

Abstract
Interleukin 17A IL-17A is a crucial immunomodulator in various chronic immunological diseases including rheumatoid arthritis and inflammatory bowel disease. The cytokine has also been demonstrated to control the pathogenesis of the Mycobacterium tuberculosis by dysregulating production of cytokines and chemokines and promoting granuloma formation. Whether IL-17A regulates innate defence mechanisms of macrophages in response to mycobacterial infection remains to be elucidated. In the current report, we investigated the effects of IL-17A on modulating the intracellular survival of Mycobacterium bovis bacillus Calmette–Guérin (BCG) in RAW264.7 murine macrophages. We observed that IL-17A pre-treatment for 24 hr was able to synergistically enhance BCG-induced nitric oxide (NO) production and inducible nitric oxide synthase expression in dose- and time-dependent manners. We further delineated the mechanisms involved in this synergistic reaction. IL-17A was found to specifically enhanced BCG-induced phosphorylation of Jun N-terminal kinase (JNK), but not of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase. By using a specific JNK inhibitor (SP600125), we found that the production of NO in BCG-infected macrophages was significantly suppressed. Taken together, we confirmed the involvement of the JNK pathway in IL-17A-enhanced NO production in BCG-infected macrophages. We further demonstrated that IL-17A significantly enhanced the clearance of intracellular BCG by macrophages through an NO-dependent killing mechanism. In conclusion, our study revealed an anti-mycobacterial role of IL-17A through priming the macrophages to produce NO in response to mycobacterial infection.
Keywords: innate immunity, nitric oxide, tuberculosis
Introduction
Mycobacterium tuberculosis, the causative agent of tuberculosis, remains a major worldwide health threat as it causes approximately 2 millions deaths each year.1 Although Mycobacterium bovis bacillus Calmette–Guérin (BCG) is available as a vaccine for protecting infants and children against M. tuberculosis infection, this vaccine has been demonstrated to have limited protective efficacy in the adults.2 Moreover, failure to comply with the long anti-tubercular regimen (about 6 months) results in the emergence of drug-resistant M. tuberculosis.3 Therefore, understanding the immunological interaction between host and mycobacteria will be crucial for the development of novel therapeutic regimens.
The interleukin-17 (IL-17) family consists of six members known as IL-17A, IL-17B, IL-17C, IL-17D, IL-17E and IL-17F.4 Of these, IL-17A, which can be produced by T helper type 17 (Th17) cells, γδ T cells and natural killer cells, has been recently identified as an important pro-inflammatory cytokine and dysregulation of its production results in pathogenesis of a variety of diseases including autoimmune diseases, tumour development and infections.5 The roles of IL-17A in host defence against mycobacterial infection have been examined by other groups. Following mycobacterial infection, a proportion of CD4+ T cells differentiate into Th17 cells, which subsequently produce IL-17A.6 It has been shown that IL-17A is required to induce the formation of mature granuloma after M. tuberculosis infection. Mice deficient in IL-17A exhibit impaired granuloma formation and weakened protective immunity against M. tuberculosis infection.7–9 Furthermore, IL-17A promotes the production of chemokines in mice during M. tuberculosis challenge, leading to recruitment of neutrophils and interferon-γ (IFN-γ) -producing CD4+ T cells, which subsequently contribute to restriction of M. tuberculosis growth in the lung.10 Despite these studies demonstrating that IL-17A has a protective role against M. tuberculosis infection, whether IL-17A regulates innate defence mechanisms of macrophage in response to mycobacterial infection remains to be investigated.
Macrophages are key phagocytic cells that control the pathogenesis of M. tuberculosis. Upon mycobacterial infection, macrophages are activated and express inducible nitric oxide synthase (iNOS), leading to production of nitric oxide (NO), a free radical that has been recognized as the most critical factor directly affecting the pathogenesis of M. tuberculosis in the host.11 The importance of NO in host defence against M. tuberculosis is supported by in vivo models that iNOS-deficient mice exhibit increased bacterial burden and higher mortality rate than wild-type mice after M. tuberculosis challenge.12 Furthermore, injection or feeding iNOS inhibitor into mice harbouring latent tuberculosis results in reactivation of M. tuberculosis.13,14 The expression of iNOS in activated macrophage is regulated by various mitogen-activated protein kinases (MAPKs) including Jun N-terminal kinase (JNK), extracellular signal-regulated kinase 1/2 (ERK1/2) and p38 MAPK and also by transcription factors including nuclear factor-κB (NF-κB).15,16 Moreover, pro-inflammatory cytokines such as IFN-γ and tumour necrosis factor-α have been shown to enhance both iNOS expression and NO production in mycobacteria-infected macrophages.17,18 These studies suggest the participation of pro-inflammatory cytokines in modulating innate defence mechanism of macrophages in response to mycobacterial infection.
Previously, our group showed that IL-17A is able to enhance the production of IL-6, which is required for the differentiation of Th17 cells, in human macrophages during BCG infection. Our study suggests a role for IL-17A in modulating macrophage cytokine production and overall immune responses towards mycobacterial infection.19 In the current study, we focus on the role of IL-17A in modulating intracellular survival of BCG in macrophages. Given that NO has a potent bactericidal effect towards mycobacteria and the production of NO can be modulated by pro-inflammatory cytokines, we are interested in examining whether IL-17A can also augment NO production and therefore achieve enhanced clearance of intracellular BCG. Our data reveal an anti-mycobacterial role of IL-17A towards intracellular BCG through an NO-dependent killing mechanism.
Materials and methods
Reagents and antibodies
Recombinant mouse IL-17A and recombinant human IL-17A were purchased from R & D Systems (Minneapolis, MN). Antibody against iNOS (clone NOS-IN) was purchased from Sigma-Aldrich (St Louis, MO). Antibodies against phospho-JNK, JNK, phospho-p38 MAPK, p38 MAPK, phospho-ERK1/2 and ERK were purchased from Cell Signaling Technology (Beverly, MA). Antibody against NF-κB p65 was purchased from Calbiochem (San Diego, CA). Antibodies against IκBα, actin and lamin B were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Horseradish peroxidase (HRP) -conjugated goat anti-rabbit antibody was purchased from BD Biosciences (San Jose, CA) and HRP-conjugated rabbit anti-goat antibody was purchased from Invitrogen (Carlsbad, CA). The JNK inhibitor SP600125 was purchased from Calbiochem and the iNOS inhibitor aminoguanidine (AG) was purchased from Sigma-Aldrich.
Cell line and bacteria
Murine macrophage cell line RAW264.7 was obtained from the American Type Culture Collection (Rockville, MD). The cell line was maintained in Dulbecco's modified Eagle's medium (Gibco, Invitrogen, Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum (Gibco, Invitrogen), 100 units/ml penicillin and 100 μg/ml streptomycin. A lyophilized form of M. bovis BCG Danish strain 1331 was purchased from Statens Serum Institut (Copenhagen, Denmark). According to the manufacturer's specification, the vaccine strain was free from contamination by M. tuberculosis antigens. The lyophilized bacteria were freshly reconstituted with vaccine diluent before being added to the macrophages.
Isolation of human monocyte-derived macrophages
Human monocyte-derived macrophages (MDM) from buffy coats of healthy donors were isolated by density-gradient centrifugation as described previously.19 Briefly, buffy coats were layered on Ficoll-Paque PLUS (GE Healthcare, Piscataway, NJ), followed by centrifugation at 1000 g for 20 min. Mononuclear cells were collected and plated onto Petri dishes and incubated at 37° for 1 hr. Non-adherent cells were removed by extensive washes with RPMI-1640. Isolated MDM were seeded into 24-well plates at a density of 5 × 105 cells/well and were cultured in RPMI-1640 supplemented with 5% heat-inactivated autologous plasma, 100 units/ml penicillin and 100 μg/ml streptomycin for 7–10 days. One day before treatment, the culture medium was replaced by antibiotic-free Macrophage Serum Free Medium (Gibco, Invitrogen).
