On the other hand, the bacterial system forms two distinct clusters, with each cluster sparsely connecting the two subunits. Greater connectivity between the two subunits spanning across the interface in archeal and yeast TyrRS facilitates better communication between their active sites in contrast to a point connection at the interface in bacterial TyrRS. On increasing the Imin to 7%, the sole connection between the two clusters completely vanishes in bacterial TyrRS, while no such breakage happens in the archael/yeast counterparts.
Figure 8.
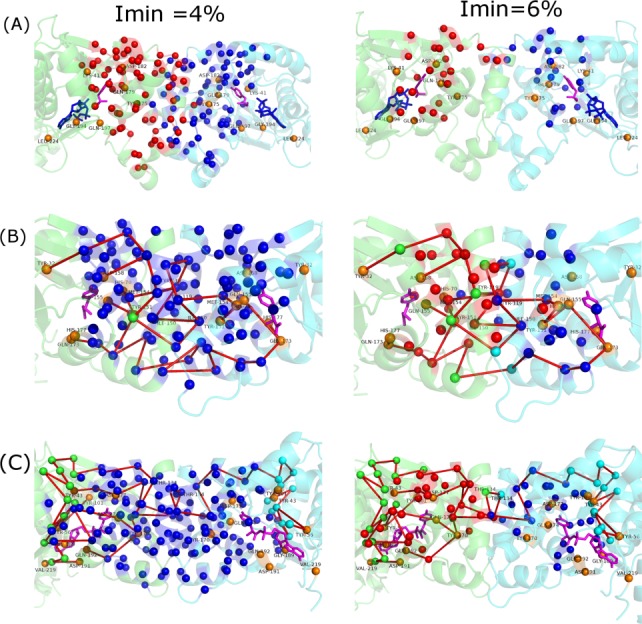
Pictorial representation of interface clusters in TyrRS from (a) Thermus thermophilus (bacteria), (b) Methanococcus janachsii (archea), and (c) Saccharomyces cerevisiae (yeast) at Imin of 4% and 6%. TyrRS backbone is shown in cartoon representation in green (subunit A) and cyan (subunit B). Tyrosine and ATP molecules are shown in stick representation in pink and blue, respectively. The interface cluster residues are depicted as van der Waals spheres in blue and red. Source residues and residues not part of the interface cluster (which are present in paths between active sites in the two subunits) are depicted as van der Waals spheres in orange and green (in subunit A)/cyan (subunit B), respectively. The intra subunit paths between the active sites are shown in red. Presence of a single large interface cluster at 4% and two distinct interface clusters spanning the interface at 6% ensures effective communication between the two subunits in archeal and yeast TyrRS which is absent in bacterial TyrRS. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The paths of communication obtained at Imin = 3% between active sites of the two subunits are shown on the interface clusters obtained at 4% and 6%. Most of the residues important for communication in the interface clusters at 4% are retained at 6% in both archeal and yeast system. The paths in archeal TyrRS [Fig. 8(b)] span throughout the interface explaining the strong interaction/communication between the two subunits. It is remarkable to note that in the archeal system, all the residues (except for I153, shown as green van der Waal sphere) involved in communication across subunits feature in the interface cluster at 4%. At 6%, the number of residues important for communication not featuring in the interface cluster increases to 7 (shown in green/cyan). Most of the paths from the active site in subunit A to the active site in subunit B pass through one or more of these green/cyan colored residues which do not feature in the interface clusters, hence, these paths (which exist at 3%) do not exist at 6%. Only four paths from subunit A manage to pass through the interface to subunit B thorough the residues A-154 C-119 A-150 C-150 A-119 C-154 (labeled in the figure) which come up in the interface clusters even at 6%. Although the paths in the yeast TyrRS [Fig. 8(c)] do not span throughout the interface as in the archeal system, they do exist across subunits with relatively fewer residues in the interface at 4% being involved in communication. The key residues Thr 134 (labeled in the figure) from both subunits through which all the paths from subunit A pass to reach subunit B are absent from the interface clusters at 6%. This being the case, there would be no paths from subunit A to subunit B at this Imin. Nevertheless, it is clearly evident that the connectivity between different subunits through the interface clusters is greater in archeal/yeast TyrRS than bacterial TyrRS.
