
Figure 2.
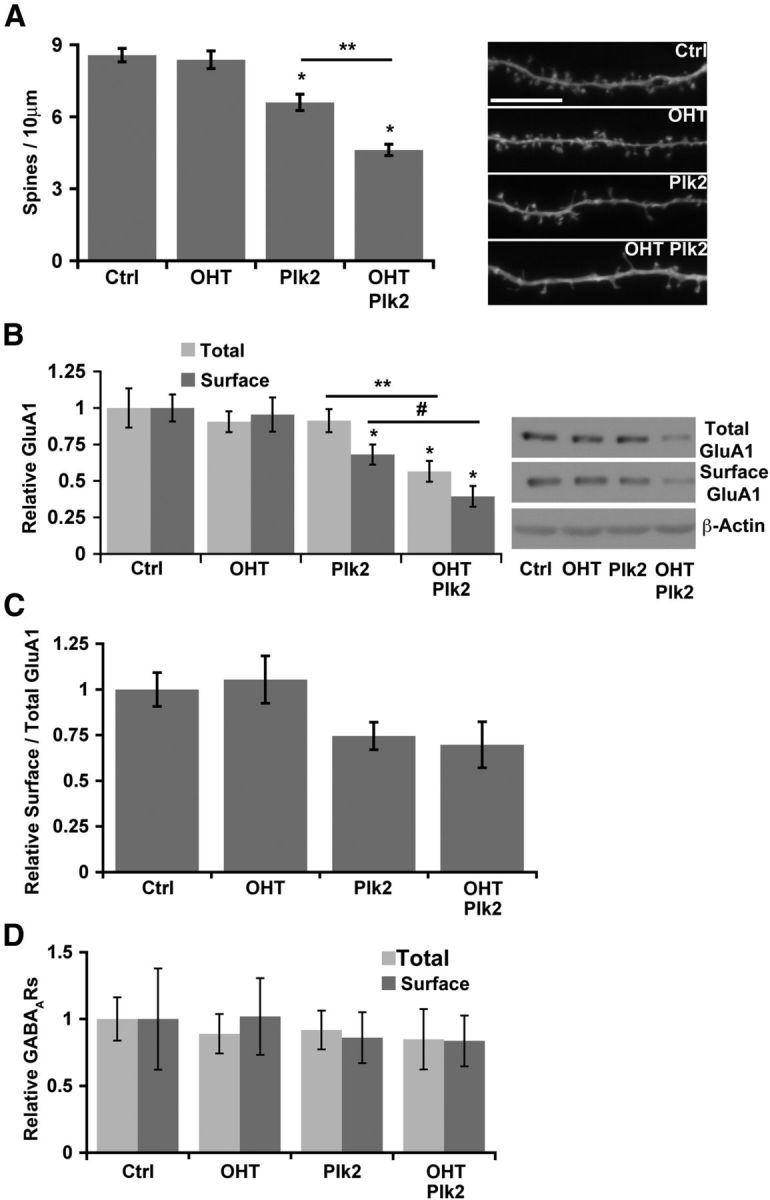
NF-κB and Plk interaction determines neuronal spine density and excitatory neurotransmitter receptor composition. A, Neuronal NF-κB opposes and limits Plk2-induced spine loss. Average spine densities (left) quantitated from confocal projections of dendrites (representative segments; right) from DIV21 RelAF/F murine cultured hippocampal neurons transduced with CreERT2 by lentivirus infection and treated with vehicle or OHT to induce p65-deficiency. Plk2 (either wild-type or kinase-active with data pooled) was expressed for 24 h (*p ≤ 1.2 × 10−5 relative to Ctrl; **p = 1.83 × 10−6, ANOVA; n ≥ 44 dendritic segments from ≥10 independent experiments). Scale bar, 10 μm. B, NF-κB limits Plk2-induced loss of surface GluA1 AMPA receptor subunits in hippocampal neurons. Averages of normalized total and surface GluA1 quantitated by densitometry (left) from immunoblots (representative blots; right) after surface biotinylation from DIV21 neurons in the presence (Ctrl) or absence of p65 (OHT) are shown, and wild-type Plk2 was transduced by lentiviral infection. β-Actin was used as the loading control (*p ≤ 0.012 relative to Ctrl; **p = 0.004; #p = 0.009, ANOVA; n ≥ 7 independent experiments). C, Surface to total GluA1 ratio does not significantly change with Plk2 overexpression in wild-type or p65 deficient neurons (data from same experiments as in B). D, NF-κB and Plk2 do not regulate inhibitory GABAA receptors in neurons (biotinylation assay as is Fig. 1B). Densitometric quantification of average surface and total GABAARs. All error bars indicate SEM.