Treatment of macrophages
RAW264.7 macrophages were seeded into 24-well plates at a density of 5 × 104 cells/well in antibiotic-free Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum and incubated overnight. Murine macrophages or human MDM were pre-treated with recombinant mouse IL-17A or recombinant human IL-17A, respectively, for 24 hr before BCG infection at a multiplicity of infection of 1. Vaccine diluent was used as mock infection control in all experiments. For experiments involving the use of chemical inhibitors [SP600125 (10 μm) or AG (100 μg/ml)], the inhibitors were added 1 hr before IL-17A pre-treatment. DMSO at 0·2% concentration was added as solvent control for SP600125.
Measurement of NO production
Culture supernatants from treated macrophages were harvested, followed by centrifugation at 16 000 g for 5 min to remove cell debris. The culture supernatants were mixed with equal volumes of modified Griess reagent (Sigma-Aldrich) and incubated in the dark for 10 min. Absorbance readings at 570 nm were taken.
Cell viability assay
Culture supernatants from treated macrophages were harvested, followed by centrifugation at 16 000 g for 5 min to remove cell debris. The culture supernatants were mixed with lactate dehydrogenase (LDH) assay reagents (Sigma-Aldrich) at a volume ratio of 1 : 2 and incubated in the dark for 30 min. Absorbance readings at 490 nm with reference wavelength of 655 nm were taken.
RNA extraction and quantitative PCR
Total RNA from treated macrophages was extracted using TRIzol reagent (Invitrogen) as previously described.19,20 Equal amounts of RNA were reverse transcribed to complementary DNA by using SuperScript II (Invitrogen) according to the manufacturer's instruction. The expression level of iNOS mRNA was determined by using a gene-specific probe (Roche Applied Science, Penzberg, Germany). Mouse β-actin was used as a reference gene for quantitative PCR (qPCR) analysis. All qPCR assays were performed in duplicate in a LightCycler 480 System (Roche Applied Science). The Ct value of target gene in each sample was normalized to that of reference gene, giving ΔCt. Then the ΔCt values of treated macrophages were compared with that of untreated ones, giving ΔΔCt. The logarithm  was used to calculate the relative expression of the target gene.
was used to calculate the relative expression of the target gene.
Inducible NOS mRNA stability assay
The macrophages were pre-treated with recombinant mouse IL-17A for 24 hr before BCG infection at a multiplicity of infection of 1. After 3 hr of BCG infection, infected macrophages were washed with PBS and replenished with fresh medium containing 1 μg/ml actinomycin D (Sigma-Aldrich). At the indicated time-points, total RNA from infected macrophages was extracted by using TRIzol reagent and reverse transcribed to complementary DNA. The relative expression level of iNOS mRNA was determined by qPCR.
Phagocytosis and bacterial survival assay
After 2 hr (phagocytosis assay) or 48 hr (bacteria survival assay) of BCG infection, the intracellular bacteria were recovered based on the methods described previously.21 Briefly, the infected macrophages were washed thrice with PBS. The cells were then lysed by lysis buffer (PBS, 0·5% Triton X-100) to recover intracellular bacteria. The cell lysates were appropriately diluted in PBS containing 0·05% Tween-80 and were plated onto Middlebrook 7H10 agar (BD Biosciences). The agar plates were incubated at 37° supplemented with 5% CO2. Colony-forming units (CFU) were enumerated after 3 weeks of incubation.
Preparation of whole cell lysates
To collect whole cell lysates, the macrophages were washed once with PBS and lysed by ice-cold whole cell lysis buffer (10 mm Tris–HCl, pH 7·4, 50 mm NaCl, 50 mm NaF, 10 mm β-glycerophosphate, 0·1 mm EDTA, 10% glycerol, 1% Triton X-100, 2 μg/ml aprotinin, 1 mm sodium orthovanadate, 2 μg/ml leupeptin, 2 μg/ml pepstatin and 1 mm PMSF). Soluble proteins were harvested after centrifugation at 16 000 g for 5 min. The protein concentrations in the whole cell lysates were quantified by bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions.
Preparation of cytoplasmic proteins and nuclear proteins
The extraction of cytoplasmic proteins and nuclear proteins was based on the methods described previously.22 Briefly, the macrophages were washed twice with cold 1 × PBS, followed by incubation with buffer A (10 mm HEPES, pH 7·9, 10 mm KCl, 0·1 mm EDTA, 0·1 mm EGTA, 1 mm dithiothreitol, 2 μg/ml aprotinin, 1 mm sodium orthovanadate, 2 μg/ml leupeptin, 2 μg/ml pepstatin and 1 mm PMSF) on ice for 15 min. The cells were lysed by adding nonidet P-40 to a final concentration of 0·625%. The lysates were centrifuged at 16 000 g for 5 min at 4°. The supernatant containing cytoplasmic proteins was harvested. The pellets were washed once with buffer A and then lysed in buffer C (20 mm HEPES, pH 7·9, 0·4 mm NaCl, 50 mm NaF, 1 mm EDTA, 0·1 mm EGTA, 1 mm dithiothreitol, 2 μg/ml aprotinin, 1 mm sodium orthovanadate, 2 μg/ml leupeptin, 2 μg/ml pepstatin and 1 mm PMSF). The lysates were centrifuged at 16 000 g for 5 min at 4°. The supernatants containing nuclear proteins were harvested. The protein concentrations in the cytoplasmic fraction and nuclear fraction were quantified by BCA protein assay kit (Thermo Fisher Scientific) according to the manufacturer's instructions.
Western blot analysis
The proteins were denatured with 4× sample loading buffer (100 mm Tris–HCl, pH 6·8, 200 mm dithiothreitol, 4% SDS, 20% glycerol and 0·2% bromophenol blue) at 95° for 5 min. Equal amounts of proteins were resolved in 10% SDS–PAGE and then transferred onto nitrocellulose membrane (Whatman, Maidstone, UK). The membranes were blocked and then incubated with primary antibodies against iNOS, phospho-JNK, JNK, phospho-p38 MAPK, p38 MAPK, phospho-ERK1/2, ERK, IκBα, NF-κB p65, actin or lamin B overnight at 4°, followed by incubation with corresponding HRP-conjugated secondary antibodies for 1 hr. The protein bands were visualized using enhanced chemiluminescence solutions (GE Healthcare, Little Chalfont, UK).
Statistical analysis
Statistical analysis was assisted by GraphPad Prism 5 (GraphPad Software Inc., La Jolla, CA). Student's t-test or one-way analysis of variance with Newman–Keuls post-hoc test was adopted when appropriate. P < 0·05 was considered statistically significant.
Results
IL-17A enhances NO production in BCG-infected macrophages by up-regulating iNOS expression
To investigate whether IL-17A affects NO production in BCG-infected macrophages, we first investigated the effects of various doses of IL-17A on BCG-induced NO production in human MDM. The macrophages were pre-treated with recombinant human IL-17A at 5, 25 or 100 ng/ml for 24 hr, followed by BCG infection for 24–72 hr. We observed that human MDM failed to produce substantial amounts of NO in response to BCG infection. The level of NO in BCG-infected macrophages was comparable to that in untreated cells (Table 1). Moreover, the addition of human IL-17A did not augment the production of NO in infected human MDM (Table 1).