The strength of PSN-Ensemble to effectively compare various network parameters of proteins performing the same function across species is illustrated using the above example. The absence of long-range inter-subunit communication pathways in TyrRS from Thermus thermophils is analyzed efficiently using the package. The results presented here for TyrRS and from GPCR structures are obtained effortlessly within a few seconds and with practically no computational cost. Thus, it serves as an extremely useful module to quickly compare allosteric communication from a large number of structures from diverse proteins. Furthermore, the analysis of ensemble structures generated from MD simulations on proteins selected from the set, can be performed (as demonstrated in the case of UbcH5b system) to obtain detailed information.
Methods
Description of the PSN-Ensemble package
A comprehensive blueprint of PSN-Ensemble is provided in Figure 1. PSN-Ensemble is a freely distributed, standalone package. The input to the program comprises of MD snapshots/NMR models/single crystal structure or multiple crystal structures of the same protein (in PDB format). The program computes various network parameters that are dynamically stable (i.e., occurs in greater than a user-defined fraction of the structure ensemble) and stores them in relevant folders (see PSN-Ensemble documentation for details). A set of well-defined user options are available at each step of the program execution. The user has to choose the strength of noncovalent interaction (denoted by the parameter Imin as described in a later section) to define the edge in the construction of PScN. The various network parameters that can be evaluated using PSN-Ensemble are hubs, clusters, cliques, communities, paths of communication between a given pair of important residues (user defined) and “junction residues” that flux communications between domains/modules (user defined). The network parameters related to long-range communication can be evaluated using various weighing schemes, like dynamic stability, dynamic cross-correlation and interaction energies (the cross-correlation/interaction energy matrices has to be submitted by the user).10,13 The main objective of the program is to evaluate the network properties of protein structures at the detailed side-chain interaction level from an ensemble perspective. It is to be noted that many previous studies which focus on allosteric communication have concentrated on the PBNs.42–44 Hence, for the communication related modules in PSN-Ensemble, the option for using PBN is additionally made available to the users for ease of comparison, along with PScN.
To summarize, the user can interactively choose a suitable combination of input options from the package (Fig. 1) based on the questions being addressed. Furthermore, the output network parameters thus generated are automatically mapped back by the package to the original/starting PDB structure. For multimeric proteins, the chain details are preserved in the final output. These features facilitate an easy interpretation and comparison of the results with existing data in the literature. An additional feature of the program is the storage of precomputed adjacency matrices for PScN in appropriate folders for a given dataset at a given Imin value. This can always be reused to obtain the desired network parameters for a given dataset barring the requirement of re-evaluating the adjacency matrix representation for PScN, which is a computationally expensive step. All these features make PSN-Ensemble user-friendly and effective.
Input/output formats in PSN-Ensemble
The user has to feed in the structural ensemble in PDB format (from MD snapshots, NMR models, multiple crystal structures of the same protein). Water and other ions must be stripped off from the MD snapshots before input to PSN-Ensemble. The user has to also key in the desired Imin, cutoff percentage for dynamic stability and the full path to the folder containg the structure files. For an MD ensemble, the user has an option of either using the numbering scheme in the MD snapshots (here the residues are renumbered continuously, which leads to a change in the residue number and the chain information is lost in some packages, particularly in the context of multimers) or the original/starting structure for MD (X-ray or modeled structures; the user has to provide the full path for the folder containing this structure.) for representing the results from network analysis. A set of options (1–7) are provided to the user to appropriately choose the network parameter of their interest. Options 1–5 deal with parameters like hubs, clusters, cliques, and k-1/k-2 communities, respectively, which are broadly associated with conformational organization in proteins, from a sidechain noncovalent interaction perspective (i.e., using PScN). Options 6–7 are relevant to characterizing long-range communications in proteins, and the user has to define a set of source and sink residues/domains between which the signal transfer has to be evaluated. Herein, apart from using dynamically stable PScN, additional features that include weighing the network using average cross-correlation/interaction energy matrix and the flexibility to use PBN is introduced. The user has to provide the average cross-correlation/interaction energy matrix for weighing and these matrices can be obtained from multiple packages based on the nature of the structural ensemble and the questions at hand (see later section on weighing schemes).
