

Abstract
Fibroblast growth factor-inducible 14 (Fn14) is a member of the tumour necrosis factor (TNF) receptor family that is induced in a variety of cell types in situations of tissue injury. Fn14 becomes activated by TNF-like weak inducer of apoptosis (TWEAK), a typical member of the TNF ligand family. TWEAK is constitutively expressed by monocytes and some tumour cell lines and also shows cytokine inducible expression in various other cell types. Fn14 activation results in stimulation of signalling pathways culminating in the activation of NFκB transcription factors and various MAPKs but might also trigger the PI3K/Akt pathway and GTPases of the Rho family. In accordance with its tissue damage-associated expression pattern and its pleiotropic proinflammatory signalling capabilities, the TWEAK-Fn14 system has been implicated in a huge number of pathologies. The use of TWEAK- and Fn14-knockout mice identified the TWEAK-Fn14 system as a crucial player in muscle atrophy, cerebral ischaemia, kidney injury, atherosclerosis and infarction as well as in various autoimmune scenarios including experimental autoimmune encephalitis, rheumatoid arthritis and inflammatory bowel disease. Moreover, there is increasing preclinical evidence that Fn14 targeting is a useful option in tumour therapy. Based on a discussion of the signalling capabilities of TWEAK and Fn14, this review is focused on two major issues. On the one hand, on the molecular and cellular basis of the TWEAK/Fn14-related pathological outcomes in the aforementioned diseases and on the other hand, on the preclinical experience that have been made so far with TWEAK and Fn14 targeting drugs.
Keywords: antibodies, autoimmune disease, cancer, fibrosis, Fn14, ischaemia, nuclear factor of kappaB, TWEAK, tumour necrosis factor ligand and receptor family
Introduction
Tumour necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK) is a typical member of the TNF ligand family and is as such initially expressed as type II class transmembrane glycoprotein from which a soluble ligand form can be released by proteolytic processing (Bodmer et al., 2002). The 249 amino acid-comprising membrane-bound form of TWEAK consists extracellularly of the characteristic C-terminally located TNF homology domain (THD), which is separated from the transmembrane domain by a stalk region of about 55 amino acids (Figure 1A). As in other ligands of the TNF family, the THD of TWEAK mediates assembly into homotrimeric proteins and receptor binding while the stalk region contains the recognition site for proteases involved in ligand processing, in case of TWEAK, serine proteases of the furin family. The short intracellular N-terminal domain of TWEAK is highly conserved like the rest of the molecule but its functional relevance is still unclear. Although TWEAK expression has been documented at the mRNA level in a variety of cell types and cell lines, cell surface expression has so far only be reported for monocytes, dendritic cells, interferon-γ stimulated NK cells (Nakayama et al., 2000; Felli et al., 2005; Maecker et al., 2005), multiple sclerosis patient-derived monocytes (Desplat-Jego et al., 2009), a few colonic adenocarcinoma (Kawakita et al., 2005) and hepatocellular carcinoma (Kawakita et al., 2004) cell lines and the MDA-MB-231 breast cancer cell line (Willis et al., 2008). Induction of TWEAK expression has frequently reported in scenarios of tissue damage and hypoxia. In line with this, the hypoxia regulated forkhead transcription factor FOXO3a and interferon-γ have been implicated in TWEAK up-regulation (Baxter et al., 2006). Otherwise the transcriptional regulation of TWEAK has been poorly investigated.
Figure 1.
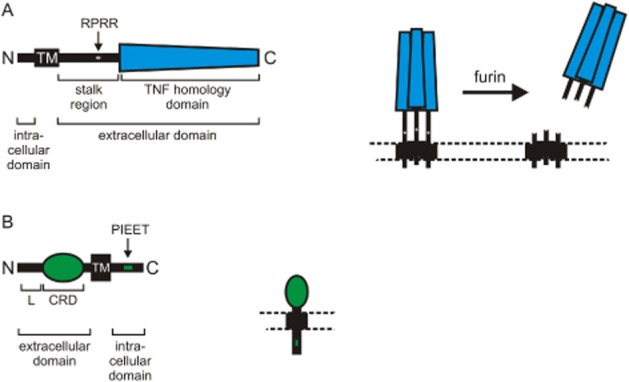
Domain architecture of TWEAK (A) and Fn14 (B). The amino acid sequences of the furin recognition site of TWEAK and the TRAF-binding motif of Fn14 are indicated in the single-letter code.
With a molecular weight of about 14 kDa, fibroblast growth factor inducible (Fn14) is an unusual small member of the TNF receptor family (Wiley et al., 2001). Fn14 and B cell maturation, BaffR and the short isoform of transmembrane activator and calcium-modulator and cytophilin ligand interactor are the only TNF receptors which only contain a single cystein-rich domain in their ectodomain while all other TNF receptors have two to six copies of this characteristic motif (Figure 1B, Locksley et al., 2001). Moreover, the cytoplasmic domain of Fn14 only comprises 28 amino acid residues. TNF receptors can be categorized into three classes: (i) death receptors that contain a conserved protein-protein interaction domain, called the death domain, in their cytoplasmic part and which are often able to trigger apoptosis, (ii) TNF receptor associated factor (TRAF)-interacting receptors that harbour one or more binding motifs for adapter proteins of the TRAF family in their cytoplasmic part and (iii) decoy receptors without own signalling capacity that occur either as soluble or membrane-attached proteins. Fn14 is only equipped with a single TRAF binding motif and is thus a typical TRAF-interacting TNF receptor. Fn14 is a single spanning transmembrane type I receptor that has been initially identified as a transcriptional target of fibroblast growth factor-1 in NIH 3T3 fibroblasts (Meighan-Mantha et al., 1999). Only later, it has been recognized as a TNF receptor family member able to transduce TWEAK signals (Wiley et al., 2001). Fn14 is found to been highly expressed on most tumour cell lines of non-lymphoid origin. Immunohistochemical analysis of murine tissue samples and human biopsy specimens, however, revealed a more differentiated expression pattern of Fn14 in vivo. Low expression of Fn14 has been reported for fibroblasts and endothelial and epithelial cells of healthy homeostatic tissue. In contrast, high Fn14 expression was regularly found on epithelial and mesenchymal progenitor cells and especially in contexts of tissue damage caused by different insults including hypoxia, oxidative stress, chemical and mechanical injuries, inflammation and tumour growth (Winkles, 2008; Burkly et al., 2011). It is therefore not only surprising that induction of Fn14 expression has been reported for a variety of immune diseases, including hepatitis, glomerulonephritis, rheumatic arthritis, multiple sclerosis and bowels disease but also in atherosclerosis and after stroke and heart attack. Furthermore, up-regulation of Fn14 has been found in melanoma, glioma and non-small cell lung, breast, liver and prostate cancer (Feng et al., 2000; Tran et al., 2006; Sanz et al., 2012; Whitsett et al., 2012; Chao et al., 2013; Zhou et al., 2013). In accordance with the tissue damage-associated expression pattern of Fn14, several growth factors, including EGF and VEGF, as well as cytokines, such as IFN-γ, TNF and TGF-β, that are released by immune cells associated with tissue repair processes, have found to be able to induce Fn14 expression (Donohue et al., 2003; Hosokawa et al., 2006; Ebihara et al., 2009; Sanz et al., 2012; Whitsett et al., 2012).
Besides Fn14, the TNF receptor family member DR3 and CD163, a cysteine rich haemoglobin scavenger receptor expressed on monocytes and macrophages, have been claimed to be TWEAK receptors (Marsters et al., 1998; Bover et al., 2007). However, both the TWEAK-DR3 interaction as well as the TWEAK-CD163 interaction failed to be confirmed in other studies (Schneider et al., 1999; Kaptein et al., 2000; Fick et al., 2012).
TWEAK/Fn14-stimulated signalling pathways and resulting cellular responses
In the initial study, describing its cloning and preliminary characterization, TWEAK got its name due to the observation that it induces cell death in IFN-γ sensitized HT29 cells but not in a panel of other cell lines (Chicheportiche et al., 1997). Although it has been found in recent years that TWEAK also efficiently induces cell death in a few other cell lines (e.g. SKOV-3, OVCAR-4, Kym-1), this quality of cellular response remains still the rare exception for the TWEAK-Fn14 system. Other ligands of the TNF family triggering robust cell death responses, such as TNF, TNF-related apoptosis inducing ligand (TRAIL) and CD95L, do this by stimulation of receptors belonging to the above mentioned death receptor subgroup of the TNF receptor family. The characteristic death domain of death receptors either serves directly as the hub for the formation of a complex catalyzing the activation of caspase-8 or triggers the formation of a cytoplasmic caspase-8 activating protein complex (Dickens et al., 2012). Activated caspase-8 then further propagates the apoptotic signal. Death receptors also interact with the death domain-containing serine/threonine kinase RIP1 which allows them to trigger, under defined circumstances, an alternative necrotic form of programmed cell death (Dickens et al., 2012). Now, Fn14 belongs to the TRAF-interacting subgroup of TNF receptors raising the obvious question how it accomplishes cell death induction without having a death domain. At least for the most sensitive cell lines Kym-1, SKOV-3 and OVCAR-4, the counterintuitive apoptotic signalling capabilities of the TWEAK-Fn14 system could be traced back to an indirect mechanism, namely the induction of TNF and subsequent stimulation of the prototypic death receptor TNFR1 (Schneider et al., 1999; Vince et al., 2008; Figure 2). In experiments where TNFR1 stimulation has been uncoupled from the activity of TWEAK-induced TNF by the use of TNF-specific antibodies to neutralize TWEAK-induced TNF and agonistic TNFR1-specific antibodies to trigger TNFR1, TWEAK treatment enabled per se non-toxic concentrations of the latter to efficiently induce cell death (Schneider et al., 1999). Thus, TWEAK-induced apoptosis not only relies on the induction of TNF but also involves a second per se non-toxic sensitizing mechanism. TNF-induced cell death is also strongly enhanced by TWEAK and Fn14 in per se TWEAK resistant cell lines suggesting that this sensitizing mechanism reflects a common feature of Fn14 activation (Wicovsky et al., 2009). Apoptosis induction by the TNFR1-related TRAIL death receptors is not or only poorly enhanced by TWEAK treatment further indicating that Fn14 signalling affects an apoptosis-relevant issue of special relevance in TNFR1 signalling (Wicovsky et al., 2009). In fact, enhancement of TNFR1-mediated apoptosis by TWEAK has been ascribed to the capability of activated Fn14 to recruit TRAF2-cIAP1/2 complexes. TRAF2 is a typical member of the aforementioned TRAF adapter protein family and possesses a really interesting new gene (RING) domain possibly with E3 ligase activity (Gonzalvez et al., 2012). Cellular inhibitor of apoptosis-1 (cIAP1) and cIAP2 are also RING domain E3 ligases which possess besides the RING domain also a caspase recruitment domain and three baculovirus inhibitor of apoptosis (IAP) repeat domains involved in substrate recognition. Now, TRAF2-cIAP1/2 complexes are efficiently recruited to TNFR1 and are also part of the secondarily formed TNFR1-induced cytosolic caspase-8 activating complex but do not (or only poorly) interact with the caspase-8 activating plasma membrane-associated TRAIL death receptor signalling complex. As the expression levels of TRAF2-cIAP1/2 complexes are rather low compared to Fn14 expression in most cells, TWEAK-induced recruitment of TRAF2-cIAP1/2 complexes to Fn14 results in depletion of the cytosolic pool of these molecules and restricts so their availability for TNFR1 (Vince et al., 2008; Wicovsky et al., 2009). As a consequence, TNFR1 activation leads to enhanced apoptosis induction provided caspase-8 activation is not inhibited by other independently acting anti-apoptotic proteins, for example FLIP or Bcl2. It is worth mentioning that other TRAF2 interacting TNF receptors can substitute for Fn14 in the TNF-Fn14 crosstalk. Indeed, TRAF2 depletion-mediated enhancement of TNFR1-induced cell death has originally been described for TNFR2 and has also been demonstrated for CD30 and CD40 (Duckett and Thompson, 1997; Weiss et al., 1997; 1998; Grell et al., 1999; Fotin-Mleczek et al., 2002). Moreover, SMAC mimetics, a novel group of anticancer drugs currently under investigation in clinical trials trigger proteasomal degradation of cIAP1 and cIAP2 (for review see Fulda and Vucic, 2012) and modulate TNFR1 signalling in a similar fashion to TWEAK and Fn14. SMAC mimetics, however, not only trigger cIAP1/2 degradation but also inhibit the anti-apoptotic xIAP protein (Fulda and Vucic, 2012) and thus let expect a broader apoptosis sensitizing activity than TWEAK. Last but not least, cell death induction by the non-death receptor TNFR2 and SMAC mimetics has also been traced back to the cooperative activity of endogenous TNF and depletion of cIAP1/2 and TRAF2 (Grell et al., 1999; Fulda and Vucic, 2012). Thus, the TRAF2-cIAP1/2 based mechanisms underlying the Fn14-TNFR1 crosstalk are of general nature and are also of relevance for other TRAF2 interacting TNF receptors and SMAC mimetics.