Table 1.
Effect of human interleukin-17A pre-treatment on nitric oxide production in bacillus Calmette–Guérin-infected human monocyte-derived macrophages
| Nitrite (μm) (mean ± SE) | |||
|---|---|---|---|
| 24 hr p.i. | 48 hr p.i. | 72 hr p.i. | |
| Untreated | 1·76 ± 0·57 | 4·17 ± 0·67 | 4·82 ± 1·08 |
| BCG | 0·18 ± 0·11 | 2·56 ± 0·78 | 3·10 ± 0·95 |
| hIL-17A (5 ng/ml) + BCG | 1·31 ± 0·59 | 3·15 ± 0·36 | 3·47 ± 0·67 |
| hIL-17A (25 ng/ml) + BCG | 0·82 ± 0·43 | 3·63 ± 1·69 | 2·55 ± 0·55 |
| hIL-17A (100 ng/ml) + BCG | 1·42 ± 0·76 | 2·66 ± 0·94 | 2·34 ± 0·66 |
| hIL-17A (5 ng/ml) | 0·23 ± 0·22 | 2·61 ± 1·03 | 1·86 ± 0·61 |
| hIL-17A (25 ng/ml) | 0·11 ± 0·11 | 2·51 ± 1·10 | 1·37 ± 0·37 |
| hIL-17A (100 ng/ml) | 0·34 ± 0·15 | 1·86 ± 0·66 | 2·18 ± 0·52 |
Isolated human monocyte-derived macrophages were pre-treated with indicated doses of recombinant human interleukin-17A (hIL-17A) (5 ng/ml, 25 ng/ml or 100 ng/ml) for 24 hr, followed by bacillus Calmette–Guérin (BCG) infection at a multiplicity of infection = 1 for 24–72 hr. The culture supernatants were harvested for determination of NO production by Griess reaction. The data represented mean ± SE of six healthy donors.
As human MDM did not produce NO in response to BCG infection, we decided to use RAW264.7 murine macrophages, which readily produce NO upon infection or stimulation,15 as a model to study the effects of IL-17A on NO production in BCG-infected macrophages. We observed that IL-17A was able to synergistically enhance BCG-induced NO in a dose-dependent manner. The production of NO in macrophages was enhanced by 20%, 43% or 31% when pre-treated with 5 ng/ml, 25 ng/ml or 100 ng/ml of IL-17A, respectively. The IL-17A alone did not induce NO production in macrophages at all doses being tested (Fig. 1a). As IL-17A at 25 ng/ml had the greatest enhancing effect on BCG-induced NO production, we chose to use this concentration of IL-17A in all subsequent experiments. Next, we studied the kinetics of NO production and iNOS expression in BCG-infected macrophages. The macrophages were pre-treated with IL-17A for 24 hr, followed by BCG infection. The culture supernatants were collected at the indicated time-points for determination of NO production. The results showed that with IL-17A pre-treatment, the NO production in BCG-infected macrophages was significantly enhanced in a time-dependent manner (Fig. 1b). We also examined the kinetics of iNOS expression in BCG-infected macrophages with IL-17A pre-treatment by qPCR and Western blot analysis. From qPCR analysis, we observed that the expression level of iNOS mRNA in BCG-infected macrophages was enhanced by IL-17A over a time course of 24 hr (Fig. 1c). Similar observations could be obtained using Western blot analysis. The production of iNOS protein in BCG-infected macrophages was enhanced by IL-17A as early as 3 hr post-infection and the enhancing effect continued to 12 hr post-infection (Fig. 1d). At 24 hr post-infection, we observed that the protein levels of iNOS were comparable between BCG-infected macrophages with or without IL-17A pre-treatment. Interleukin-17A alone did not induce detectable level of iNOS protein in macrophages at all time-points being tested (Fig. 1d). Taken together, our data suggest that IL-17A is able to enhance NO production in macrophages by up-regulating iNOS expression during BCG infection.
Figure 1.

Interleukin-17A (IL-17A) enhances nitric oxide (NO) production by up-regulating inducible nitric oxide synthase (iNOS) expression in bacillus Calmette–Guérin (BCG) -infected macrophages. (a) Macrophages were pre-treated with indicated doses of IL-17A (5 ng/ml, 25 ng/ml or 100 ng/ml) for 24 hr, followed by BCG infection at a multiplicity of infection (MOI) = 1 for another 24 hr. The culture supernatants were harvested for determination of NO production by Griess reaction. The data represented mean ± SE of three independent experiments. *P < 0·05. (b) Kinetics of NO production in BCG-infected macrophages. Macrophages were pre-treated with IL-17A (25 ng/ml) for 24 hr, followed by BCG infection at an MOI = 1. At the indicated time-points, the culture supernatants were harvested for determination of the NO production by Griess reaction. The data represented mean ± SE of three independent experiments. *P < 0·05. (c) Kinetics of iNOS mRNA expression. Macrophages were treated as described in (b). At the indicated time-points, total RNA from macrophages was extracted for reverse transcription and quantitative PCR analysis of iNOS mRNA expression. The data represented mean ± SE of two independent experiments. (d) Western blot analysis of kinetics of iNOS expression. Macrophages were treated as described in (b). At indicated time-points, whole cell lysates were harvested for Western blot analysis of iNOS. Actin was used as a loading control. The data were representative of three independent experiments.
IL-17A specifically enhances activation of JNK, but not ERK1/2 and p38 MAPK, during BCG infection
Signalling pathways of MAPK, including JNK, ERK1/2 and p38 MAPK, are activated in macrophages in response to mycobacterial infection, leading to production of pro-inflammatory cytokines.19,21,23 The expression of iNOS has also been shown to be regulated by those MAPK pathways.15,24 To investigate whether IL-17A pre-treatment affects BCG-activated MAPK pathways, we analysed the phosphorylations of various MAPKs. We pre-treated the macrophages with IL-17A for 24 hr, followed by BCG infection for 60, 90, 120 and 150 min. Total cell lysates were harvested for Western blot analysis of phosphorylation of JNK, p38 MAPK and ERK1/2. Our results showed that phosphorylation of JNK, p38 MAPK and ERK1/2 in macrophages was strongly induced by BCG at 60 and 90 min post-infection (Fig. 2a, lane 2 and lane 6) and became diminished at 120 and 150 min post-infection (Fig. 2a, lane 10 and lane 14). The levels of phosphorylated JNK at 60 min post-infection were found to be similar between BCG-infected macrophages with or without IL-17A pre-treatment (Fig. 2a, lane 2 versus lane 3). However, we observed that in the presence of IL-17A, the BCG-induced phosphorylation of JNK was enhanced at 90, 120 and 150 min (Fig. 2a, lane 7, land 11 and lane 15, respectively). The data suggest that IL-17A is able to prolong BCG-induced phosphorylation of JNK. On the other hand, IL-17A had no effects on BCG-activated ERK1/2 and p38 MAPK at all time-points being tested (Fig. 2a).
Figure 2.