The corresponding output files for various network parameters are stored in the folder PSN-Ensemble/Results/Adj_Imin/representative_PDB_id. The network parameters computed are also automatically visualized on the three-dimensional structure of a representative structure using Pymol. All the hub residues, cliques and “junction residues” and top three largest clusters and k-1/k-2 communities are plotted. The largest, second largest, and the third largest clusters and communities are color coded in dark blue, red, and yellow, respectively. For paths of communication, both OP (in red) and top six SOPs are plotted. Additionally, the cost of communication between a pair of residues or between user-specified domains/modules is also plotted using Matlab, and the plots are stored in PNG format within the PSN-Ensemble/Results/Adj_Imin/representative_PDB_id folder. The individual chains in multimeric proteins are colored differently in the PyMol representation of the results. The various output files also contain the chain details from the original PDB structure on which the results are being mapped back. A detailed stepwise description for the execution of the program is provided in Supporting Information Figure S1(a,b).
Cleaning of input PDB files
A continuous, unique numbering scheme is required to generate the connectivity matrix (adjacency matrix), representing the noncovalent interactions between pairs of residues in protein structures. This requires preprocessing of the X-ray and NMR structures to address problems such as the removal of heteroatoms that are not part of the protein sequence, choosing one set of coordinates for residues having multiple occupancy and so on. This is internally handled by PSN-Ensemble. Furthermore, the NMR model can be entered in the PDB format and the individual models are extracted automatically by the program. For multiple crystal structures of the same protein, the input list is also filtered to bin structures of equal length. The bin with the largest number of entries is chosen for further analysis. On the other hand, MD snapshots are cleaned to remove hydrogen atoms. This cleaning process of the input structure files are performed for internal execution of the programs, and all the results obtained are mapped back onto the starting X-ray/NMR structure or the starting structure for MD simulation (the flexibility of obtaining results based on MD snapshots is also provided) (Supporting Information Fig. S2). The processed structure/s are subjected to further analysis.
Construction of protein sidechain network
Protein sidechain networks (PScN) are constructed by considering amino acid residues as nodes and edges are constructed between the nodes on the basis of noncovalent interactions between them (as evaluated from the normalized number of contacts between them) for each system. The details of the construction of such a graph at a particular interaction cut-off (Imin) and the implications of such graphs have been previously described.6,11 The noncovalent interaction between side chain atoms of amino acid residues (with the exception of Gly where Cα atom) are considered, ignoring the interaction between sequence neighbors (±2 immediate neighbors). The interaction between two residues i and j has been quantified previously in our laboratory as
where nij is number of distinct atom pairs between the side chains of amino acid residues i and j, which come within a distance of 4.5 Å and Ni and Nj are the normalization factors for residues i and j.6 The pair of amino acid residues having interaction strength (Iij) greater than a user-defined cut-off (Imin) are connected by edges to give a PScN for a given interaction strength Imin. Generally, Imins in the PScNs vary from 1% to 15%. We construct an adjacency matrix (a mathematical representation of the PScN) based on the noncovalent side-chain interactions at a given cut-off for Imin.
Imin is a measure of the extent of connectivity in the PScNs. A lower Imin is associated with higher connectivity and vice versa. Several previous reports from our group have shown that the optimal interaction strength in a protein structure is exhibited at an Imin at which the size of the largest noncovalently connected cluster (LClu) undergoes a transition.7 Earlier studies have pointed out that a transition in the size of the LClu is noted between an Imin of 2%–5%. Additionally, largest community [assemblage of cliques as discussed below] (LComm) profile as a function of different Imin values also indicated a transition in the Imin range of about 2%–4%. At lower Imin values (pretransition region), the network is too densely connected, whereas at higher Imin values (post-transition region) the network is very sparse, marking the two extremes of the Imin range. As a consequence, the transition regions in LClu and LComm profiles have been shown to highlight the meaningful connections in the network.7,11 In this article, investigations on UbcH5b and β2-adrenergic receptor are reported at Imin = 2% and 3%, respectively. The analysis on paths of communication in the TyrRS systems is done at Imin = 3%, while the analysis on interface clusters in the same system is done at an Imin of 4% and 6%.
Construction of PBN
PBN is constructed by considering interactions between the backbone-Cα atoms of the amino acid residues. An edge is constructed between residue pairs (with the exception of two immediate sequence neighbors) in PBN if their respective Cα atoms come within a distance of 6.5 Å. PBN is implemented only for the paths of communication and “junction residues” modules (Option: 6–7) in the program.