Figure 2.
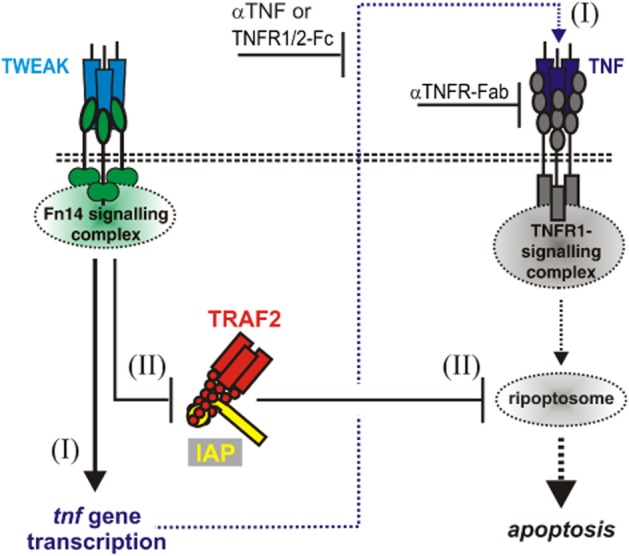
Mechanisms of TWEAK/Fn14-induced apoptosis. Binding of soluble or membrane TWEAK to Fn14 results in the recruitment of TRAF2-cIAP1/2 complexes and their depletion from the cytosol. This triggers two events that in a concerted action stimulate cell death in apoptosis-proficient cells: NFκB-mediated induction of TNF (I) and sensitization for TNFR1-induced ripoptosome activity (II). Thus, TWEAK-induced apoptosis eventually rely on activation of the death receptor TNFR1 and is thus inhibitable by TNF and TNFR1 blocking antibodies.
A few studies also describe TNF-independent modes of TWEAK/Fn14-induced cell death in cortical neurons and normal human keratinocytes. The underlying signalling mechanisms, however, have been poorly analysed so far (Haile et al., 2010a; Sabour Alaoui et al., 2012).
Depletion of the cytoplasmic TRAF2-cIAP1/2 pool and enhancement of TNFR1-induced caspase-8 activation are not the only consequences of TWEAK-induced binding of the TRAF2-cIAP1/2 complex to Fn14. TRAF2, cIAP1 and cIAP2 are also crucially involved in the regulation of non-apoptotic signalling pathways triggered by pattern recognition receptors and members of the TNF receptor family. Consequently, these molecules are also of central relevance in TWEAK/Fn14-induced activation of various MAPK family members and transcription factors of the NFκB family (Vince et al., 2008). Fn14-mediated activation of NFκBs is comparably well understood and account for many of the proinflammatory effects that have been described for the TWEAK-Fn14 system (Winkles, 2008; Burkly et al., 2011). The NFκBs comprise homo- and heterodimers formed by the five members of the Rel transcription factor family (Hayden and Ghosh, 2012). NFκBs substantially differ in the way they become activated and also in their target genes. There are two prototypic signalling pathways that lead to activation of distinct NFκB dimers, the classical and the alternative NFκB pathway (Figure 3, (Hayden and Ghosh, 2012). TWEAK and Fn14 are principally able to stimulate both pathways. Central step in the alternative NFκB pathway is the inhibitor of kappaB kinase (IKK)1-mediated phosphorylation of the p100 precursor form of the p52 NFκB subunit. This results in K48-ubiquitination and proteasomal processing of p100 to p52 and nuclear translocation of p52-containing NFκB dimers (Hayden and Ghosh, 2012). Now, IKK1 activation requires phosphorylation by NIK, a cytosolic MAP3-kinase that is constitutively degraded in non-stimulated cells by a TRAF2-cIAP1/2 complex-dependent mechanism. TRAF2 recruits NIK by interaction with the NIK-binding protein TRAF3 and subjected it so to K48-ubiquitination by the TRAF2-associated IAPs and subsequent proteasomal degradation (Hayden and Ghosh, 2012). Thus, in context of the alternative NFκB pathway cytosolic TRAF2-cIAP1/2 complexes fulfil an inhibitory function that is antagonized by their TWEAK-induced recruitment to membrane-bound Fn14. In contrast, activation of the classical NFκB pathway by TWEAK and various other ligands of the TNF family requires stimulatory activities of TRAF2 and the cIAPs (Hayden and Ghosh, 2012). NFκB dimers activated via the classical NFκB pathway are retained in the cytoplasm of unstimulated cells by forming a ternary complex with inhibitory proteins, the IκBs. Central step in the classical NFκB pathway is the activation of the IKK complex, which contains the scaffolding protein NEMO, IKK2 and IKK1 whereby the latter appears largely dispensable for the function of the IKK complex in classical NFκB signalling. The IKK complex phosphorylates IκB proteins triggering their proteasomal degradation and nuclear translocation of the released NFκB dimers. The TRAF2-cIAP1/2 complex plays a dual role in activation of the classical NFκB pathway. While TRAF2 acts as an adapter protein that recruits the IKK complex to activated TNF receptors, the cIAPs K63-ubiquitinate the IKK subunit NEMO, and in context of TNFR1 signalling also RIP, creating docking sites for a variety of ubiquitin binding proteins. The latter are involved in IKK activation, for example, the IKK2 phosphorylating TAB2-TAB3-TAK1 complex or the linear ubiquitin chain assembly complex (LUBAC), which creates additional docking sites for ubiquitin binding proteins, but also comprises antagonistic factors such as A20 and Cyld, two deubiquitinating enzymes. The relevance of the IKK2, TRAF2, TAK1 and the cIAPs for Fn14-mediated activation of the classical NFκB pathway has been recently demonstrated by help of knockout mice-derived fibroblasts, siRNA experiments, dominant-negative ubiquitin binding domains and IAP antagonists (Saitoh et al., 2003; Kumar et al., 2009; Sims et al., 2012; Varfolomeev et al., 2012). The role of most of the ubiquitin binding proteins interacting with ubiquitinated NEMO or TRAF2, however, has so far only been studied in detail in context of TNFR1 signalling.
Figure 3.
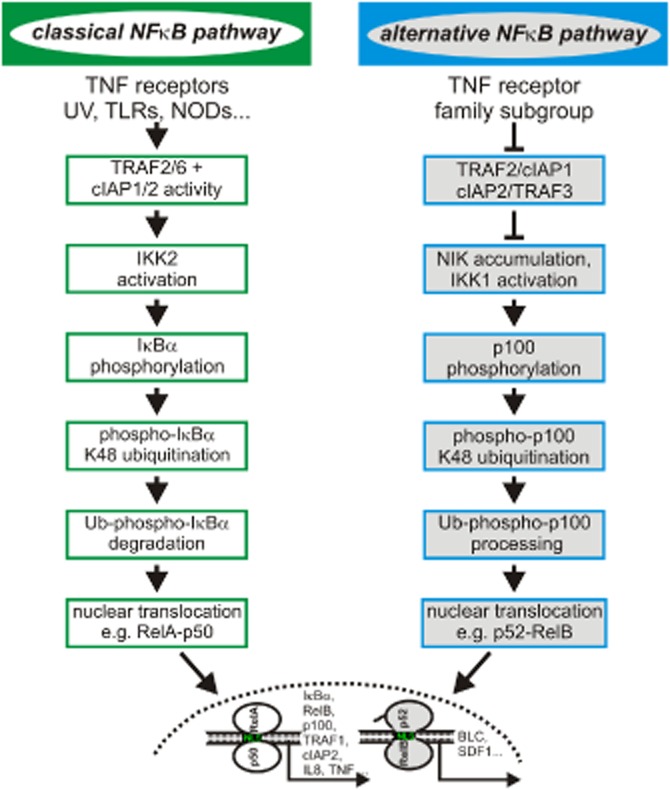
Activation of NFκB transcription factors. Two signalling pathways control nuclear translocation of the dimeric transcription factors of the NFκB family: the classical and alternative NFκB pathway. Noteworthy, the proteins TRAF2, cIAP1 and cIAP2 act as positive mediators in the classical pathway but as inhibitors in the alternative pathway. Please be aware that there can be significant crosstalk between the two pathways in a cell type-specific manner by various mechanisms, including NIK-mediated activation of the IKK complex, inhibition of p65-p50 dimers by p100 and transcriptional upregulation of p100 and RelB via the classical pathway.
Notably, soluble TWEAK only triggers weak and delayed activation of the classical NFκB pathway despite concomitant strong stimulation on the alternative NFκB pathway while membrane TWEAK expressing cells trigger both pathways efficiently. Likewise, artificial immobilization on a cell surface transforms soluble TWEAK from a weak to a strong activator of classical NFκB signalling while having no major effect on the efficacy of activation of the alternative NFκB pathway (Roos et al., 2010). Thus, the enhanced efficacy with which membrane bound TWEAK stimulates classical NFκB signalling seems to be primarily related to the spatially restricted way of how membrane TWEAK is presented to Fn14 and does not reflect a need for sequence information that is only available in membrane TWEAK. Robust activation of the classical NFκB pathway can also be achieved with dimerized or oligomerized trimers of soluble TWEAK. This has, however, no major effect on the dose dependency of TWEAK-induced activation of the alternative NFκB pathway or on Fn14 occupation by TWEAK (Fick et al., 2012). It is therefore tempting to speculate that the secondary interaction of two or more initially formed TWEAK-Fn14-TRAF2-cIAP1/2 complexes is required for activation of the classical NFκB pathway while the sole recruitment of the TRAF2-cIAP1/2 complex is already fully sufficient for triggering of alternative NFκB signalling. This hypothesis is also in good accordance with the stoichiometry of the TRAF2-cIAP1/2 complex and the mechanisms of activation of the E3 ligase activity of cIAP1 and cIAP2. On the one hand TRAF2 forms homotrimeric molecules that can interact with three Fn14 molecules but only bind a single molecule of cIAP1 or cIAP2 (Zheng et al., 2010). On the other hand, there is evidence that activation of the E3 ligase activity of cIAPs requires trans-activation of two molecules (Dueber et al., 2011; Feltham et al., 2011). Binding of three molecules of Fn14 by soluble TWEAK trimers is therefore sufficient to recruit TRAF2-cIAP1/2 complexes and to keep them from triggering degradation of NIK in the cytosol while Fn14-mediated activation of the classical NFκB pathway needs additionally secondary interaction of two TWEAK-Fn14-TRAF2-cIAP1/2 complexes to ensure trans-activation of two cIAP molecules. Thus, the parallel orientation and the limited mobility of membrane TWEAK in the plasma membrane seem to facilitate by yet poorly understood mechanisms the secondary interaction of TWEAK induced Fn14 signaling complexes and this secondary interaction is required to fully unleash all Fn14 signaling activities (Figure 4).
Figure 4.
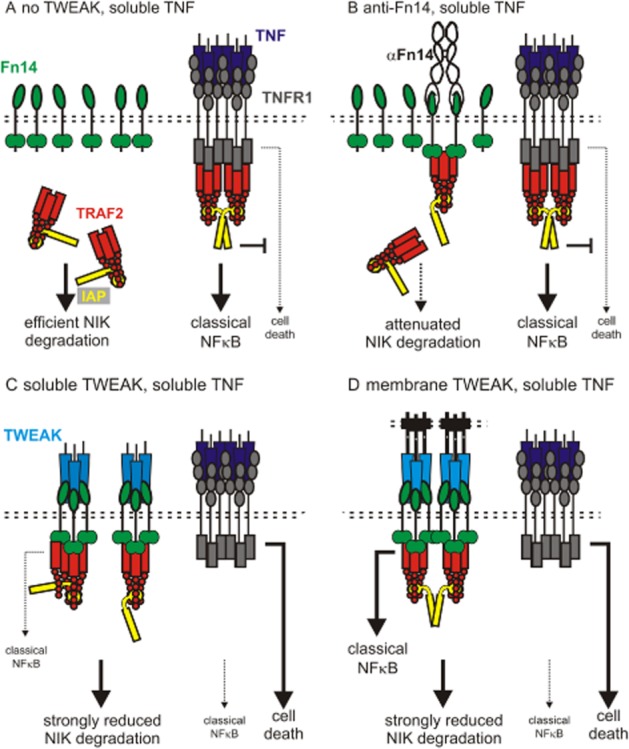
Activation of Fn14-associated signalling pathways. (A) In non-stimulated cells TRAF2-cIAP1/2 are localized in the cytoplasm and trigger NIK degradation. TNFR1 signalling is not affected and dominated by classical NFκB activation. (B) Fn14 antibodies induce Fn14 dimers which poorly recruit TRAF2-cIAP1/2 complexes. This results in weak depletion of the cytosolic TRAF2-cIAP1/2 pool, which can be amplified by an enzymatic cascade leading to p100 processing but is insufficient to relief the ripoptosome from its inhibition by TRAF2-cIAP1/2 complexes. (C) Binding of soluble TWEAK trimers to Fn14 results in recruitment and depletion of cytosolic TRAF2-cIAP1/2 but not in the transactivation of the single inhibitor of apoptosis (IAP) molecules associated with a TRAF2 trimer. Soluble TWEAK thus sensitizes for ripoptosome-dependent cell death signalling and triggers the alternative NFκB pathway, responses otherwise suppressed by cytosolic TRAF2-cIAP1/2. (D) Binding of membrane TWEAK (or hexameric/oligomeric soluble TWEAK or immobilized Fn14 antibodies) to Fn14 not only result in recruitment and depletion of cytosolic TRAF2-cIAP1/2 but also in the transactivation of TRAF2-associated IAP molecules. This triggers the signalling events mentioned under ‘C’ and in addition in signalling pathways dependent on cIAP activity, such as the classical NFκB pathway.