Interleukin-17A (IL-17A) specifically enhances activation of Jun N-terminal kinase (JNK) pathway, but not extracellular signal-regulated kinase 1/2 (ERK1/2) and p38 mitogen-activated protein kinase (MAPK) pathways, during BCG infection. (a) Macrophages were pre-treated with IL-17A (25 ng/ml) for 24 hr, followed by BCG infection at a multiplicity of infection (MOI) = 1. At indicated time-points, whole cell lysates were collected for Western blot analysis of JNK, ERK1/2 or p38 MAPK. The data were representative of three independent experiments. (b) Lactate dehydrogenase (LDH) assay for cell viability in the presence of SP600125. Macrophages were incubated with JNK inhibitor SP600125 (10 μm) for 1 hr, followed by IL-17A (25 ng/ml) pre-treatment for 24 hr. The macrophages were then infected by bacillus Calmette–Guérin (BCG) at an MOI = 1 for another 24 hr. The culture supernatants were collected for determination of LDH release. The data represented mean ± SE of three independent experiments. (c) Inhibition of JNK pathway suppresses IL-17-enhanced nitric oxide (NO) production in BCG-infected macrophages. Macrophages were treated as described in (b).The culture supernatants were collected for determination of NO by Griess reaction. The data represented mean ± SE of three independent experiments. *P < 0·05. (d) Analysis of specificity of SP600125. Macrophages were incubated with JNK inhibitor SP600125 (10 μm) for 1 hr, followed by IL-17A (25 ng/ml) pre-treatment for 24 hr. The macrophages were then infected by BCG at an MOI = 1 for another 60 min. Whole cell lysates were collected for Western blot analysis of JNK, ERK1/2 or p38 MAPK. The data were representative of three independent experiments. The determination of the intensities of protein bands was assisted by imageJ software (National Institutes of Health, Bethesda, MD). The intensities of phosphorylated proteins were normalized to the corresponding total proteins. The values in parentheses were relative intensities compared with untreated control.
For verification that JNK was involved in the enhancement of BCG-induced NO production by IL-17A, we blocked the activation of the JNK pathway by using SP600125, which is a reversible ATP competitive inhibitor specific to JNK.25 Previous studies reported by other groups have shown that the JNK inhibitor SP600125 is able to suppress NO production in macrophages being stimulated by Toll-like receptor agonists including BCG and lipopolysaccharide.24,26 We incubated the macrophages with SP600125 or DMSO for 1 hr before IL-17A pre-treatment for 24 hr. The macrophages were then infected by BCG for 24 hr. We used an LDH assay to analyse the viability of macrophages in the presence of SP600125. The data revealed that there was no significant difference in LDH release among the groups, suggesting that the viabilities of macrophages among the groups were similar (Fig. 2b). Consistent with previous studies, with the addition of SP600125, NO production in BCG-infected macrophages was significantly reduced by about 74% when compared with solvent control. The inhibitor also significantly reduced IL-17A-enhanced NO production by about 66% (Fig. 2c). The specificity of SP600125 towards JNK, ERK1/2 and p38 MAPK was also analysed. It was observed that only the BCG-induced phosphorylation of JNK, but not ERK1/2 or p38 MAPK, was inhibited by SP600125 (Fig. 2d, lane 2 versus lane 6; lane 3 versus lane 7). The data suggested that SP600125 was able to specifically block the activation of JNK. Taken together, we confirmed the involvement of JNK in IL-17A-enhanced NO production in BCG-infected macrophages.
IL-17A enhances the stability of BCG-induced iNOS mRNA
The expression of iNOS has been shown to be regulated at the post-transcriptional level via the JNK signalling pathway, which contributes to stabilization of iNOS mRNA.27 Our data showed that IL-17 was able to enhance BCG-induced phosphorylation of JNK (Fig. 2a). Therefore, we are interested to assess whether IL-17A is able to affect the stability of BCG-induced iNOS mRNA. Using qPCR analysis, our data showed that the half-life of iNOS mRNA in BCG-infected macrophages was about 101 min. In the presence of IL-17A, the half-life of BCG-induce iNOS mRNA was prolonged to about 227 min (Fig. 3). Our results indicated that IL-17A was able to enhance the stability of BCG-induced iNOS mRNA, thereby allowing for increased NO production.
Figure 3.
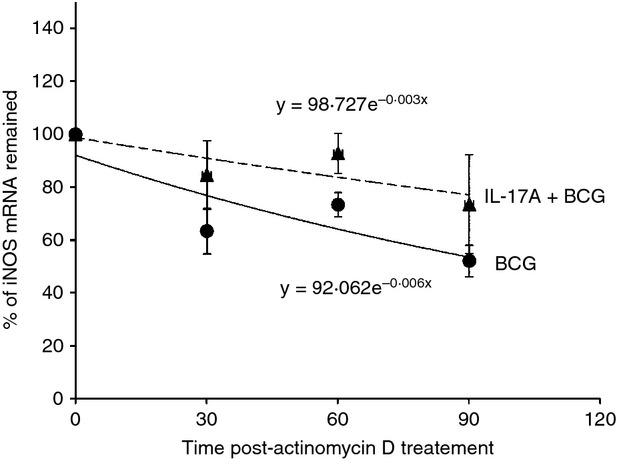
Interleukin-17A (IL-17A) enhances the stability of bacillus Calmette–Guérin (BCG) -induced inducible nitric oxide synthase (iNOS) mRNA. Macrophages were pre-treated with IL-17A (25 ng/ml) for 24 hr, followed by BCG infection at a multiplicity of infection (MOI) = 1 for 3 hr. Then, the infected macrophages were washed with PBS and replenished with fresh medium containing 1 μg/ml actinomycin D. At indicated time-points, total RNA from macrophages were extracted for reverse transcription and subjected to quantitative PCR analysis of iNOS mRNA expression. The iNOS mRNA expression level at 0 min post-actinomycin D treatment was set as 100%. The half-life of iNOS mRNA was defined as the time required for iNOS mRNA to degrade until 50% remained. The data represented mean ± SE of seven independent experiments.
IL-17A does not affect activation of NF-κB pathway during BCG-infection
Nuclear factor-κB is a key transcription factor that drives the expression of iNOS.28,29 The BCG-induced activation of the NF-κB pathway in macrophages requires degradation of IκBα in the cytoplasm, which allows the release of NF-κB and subsequent translocation of NF-κB into the nucleus for initiation of gene expression.19,30,31 To investigate whether IL-17A pre-treatment affects BCG-activated NF-κB pathways, we analysed the degradation of IκBα in the cytoplasm and translocation of NF-κB p65 into the nucleus. We pre-treated the macrophages with IL-17A for 24 hr, followed by BCG infection for 15 min. Cytoplasmic proteins and nuclear proteins were extracted for Western blot analysis of IκBα and NF-κB p65, respectively. Our results showed that infection of macrophages by BCG caused degradation of IκBα and also translocation of NF-κB p65 into the nucleus (Fig. 4, lane 2). However, neither process was affected by IL-17A pre-treatment (Fig. 4, lane 3). Our results suggested that IL-17A had no effects on the activation of the NF-κB pathway during BCG infection.
Figure 4.
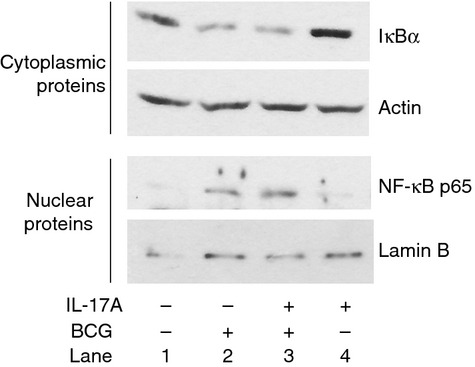
Interleukin-17A (IL-17A) does not affect activation of nuclear factor-κB (NF-κB) pathway during bacillus Calmette–Guérin (BCG) infection. Macrophages were pre-treated with IL-17A (25 ng/ml) for 24 hr, followed by BCG infection at a multiplicity of infection (MOI) = 1 for 15 min. Cytoplasmic proteins and nuclear proteins were extracted for Western analysis of IκBα and NF-κB p65, respectively. Actin and lamin B were used as loading controls for cytoplasmic proteins and nuclear proteins, respectively. The data were representative of three independent experiments.