Weighing schemes for network and normalization of weights
The option of evaluating network parameters related to allosteric communication from weighted PScN/PBN is included in PSN-Ensemble. In this case, the user is prompted to input the residuewise cross-correlation or pairwise interaction energy matrices. The analysis of MD simulation trajectories to obtain cross-correlation or residue pairwise interaction energies (e.g., using MM-PBSA in AMBER,45,46 or related modules in GROMACS,47 CHARMM,48 and so on) is fairly straightforward. One can also obtain the residuewise cross-correlation values from elastic network models (ENM/GNM)49 or knowledge based pairwise interaction energies50 among residues to weigh the network. The cross-correlation/interaction energies between two residues is a measure of the ease of information transfer between them. We have used Dijkstra's algorithm for the shortest path computation (detailed in a later subsection) which is based on an efficient optimization of cost of communication. So, we normalize the edge weights in our package as cost by a simple inverse linear transformation such that the higher cross-correlation/interaction energy connection (Cij/Eij) is read as low cost (lower Wij) and vice versa (for details, see Ref. 10).
Network parameters
Various network parameters are characterized to unravel the topological features of PScN. The parameters which appear in greater than a user-defined fraction of the structure ensemble (default is 50%) are termed as dynamically stable. A brief definition of each parameter and its general physical significance follows (a detailed pictorial description is given in Fig. 1).
Hubs
Highly connected nodes are defined as hubs. In the context of protein structures, there is a limit to the possible number of noncovalent connections made by an amino acid due to steric constraints. Consequently, hubs are defined as residues/nodes connected by four or more edges to its neighbors. The hub residues are suggested as potential structural and/or functional “hot spots” in proteins.
Cluster
A connected set of amino acids in a graph is defined as a cluster. The size of the largest cluster (SLClu) varies as a function of the Imin and provides an estimate of the noncovalent connectivity in a network.7 Protein–protein interaction between the constituent subunits in a multimeric protein is captured by the interface cluster (IntClu). Of particular relevance is the investigation of interface clusters that are comprised of nodes from two different subunits, which give insights into protein–protein association. For identifying meaningful interface clusters, Imin values of 5%–6% have been shown to be appropriate in previous studies36,41 (lower Imin leads to SLClu ∼ equal to the length of the protein, thus merging the subtle interface specific clusters into one large cluster).
K-cliques
A k-clique is defined as a set of k nodes in which each node is connected to every other node. The algorithm used for the construction of cliques is proposed by Palla et al.51 The cliques indicate regions of rigidity in protein structures in terms of higher order connectivity. Again, the number and the size of the cliques in a protein structure are a function of Imin. However, cliques of size three or four are found most often even at a greater connectivity level (i.e., lower Imin).
Community
A community is an assemblage of smaller k-cliques that share node/s. In mathematical literature, a community is defined as set of k-cliques sharing k-1 nodes (k-1 community). However, for PScN, a variation of this definition is introduced by our laboratory in terms of k-2 communities, where a set of k-cliques share k-2 nodes. CFinder33 is used to obtain k-1 community. In house codes are used to obtain k-2 community using outputs from CFinder.33 Communities are percolating rigid units in PScN and indicate highly connected structural features. The cliques/communities are ideal parameters for examining rigidity/flexibility in protein structures. These parameters are also apt in capturing ligand induced conformational reorientations in terms of rigidity/flexibility/percolation.8,52
Paths and cost of communication
Dijkstra's algorithm is used to determine the shortest paths or OPs of communication between two residues in the PScN/PBN. The cost of path between pairs of residues of interest, i and j (termed termini), lacking direct noncovalent interaction is the sum of the edge weights (Wkl) between the consecutive interacting nodes (kl) constituting the path and is defined as (costij) = ΣWkl. The lower the cost, the higher is the efficiency of communication along a path. The SOPs are the alternate routes of communication with costs greater than those of the OPs. SOPs are identified in this study by systematically removing all interactions of OP node(s) in the network, thus forcing the traversal of a less than OP. The paths and cost of communication are ideal parameters to investigate allosteric crosstalk between distant pair of termini. The path cost in an unweighted graph is the sum of edges connecting the termini residues. Edge weights (Wkl) can be further introduced by various methods. Our program provides options to choose Wkl by following ways: using dynamic cross-correlation or average pairwise interaction energies obtained from MD simulations to weigh the edges constituting the paths of communication. In the later case, the path cost is the sum of edges weights.