Activation of the MAPKs ERK, JNK and p38 by TWEAK and Fn14 has also been repeatedly reported and appears to involve, at least in case of JNK and p38 signalling, again TRAF2, cIAPs and TAK1 (Kumar et al., 2009; Varfolomeev et al., 2012). Regulation of the PI3K/Akt pathway by TWEAK has also been described. On the one hand, an inhibitory effect of TWEAK/Fn14 signalling on Akt activity has been shown in C2C12 myotubes (Dogra et al., 2007a,b) and hepatocarcinoma cell lines (Feng et al., 2008); on the other hand, activation of Akt by TWEAK has been reported in renal tubular cells (Sanz et al., 2009), gingival fibroblasts (Hosokawa et al., 2012), glioma cell lines (Fortin et al., 2009) and osteoblastic cells (Ando et al., 2006). The molecular basis of the opposing effects of TWEAK on the PI3K/Akt pathway is currently not understood. Rather unusual for members of the TNF receptor/ligand family, there are reports showing TWEAK/Fn14-induced activation of the Rho GTPases Cdc42 and Rac1 (Tran et al., 2006; Fortin et al., 2012) and stimulation of the JAK2 kinase (Ucero et al., 2013). TWEAK-induced activation of JAK2 in the latter study took place very rapidly within a couple of minutes and in so far clearly differs from another recent report demonstrating a delayed indirect activation of the JAK/STAT pathway by TWEAK-induced upregulation of interferon-β (Chapman et al., 2013). Ligand-induced recruitment to Fn14 has been shown for Rac1 as well as for Cdc42 but epistatic siRNA experiments placed Cdc42 upstream of Rac1. To fulfil their function in Fn14 signal transduction, the two GTPases depend on the support of two guanine nucleotide exchange factors, Ect2 that stimulates Cdc42 and Tio, which activates Rac1 (Fortin et al., 2012). TWEAK-induced activation of Cdc42/Ect2 and Rac1/Tio results in glioma cells in enhanced cellular migration and lamellipodia formation.
TWEAK and Fn14 in autoimmune diseases
TWEAK closely resembles TNF in so far that it also stimulates in a variety of cell types proinflammatory pathways. In view of the overwhelming importance of TNF in autoimmune diseases, several groups addressed therefore in animal models a possible similar role of the TWEAK-Fn14 system. Analysis of sera of mice suffering on collagen-induced arthritis and human specimens revealed increased TWEAK levels (Perper et al., 2006; Park et al., 2008). In collagen-induced arthritis (CIA), neutralizing TWEAK antibodies reduced disease score and histological observable defects but failed to interfere with the disease causing immune response to collagen (Kamata et al., 2006; Perper et al., 2006). In accordance with the well-established proangiogenic function of the TWEAK-Fn14 system, TWEAK blockade further inhibited angiogenesis in the synovium of CIA mice (Kamata et al., 2006; Perper et al., 2006). A disease-driving role of TWEAK and Fn14 in rheumatoid arthritis (RA) is also in line with in vitro data showing that TWEAK induces expression of cytokines and adhesion molecules in synovial fibroblasts (Kamijo et al., 2008; Yamana et al., 2012). It is noteworthy that one of the studies reporting TWEAK-induced production of proinflammatory cytokines showed furthermore that TWEAK concomitantly inhibits the much stronger proinflammatory effect of TNF on RA synovial fibroblasts (Yamana et al., 2012). The latter may reflect the antagonizing effect of soluble TWEAK-induced depletion of TRAF2-cIAP1/2 complexes on TNFR1 signalling discussed above and may require attention in the development of TWEAK neutralizing treatment regimes for RA (Yamana et al., 2012).
A pivotal role of the TWEAK-Fn14 system has also been observed in models of inflammatory bowel diseases (IBD). Both TWEAK and Fn14 deficiency protect from trinitrobenzene sulfonic acid-induced colitis and interleukin (IL)10 deficiency-triggered spontaneous colitis (Dohi et al., 2009; Kawashima et al., 2011). These results are in accordance with complementary experiments demonstrating a crucial role of the TWEAK-Fn14 system in immune stimulation and cell death induction in intestinal epithelial cells (IECs) by γ-irradiation and IL13 (Dohi et al., 2009; Kawashima et al., 2011). Cell death induction in IECs represents an early key event in IBD pathogenesis and leads to breakdown of the mucosal epithelial barrier, subsequent activation of immune cells by luminal bacteria-derived pathogen associated molecular patterns and propagation and persistence of inflammatory processes. Now, γ-irradiation induces cell death in IECs and this is inhibited in TWEAK and Fn14-knockout mice (Dohi et al., 2009). In accordance with data showing a pivotal function of IL13 in γ-irradiation-induced IEC cell death, it has furthermore observed that IL13 induces IEC death in primary jejunum cultures by a TWEAK- and Fn14-dependent mechanism (Kawashima et al., 2011). Noteworthy, the cytotoxic effect of IL13 on IECs appears not only to be dependent on TWEAK but also on TNF. IL13-induced IEC apoptosis was blocked by a soluble TNF receptor fusion protein (Table 1) and apoptosis induction in IECs by TNF was reduced in Fn14-knockout cells. Indeed, inhibition of TNF shedding in jejunum tissue reduced IL13-induced caspase activation. Moreover, in the IL10-knockout model of spontaneous colitis, where IL13 plays a major role, TWEAK and Fn14 deficiency not only inhibit colitis pathogenesis but also up-regulation of TNF. It is tempting to speculate that the concerted action of TNF and TWEAK observed in IL13-induced IEC cell death mirrors the in vitro data discussed before showing sensitization for TNFR1-induced cell death by Fn14-mediated depletion of protective TRAF2-cIAP1/2 complexes.
Table 1.
Therapeutic effects of Fn14 or TWEAK targeting proteins in preclinical models
| Reagent | Model | Effect | Reference |
|---|---|---|---|
| Hamster anti-TWEAK | CIA | 50% reduction in cumulative disease score Reduced synovial angiogenesis, | (Perper et al., 2006) |
| Rat anti-TWEAK | CIA | 50% reduction in cumulative disease score Reduced synovial angiogenesis, | (Kamata et al., 2006) |
| anti-TWEAK | TNBS colitis | ∼40% reduction in mean clinical score | (Dohi et al., 2009) |
| Fn14-TRAIL | EAE | ∼50% reduction in mean clinical score, superior to side by side treatment with Fn14-Fc and TRAIL | (Razmara et al., 2009) |
| anti-TWEAK | EAE | ∼40–80% reduction in mean clinical score | (Desplat-Jego et al., 2005) |
| TWEAK vaccination | EAE | ∼20–60% reduction in mean clinical score | (Mueller et al., 2005) |
| anti-TWEAK | MACO | ∼20% reduction in mean infarct size | (Potrovita et al., 2004) |
| Fn14-Fc | MACO | ∼33% reduction in mean infarct size | (Yepes et al., 2005) |
| Fn14-Fc | MACO | ∼70% inhibition of MACO-induced BBB leakiness | (Polavarapu et al., 2005) |
| Fn14-Fc | MACO | ∼75% inhibition of MACO-induced BBB leakiness ∼50% reduction in mean infarct size | (Zhang et al., 2007) |
| TWEAK | MACO | ∼33% reduction in MACO-induced mean infarct size by TWEAK pretreatment | (Echeverry et al., 2012) |
| anti-TWEAK | Denervation induced muscle atrophy | Less reduction in muscle fibre cross section area | (Mittal et al., 2010a) |
| anti-TWEAK | atherosclerosis | Reduced renal damage (∼50%) Reduced RANTES and MCP1 expression in atherosclerotic plaques and kidney (>50%) | (Munoz-Garcia et al., 2009) |
| Fn14-Fc | atherosclerosis | Attenuated plaque progression Reduced collagen deposition in advance plaques | (Schapira et al., 2009) |
| anti-TWEAK | NTN | >80% reduction in proteinuria Reduced expression of MCP-1, RANTES, IP-10 | (Xia et al., 2012) |
| anti-TWEAK | cGvHD | >50% reduction in proteinuria Reduced expression of MCP-1, RANTES, IP-10 | (Zhao et al., 2007) |
| anti-Fn14 | IRI1 | Reduced inflammation >70% reduction in tubular damage score | (Hotta et al., 2011) |
| anti-TWEAK | Folic acid-induced nephropathy | Reduced inflammation and T-cell infiltration ∼30% reduction in tubular damage score ∼50% reduced serum creatinine levels | (Sanz et al., 2008; 2010) |
IRI, ischaemia reperfusion injury.
Based on in vitro data showing induction of proinflammatory genes in astrocytes and microglia (Saas et al., 2000; Desplat-Jego et al., 2002), early on a role of the TWEAK-Fn14 system has been addressed in animal models of demyelinating disorders. Both, in the microglia-driven cuprizone model and the CD4+ T-cell driven myelin oligodendrocyte glycoprotein (MOG)-induced experimental autoimmune encephalomyelitis (EAE) model, increased expression of TWEAK and Fn14 has been reported (Desplat-Jego et al., 2002; 2005; Iocca et al., 2008). More important, disease severity in these models were reduced by various means including TWEAK deficiency, vaccination with recombinant TWEAK or Fn14 and TWEAK-specific antibodies but were enhanced in TWEAK transgenic animals. Similar to the role of the TWEAK-Fn14 system in RA, TWEAK blockade failed in the EAE model to inhibit the disease-inducing anti-MOG T-cell response and rather affected production of proinflammatory proteins involved in immune cell recruitment and control of the permeability of the blood brain barrier (BBB; (Desplat-Jego et al., 2005; Razmara et al., 2009). In accordance with an involvement of TWEAK and Fn14 in multiple sclerosis, increased expression of these molecules has recently also been reported for brain tissue derived from diseased patients and TWEAK-induced leakiness has been observed in an in vitro model of the BBB using human cerebral microvascular endothelial cells (Serafini et al., 2008; Stephan et al., 2013).
Morbidity and mortality of patients suffering on systemic lupus erythematous (SLE) often result from the secondary development of kidney disease (lupus nephritis). The TWEAK-Fn14 system has been implicated in several aspects of lupus nephritis that eventually base again on its capability to trigger concerted cellular responses culminating in inflammation, angiogenesis, proliferation, fibrosis and cell death. Higher levels of urinary TWEAK are found in SLE patients suffering on lupus nephritis than in SLE patients without renal involvement (Schwartz et al., 2006; 2009; El-Shehaby et al., 2011). TWEAK expression was also higher in peripheral blood mononuclear cells derived of patients with lupus nephritis compared to patients without renal involvement and, moreover, there is also enhanced TWEAK-induced production of IL-10 and MCP-1 in in vitro cultures of lupus nephritis patients (Zhi-Chun et al., 2012). In accordance with this clinical evidence, animal models, where autoantibody-mediated glomerulonephritis was triggered by MHCII mismatched spleen cell-induced chronic graft versus host disease or by treatment with rabbit antisera raised against glomerular basement membrane components (nephrotoxic serum transfer nephritis), revealed reduced disease activity in Fn14-knockout mice (Zhao et al., 2007; Xia et al., 2012). Furthermore, TWEAK neutralizing antibodies reduce renal inflammation and diminish proteinuria in both models without affecting the formation of autoantibodies (Zhao et al., 2007; Xia et al., 2012).
TWEAK and Fn14 in acute kidney injury
The TWEAK-Fn14 system also critical determines various aspects of acute kidney injury. According to its pleiotropic mode of action, practically each cell type of relevance in kidney injury is in some way regulated by the activities of TWEAK and Fn14. This reaches from the up-regulation of proinflammatory gene expression programmes in glomerular mesangial cells, tubular cells and podocytes, over induction of proliferation in tubular and mesangial cells to cell death induction in TNF/IFNγ exposed tubular epithelial cells (Campbell et al., 2006; Justo et al., 2006; Sanz et al., 2008; 2009; Gao et al., 2009). In the model of folic acid overdose-induced acute kidney injury inflammation, histological signs of tissue injury and renal failure are reduced in TWEAK-deficient mice or upon treatment with anti-TWEAK (Sanz et al., 2008; 2009). The proinflammatory effects of TWEAK in this model have been traced back to activation of the classical and alternative NFκB pathway whereby the latter is of special relevance for the induction of CCL19 and CCL21 and recruitment of T-cells (Sanz et al., 2010). In the model of unilateral nephrectomy, where compensatory kidney growth occurs independently of inflammation, there was significant less tubular cell proliferation in TWEAK-deficient mice (Sanz et al., 2009). The role of TWEAK and Fn14 has also been addressed in ischaemia reperfusion injury. Here, antagonistic Fn14-specific antibodies not only blocked the up-regulation of proinflammatory cytokines and chemokines and recruitment of inflammatory cells but also reduced apoptosis induction in tubular cell (Hotta et al., 2011). Especially, this translated into reduced chronic fibrosis and prolonged survival.