IL-17A enhances the clearance of intracellular BCG
The intracellular survival of BCG in macrophages is largely affected by NO being produced inside the host cells.13 We have demonstrated that IL-17A can enhance NO production in BCG-infected macrophages (Fig. 1). As a result, we are also interested in whether IL-17A can enhance the clearance of intracellular BCG by macrophages. We pre-treated the macrophages with IL-17A for 24 hr, followed by BCG infection. Intracellular BCG was recovered after 2 hr or 48 hr of infection and plated onto 7H10 agar for the determination of phagocytosis or intracellular survival of BCG, respectively. Our data revealed that IL-17A had no effects on phagocytosis of BCG by macrophage (Fig. 5a). On the other hand, the survival of intracellular BCG in macrophages with IL-17A pre-treatment was significantly reduced by 30% after 48 hr of infection (Fig. 5b). The results indicated that IL-17A has anti-mycobacterial effects towards intracellular BCG.
Figure 5.
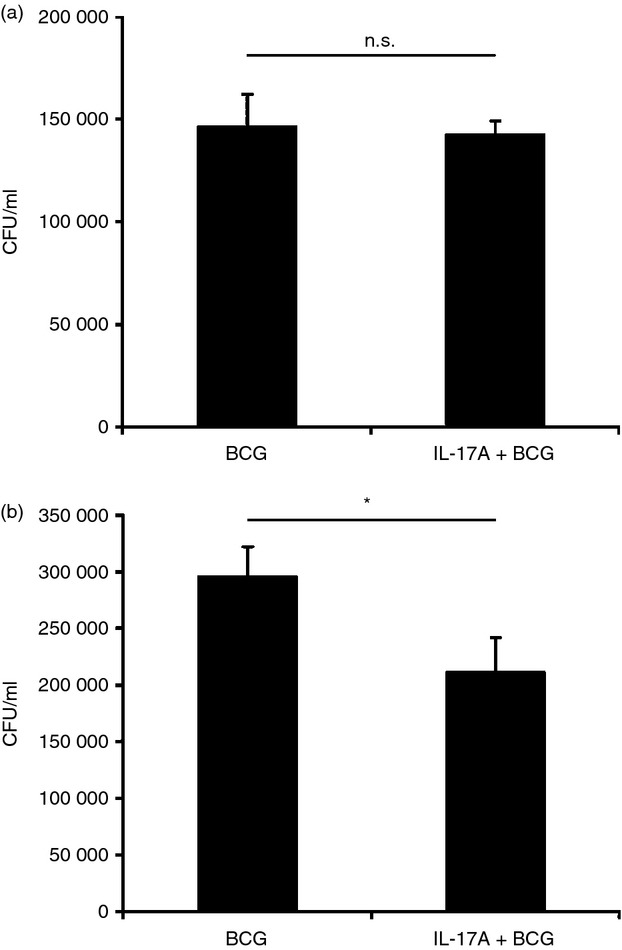
Interleukin-17A (IL-17A) enhances the clearance of intracellular bacillus Calmette–Guérin (BCG). (a) Phagocytosis of BCG by macrophages with or without IL-17A pre-treatment. The macrophages were pre-treated with IL-17A (25 ng/ml) for 24 hr, followed by BCG infection at a multiplicity of infection (MOI) = 1. After 2 hr of BCG infection, non-phagocytosed bacteria were washed. Intracellular BCG was recovered by lysis of the macrophages and plated onto 7H10 agar. Colony-forming units (CFU) were enumerated after 3 weeks of incubation. The data represented mean ± SE of three independent experiments. N.S., not significant. (b) Intracellular survival of BCG in macrophages with or without IL-17A pre-treatment. The macrophages were pre-treated same as (a). After 48 hr of BCG infection, non-phagocytosed bacteria were washed. Intracellular BCG was recovered by lysis of the macrophages and plated onto 7H10 agar. CFU were enumerated after 3 weeks of incubation. The data represented mean ± SE of five independent experiments. *P < 0·05.
IL-17A-enhanced clearance of intracellular BCG is dependent on NO
To investigate whether enhanced clearance of intracellular BCG in IL-17A-treated macrophages correlates with NO production, we used AG, a specific iNOS inhibitor, to suppress the enzymatic activity of iNOS.32 The macrophages were incubated with AG for 1 hr, followed by IL-17A pre-treatment for 24 hr and then BCG infection. The viabilities of macrophages in the presence of AG were analysed by LDH assay. We observed that there was no significant difference in LDH release among the groups, suggesting that the viabilities of macrophages among the groups were similar (Fig. 6a). With the addition of AG, we observed that the production of NO in infected macrophages was abolished, regardless of the presence of IL-17A (Fig. 6b). Incubation of macrophages with AG resulted in a 24% increase of intracellular BCG. The same inhibitor also abolished IL-17A-enhanced clearance of intracellular BCG, producing a c.49% increase in intracellular BCG (Fig. 6c). Our data confirmed that IL-17A-enhanced clearance of intracellular BCG is mediated through an NO-dependent mechanism.
Figure 6.

Interleukin-17A (IL-17A) -enhanced clearance of intracellular bacillus Calmette–Guérin (BCG) is dependent on nitric oxide (NO). (a) Lactate dehydrogenase (LDH) assay for cell viability in the presence of aminoguanidine (AG). Macrophages were incubated with Jun N-terminal kinase (JNK) inhibitor AG (100 μg/ml) for 1 hr, followed by IL-17A (25 ng/ml) pre-treatment for 24 hr. The macrophages were then infected by BCG at a multiplicity of infection (MOI) = 1 for another 48 hr. The culture supernatants were collected for determination of LDH release. The data represented mean ± SE of three independent experiments. (b) NO production in BCG-infected macrophages in the presence of inducible nitric oxide synthase (iNOS) inhibitor. Macrophages were incubated with AG (100 μg/ml) for 1 hr, followed by IL-17A (25 ng/ml) pre-treatment for 24 hr. The macrophages were then infected by BCG at MOI = 1. The culture supernatants were collected after 24 hr of BCG infection for determination of NO by Griess reaction. The data represented mean ± SE of four independent experiments. *P < 0·05. (c) Intracellular survival of BCG in macrophages in the presence of iNOS inhibitor. The macrophages were treated as described in (b). After 48 hr of BCG infection, non-phagocytosed bacteria were washed. Intracellular BCG was recovered by lysis of the macrophages and plated onto 7H10 agar. Colony-forming units (CFU) were enumerated after 3 weeks of incubation. The data represented mean ± SE of four independent experiments. *P < 0·05.
Discussion
In response to microbial infection, macrophages eliminate the phagocytosed pathogens through innate defence mechanisms. The bactericidal effects of NO on intracellular mycobacteria have long been appreciated in murine models.12,33,34 Unlike murine macrophages, which readily produce NO in response to infections or stimulation, activated human macrophages fail to produce detectable levels of NO in vitro despite iNOS protein expression.35 Such observations were controversial when regarding the use of NO by human macrophages as an anti-mycobacterial effector. However, several lines of evidence have demonstrated that macrophages isolated from patients with tuberculosis, but not healthy donors, express iNOS and release substantial amounts of NO.36–38 Further support is provided by studies that use iNOS inhibitors to abolish the killing effects of human macrophages isolated from patients towards intracellular pathogens including BCG and Klebsiella pneumoniae.37,39 It is now compelling that NO is also used by human macrophages as an innate defence mechanism against mycobacterial infection.40,41 The dependence of both human and murine macrophages on NO to control the pathogenesis of mycobacteria inside the host suggests that adequate activation of macrophages to produce this free radical is critical for host defence.