“Junction residues” between domains
Herein, we define the term “junction nodes” as the residues that flux information between domains/modules. This is similar to the concept of node betweenness (NB) in network terminology, where the nodes with high NB are responsible for signal transmission within the network. The “junction residues” are important for capturing the key players/nodes that are critical for information transfer between domains/modules, much like the “sociometric superstars” in Milgram's experiment.53 These residues can be identified by constructing OPs/SOPs from every residue in the source domain to every residue in the sink domain from the PScN/PBN. The residues which appear in more than 10% of the communication paths in greater than a user-defined fraction of the structure ensemble are considered to be important for transmission of information between the chosen domain/modules in the protein. The average cost of information transfer between a pair of domains/modules (costflux), is the average cost of communication through all the constituent paths (OPs and SOPs) between the two domains. A lower value of costflux indicates higher efficacy of interdomain communication. Again the weighing schemes that are described in the previous section can be implemented in this module.
To summarize, network parameters such as hubs, clusters, cliques/communities (k-1/k-2), computed at a user-defined Imin and dynamic stability cutoff, capture the detailed topological features in proteins. On the other hand, allosteric communication in proteins can be probed by evaluating the paths/cost of communication between important residues or by identifying “junction residues” that are responsible for signal transfer across domains. The user is given an option of calculating these later parameters not only from a dynamic stability perspective but also applying weighing schemes as described previously on PScN/PBN.
System requirements, benchmarking, and application
The package PSN-Ensemble is freely available for Linux platforms. The system requirements are minimal. Basic packages like Perl, Matlab (MatlabBGL: http://www.cs.purdue.edu/homes/dgleich/packages/matlab_bgl/), and Pymol are the only software requirements, apart from CFinder,33 for running PSN-Ensemble. The application of PSN-Ensemble to probe biologically relevant problems is demonstrated using three examples. The key feature of the program, in terms of handling and examining various structural ensembles, is exemplified by using long MD trajectories of UbcH5b (PDB_id: 2ESK), an ubiquitin conjugating enzyme and an NMR ensemble (PDB_id: 1W4U) of the same.27,54 The strength of the package in providing important insights even from a single crystal structure is further manifested by using examples from one of the largest family of membrane proteins, the G-protein coupled receptors,35 and a tRNA binding protein, tyrosyl-tRNA synthetase from three different organisms. Herein, we have chosen the β2-adrenergic receptor (PDB_id: 3SN6/2RH1) in its active and inactive states for our analysis. The structure of the inactive form is determined in the presence of an inverse agonist (carazolol) and the active form is bound to Gs protein (a heterotrimer comprised of Gα and Gβγ subunits) and an agonist.28,29 In the active state, only the Gα subunit has been found to interact directly with the β2-AR.29 Consequently, for our analysis of the active state, we have considered only the β2-AR complexed with the Gα subunit (Chain A and Chain R in 3SN6). Also we have removed all the components that facilitate crystallogenesis (such as lysozyme, antibody, etc.) in both the structures for ease of comparison between the active and inactive states and visualization. For our analysis on the ternary complex of tyrosyl tRNA synthetases, the coordinates of the dimers are provided as input to PSN-Ensemble after removing tRNA from the structure.
The total runtime depends on the size of the protein, the number of structures in the ensemble, and the number of network parameters being computed. However, the package is optimal in terms of time and gives results within few a minutes to a few hours. It is worth mentioning that the construction of PscN/PBN is the rate determining step during the computation.
MD simulations protocols
Explicit solvent MD simulations (150 ns) are performed on apo UbcH5b (PDB_id: 2ESK) at 300 K using AMBER945 suit of programs with ff03 force field and parm99 parameters. The MD simulations are performed in aqueous medium using the TIP3P water model. The solvation box is 12 Å from the farthest atom along any axis. Na+ ions are added to neutralize the net charges on UbcH5b using the Leap module in AMBER9. The MD simulations are performed under NPT conditions using the Berendsen thermostat and periodic boundary conditions. Particle Mesh Ewald summation is used for long-range electrostatics and the van der Waals cut-off used is 10 Å. The pressure and temperature relaxations are set to 0.5 ps−1. SHAKE constraints are applied to all bonds involving H atoms. A time step of 2 fs is used with the integration algorithm, and the structures are stored every 1 ps. All the simulations are implemented and analyzed using a 264 core Intel Xeon HPC cluster.