The TWEAK-Fn14 system in cerebral ischemia
In line with their tissue repair/remodelling-associated expression pattern and their proangiogenic activity, TWEAK and Fn14 have been found to be up-regulated after cerebral ischemia in the middle cerebral artery occlusion (MACO) model (Potrovita et al., 2004; Yepes et al., 2005; Haile et al., 2010a) and Fn14 expression is increased in infarcted and peri-infarct tissue of stroke patients (Inta et al., 2008). Moreover, several lines of evidence suggest that the TWEAK-Fn14 system plays a role in the development of tissue damage secondarily arising from cerebral ischaemia. Fn14-knockout mice displayed significantly reduced infarct size and less degradation of the basement membrane after MACO (Zhang et al., 2007) and various reagents interfering with TWEAK-Fn14 interaction elicited corresponding therapeutic effects in the MACO model (Table 1). At the cellular level various TWEAK/Fn14-mediated effects may contribute to cerebral ischaemia. Firstly, TWEAK may induce cell death in neurons of the infarct area because oxygen-glucose deprivation experiments, which mimic some aspects of ischemia in vitro, revealed TWEAK/Fn14-mediated cell death in stressed cortical neurons (Potrovita et al., 2004; Haile et al., 2010a). Secondly, TWEAK increases the permeability of the BBB in vitro and in vivo by at least two mechanisms, on the one hand by classical NFκB-mediated induction of the basement degrading MMP9 protease (Polavarapu et al., 2005) and on the other hand by stimulating transcellular transport of neutrophils through the interface of astrocytes and endothelial cells (Haile et al., 2010a). Thirdly, TWEAK-induced activation of microglia and astrocytes results in the production of proinflammatory cytokines and chemokines (Haile et al., 2010b) including TGFα, which can act as an autocrine inducer of astrocyte proliferation via EGFR stimulation (Rousselet et al., 2012). For example, immunohistochemical analysis of the necrotic core of MACO-induced ischaemia revealed a strong increase of activated microglia and this was found to been reduced in animals with intracerebroventricular injection of Fn14-Fc (Yepes et al., 2005). TWEAK and Fn14 appear not only to contribute to the devastating effects induced by ischaemic stroke but also surprisingly elicit a neuroprotective activity. Sublethal exposure to hypoxia protects cortical neurons against a subsequent lethal hypoxic challenge. The protective effect of such a preconditioning treatment has found to be significantly reduced in Fn14- and TWEAK-deficient neurons and is furthermore mimicked by treatment with recombinant TWEAK instead of sublethal hypoxia (Echeverry et al., 2012). Indeed, intraperitoneal injection of recombinant TWEAK 1 day before induction of cerebral ischaemia by MACO resulted in significant reduction in the volume of the infarcted tissue (Echeverry et al., 2012). It is worth mentioning that the protective effect of TWEAK preconditioning on cortical neurons was found to be dependent on TNF induction (Echeverry et al., 2012) while the cytotoxic effect of TWEAK produced in course of oxygen-glucose deprivation was TNF independent (Haile et al., 2010a).
The TWEAK-Fn14 system regulates various aspects of muscle biology
Early on it has been recognized that TWEAK stimulates proliferation of mesenchymal progenitor cells. This aspect of TWEAK-Fn14 biology has been most intensively studied in context of myotube formation from satellite cells during regeneration of skeletal muscle. However, TWEAK and Fn14 have also been implicated in muscle atrophy. Myoblasts express Fn14 and respond in vitro to TWEAK dependent on the culture conditions. In growth medium, where myoblasts proliferate and do no undergo differentiation in myotubes, TWEAK enhances proliferation of the mononuclear myoblasts (Girgenrath et al., 2006). In contrast, in differentiation medium TWEAK treatment of murine myoblasts and human mesoangioblasts results in reduced myotube formation and cell death (Dogra et al., 2006; Girgenrath et al., 2006; Morosetti et al., 2012). From studies with the myoblastic C2C12 cell line, there is further evidence that TWEAK-mediated inhibition of myoblast differentiation is due to NFκB-mediated inhibition of expression of MyoD, a transcription factor crucially involved in myogenic differentiation (Girgenrath et al., 2006). There is further evidence that also endogenous TWEAK contributed to myoblast formation. Lower numbers of myoblasts are generated in cultures derived from Fn14-deficient mice although these cells are still capable to differentiate to myotubes (Girgenrath et al., 2006) and inhibition of TWEAK leads to improved myotube formation in human mesoangioblast cultures (Morosetti et al., 2012). However, there is evidence that the role of TWEAK and Fn14 in myotube formation is more complex. Myotubes derived from Fn14-deficient mice are thicker than their wild type counterparts (Girgenrath et al., 2006) and myotubes derived from C2C12 cells in the presence of TWEAK showed a reduced diameter (Dogra et al., 2007a). Moreover, a recent report even demonstrated that TWEAK induces myotube formation by activation of the alternative NFκB pathway (Enwere et al., 2012). The disparate effects of TWEAK on myoblast differentiation in the various studies cited remain to be solved. However, it is tempting to speculate that the different responsiveness of Fn14 to different agonists together with differences in the dose response of how various cellular signalling pathways are linked to Fn14 activation is here decisive. Indeed, in the study of Enwere et al. (2012), low concentrations of trimeric TWEAK, that presumably only activated the alternative NFκB pathway, have been used while in the other studies much higher concentrations or hexameric Fc-TWEAK have been used that activate both NFκB pathways (see above). In line with this idea, recent studies give evidence for an inhibitory role of the classical NFκB pathway on myoblast differentiation and a mitochondrial biogenesis-stimulating function of the alternative NFκB pathway, which involves induction of PPARγ co-activator 1β and support of myotube maintenance (Bakkar et al., 2008; 2012).
The complex effects of TWEAK and Fn14 on myoblasts differentiation observed in vitro might contribute to the crucial role of the TWEAK-Fn14 system in muscle regeneration that has been deduced from the cardiotoxin-induced model of muscle injury. Injection of cardiotoxin in the tibialis anterior muscle results in muscle fibre damage and subsequent robust muscle regeneration by activation of quiescent satellite cells, muscle precursor cells of skeletal muscles. While expression of TWEAK and Fn14 is low in healthy skeletal muscles, cardiotoxin injured muscles display high Fn14 expression and induction of TWEAK whereby the latter seems to be primarily expressed from infiltrating macrophages (Girgenrath et al., 2006; Mittal et al., 2010b). Fn14 deficient mice showed significantly delayed muscle regeneration in response to cardiotoxin-induced muscle injury and this delayed regenerative response came along with reduced infiltration of immune cells and lowered cell proliferation in the affected muscle area (Girgenrath et al., 2006). However, as on the one side TWEAK not only stimulates myoblast proliferation but also production of chemotactic cytokines and as on the other side the inflammatory microenvironment contributes to muscle regeneration, it is not unequivocally possible to specify the contribution of direct TWEAK-induced myoblast proliferation to the regenerative response. The situation becomes even more complex by findings showing essentially the opposite, namely improved muscle regeneration in the cardiotoxin model in TWEAK-deficient mice (Mittal et al., 2010b). Moreover, in various models of skeletal muscle atrophy (Table 2), TWEAK and Fn14 drive the latter by induction of the E3 ubiquitin ligases MuRF1 and MAFbx, which mark myosin heavy chain for degradation by the autophagosome and the proteasome.
Table 2.
Muscle atrophy in genetic models of TWEAK and Fn14
| TWEAK/Fn14 model | Atrophy inducer | Effect | Ref. |
|---|---|---|---|
| TWEAK knockout | Denervation | Reduced loss in muscle mass, less reduction in muscle fibre cross section area, reduced fibrosis | (Mittal et al., 2010a) |
| TWEAK tg | Denervation | Enhanced loss in muscle mass, enhanced reduction in muscle fibre cross section area, more fibrosis | (Mittal et al., 2010a) |
| TWEAK tg | – | Reduced muscle mass, reduced muscle fibre cross section area | (Dogra et al., 2007a) |
| TWEAK tg | Cardiotoxin | Enhanced fibrosis | (Mittal et al., 2010b) |
| TWEAK knockout | Starvation | Less reduction in muscle fibre cross section area | (Paul et al., 2012) |
TWEAK/Fn14 and heart-related diseases
In view of its multiple functions in immune regulation and tissue remodelling/repair, it is no surprise that the TWEAK-Fn14 system plays also a role in the development of atherosclerosis and some of its most severe possible consequences myocardial infarction and renal damage. TWEAK is expressed on macrophages recruited to atherosclerotic plaques and renal lesions but also by associated smooth muscle cells (SMCs) (Munoz-Garcia et al., 2006; Schapira et al., 2009). In the vascular and renal lesions TWEAK induces production of NFκB-regulated chemokines, such as RANTES and MCP1 (Munoz-Garcia et al., 2009). Furthermore, TWEAK triggers production of thrombosis promoting factors like tissue factor and plasminogen inhibitor-1 in human aortic SMCs in vitro and in the aortic root of ApoE-deficient mice in vivo. Moreover, induction of chemokines, tissue factor and plasminogen inhibitor-1 is reduced when endogenous TWEAK is blocked in ApoE-deficient mice by help of TWEAK neutralizing antibodies or soluble Fn14-Fc (Munoz-Garcia et al., 2009). Especially, Atorvastin, a statin broadly used for the prophylaxis against myocardial infarction and stroke, has been found to inhibit cytokine-induced up-regulation of Fn14 in aortic SMCs by interfering with RhoA and ROCKI activity (Munoz-Garcia et al., 2006). RhoA/ROCKI-mediated up-regulation of Fn14 has also been observed in cultured cardiomyocytes in response to ventricular hypertrophy-stimulating triggers such as stretch-stress, angiotensin II and norepinephrine (Chorianopoulos et al., 2010a). In fact, mice overexpressing soluble TWEAK suffer on dilated cardiomyopathy and die from cardiac dysfunction and Fn14-deficiency completely abrogated this pathology (Jain et al., 2009). Last but not least, like in many other tissue injury-related pathologies, TWEAK and Fn14 are also of importance for fibrosis in the heart. TWEAK stimulates proliferation and NFκB-mediated collagen synthesis in rat neonatal cardiac fibroblasts in vitro (Chen et al., 2012). Moreover, in an in vivo model of pulmonary arterial hypertension-induced right ventricular failure Fn14-deficiency reduces collagen expression and myofibroblast differentiation (Novoyatleva et al., 2013). Clinical data from patients with acute ST-elevation myocardial infarction or idiopathic dilated cardiomyopathy revealed increased soluble TWEAK levels while patients with stable disease after non-ischaemic heart failure showed reduced levels of soluble TWEAK (Table 3). Noteworthy, in the latter case soluble TWEAK represents an independent adverse prognostic factor (Richter et al., 2010).
Table 3.