In the present study, we demonstrated that IL-17A synergistically enhanced NO production and iNOS expression in BCG-infected macrophages in dose- and time-dependent manners. Kinetics study revealed that IL-17A enhanced iNOS expression at early time-points after BCG infection. Incubation of IL-17A did not further enhance iNOS expression in macrophages after 24 hr of BCG infection (Fig. 1c). Such observation can be explained by negative feedback regulation on iNOS to prevent over-production of NO.28,29 Under the conditions we have tested, we observed that IL-17A alone did not induce detectable levels of iNOS protein and NO production in macrophages. Our data suggest that IL-17A is able to prime macrophages to produce NO in response to mycobacterial infection. Similar observations have been reported by Kawanokuchi et al.42 – that IL-17A is able to enhance both iNOS expression and NO production in lipopolysaccharide-stimulated microglia, whereas IL-17A by itself has no effect on either product. In another study, IL-17A has been shown to induce iNOS expression and NO production in articular chondrocytes.43 Interleukin-17A also induces NO production in cartilage explants from osteoarthritis patients.44 The differences between observations among these studies may implicate differential effects of IL-17A on NO production in specific cell types.
Binding of the cell wall components (e.g. lipoarabinomannan and peptidoglycan) and secretory proteins (e.g. 38 000 molecular weight glycolipoprotein) of mycobacteria to Toll-like receptor 2 triggers the activation of multiple MAPKs in macrophages.15,45,46 Consistent with our previous studies,19,21,23 our results demonstrated that BCG is able to induce the phosphorylation of JNK, ERK1/2 and p38 MAPK and also translocation of NF-κB p65 in macrophages. Our results revealed that IL-17A specifically enhanced BCG-induced phosphorylation of JNK in macrophages. Neither BCG-induced phosphorylation of ERK1/2 nor p38 MAPK was affected by IL-17A. Moreover, our data suggest that the enhanced iNOS expression in IL-17A-pre-treated, BCG-infected macrophages can be explained by enhanced iNOS mRNA stability in these macrophages. Korhonen et al.27 showed that cytokine-induced iNOS mRNA can be stabilized by a JNK signalling pathway through a tristetraprolin-dependent mechanism. The study may provide insights into the mechanism regarding our finding that IL-17A can enhance the stability of BCG-induced iNOS mRNA. Although our data indicate that NF-κB is not involved in IL-17A-enhanced iNOS expression in BCG-infected macrophages, other activated transcription factors may have been involved. The activation of JNK follows the conventional MAPK signalling cascades which involve sequential activations of MAPKK kinases (e.g. MEKK and TAK1) and MAPK kinases (e.g. MKK4 and MKK7). Following phosphorylation by its upstream MAPK kinases, JNK activates its downstream transcription factors such as Elk1 and AP-1.47,48 Of these, AP-1 has been shown to mediate the expression of iNOS in macrophages and epithelial cells stimulated by lipopolysaccharide.49,50 Therefore, it will be interesting to assess, in the presence of IL-17A, whether JNK is able to up-regulate the activity of AP-1, which eventually leads to enhancement of iNOS expression in BCG-infected macrophages.
Pro-inflammatory cytokines such as IFN-γ and tumour necrosis factor-α have been demonstrated to facilitate the clearance of intracellular mycobacteria in macrophages through NO-dependent killing.13,18,33 Our results indicated that the survival of BCG was significantly reduced in macrophages in the presence of IL-17A. Such a reduction was not associated with phagocytosis because we showed that in the presence of IL-17A, phagocytosis of BCG by macrophages was not affected. By using a specific iNOS inhibitor, we confirmed that IL-17A-enhanced clearance of intracellular BCG is NO-dependent. Our results show agreement with previous studies showing that inhibition of NO production using iNOS inhibitors is beneficial to intracellular survival of mycobacteria in macrophages.13,33 More importantly, our data revealed that IL-17A, similar to IFN-γ and tumour necrosis factor-α, can also prime the macrophages to produce NO in response to mycobacterial infection, leading to enhanced clearance of the intracellular mycobacteria. In addition to mediating NO-dependent clearance of intracellular mycobacteria, pro-inflammatory cytokines also activate other innate defence mechanisms in macrophages during mycobacterial infection. Recently, our group has demonstrated that treatment of primary human macrophages with IFN-γ results in the induction of autophagy,51 a self-digestion process that not only controls the homeostasis of cellular organelles but also contributes to the inhibition of intracellular survival of mycobacteria.52–54 Although our current data suggest that IL-17A is not involved in the initial phagocytosis during BCG infection, the intracellular processing (e.g. formation of autophagosome) of phagocytosed bacteria in the presence of IL-17A remains to be elucidated. Furthermore, a study carried out by Herbst et al.55 has demonstrated that NO is required for the induction of apoptosis in IFN-γ-activated macrophages derived from the bone marrow of mouse. The NO-dependent induction of apoptosis contributes to growth restriction of both BCG and M. tuberculosis inside the macrophages. It will be interesting to investigate if IL-17A can mediate similar mechanisms in macrophages during mycobacterial infection.
In summary, our present study has described the role of IL-17A in modulating the innate defence mechanism of macrophages. We showed that both BCG-induced iNOS expression and NO production in macrophages can be synergistically enhanced by IL-17A. The underlying mechanism regarding such enhancement involves specific up-regulation on JNK phosphorylation by IL-17A. Most importantly, our study confirmed a role for IL-17A in enhancing the clearance of intracellular mycobacteria by macrophages through an NO-dependent killing mechanism (summarized in Fig. 7). Given that NO is a potent innate defence mechanism against not only mycobacteria but also other intracellular pathogens including Klebsiella pneumoniae, Salmonella typhimurium and Leishmania major,39,56,57 it is possible that IL-17A may contribute to control of pathogenesis of these pathogens.
Figure 7.
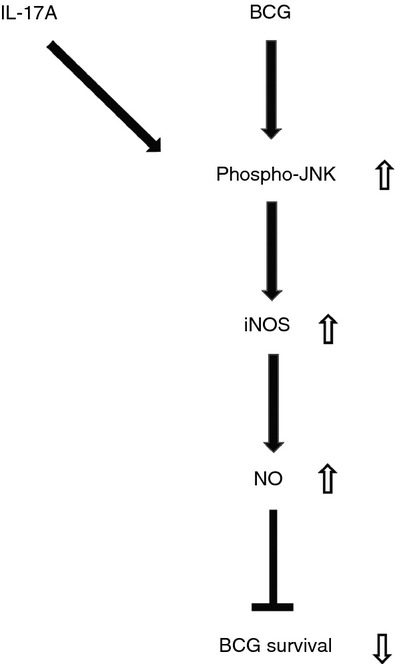
Schematic diagram representing anti-mycobacterial mechanism of interleukin-17A (IL-17A). In response to bacillus Calmette–Guérin (BCG) infection, activated macrophages express of inducible nitric oxide synthase (iNOS) and produce nitric oxide (NO). We propose that IL-17A enhances BCG-induced phosphorylation of Jun N-terminal kinase (JNK), which in turn up-regulates the expression of iNOS and hence production of NO. Enhanced NO production eventually facilitates the clearance of intracellular survival of BCG.