Conclusion
PSN-Ensemble is a standalone, extensive, robust, time-optimized, and easy-to-use program, which is developed with the perception of using a generalized package for probing the network organization of structure ensembles from MD simulations, NMR, and multiple X-ray structures. The package can efficiently handle monomeric as well as multimeric proteins. At a simpler level, the program can be used to extract answers to biological questions of consequence even from single crystal structures. The multiple modules available in the package for computing various network parameters empowers users to make a judicious choice from an array of options based on the biological question being addressed. A flexible weighing scheme permits inclusion of residue pairwise cross-correlation or interaction energy edge weights into the topology based network, thereby facilitating introduction of dynamical/chemical knowledge into the communication related calculations, respectively. Additionally, PSN-Ensemble offers a convenient and user-friendly output format in terms of automatic representation of the evaluated network parameters on the three-dimensional structure, along with the raw data being stored in appropriately labeled folders. All the results are mapped back onto the original input structure for ease of comparison with complementary experimental and theoretical data. To summarize, the package is a unique tool that can cater to the understanding of protein structures in terms of their spatial three-dimensional organizations, including the intricate details of side-chain conformations, keeping in mind the biological questions that are widely relevant. The applicability of this package (which is freely available from http://vishgraph.mbu.iisc.ernet.in/PSN-Ensemble/psn_index.html) is demonstrated by investigating three examples pertaining to different biological problems.
Acknowledgments
S.V. and M.B. thanks CSIR for emeritus professorship and fellowship, respectively. The authors also thank Vasundhara Gadiyaram for the help provided in creating the webpage for PSN-Ensemble. The fellowship for CRB was supported by Microsoft-Research grant.
Supplementary material
Additional Supporting Information may be found in the online version of this article
References
- 1.Newman M. The structure and function of complex networks. SIAM Rev. 2003;45:167–256. [Google Scholar]
- 2.Newman M, Barabasi A-L, Watts DJ. The structure and dynamics of networks. Princeton University Press; 2006. [Google Scholar]
- 3.Vishveshwara S, Brinda KV, Kannan N. Protein structure: insights from graph theory. J Theor Comput Chem. 2002;1:187–211. [Google Scholar]
- 4.Atilgan AR, Atilgan C. Local motifs in proteins combine to generate global functional moves. Brief Funct Genomics. 2012;11:479–488. doi: 10.1093/bfgp/els027. [DOI] [PubMed] [Google Scholar]
- 5.Greene LH, Higman VA. Uncovering network systems within protein structures. J Mol Biol. 2003;334:781–791. doi: 10.1016/j.jmb.2003.08.061. [DOI] [PubMed] [Google Scholar]
- 6.Kannan N, Vishveshwara S. Identification of side-chain clusters in protein structures by a graph spectral method. J Mol Biol. 1999;292:441–464. doi: 10.1006/jmbi.1999.3058. [DOI] [PubMed] [Google Scholar]
- 7.Sukhwal A, Bhattacharyya M, Vishveshwara S. Network approach for capturing ligand-induced subtle global changes in protein structures. Acta Cryst D. 2011;67:429–439. doi: 10.1107/S0907444911007062. [DOI] [PubMed] [Google Scholar]
- 8.Bhattacharyya M, Ghosh A, Hansia P, Vishveshwara S. Allostery and conformational free energy changes in human tryptophanyl-tRNA synthetase from essential dynamics and structure networks. Proteins. 2009;78:506–517. doi: 10.1002/prot.22573. [DOI] [PubMed] [Google Scholar]
- 9.Ghosh A, Vishveshwara S. A study of communication pathways in methionyl-tRNA synthetase by molecular dynamics simulations and structure network analysis. Proc Natl Acad Sci USA. 2007;104:15711–15716. doi: 10.1073/pnas.0704459104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhattacharyya M, Vishveshwara S. Probing the allosteric mechanism in pyrrolysyl-tRNA synthetase using energy-weighted network formalism. Biochemistry. 2011;50:6225–6236. doi: 10.1021/bi200306u. [DOI] [PubMed] [Google Scholar]