TWEAK in human patients
| Compared samples | TWEAK levels | Remarks | Ref. |
|---|---|---|---|
| SLE with and without LN | 16.3 versus 5.5 pg·mg−1 creatinine | Urinary TWEAK uTWEAK correlated with renal disease activity index | (Schwartz et al., 2006) |
| SLE with and without LN SLE with LN and healthy volunteers | 12.2 versus 6.6 pg·mg−1 creatinine 12.2 versus 5.7 pg·mg−1 creatinine | Urinary TWEAK uTWEAK correlated with renal disease activity index | (El-Shehaby et al., 2011) |
| Peripheral artery disease | 134 versus 147 pg·mL−1 | 161 ± 35 versus 100 ± 6 | (Moreno et al., 2010) |
| Acute ST-elevation myocardial infarction versus healthy or stable coronary artery disease | 524 pg·mL−1 (healthy) 377 pg·mL−1 (CAD) 1393 pg·mL−1 (STEMI) | TWEAK increased in patients with acute myocardial infarction Association with adverse short-term outcome | (Chorianopoulos et al., 2010b) |
| Stable heart failure | 217 versus 325 pg·mL−1 | sTWEAK reduced in stable heart failure patients TWEAK level below 227 pg·mL−1 correlate with significantly higher 4-year mortality rate | (Chorianopoulos et al., 2009) |
| Non-ischaemic versus ischaemic heart failure | 332 versus 362 pg·mL−1 | Levels not significantly different between non-ischaemic and ischaemic HF Inverse correlation with mortality in non-ischaemic HF patients | (Richter et al., 2010) |
| Orthotopic heart transplant recipient versus healthy | 229 versus 418 pg·mL−1 | Significant reduced sTWEAK levels | (Przybylowski et al., 2011) |
| Chronic kidney disease (CKD) patients | 450 versus 161 (stage 3) to 380 pg·mL−1 (stage 1) | sTWEAK levels significantly lower in CKD patients | (Hassan et al., 2012) |
TWEAK and Fn14 in tumour development and metastasis
Tumourigenesis is often tightly linked to chronic inflammatory processes and associated with repetitive tissue damage and the corresponding repair processes. The tumour microenvironment therefore typically contains many factors implicated in upregulation of Fn14 expression. Furthermore, tumour infiltrating immune cells represent a bona fide source of TWEAK and expression of the latter might also be induced in activated resident cells of the tumour microenvironment or in the transformed cells itself. Indeed, Fn14 expression is frequently found to be strongly enhanced in tumour tissue compared to non-transformed tissue in specimens of non-small cell lung carcinoma, breast cancer, hepatocellular cancer, pancreatic cancer, malignant melanoma as well as glioblastoma multiforme and is also regularly detectable on tumour cell lines (Feng et al., 2000; Tran et al., 2003; 2006; Willis et al., 2008; Dai et al., 2009; Culp et al., 2010; Kwon et al., 2012; Zhou et al., 2013). TWEAK has been detected at the mRNA level in various tumour entities and tumour cell lines and is also found to be up-regulated in tumour tissue by immunohistochemistry but cell surface expression of membrane TWEAK has only be reported for a very few hepatocellular carcinoma and breast cancer cell lines, so far (Kawakita et al., 2004). With exception of the proapoptotic TWEAK-TNF crosstalk, which appears to be only relevant in rare cases, the huge majority of cellular effects elicited by Fn14 and TWEAK are obviously of potential advantage for tumour development. Indeed, TWEAK/Fn14-induced cell migration and invasion have been demonstrated for glioblastoma cells, non-small cell lung cancer, ovarian cancer cells, breast cancer cell lines and androgen-independent prostate cancer cells (Tran et al., 2003; Willis et al., 2008; Dai et al., 2009; Huang et al., 2011). Moreover, Fn14 overexpressing A549 adenoma carcinoma cells display enhanced lung metastasis upon tail vein injection in mice (Whitsett et al., 2012) and TWEAK-expressing matrigel encapsulated Hek293 cells showed enhanced angiogenesis in nude mice (Ho et al., 2004). There is also clinical evidence for protumoural functions of TWEAK and Fn14. High Fn14 expression correlates with clinical indicators of poor prognosis in breast cancer patients (Willis et al., 2008). Likewise, Fn14 expression has been found to be increased in glioma specimens and again correlate with poor prognosis (Tran et al., 2006). High Fn14 level late stage patients suffering on gastric cancer have also a reduced survival rate (Kwon et al., 2012) and strong Fn14 expression is also associated with lower prostate specific antigen-free survival in prostate cancer patients (Huang et al., 2011). It is worth mentioning that high levels of TWEAK and Fn14 might not ever been connected with poor patient outcome. In colorectal cancer, high TWEAK expression correlates with increased overall and disease-free survival and this correspond to an inhibitory effect of TWEAK on the invasiveness of colon cancer cell lines in vitro (Lin et al., 2012).
In view of its broad and high tumour-associated expression and its multiple protumoural functions, Fn14 is an attractive anti-tumour target on two counts. First, blockade of Fn14 or TWEAK might deprive tumour cells from beneficial Fn14-mediated activities and secondly, Fn14 might serve as a tumour-associated target for delivering of antibodies or antibody drug conjugates, irrespective of its tumour-related functions. In line with the latter, recent reports demonstrated antitumor effects of Fn14 antibodies that based on the activation of antibody effector functions, such as antibody-dependent cellular cytotoxicity (ADCC), or on the activity of antibody conjugated drugs (Culp et al., 2010; Michaelson et al., 2011; Zhou et al., 2011; 2013). In some cases, the apoptotic TWEAK-TNF crosstalk might also be exploited for tumour therapy by triggering tumour cell death using recombinant TWEAK or agonistic Fn14-antibodies. In view of the poor serum half-life of recombinant TNF ligands and the additional Fc effector functions delivered by agonistic antibodies, the latter represents in this respect presumably the more practicable approach (Culp et al., 2010; Michaelson et al., 2011).
TWEAK and Fn14 targeting drugs
Dependent on the concrete pathology considered, blockade and stimulation of Fn14 could be a therapeutic option (Figure 5). Due to their extracellular accessibility, TWEAK and Fn14 are straight forwardly druggable with high selectivity by help of recombinant soluble variants of these molecules or antibodies. Indeed, in a variety of the disease models discussed above, it has been already demonstrated that pronounced therapeutic effects are achievable with soluble Fn14 and TWEAK- and Fn14-specific antibodies (Table 1). It is thus no surprise that TWEAK- and Fn14-specific antibodies are currently under investigation in phase I studies in patients suffering on RA, lupus or solid tumours (http://clinicaltrials.gov/; NCT00771329, NCT01499355 and NCT00738764).
Figure 5.
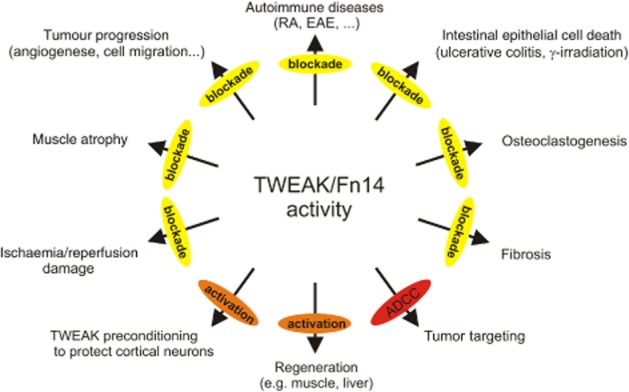
Targeting of TWEAK/Fn14-related pathologies.
Recombinant proteins and antibodies targeting Fn14 and TWEAK elicit in vivo effects by complex mechanisms going beyond simple blockade or activation of Fn14 and could therefore vary with the type of agonists or antagonist used. The most important TWEAK and Fn14 targeting drug formats and their molecular mode of action are:
Anti-TWEAK antibodies – TWEAK-specific antibodies that block binding to Fn14 have been described (Table 1). Blocking TWEAK antibodies may predominately act as inhibitors of the TWEAK-Fn14 system but as TWEAK is also expressed as a membrane-bound molecule in some cell types, Fc domain-mediated effects, for example, ADCC or complement activation, cannot be completely ruled out.
Fn14-Fc – A fusion protein of the ectodomain of Fn14 with the Fc domain of human IgG1 has been successfully used in various preclinical studies to block TWEAK-mediated effects (Table 1). As in case of blocking TWEAK antibodies, effector function emanating from the Fc domain must be taken into consideration in vivo for membrane TWEAK expressing cells.
Anti-Fn14 antibodies – Fn14-specific antibodies can elicit quite different effects dependent on their isotype, their idiotype and the availability of Fcγ-receptor expressing cells. For example, P4A8 and PDL192, two Fn14-specific IgG1 antibodies under investigation in clinical trials (http://clinicaltrials.gov/), strongly differ in their capability to block TWEAK-Fn14 interaction but act both as potent Fn14 agonists upon binding to Fcγ receptors or oligomerization by protein G (Salzmann et al., 2013). Noteworthy, without oligomerization or FcγR-binding, both antibodies significantly trigger p100 processing at moderate concentrations without showing a relevant effect on other Fn14-mediated cellular effects. In case of the Fn14-blocking P4A8 antibody, this has the strange consequence that it act as agonist for Fn14-mediated p100 processing but concomitantly as an antagonist for other Fn14-mediated responses induced by soluble or membrane TWEAK. Thus, as long not bound to Fcγ receptors Fn14-specific IgG1 antibodies trigger a state of Fn14 activity that neither reflects the activity of soluble TWEAK nor that of membrane TWEAK (Salzmann et al., 2013). Although the molecular mechanisms that constitute the different Fn14 signalling capabilities in response to IgG1 antibodies and TWEAK are yet not fully elucidated, there is evidence for a crucial role of two parameters, the stoichiometry of the antibody/ligand interaction with Fn14 and the differential relevance of amplification mechanisms in distinct Fn14-associated signalling pathways.
Soluble TWEAK – Soluble recombinant trimeric TWEAK mimicking the natural occurring soluble TWEAK generated by furin processing of membrane TWEAK is straight forwardly available by expression of corresponding DNA fragments in Escherichia coli or eukaryotic cells. Two major issues must be considered in concepts using soluble TWEAK. First, as discussed before, soluble TWEAK only triggers robust activation of a subset of the Fn14-associated effects that can be induced by membrane TWEAK. Secondly, like other soluble TNF ligands, soluble TWEAK has a very short half-life in vivo. However, this limitation can be overcome using fusion proteins of soluble TWEAK with proteins having long half-life in vivo, such as serum albumin (Muller et al., 2010).
Fc-TWEAK – Linking soluble TWEAK to the C-terminal end of the Fc domain of human IgG1 results in a fusion protein (Fc-TWEAK) that assembles into hexameric molecules. Fc-TWEAK efficiently activates the full set of Fn14-associated signalling pathways and in so far functionally mimics membrane TWEAK rather than soluble TWEAK (Roos et al., 2010). Especially, one has to consider in vivo that this protein might trigger Fc domain-dependent immune cell responses upon binding to Fn14-expressing cells. As different amino acids of the Fc-domain are involved in dimerization and FcγR binding, it is, however, possible to generate hexameric Fc-TWEAK variants with a mutated Fc domain showing strongly reduced FcγR binding but unchanged Fn14 stimulatory activity.
Conclusions
Inhibition of TWEAK/Fn14 activities has a broad potential for the therapy of tissue damage-associated pathologies and cancer. Indeed, therapeutic concepts that base on the exogenous inhibition of TWEAK or Fn14 haven already been successfully tested in preclinical models and are currently in clinical trials. The latter must now clarify whether the hopes resulting from the impressive preclinical data on TWEAK/Fn14 inhibition become real. Although, the TWEAK-Fn14 system has so far primarily recognized as a ‘pathological’ factor, it is obvious that its direct cellular effects, such as induction of angiogenic and proinflammatory proteins or stimulation of proliferation might contribute to the control of tissue homeostasis and wound healing in the healthy organism. It is thus tempting to speculate that the disease-associated activities of TWEAK and Fn14 reflect the conversion of initially beneficial functions in the repair of acute tissue damage into an exaggerated and chronic pathological repair response. In view of the potential dichotomous role of the TWEAK-Fn14 system, it appears possible that treatment modalities (e.g. timing of treatment) considerably influence the success of TWEAK/Fn14-targeting therapeutic concepts. Moreover, besides inhibition of the TWEAK-Fn14 system, its activation, at least during a certain stage of a disease, might become a therapeutic option and should be addressed in future preclinical studies.
Acknowledgments
This work was supported by Deutsche Forschungsgemeinschaft (DFG Wa 1025/24-1) and Deutsche Krebshilfe (project number 109922)
Glossary
- CIA
collagen-induced arthritis
- EAE
experimental autoimmune encephalitis
- Fn14
fibroblast growth factor inducible
- IBD
inflammatory bowel diseases
- IECs
intestinal epithelial cells
- MACO
middle cerebral artery occlusion
- RA
rheumatoid arthritis
- SLE
systemic lupus erythematous
- SMCs
smooth muscle cells
- TWEAK
tumour necrosis factor (TNF)-like weak inducer of apoptosis
Conflict of interest
No conflict of interest.