Acknowledgments
This work was supported by grants to JCBL and ASYL from the Research Fund for the Control of Infectious Disease (09080542), Department of Health and Welfare Bureau (Hong Kong). WLL is the recipient of a postgraduate studentship from the University of Hong Kong. We thank Ms Mei Fang for her technical support.
Authorship
WLL designed and performed the experiments, analysed the data and wrote the manuscript. WLL, LJW, JCHP and JCBL contributed significantly to experimental design, interpretation of the data and revision of the manuscript. JCBL and ASYL initiated the study, supervised the team, designed experiments and critically revised the manuscript. All authors have read and approved the final version of the manuscript.
Disclosure
The authors declare no conflict of interest.
References
- 1.Checkley AM, McShane H. Tuberculosis vaccines: progress and challenges. Trends Pharmcol Sci. 2011;32:601–6. doi: 10.1016/j.tips.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 2.Rodrigues LC, Mangtani P, Abubakar I. How does the level of BCG vaccine protection against tuberculosis fall over time? BMJ. 2011;343:5974. doi: 10.1136/bmj.d5974. [DOI] [PubMed] [Google Scholar]
- 3.Johnson R, Streicher EM, Louw GE, Warren RM, van Helden PD, Victor TC. Drug resistance in Mycobacterium tuberculosis. Curr Issues Mol Biol. 2006;8:97–111. [PubMed] [Google Scholar]
- 4.Onishi RM, Gaffen SL. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology. 2010;129:311–21. doi: 10.1111/j.1365-2567.2009.03240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pappu R, Ramirez-Carrozzi V, Sambandam A. The interleukin-17 cytokine family: critical players in host defence and inflammatory diseases. Immunology. 2011;34:8–16. doi: 10.1111/j.1365-2567.2011.03465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Torrado E, Cooper AM. IL-17 and Th17 cells in tuberculosis. Cytokine Growth Factor Rev. 2010;21:455–62. doi: 10.1016/j.cytogfr.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okamoto Yoshida Y, Umemura M, Yahagi A, et al. Essential role of IL-17A in the formation of a mycobacterial infection-induced granuloma in the lung. J Immunol. 2010;184:4414–22. doi: 10.4049/jimmunol.0903332. [DOI] [PubMed] [Google Scholar]
- 8.Cruz A, Fraga AG, Fountain JJ, et al. Pathological role of interleukin 17 in mice subjected to repeated BCG vaccination after infection with Mycobacterium tuberculosis. J Exp Med. 2010;207:1609–16. doi: 10.1084/jem.20100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Umemura M, Yahagi A, Hamada S, et al. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette–Guérin infection. J Immunol. 2007;178:3786–96. doi: 10.4049/jimmunol.178.6.3786. [DOI] [PubMed] [Google Scholar]
- 10.Khader SA, Bell GK, Pearl JE, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–77. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 11.North RJ, Jung YJ. Immunity to tuberculosis. Annu Rev Immunol. 2004;22:599–623. doi: 10.1146/annurev.immunol.22.012703.104635. [DOI] [PubMed] [Google Scholar]
- 12.MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci USA. 1997;94:5243–8. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flesch IE, Kaufmann SH. Mechanisms involved in mycobacterial growth inhibition by γ interferon-activated bone marrow macrophages: role of reactive nitrogen intermediates. Infect Immun. 1991;59:3213–8. doi: 10.1128/iai.59.9.3213-3218.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bekker LG, Freeman S, Murray PJ, Ryffel B, Kaplan G. TNF-α controls intracellular mycobacterial growth by both inducible nitric oxide synthase-dependent and inducible nitric oxide synthase-independent pathways. J Immunol. 2001;166:6728–34. doi: 10.4049/jimmunol.166.11.6728. [DOI] [PubMed] [Google Scholar]
- 15.Chan ED, Morris KR, Belisle JT, Hill P, Remigio LK, Brennan PJ, Riches DW. Induction of inducible nitric oxide synthase-NO* by lipoarabinomannan of Mycobacterium tuberculosis is mediated by MEK1-ERK, MKK7-JNK, and NF-κB signaling pathways. Infect Immun. 2001;69:2001–10. doi: 10.1128/IAI.69.4.2001-2010.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan ED, Riches DW. IFN-γ+ LPS induction of iNOS is modulated by ERK, JNK/SAPK, and p38(MAPK) in a mouse macrophage cell line. Am J Physiol Cell Physiol. 2001;280:C441–50. doi: 10.1152/ajpcell.2001.280.3.C441. [DOI] [PubMed] [Google Scholar]
- 17.Roach TI, Barton CH, Chatterjee D, Liew FY, Blackwell JM. Opposing effects of interferon-gamma on iNOS and interleukin-10 expression in lipopolysaccharide- and mycobacterial lipoarabinomannan-stimulated macrophages. Immunology. 1995;85:106–13. [PMC free article] [PubMed] [Google Scholar]
- 18.Flesch IE, Kaufmann SH. Activation of tuberculostatic macrophage functions by γ interferon, interleukin-4, and tumor necrosis factor. Infect Immun. 1990;58:2675–7. doi: 10.1128/iai.58.8.2675-2677.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang JW, Li JC, Au KY, Yim HC, Lau AS. Interleukin-17A differentially modulates BCG induction of cytokine production in human blood macrophages. J Leukoc Biol. 2011;90:333–41. doi: 10.1189/jlb.0510311. [DOI] [PubMed] [Google Scholar]
- 20.Chan MM, Cheung BK, Li JC, Chan LL, Lau AS. A role for glycogen synthase kinase-3 in antagonizing mycobacterial immune evasion by negatively regulating IL-10 induction. J Leukoc Biol. 2009;86:283–91. doi: 10.1189/jlb.0708442. [DOI] [PubMed] [Google Scholar]
- 21.Yim HC, Li JC, Pong JC, Lau AS. A role for c-Myc in regulating anti-mycobacterial responses. Proc Natl Acad Sci USA. 2011;108:17749–54. doi: 10.1073/pnas.1104892108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan LL, Cheung BK, Li JC, Lau AS. A role for STAT3 and cathepsin S in IL-10 down-regulation of IFN-γ-induced MHC class II molecule on primary human blood macrophages. J Leukoc Biol. 2010;88:303–11. doi: 10.1189/jlb.1009659. [DOI] [PubMed] [Google Scholar]
- 23.Cheung BK, Yim HC, Lee NC, Lau AS. A novel anti-mycobacterial function of mitogen-activated protein kinase phosphatase-1. BMC Immunol. 2009;10:64–73. doi: 10.1186/1471-2172-10-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Darieva Z, Lasunskaia EB, Campos MN, Kipnis TL, Da Silva WD. Activation of phosphatidylinositol 3-kinase and c-Jun-N-terminal kinase cascades enhances NF-κB-dependent gene transcription in BCG-stimulated macrophages through promotion of p65/p300 binding. J Leukoc Biol. 2004;75:689–97. doi: 10.1189/jlb.0603280. [DOI] [PubMed] [Google Scholar]
- 25.Bennett BL, Sasaki DT, Murray BW, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA. 2001;98:13681–6. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kwak HJ, Song JS, Heo JY, Yang SD, Nam JY, Cheon HG. Roflumilast inhibits lipopolysaccharide-induced inflammatory mediators via suppression of nuclear factor-κB, p38 mitogen-activated protein kinase, and c-Jun NH2-terminal kinase activation. J Pharmacol Exp Ther. 2005;315:1188–95. doi: 10.1124/jpet.105.092056. [DOI] [PubMed] [Google Scholar]