- 11.Bhattacharyya M, Upadhyay R, Vishveshwara S. Interaction signatures stabilizing the NAD(P)-binding Rossmann fold: a structure network approach. PLoS One. 2012;7:e51676. doi: 10.1371/journal.pone.0051676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh A, Vishveshwara S. Variations in clique and community patterns in protein structures during allosteric communication: investigation of dynamically equilibrated structures of methionyl tRNA synthetase complexes. Biochemistry. 2008;47:11398–11407. doi: 10.1021/bi8007559. [DOI] [PubMed] [Google Scholar]
- 13.Sethi A, Eargle J, Black AA, Luthey-Schulten Z. Dynamical networks in tRNA: protein complexes. Proc Natl Acad Sci USA. 2009;106:6620–6625. doi: 10.1073/pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhattacharyya M, Vishveshwara S. Quantum clustering and network analysis of MD simulation trajectories to probe the conformational ensembles of protein-ligand interactions. Mol BioSyst. 2011;7:2320–2330. doi: 10.1039/c1mb05038a. [DOI] [PubMed] [Google Scholar]
- 15.Chatterjee S, Bhattacharyya M, Vishveshwara S. Network properties of protein-decoy structures. J Biomol Struct Dyn. 2012;29:1110–1126. doi: 10.1080/07391102.2011.672625. [DOI] [PubMed] [Google Scholar]
- 16.Soundararajan V, Raman R, Raguram S, Sasisekharan V, Sasisekharan R. Atomic interaction networks in the core of protein domains and their native folds. PLoS One. 2010;5:e9391. doi: 10.1371/journal.pone.0009391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor WR. Protein structure comparison using bipartite graph matching and its application to protein structure classification. Mol Cell Proteomics. 2002;1:334–339. doi: 10.1074/mcp.t200001-mcp200. [DOI] [PubMed] [Google Scholar]
- 18.Vijayabaskar MS, Vishveshwara S. Insights into the fold organization of TIM barrel from interaction energy based structure networks. PLoS Comput Biol. 2012;8:e1002505. doi: 10.1371/journal.pcbi.1002505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J, Nussinov R. Allosteric effects in the marginally stable von Hippel–Lindau tumor suppressor protein and allostery-based rescue mutant design. Proc Natl Acad Sci USA. 2008;105:901–906. doi: 10.1073/pnas.0707401105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nussinov R, Tsai C-J, Csermely P. Allo-network drugs: harnessing allostery in cellular networks. Trends Pharmacol Sci. 2011;32:686–693. doi: 10.1016/j.tips.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L, Martin B, Brenneman R, Luttrell LM, Maudsley S. Allosteric modulators of G protein-coupled receptors: future therapeutics for complex physiological disorders. J Pharmacol Exp Ther. 2009;331:340–348. doi: 10.1124/jpet.109.156380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Assenov Y, Ramírez F, Schelhorn S-E, Lengauer T, Albrecht M. Computing topological parameters of biological networks. Bioinformatics. 2008;24:282–284. doi: 10.1093/bioinformatics/btm554. [DOI] [PubMed] [Google Scholar]
- 23.Doncheva NT, Klein K, Domingues FS, Albrecht M. Analyzing and visualizing residue networks of protein structures. Trends Biochem Sci. 2011;36:179–182. doi: 10.1016/j.tibs.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 24.Eargle J, Luthey-Schulten Z. NetworkView: 3D display and analysis of protein·RNA interaction networks. Bioinformatics. 2012;28:3000–3001. doi: 10.1093/bioinformatics/bts546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pasi M, Tiberti M, Arrigoni A, Papaleo E. xPyder: a PyMOL plugin to analyze coupled residues and their networks in protein structures. J Chem Inf Model. 2012;52:1865–1874. doi: 10.1021/ci300213c. [DOI] [PubMed] [Google Scholar]
- 26.Vijayabaskar MS, Niranjan V, Vishveshwara S. GraProStr—graphs of protein structures: a tool for constructing the graphs and generating graph parameters for protein structures. Open Bioinform J. 2011;5:53–58. [Google Scholar]
- 27.Özkan E, Yu H, Deisenhofer J. Mechanistic insight into the allosteric activation of a ubiquitin-conjugating enzyme by RING-type ubiquitin ligases. Proc Natl Acad Sci USA. 2005;102:18890–18895. doi: 10.1073/pnas.0509418102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. Crystal structure of the [bgr]2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi H-J, Kuhn P, Weis WI, Kobilka BK, et al. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kobayashi T, Nureki O, Ishitani R, Yaremchuk A, Tukalo M, Cusack S, Sakamoto K, Yokoyama S. Structural basis for orthogonal tRNA specificities of tyrosyl-tRNA synthetases for genetic code expansion. Nat Struct Mol Biol. 2003;10:425–432. doi: 10.1038/nsb934. [DOI] [PubMed] [Google Scholar]