References
- Ando T, Ichikawa J, Wako M, Hatsushika K, Watanabe Y, Sakuma M, et al. TWEAK/Fn14 interaction regulates RANTES production, BMP-2-induced differentiation, and RANKL expression in mouse osteoblastic MC3T3-E1 cells. Arthritis Res Ther. 2006;8:R146. doi: 10.1186/ar2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkar N, Wang J, Ladner KJ, Wang H, Dahlman JM, Carathers M, et al. IKK/NF-kappaB regulates skeletal myogenesis via a signaling switch to inhibit differentiation and promote mitochondrial biogenesis. J Cell Biol. 2008;180:787–802. doi: 10.1083/jcb.200707179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkar N, Ladner K, Canan BD, Liyanarachchi S, Bal NC, Pant M, et al. IKKalpha and alternative NF-kappaB regulate PGC-1beta to promote oxidative muscle metabolism. J Cell Biol. 2012;196:497–511. doi: 10.1083/jcb.201108118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter FO, Came PJ, Abell K, Kedjouar B, Huth M, Rajewsky K, et al. IKKbeta/2 induces TWEAK and apoptosis in mammary epithelial cells. Development. 2006;133:3485–3494. doi: 10.1242/dev.02502. [DOI] [PubMed] [Google Scholar]
- Bodmer JL, Schneider P, Tschopp J. The molecular architecture of the TNF superfamily. Trends Biochem Sci. 2002;27:19–26. doi: 10.1016/s0968-0004(01)01995-8. [DOI] [PubMed] [Google Scholar]
- Bover LC, Cardó-Vila M, Kuniyasu A, Sun J, Rangel R, Takeya M, et al. A previously unrecognized protein-protein interaction between TWEAK and CD163: potential biological implications. J Immunol. 2007;178:8183–8194. doi: 10.4049/jimmunol.178.12.8183. [DOI] [PubMed] [Google Scholar]
- Burkly LC, Michaelson JS, Zheng TS. TWEAK/Fn14 pathway: an immunological switch for shaping tissue responses. Immunol Rev. 2011;244:99–114. doi: 10.1111/j.1600-065X.2011.01054.x. [DOI] [PubMed] [Google Scholar]
- Campbell S, Burkly LC, Gao HX, Berman JW, Su L, Browning B, et al. Proinflammatory effects of TWEAK/Fn14 interactions in glomerular mesangial cells. J Immunol. 2006;176:1889–1898. doi: 10.4049/jimmunol.176.3.1889. [DOI] [PubMed] [Google Scholar]
- Chao DT, Su M, Tanlimco S, Sho M, Choi D, Fox M, et al. Expression of TweakR in breast cancer and preclinical activity of enavatuzumab, a humanized anti-TweakR mAb. J Cancer Res Clin Oncol. 2013;139:315–325. doi: 10.1007/s00432-012-1332-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman MS, Wu L, Amatucci A, Ho SN, Michaelson JS. TWEAK signals through JAK-STAT to induce tumor cell apoptosis. Cytokine. 2013;61:210–217. doi: 10.1016/j.cyto.2012.09.020. [DOI] [PubMed] [Google Scholar]
- Chen HN, Wang DJ, Ren MY, Wang QL, Sui SJ. TWEAK/Fn14 promotes the proliferation and collagen synthesis of rat cardiac fibroblasts via the NF-small ka, CyrillicB pathway. Mol Biol Rep. 2012;39:8231–8241. doi: 10.1007/s11033-012-1671-3. [DOI] [PubMed] [Google Scholar]
- Chicheportiche Y, Bourdon PR, Xu H, Hsu YM, Scott H, Hession C, et al. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem. 1997;272:32401–32410. doi: 10.1074/jbc.272.51.32401. [DOI] [PubMed] [Google Scholar]
- Chorianopoulos E, Rosenberg M, Zugck C, Wolf J, Katus HA, Frey N. Decreased soluble TWEAK levels predict an adverse prognosis in patients with chronic stable heart failure. Eur J Heart Fail. 2009;11:1050–1056. doi: 10.1093/eurjhf/hfp139. [DOI] [PubMed] [Google Scholar]
- Chorianopoulos E, Heger T, Lutz M, Frank D, Bea F, Katus HA, et al. FGF-inducible 14-kDa protein (Fn14) is regulated via the RhoA/ROCK kinase pathway in cardiomyocytes and mediates nuclear factor-kappaB activation by TWEAK. Basic Res Cardiol. 2010a;105:301–313. doi: 10.1007/s00395-009-0046-y. [DOI] [PubMed] [Google Scholar]
- Chorianopoulos E, Jarr K, Steen H, Giannitsis E, Frey N, Katus HA. Soluble TWEAK is markedly upregulated in patients with ST-elevation myocardial infarction and related to an adverse short-term outcome. Atherosclerosis. 2010b;211:322–326. doi: 10.1016/j.atherosclerosis.2010.02.016. [DOI] [PubMed] [Google Scholar]
- Culp PA, Choi D, Zhang Y, Yin J, Seto P, Ybarra SE, et al. Antibodies to TWEAK receptor inhibit human tumor growth through dual mechanisms. Clin Cancer Res. 2010;16:497–508. doi: 10.1158/1078-0432.CCR-09-1929. [DOI] [PubMed] [Google Scholar]
- Dai L, Gu L, Ding C, Qiu L, Di W. TWEAK promotes ovarian cancer cell metastasis via NF-kappaB pathway activation and VEGF expression. Cancer Lett. 2009;283:159–167. doi: 10.1016/j.canlet.2009.03.036. [DOI] [PubMed] [Google Scholar]
- Desplat-Jego S, Varriale S, Creidy R, Terra R, Bernard D, Khrestchatisky M, et al. TWEAK is expressed by glial cells, induces astrocyte proliferation and increases EAE severity. J Neuroimmunol. 2002;133:116–123. doi: 10.1016/s0165-5728(02)00368-5. [DOI] [PubMed] [Google Scholar]
- Desplat-Jego S, Creidy R, Varriale S, Allaire N, Luo Y, Bernard D, et al. Anti-TWEAK monoclonal antibodies reduce immune cell infiltration in the central nervous system and severity of experimental autoimmune encephalomyelitis. Clin Immunol. 2005;117:15–23. doi: 10.1016/j.clim.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Desplat-Jego S, Feuillet L, Creidy R, Malikova I, Rance R, Khrestchatisky M, et al. TWEAK is expressed at the cell surface of monocytes during multiple sclerosis. J Leukoc Biol. 2009;85:132–135. doi: 10.1189/jlb.0608347. [DOI] [PubMed] [Google Scholar]
- Dickens LS, Powley IR, Hughes MA, MacFarlane M. The ‘complexities’ of life and death: death receptor signalling platforms. Exp Cell Res. 2012;318:1269–1277. doi: 10.1016/j.yexcr.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Dogra C, Changotra H, Mohan S, Kumar A. Tumor necrosis factor-like weak inducer of apoptosis inhibits skeletal myogenesis through sustained activation of nuclear factor-kappaB and degradation of MyoD protein. J Biol Chem. 2006;281:10327–10336. doi: 10.1074/jbc.M511131200. [DOI] [PubMed] [Google Scholar]
- Dogra C, Changotra H, Wedhas N, Qin X, Wergedal JE, Kumar A. TNF-related weak inducer of apoptosis (TWEAK) is a potent skeletal muscle-wasting cytokine. FASEB J. 2007a;21:1857–1869. doi: 10.1096/fj.06-7537com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogra C, Hall SL, Wedhas N, Linkhart TA, Kumar A. Fibroblast growth factor inducible 14 (Fn14) is required for the expression of myogenic regulatory factors and differentiation of myoblasts into myotubes. Evidence for TWEAK-independent functions of Fn14 during myogenesis. J Biol Chem. 2007b;282:15000–15010. doi: 10.1074/jbc.M608668200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohi T, Borodovsky A, Wu P, Shearstone JR, Kawashima R, Runkel L, et al. TWEAK/Fn14 pathway: a nonredundant role in intestinal damage in mice through a TWEAK/intestinal epithelial cell axis. Gastroenterology. 2009;136:912–923. doi: 10.1053/j.gastro.2008.11.017. [DOI] [PubMed] [Google Scholar]
- Donohue PJ, Richards CM, Brown SA, Hanscom HN, Buschman J, Thangada S, et al. TWEAK is an endothelial cell growth and chemotactic factor that also potentiates FGF-2 and VEGF-A mitogenic activity. Arterioscler Thromb Vasc Biol. 2003;23:594–600. doi: 10.1161/01.ATV.0000062883.93715.37. [DOI] [PubMed] [Google Scholar]
- Duckett CS, Thompson CB. CD30-dependent degradation of TRAF2: implications for negative regulation of TRAF signaling and the control of cell survival. Genes Dev. 1997;11:2810–2821. doi: 10.1101/gad.11.21.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dueber EC, Schoeffler AJ, Lingel A, Elliott JM, Fedorova AV, Giannetti AM, et al. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science. 2011;334:376–380. doi: 10.1126/science.1207862. [DOI] [PubMed] [Google Scholar]
- Ebihara N, Nakayama M, Tokura T, Ushio H, Murakami A. Expression and function of fibroblast growth factor-inducible 14 in human corneal myofibroblasts. Exp Eye Res. 2009;89:256–262. doi: 10.1016/j.exer.2009.03.014. [DOI] [PubMed] [Google Scholar]
- Echeverry R, Wu F, Haile WB, Wu J, Yepes M. The cytokine tumor necrosis factor-like weak inducer of apoptosis and its receptor fibroblast growth factor-inducible 14 have a neuroprotective effect in the central nervous system. J Neuroinflammation. 2012;9:45. doi: 10.1186/1742-2094-9-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Shehaby A, Darweesh H, El-Khatib M, Momtaz M, Marzouk S, El-Shaarawy N, et al. Correlations of urinary biomarkers, TNF-like weak inducer of apoptosis (TWEAK), osteoprotegerin (OPG), monocyte chemoattractant protein-1 (MCP-1), and IL-8 with lupus nephritis. J Clin Immunol. 2011;31:848–856. doi: 10.1007/s10875-011-9555-1. [DOI] [PubMed] [Google Scholar]
- Enwere EK, Holbrook J, Lejmi-Mrad R, Vineham J, Timusk K, Sivaraj B, et al. TWEAK and cIAP1 regulate myoblast fusion through the noncanonical NF-kappaB signaling pathway. Sci Signal. 2012;5:ra75. doi: 10.1126/scisignal.2003086. [DOI] [PubMed] [Google Scholar]
- Felli N, Pedini F, Zeuner A, Petrucci E, Testa U, Conticello C, et al. Multiple members of the TNF superfamily contribute to IFN-gamma-mediated inhibition of erythropoiesis. J Immunol. 2005;175:1464–1472. doi: 10.4049/jimmunol.175.3.1464. [DOI] [PubMed] [Google Scholar]
- Feltham R, Bettjeman B, Budhidarmo R, Mace PD, Shirley S, Condon SM, et al. Smac mimetics activate the E3 ligase activity of cIAP1 protein by promoting RING domain dimerization. J Biol Chem. 2011;286:17015–17028. doi: 10.1074/jbc.M111.222919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng F, Wang L, Albanese N, Holmes A, Xia P. Tumor necrosis factor-like weak inducer of apoptosis attenuates the action of insulin in hepatocytes. Endocrinology. 2008;149:1505–1513. doi: 10.1210/en.2007-1119. [DOI] [PubMed] [Google Scholar]
- Feng SL, Guo Y, Factor VM, Thorgeirsson SS, Bell DW, Testa JR, et al. The Fn14 immediate-early response gene is induced during liver regeneration and highly expressed in both human and murine hepatocellular carcinomas. Am J Pathol. 2000;156:1253–1261. doi: 10.1016/S0002-9440(10)64996-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fick A, Lang I, Schafer V, Seher A, Trebing J, Weisenberger D, et al. Studies of binding of tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK) to fibroblast growth factor inducible 14 (Fn14) J Biol Chem. 2012;287:484–495. doi: 10.1074/jbc.M111.287656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin SP, Ennis MJ, Savitch BA, Carpentieri D, McDonough WS, Winkles JA, et al. Tumor necrosis factor-like weak inducer of apoptosis stimulation of glioma cell survival is dependent on Akt2 function. Mol Cancer Res. 2009;7:1871–1881. doi: 10.1158/1541-7786.MCR-09-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin SP, Ennis MJ, Schumacher CA, Zylstra-Diegel CR, Williams BO, Ross JT, et al. Cdc42 and the guanine nucleotide exchange factors Ect2 and trio mediate Fn14-induced migration and invasion of glioblastoma cells. Mol Cancer Res. 2012;10:958–968. doi: 10.1158/1541-7786.MCR-11-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotin-Mleczek M, Henkler F, Samel D, Reichwein M, Hausser A, Parmryd I, et al. Apoptotic crosstalk of TNF receptors: TNF-R2-induces depletion of TRAF2 and IAP proteins and accelerates TNF-R1-dependent activation of caspase-8. J Cell Sci. 2002;115:2757–2570. doi: 10.1242/jcs.115.13.2757. [DOI] [PubMed] [Google Scholar]
- Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11:109–124. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]
- Gao HX, Campbell SR, Burkly LC, Jakubowski A, Jarchum I, Banas B, et al. TNF-like weak inducer of apoptosis (TWEAK) induces inflammatory and proliferative effects in human kidney cells. Cytokine. 2009;46:24–35. doi: 10.1016/j.cyto.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Girgenrath M, Weng S, Kostek CA, Browning B, Wang M, Brown SA, et al. TWEAK, via its receptor Fn14, is a novel regulator of mesenchymal progenitor cells and skeletal muscle regeneration. EMBO J. 2006;25:5826–5839. doi: 10.1038/sj.emboj.7601441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalvez F, Lawrence D, Yang B, Yee S, Pitti R, Marsters S, et al. Pham VC, Stephan JP, Lill J, Ashkenazi A. TRAF2 Sets a threshold for extrinsic apoptosis by tagging caspase-8 with a ubiquitin shutoff timer. Mol Cell. 2012;48:888–899. doi: 10.1016/j.molcel.2012.09.031. [DOI] [PubMed] [Google Scholar]