- 27.Korhonen R, Linker K, Pautz A, Forstermann U, Moilanen E, Kleinert H. Post-transcriptional regulation of human inducible nitric-oxide synthase expression by the Jun N-terminal kinase. Mol Pharmacol. 2007;71:1427–34. doi: 10.1124/mol.106.033449. [DOI] [PubMed] [Google Scholar]
- 28.Aktan F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004;75:639–53. doi: 10.1016/j.lfs.2003.10.042. [DOI] [PubMed] [Google Scholar]
- 29.Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2:907–16. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 30.Cheung BK, Lee DC, Li JC, Lau YL, Lau AS. A role for double-stranded RNA-activated protein kinase PKR in Mycobacterium-induced cytokine expression. J Immunol. 2005;175:7218–25. doi: 10.4049/jimmunol.175.11.7218. [DOI] [PubMed] [Google Scholar]
- 31.Wang L, Yang CL, Or TC, Chen G, Zhou J, Li JC, Lau AS. Differential effects of Radix Paeoniae Rubra (Chishao) on cytokine and chemokine expression inducible by mycobacteria. Chin Med. 2011;6:14–27. doi: 10.1186/1749-8546-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Misko TP, Moore WM, Kasten TP, et al. Selective inhibition of the inducible nitric oxide synthase by aminoguanidine. Eur J Pharmacol. 1993;233:119–25. doi: 10.1016/0014-2999(93)90357-n. [DOI] [PubMed] [Google Scholar]
- 33.Chan J, Xing Y, Magliozzo RS, Bloom BR. Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J Exp Med. 1992;175:1111–22. doi: 10.1084/jem.175.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan J, Tanaka K, Carroll D, Flynn J, Bloom BR. Effects of nitric oxide synthase inhibitors on murine infection with Mycobacterium tuberculosis. Infect Immun. 1995;63:736–40. doi: 10.1128/iai.63.2.736-740.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weinberg JB, Misukonis MA, Shami PJ, et al. Human mononuclear phagocyte inducible nitric oxide synthase (iNOS): analysis of iNOS mRNA, iNOS protein, biopterin, and nitric oxide production by blood monocytes and peritoneal macrophages. Blood. 1995;86:1184–95. [PubMed] [Google Scholar]
- 36.Nicholson S, Bonecini-Almeida Mda G, Lapa e Silva JR, et al. Inducible nitric oxide synthase in pulmonary alveolar macrophages from patients with tuberculosis. J Exp Med. 1996;183:2293–302. [Google Scholar]
- 37.Nozaki Y, Hasegawa Y, Ichiyama S, Nakashima I, Shimokata K. Mechanism of nitric oxide-dependent killing of Mycobacterium bovis BCG in human alveolar macrophages. Infect Immun. 1997;65:3644–7. doi: 10.1128/iai.65.9.3644-3647.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang CH, Liu CY, Lin HC, Yu CT, Chung KF, Kuo HP. Increased exhaled nitric oxide in active pulmonary tuberculosis due to inducible NO synthase upregulation in alveolar macrophages. Eur Respir J. 1998;11:809–15. doi: 10.1183/09031936.98.11040809. [DOI] [PubMed] [Google Scholar]
- 39.Hickman-Davis JM, O'Reilly P, Davis IC, Peti-Peterdi J, Davis G, Young KR, Devlin RB, Matalon S. Killing of Klebsiella pneumoniae by human alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2002;282:944–56. doi: 10.1152/ajplung.00216.2001. [DOI] [PubMed] [Google Scholar]
- 40.Weinberg JB. Nitric oxide production and nitric oxide synthase type 2 expression by human mononuclear phagocytes: a review. Mol Med. 1998;4:557–91. doi: 10.1007/BF03401758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muhl H, Bachmann M, Pfeilschifter J. Inducible NO synthase and antibacterial host defence in times of Th17/Th22/T22 immunity. Cell Microbiol. 2011;13:340–8. doi: 10.1111/j.1462-5822.2010.01559.x. [DOI] [PubMed] [Google Scholar]
- 42.Kawanokuchi J, Shimizu K, Nitta A, Yamada K, Mizuno T, Takeuchi H, Suzumura A. Production and functions of IL-17 in microglia. J Neuroimmunol. 2008;194:54–61. doi: 10.1016/j.jneuroim.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 43.Shalom-Barak T, Quach J, Lotz M. Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-κB. J Biol Chem. 1998;273:27467–73. doi: 10.1074/jbc.273.42.27467. [DOI] [PubMed] [Google Scholar]
- 44.Attur MG, Patel RN, Abramson SB, Amin AR. Interleukin-17 up-regulation of nitric oxide production in human osteoarthritis cartilage. Arthritis Rheum. 1997;40:1050–3. doi: 10.1002/art.1780400609. [DOI] [PubMed] [Google Scholar]
- 45.Jung SB, Yang CS, Lee JS, et al. The mycobacterial 38-kilodalton glycolipoprotein antigen activates the mitogen-activated protein kinase pathway and release of proinflammatory cytokines through Toll-like receptors 2 and 4 in human monocytes. Infect Immun. 2006;74:2686–96. doi: 10.1128/IAI.74.5.2686-2696.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhatt KH, Sodhi A, Chakraborty R. Role of mitogen-activated protein kinases in peptidoglycan-induced expression of inducible nitric oxide synthase and nitric oxide in mouse peritoneal macrophages: extracellular signal-related kinase, a negative regulator. Clin Vaccine Immunol. 2011;18:994–1001. doi: 10.1128/CVI.00541-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22:240–73. doi: 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 49.Kiemer AK, Muller C, Vollmar AM. Inhibition of LPS-induced nitric oxide and TNF-α production by α-lipoic acid in rat Kupffer cells and in RAW 264.7 murine macrophages. Immunol Cell Biol. 2002;80:550–7. doi: 10.1046/j.1440-1711.2002.01124.x. [DOI] [PubMed] [Google Scholar]
- 50.Kristof AS, Marks-Konczalik J, Moss J. Mitogen-activated protein kinases mediate activator protein-1-dependent human inducible nitric-oxide synthase promoter activation. J Biol Chem. 2001;276:8445–52. doi: 10.1074/jbc.M009563200. [DOI] [PubMed] [Google Scholar]
- 51.Li JC, Au KY, Fang JW, Yim HC, Chow KH, Ho PL, Lau AS. HIV-1 trans-activator protein dysregulates IFN-γ signaling and contributes to the suppression of autophagy induction. AIDS. 2011;25:15–25. doi: 10.1097/QAD.0b013e328340fd61. [DOI] [PubMed] [Google Scholar]
- 52.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–66. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 54.Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–44. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Herbst S, Schaible UE, Schneider BE. Interferon γ activated macrophages kill mycobacteria by nitric oxide induced apoptosis. PLoS ONE. 2011;6:e19105–12. doi: 10.1371/journal.pone.0019105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liew FY, Millott S, Parkinson C, Palmer RM, Moncada S. Macrophage killing of Leishmania parasite in vivo is mediated by nitric oxide from l-arginine. J Immunol. 1990;144:4794–7. [PubMed] [Google Scholar]
- 57.Vazquez-Torres A, Jones-Carson J, Mastroeni P, Ischiropoulos H, Fang FC. Antimicrobial actions of the NADPH phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. I. Effects on microbial killing by activated peritoneal macrophages in vitro. J Exp Med. 2000;192:227–36. doi: 10.1084/jem.192.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]