- 31.Tsunoda M, Kusakabe Y, Tanaka N, Ohno S, Nakamura M, Senda T, Sekine M, Yokogawa T, Nishikawa K, Nakamura KT. Three-dimensional structure of the ternary complex of yeast tyrosyl-tRNA synthetase. Nucleic Acids Symp Ser. 2004;48:155–156. doi: 10.1093/nass/48.1.155. [DOI] [PubMed] [Google Scholar]
- 32.Yaremchuk A, Kriklivyi I, Tukalo M, Cusack S. Class I tyrosyl-tRNA synthetase has a class II mode of cognate tRNA recognition. EMBO J. 2002;21:3829–3840. doi: 10.1093/emboj/cdf373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adamcsek B, Palla G, Farkas I, Derényi I, Vicsek T. CFinder: locating cliques and overlapping modules in biological networks. Bioinformatics. 2006;22:1021–1023. doi: 10.1093/bioinformatics/btl039. [DOI] [PubMed] [Google Scholar]
- 34.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 35.Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 36.Brinda KV, Mitra N, Surolia A, Vishveshwara S. Determinants of quaternary association in legume lectins. Protein Sci. 2004;13:1735–1749. doi: 10.1110/ps.04651004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schimmel P. Development of tRNA synthetases and connection to genetic code and disease. Protein Sci. 2008;17:1643–1652. doi: 10.1110/ps.037242.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brick P, Bhat TN, Blow DM. Structure of tyrosyl-tRNA synthetase refined at 2.3 Å resolution: interaction of the enzyme with the tyrosyl adenylate intermediate. J Mol Biol. 1989;208:83–98. doi: 10.1016/0022-2836(89)90090-9. [DOI] [PubMed] [Google Scholar]
- 39.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hansia P, Ghosh A, Vishveshwara S. Ligand dependent intra and inter subunit communication in human tryptophanyl tRNA synthetase as deduced from the dynamics of structure networks. Mol BioSyst. 2009;5:1860–1872. doi: 10.1039/b903807h. [DOI] [PubMed] [Google Scholar]
- 42.Atilgan AR, Turgut D, Atilgan C. Screened nonbonded interactions in native proteins manipulate optimal paths for robust residue communication. Biophys J. 2007;92:3052–3062. doi: 10.1529/biophysj.106.099440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dixit A, Verkhivker GM. Computational modeling of allosteric communication reveals organizing principles of mutation-induced signaling in ABL and EGFR kinases. PLoS Comput Biol. 2011;7:e1002179. doi: 10.1371/journal.pcbi.1002179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Daily MD, Gray JJ. Allosteric communication occurs via networks of tertiary and quaternary motions in proteins. PLoS Comput Biol. 2009;5:e1000293. doi: 10.1371/journal.pcbi.1000293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, Walker RC, Zhang W, Wang B, Hayik S, Roitberg A, Seabra G, Wong KF, Paesani F, Wu X, Brozell S, Tsui V, Gohlke V, Yang L, Tan C, Mongan J, Hornak V, Cui G, Beroza P, Mathews DH, Schafmeister C, Ross WH, Kollman PA, AMBER 9 . San Francisco: University of California; 2006. [Google Scholar]
- 46.Case D, Cheatham T, Darden T, Gohlke H, Luo R, Merz K, Jr, Onufriev A, Simmerling C, Wang B, Woods R. The Amber biomolecular simulation programs. J Comp Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 48.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 49.Bahar I, Lezon TR, Yang L-W, Eyal E. Global dynamics of proteins: bridging between structure and function. Annu Rev Biophys. 2010;39:23–42. doi: 10.1146/annurev.biophys.093008.131258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sippl MJ. Knowledge-based potentials for proteins. Curr Opin Struct Biol. 1995;5:229–235. doi: 10.1016/0959-440x(95)80081-6. [DOI] [PubMed] [Google Scholar]
- 51.Palla G, Derenyi I, Farkas I, Vicsek T. Uncovering the overlapping community structure of complex networks in nature and society. Nature. 2005;435:814–818. doi: 10.1038/nature03607. [DOI] [PubMed] [Google Scholar]
- 52.Deb D, Vishveshwara S, Vishveshwara S. Understanding protein structure from a percolation perspective. Biophys J. 2009;97:1787–1794. doi: 10.1016/j.bpj.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Milgram S. The small world problem. Psychol Today. 1967;2:60–67. [Google Scholar]
- 54.Houben K, Dominguez C, van Schaik FMA, Timmers HTM, Bonvin AMJJ, Boelens R. Solution structure of the ubiquitin-conjugating enzyme UbcH5B. J Mol Biol. 2004;344:513–526. doi: 10.1016/j.jmb.2004.09.054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.