- Grell M, Zimmermann G, Gottfried E, Chen CM, Grünwald U, Huang DC, et al. Induction of cell death by tumour necrosis factor (TNF) receptor 2, CD40 and CD30: a role for TNF-R1 activation by endogenous membrane-anchored TNF. EMBO J. 1999;18:3034–3043. doi: 10.1093/emboj/18.11.3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haile WB, Echeverry R, Wu F, Guzman J, An J, Wu J, et al. Tumor necrosis factor-like weak inducer of apoptosis and fibroblast growth factor-inducible 14 mediate cerebral ischemia-induced poly(ADP-ribose) polymerase-1 activation and neuronal death. Neuroscience. 2010a;171:1256–1264. doi: 10.1016/j.neuroscience.2010.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haile WB, Echeverry R, Wu J, Yepes M. The interaction between tumor necrosis factor-like weak inducer of apoptosis and its receptor fibroblast growth factor-inducible 14 promotes the recruitment of neutrophils into the ischemic brain. J Cereb Blood Flow Metab. 2010b;30:1147–1156. doi: 10.1038/jcbfm.2009.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan SB, El-demery AB, Ahmed AI, Abukhalil RE. Soluble TWEAK and cardiovascular morbidity and mortality in chronic kidney disease patients. Arab J Nephrol Transplant. 2012;5:27–32. [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho DH, Vu H, Brown SA, Donohue PJ, Hanscom HN, Winkles JA. Soluble tumor necrosis factor-like weak inducer of apoptosis overexpression in HEK293 cells promotes tumor growth and angiogenesis in athymic nude mice. Cancer Res. 2004;64:8968–8972. doi: 10.1158/0008-5472.CAN-04-1879. [DOI] [PubMed] [Google Scholar]
- Hosokawa Y, Hosokawa I, Ozaki K, Nakae H, Matsuo T. Proinflammatory effects of tumour necrosis factor-like weak inducer of apoptosis (TWEAK) on human gingival fibroblasts. Clin Exp Immunol. 2006;146:540–549. doi: 10.1111/j.1365-2249.2006.03233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa Y, Hosokawa I, Shindo S, Ozaki K, Nakae H, Matsuo T. Tumor necrosis factor-like weak inducer of apoptosis increases CC chemokine ligand 20 production in interleukin 1beta-stimulated human gingival fibroblasts. Hum Immunol. 2012;73:470–473. doi: 10.1016/j.humimm.2012.02.021. [DOI] [PubMed] [Google Scholar]
- Hotta K, Sho M, Yamato I, Shimada K, Harada H, Akahori T, et al. Direct targeting of fibroblast growth factor-inducible 14 protein protects against renal ischemia reperfusion injury. Kidney Int. 2011;79:179–188. doi: 10.1038/ki.2010.379. [DOI] [PubMed] [Google Scholar]
- Huang M, Narita S, Tsuchiya N, Ma Z, Numakura K, Obara T, et al. Overexpression of Fn14 promotes androgen-independent prostate cancer progression through MMP-9 and correlates with poor treatment outcome. Carcinogenesis. 2011;32:1589–1596. doi: 10.1093/carcin/bgr182. [DOI] [PubMed] [Google Scholar]
- Inta I, Frauenknecht K, Dorr H, Kohlhof P, Rabsilber T, Auffarth GU, et al. Induction of the cytokine TWEAK and its receptor Fn14 in ischemic stroke. J Neurol Sci. 2008;275:117–120. doi: 10.1016/j.jns.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Iocca HA, Plant SR, Wang Y, Runkel L, O'Connor BP, Lundsmith ET, et al. TNF superfamily member TWEAK exacerbates inflammation and demyelination in the cuprizone-induced model. J Neuroimmunol. 2008;194:97–106. doi: 10.1016/j.jneuroim.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Jain M, Jakubowski A, Cui L, Shi J, Su L, Bauer M, et al. A novel role for tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in the development of cardiac dysfunction and failure. Circulation. 2009;119:2058–2068. doi: 10.1161/CIRCULATIONAHA.108.837286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justo P, Sanz AB, Sanchez-Nino MD, Winkles JA, Lorz C, Egido J, et al. Cytokine cooperation in renal tubular cell injury: the role of TWEAK. Kidney Int. 2006;70:1750–1758. doi: 10.1038/sj.ki.5001866. [DOI] [PubMed] [Google Scholar]
- Kamata K, Kamijo S, Nakajima A, Koyanagi A, Kurosawa H, Yagita H, et al. Involvement of TNF-like weak inducer of apoptosis in the pathogenesis of collagen-induced arthritis. J Immunol. 2006;177:6433–6439. doi: 10.4049/jimmunol.177.9.6433. [DOI] [PubMed] [Google Scholar]
- Kamijo S, Nakajima A, Kamata K, Kurosawa H, Yagita H, Okumura K. Involvement of TWEAK/Fn14 interaction in the synovial inflammation of RA. Rheumatology (Oxford) 2008;47:442–450. doi: 10.1093/rheumatology/ken006. [DOI] [PubMed] [Google Scholar]
- Kaptein A, Jansen M, Dilaver G, Kitson J, Dash L, Wang E, et al. Studies on the interaction between TWEAK and the death receptor WSL-1/TRAMP (DR3) FEBS Lett. 2000;485:135–141. doi: 10.1016/s0014-5793(00)02219-5. [DOI] [PubMed] [Google Scholar]
- Kawakita T, Shiraki K, Yamanaka Y, Yamaguchi Y, Saitou Y, Enokimura N, et al. Functional expression of TWEAK in human hepatocellular carcinoma: possible implication in cell proliferation and tumor angiogenesis. Biochem Biophys Res Commun. 2004;318:726–733. doi: 10.1016/j.bbrc.2004.04.084. [DOI] [PubMed] [Google Scholar]
- Kawakita T, Shiraki K, Yamanaka Y, Yamaguchi Y, Saitou Y, Enokimura N, et al. Functional expression of TWEAK in human colonic adenocarcinoma cells. Int J Oncol. 2005;26:87–93. [PubMed] [Google Scholar]
- Kawashima R, Kawamura YI, Oshio T, Son A, Yamazaki M, Hagiwara T, et al. Interleukin-13 damages intestinal mucosa via TWEAK and Fn14 in mice-a pathway associated with ulcerative colitis. Gastroenterology. 2011;141:2119–2129. doi: 10.1053/j.gastro.2011.08.040. e2118. [DOI] [PubMed] [Google Scholar]
- Kumar M, Makonchuk DY, Li H, Mittal A, Kumar A. TNF-like weak inducer of apoptosis (TWEAK) activates proinflammatory signaling pathways and gene expression through the activation of TGF-beta-activated kinase 1. J Immunol. 2009;182:2439–2448. doi: 10.4049/jimmunol.0803357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon OH, Park SJ, Kang TW, Kim M, Kim JH, Noh SM, et al. Elevated fibroblast growth factor-inducible 14 expression promotes gastric cancer growth via nuclear factor-kappaB and is associated with poor patient outcome. Cancer Lett. 2012;314:73–81. doi: 10.1016/j.canlet.2011.09.016. [DOI] [PubMed] [Google Scholar]
- Lin BR, Huang MT, Chen ST, Jeng YM, Li YJ, Liang JT, et al. Prognostic significance of TWEAK expression in colorectal cancer and effect of its inhibition on invasion. Ann Surg Oncol. 2012;19(Suppl. 3):S385–S394. doi: 10.1245/s10434-011-1825-x. [DOI] [PubMed] [Google Scholar]
- Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- Maecker H, Varfolomeev E, Kischkel F, Lawrence D, LeBlanc H, Lee W, et al. TWEAK attenuates the transition from innate to adaptive immunity. Cell. 2005;123:931–944. doi: 10.1016/j.cell.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Marsters SA, Sheridan JP, Pitti RM, Brush J, Goddard A, Ashkenazi A. Identification of a ligand for the death-domain-containing receptor Apo3. Curr Biol. 1998;8:525–528. doi: 10.1016/s0960-9822(98)70204-0. [DOI] [PubMed] [Google Scholar]
- Meighan-Mantha RL, Hsu DK, Guo Y, Brown SA, Feng SL, Peifley KA, et al. The mitogen-inducible Fn14 gene encodes a type I transmembrane protein that modulates fibroblast adhesion and migration. J Biol Chem. 1999;274:33166–33176. doi: 10.1074/jbc.274.46.33166. [DOI] [PubMed] [Google Scholar]
- Michaelson JS, Amatucci A, Kelly R, Su L, Garber E, Day ES, et al. Development of an Fn14 agonistic antibody as an anti-tumor agent. Mabs. 2011;3:362–375. doi: 10.4161/mabs.3.4.16090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal A, Bhatnagar S, Kumar A, Lach-Trifilieff E, Wauters S, Li H, et al. The TWEAK-Fn14 system is a critical regulator of denervation-induced skeletal muscle atrophy in mice. J Cell Biol. 2010a;188:833–849. doi: 10.1083/jcb.200909117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal A, Bhatnagar S, Kumar A, Paul PK, Kuang S. Genetic ablation of TWEAK augments regeneration and post-injury growth of skeletal muscle in mice. Am J Pathol. 2010b;177:1732–1742. doi: 10.2353/ajpath.2010.100335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Dejouvencel T, Labreuche J, Smadja DM, Dussiot M, Martin-Ventura JL, et al. Peripheral artery disease is associated with a high CD163/TWEAK plasma ratio. Arterioscler Thromb Vasc Biol. 2010;30:1253–1262. doi: 10.1161/ATVBAHA.110.203364. [DOI] [PubMed] [Google Scholar]
- Morosetti R, Gliubizzi C, Sancricca C, Broccolini A, Gidaro T, Lucchini M, et al. TWEAK in inclusion-body myositis muscle: possible pathogenic role of a cytokine inhibiting myogenesis. Am J Pathol. 2012;180:1603–1613. doi: 10.1016/j.ajpath.2011.12.027. [DOI] [PubMed] [Google Scholar]
- Mueller AM, Pedre X, Kleiter I, Hornberg M, Steinbrecher A, Giegerich G. Targeting fibroblast growth factor-inducible-14 signaling protects from chronic relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;159:55–65. doi: 10.1016/j.jneuroim.2004.10.001. [DOI] [PubMed] [Google Scholar]
- Muller N, Schneider B, Pfizenmaier K, Wajant H. Superior serum half life of albumin tagged TNF ligands. Biochem Biophys Res Commun. 2010;396:793–799. doi: 10.1016/j.bbrc.2010.04.134. [DOI] [PubMed] [Google Scholar]
- Munoz-Garcia B, Martin-Ventura JL, Martinez E, Sanchez S, Hernandez G, Ortega L, et al. Fn14 is upregulated in cytokine-stimulated vascular smooth muscle cells and is expressed in human carotid atherosclerotic plaques: modulation by atorvastatin. Stroke. 2006;37:2044–2053. doi: 10.1161/01.STR.0000230648.00027.00. [DOI] [PubMed] [Google Scholar]
- Munoz-Garcia B, Moreno JA, Lopez-Franco O, Sanz AB, Martin-Ventura JL, Blanco J, et al. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) enhances vascular and renal damage induced by hyperlipidemic diet in ApoE-knockout mice. Arterioscler Thromb Vasc Biol. 2009;29:2061–2068. doi: 10.1161/ATVBAHA.109.194852. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Kayagaki N, Yamaguchi N, Okumura K, Yagita H. Involvement of TWEAK in interferon gamma-stimulated monocyte cytotoxicity. J Exp Med. 2000;192:1373–1380. doi: 10.1084/jem.192.9.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoyatleva T, Schymura Y, Janssen W, Strobl F, Swiercz JM, Patra C, et al. Deletion of Fn14 receptor protects from right heart fibrosis and dysfunction. Basic Res Cardiol. 2013;108:325. doi: 10.1007/s00395-012-0325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MC, Chung SJ, Park YB, Lee SK. Relationship of serum TWEAK level to cytokine level, disease activity, and response to anti-TNF treatment in patients with rheumatoid arthritis. Scand J Rheumatol. 2008;37:173–178. doi: 10.1080/03009740801898608. [DOI] [PubMed] [Google Scholar]
- Paul PK, Bhatnagar S, Mishra V, Srivastava S, Darnay BG, Choi Y, et al. The E3 ubiquitin ligase TRAF6 intercedes in starvation-induced skeletal muscle atrophy through multiple mechanisms. Mol Cell Biol. 2012;32:1248–1259. doi: 10.1128/MCB.06351-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perper SJ, Browning B, Burkly LC, Weng S, Gao C, Giza K, et al. TWEAK is a novel arthritogenic mediator. J Immunol. 2006;177:2610–2620. doi: 10.4049/jimmunol.177.4.2610. [DOI] [PubMed] [Google Scholar]
- Polavarapu R, Gongora MC, Winkles JA, Yepes M. Tumor necrosis factor-like weak inducer of apoptosis increases the permeability of the neurovascular unit through nuclear factor-kappa B pathway activation. J Neurosci. 2005;25:10094–10100. doi: 10.1523/JNEUROSCI.3382-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potrovita I, Zhang W, Burkly L, Hahm K, Lincecum J, Wang MZ, et al. Tumor necrosis factor-like weak inducer of apoptosis-induced neurodegeneration. J Neurosci. 2004;24:8237–8244. doi: 10.1523/JNEUROSCI.1089-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybylowski P, Malyszko JS, Malyszko J. Soluble tumor necrosis factor-like weak inducer of apoptosis plasma levels as a novel biomarker of endothelial function in prevalent orthotopic heart transplant recipients. Transplant Proc. 2011;43:1900–1903. doi: 10.1016/j.transproceed.2011.03.040. [DOI] [PubMed] [Google Scholar]
- Razmara M, Hilliard B, Ziarani AK, Murali R, Yellayi S, Ghazanfar M, et al. Fn14-TRAIL, a chimeric intercellular signal exchanger, attenuates experimental autoimmune encephalomyelitis. Am J Pathol. 2009;174:460–474. doi: 10.2353/ajpath.2009.080462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter B, Rychli K, Hohensinner PJ, Berger R, Mortl D, Neuhold S, et al. Differences in the predictive value of tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in advanced ischemic and non-ischemic heart failure. Atherosclerosis. 2010;213:545–548. doi: 10.1016/j.atherosclerosis.2010.08.061. [DOI] [PubMed] [Google Scholar]
- Roos C, Wicovsky A, Muller N, Salzmann S, Rosenthal T, Kalthoff H, et al. Soluble and transmembrane TNF-like weak inducer of apoptosis differentially activate the classical and noncanonical NF-kappa B pathway. J Immunol. 2010;185:1593–1605. doi: 10.4049/jimmunol.0903555. [DOI] [PubMed] [Google Scholar]
- Rousselet E, Traver S, Monnet Y, Perrin A, Mandjee N, Hild A, et al. Tumor necrosis factor-like weak inducer of apoptosis induces astrocyte proliferation through the activation of transforming-growth factor-alpha/epidermal growth factor receptor signaling pathway. Mol Pharmacol. 2012;82:948–957. doi: 10.1124/mol.112.079608. [DOI] [PubMed] [Google Scholar]
- Saas P, Boucraut J, Walker PR, Quiquerez AL, Billot M, Desplat-Jego S, et al. TWEAK stimulation of astrocytes and the proinflammatory consequences. Glia. 2000;32:102–107. [PubMed] [Google Scholar]
- Sabour Alaoui S, Dessirier V, de Araujo E, Alexaki VI, Pelekanou V, Lkhider M, et al. TWEAK affects keratinocyte G2/M growth arrest and induces apoptosis through the translocation of the AIF protein to the nucleus. PLoS ONE. 2012;7:e33609. doi: 10.1371/journal.pone.0033609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Nakayama M, Nakano H, Yagita H, Yamamoto N, Yamaoka S. TWEAK induces NF-kappaB2 p100 processing and long lasting NF-kappaB activation. J Biol Chem. 2003;278:36005–36012. doi: 10.1074/jbc.M304266200. [DOI] [PubMed] [Google Scholar]
- Salzmann S, Seher A, Trebing J, Weisenberger D, Rosenthal A, Siegmund D, et al. Fibroblast growth factor inducible (Fn14)-specific antibodies concomitantly display signaling pathway-specific agonistic and antagonistic activity. J Biol Chem. 2013;288:13455–13466. doi: 10.1074/jbc.M112.435917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz AB, Justo P, Sanchez-Nino MD, Blanco-Colio LM, Winkles JA, Kreztler M, et al. The cytokine TWEAK modulates renal tubulointerstitial inflammation. J Am Soc Nephrol. 2008;19:695–703. doi: 10.1681/ASN.2007050577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz AB, Sanchez-Nino MD, Izquierdo MC, Jakubowski A, Justo P, Blanco-Colio LM, et al. Tweak induces proliferation in renal tubular epithelium: a role in uninephrectomy induced renal hyperplasia. J Cell Mol Med. 2009;13(9B):3329–3342. doi: 10.1111/j.1582-4934.2009.00766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz AB, Sanchez-Nino MD, Izquierdo MC, Jakubowski A, Justo P, Blanco-Colio LM, et al. TWEAK activates the non-canonical NFkappaB pathway in murine renal tubular cells: modulation of CCL21. PLoS ONE. 2010;5:e8955. doi: 10.1371/journal.pone.0008955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz AB, Sanchez-Nino MD, Carrasco S, Manzarbeitia F, Ruiz-Andres O, Selgas R, et al. Inflammatory cytokines and survival factors from serum modulate tweak-induced apoptosis in PC-3 prostate cancer cells. PLoS ONE. 2012;7:e47440. doi: 10.1371/journal.pone.0047440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira K, Burkly LC, Zheng TS, Wu P, Groeneweg M, Rousch M, et al. Fn14-Fc fusion protein regulates atherosclerosis in ApoE-/- mice and inhibits macrophage lipid uptake in vitro. Arterioscler Thromb Vasc Biol. 2009;29:2021–2027. doi: 10.1161/ATVBAHA.109.195040. [DOI] [PubMed] [Google Scholar]
- Schneider P, Schwenzer R, Haas E, Muhlenbeck F, Schubert G, Scheurich P, et al. TWEAK can induce cell death via endogenous TNF and TNF receptor 1. Eur J Immunol. 1999;29:1785–1792. doi: 10.1002/(SICI)1521-4141(199906)29:06<1785::AID-IMMU1785>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Schwartz N, Su L, Burkly LC, Mackay M, Aranow C, Kollaros M, et al. Urinary TWEAK and the activity of lupus nephritis. J Autoimmun. 2006;27:242–250. doi: 10.1016/j.jaut.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Schwartz N, Rubinstein T, Burkly LC, Collins CE, Blanco I, Su L, et al. Urinary TWEAK as a biomarker of lupus nephritis: a multicenter cohort study. Arthritis Res Ther. 2009;11:R143. doi: 10.1186/ar2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini B, Magliozzi R, Rosicarelli B, Reynolds R, Zheng TS, Aloisi F. Expression of TWEAK and its receptor Fn14 in the multiple sclerosis brain: implications for inflammatory tissue injury. J Neuropathol Exp Neurol. 2008;67:1137–1148. doi: 10.1097/NEN.0b013e31818dab90. [DOI] [PubMed] [Google Scholar]
- Sims JJ, Scavone F, Cooper EM, Kane LA, Youle RJ, Boeke JD, et al. Polyubiquitin-sensor proteins reveal localization and linkage-type dependence of cellular ubiquitin signaling. Nat Methods. 2012;9:303–309. doi: 10.1038/nmeth.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan D, Sbai O, Wen J, Couraud PO, Putterman C, Khrestchatisky M, et al. TWEAK/Fn14 pathway modulates properties of a human microvascular endothelial cell model of blood brain barrier. J Neuroinflammation. 2013;10:9. doi: 10.1186/1742-2094-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran NL, McDonough WS, Donohue PJ, Winkles JA, Berens TJ, Ross KR, et al. The human Fn14 receptor gene is up-regulated in migrating glioma cells in vitro and overexpressed in advanced glial tumors. Am J Pathol. 2003;162:1313–1321. doi: 10.1016/S0002-9440(10)63927-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran NL, McDonough WS, Savitch BA, Fortin SP, Winkles JA, Symons M, et al. Increased fibroblast growth factor-inducible 14 expression levels promote glioma cell invasion via Rac1 and nuclear factor-kappaB and correlate with poor patient outcome. Cancer Res. 2006;66:9535–9542. doi: 10.1158/0008-5472.CAN-06-0418. [DOI] [PubMed] [Google Scholar]
- Ucero AC, Berzal S, Ocana-Salceda C, Sancho M, Orzaez M, Messeguer A, et al. A polymeric nanomedicine diminishes inflammatory events in renal tubular cells. PLoS ONE. 2013;8:e51992. doi: 10.1371/journal.pone.0051992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varfolomeev E, Goncharov T, Maecker H, Zobel K, Komuves LG, Deshayes K, et al. Cellular inhibitors of apoptosis are global regulators of NF-kappaB and MAPK activation by members of the TNF family of receptors. Sci Signal. 2012;5:ra22. doi: 10.1126/scisignal.2001878. [DOI] [PubMed] [Google Scholar]
- Vince JE, Chau D, Callus B, Wong WW, Hawkins CJ, Schneider P, et al. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFalpha. J Cell Biol. 2008;182:171–184. doi: 10.1083/jcb.200801010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss T, Grell M, Hessabi B, Bourteele S, Müller G, Scheurich P, et al. Enhancement of TNF receptor p60-mediated cytotoxicity by TNF receptor p80: requirement of the TNF receptor-associated factor-2 binding site. J Immunol. 1997;158:2398–2404. [PubMed] [Google Scholar]
- Weiss T, Grell M, Siemienski K, Mühlenbeck F, Dürkop H, Pfizenmaier K, et al. TNFR80-dependent enhancement of TNFR60-induced cell death is mediated by TNFR-associated factor 2 and is specific for TNFR60. J Immunol. 1998;161:3136–3142. [PubMed] [Google Scholar]
- Whitsett TG, Cheng E, Inge L, Asrani K, Jameson NM, Hostetter G, et al. Elevated expression of Fn14 in non-small cell lung cancer correlates with activated EGFR and promotes tumor cell migration and invasion. Am J Pathol. 2012;181:111–120. doi: 10.1016/j.ajpath.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicovsky A, Salzmann S, Roos C, Ehrenschwender M, Rosenthal T, Siegmund D, et al. TNF-like weak inducer of apoptosis inhibits proinflammatory TNF receptor-1 signaling. Cell Death Differ. 2009;16:1445–1459. doi: 10.1038/cdd.2009.80. [DOI] [PubMed] [Google Scholar]
- Wiley SR, Cassiano L, Lofton T, Davis-Smith T, Winkles JA, Lindner V, et al. A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity. 2001;15:837–846. doi: 10.1016/s1074-7613(01)00232-1. [DOI] [PubMed] [Google Scholar]
- Willis AL, Tran NL, Chatigny JM, Charlton N, Vu H, Brown SA, et al. The fibroblast growth factor-inducible 14 receptor is highly expressed in HER2-positive breast tumors and regulates breast cancer cell invasive capacity. Mol Cancer Res. 2008;6:725–734. doi: 10.1158/1541-7786.MCR-08-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkles JA. The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat Rev Drug Discov. 2008;7:411–425. doi: 10.1038/nrd2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Campbell SR, Broder A, Herlitz L, Abadi M, Wu P, et al. Inhibition of the TWEAK/Fn14 pathway attenuates renal disease in nephrotoxic serum nephritis. Clin Immunol. 2012;145:108–121. doi: 10.1016/j.clim.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamana J, Morand EF, Manabu T, Sunahori K, Takasugi K, Makino H, et al. Inhibition of TNF-induced IL-6 by the TWEAK-Fn14 interaction in rheumatoid arthritis fibroblast like synoviocytes. Cell Immunol. 2012;272:293–298. doi: 10.1016/j.cellimm.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Yepes M, Brown SA, Moore EG, Smith EP, Lawrence DA, Winkles JA. A soluble Fn14-Fc decoy receptor reduces infarct volume in a murine model of cerebral ischemia. Am J Pathol. 2005;166:511–520. doi: 10.1016/S0002-9440(10)62273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Winkles JA, Gongora MC, Polavarapu R, Michaelson JS, Hahm K, et al. TWEAK-Fn14 pathway inhibition protects the integrity of the neurovascular unit during cerebral ischemia. J Cereb Blood Flow Metab. 2007;27:534–544. doi: 10.1038/sj.jcbfm.9600368. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Burkly LC, Campbell S, Schwartz N, Molano A, Choudhury A, et al. TWEAK/Fn14 interactions are instrumental in the pathogenesis of nephritis in the chronic graft-versus-host model of systemic lupus erythematosus. J Immunol. 2007;179:7949–7958. doi: 10.4049/jimmunol.179.11.7949. [DOI] [PubMed] [Google Scholar]
- Zheng C, Kabaleeswaran V, Wang Y, Cheng G, Wu H. Crystal structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 complexes: affinity, specificity, and regulation. Mol Cell. 2010;38:101–113. doi: 10.1016/j.molcel.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhi-Chun L, Qiao-Ling Z, Zhi-Qin L, Xiao-Zhao L, Xiao-xia Z, Rong T. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) mediates p38 mitogen-activated protein kinase activation and signal transduction in peripheral blood mononuclear cells from patients with lupus nephritis. Inflammation. 2012;35:935–943. doi: 10.1007/s10753-011-9396-3. [DOI] [PubMed] [Google Scholar]
- Zhou H, Marks JW, Hittelman WN, Yagita H, Cheung LH, Rosenblum MG, et al. Development and characterization of a potent immunoconjugate targeting the Fn14 receptor on solid tumor cells. Mol Cancer Ther. 2011;10:1276–1288. doi: 10.1158/1535-7163.MCT-11-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Ekmekcioglu S, Marks JW, Mohamedali KA, Asrani K, Phillips KK, et al. The TWEAK receptor Fn14 is a therapeutic target in melanoma: immunotoxins targeting Fn14 receptor for malignant melanoma treatment. J Invest Dermatol. 2013;133:1052–1062. doi: 10.1038/jid.2012.402. [DOI] [PMC free article] [PubMed] [Google Scholar]