

Abstract
7H-Pyrrolo[3,2-f]quinazoline-1,3-diamine (1) is a privileged chemical scaffold with significant biological activities. However, the currently accessible chemical space derived from 1 is rather limited. Here we expanded the chemical space related to 1 by developing efficient methods for regioselective monoacylation at N1, N3 and N7, respectively. With this novel methodology, a focused library of mono-N-acylated pyrroloquinazoline-1,3-diamines were prepared and screened for anti-breast cancer activity. The structure-activity relationship (SAR) results showed that N3-acylated compounds were in general more potent than N1-acylated compounds while N7-acylation significantly reduced their solubility. Among the compounds evaluated, 7f possessed 8-fold more potent activity than 1 in MDA-MB-468 cells. More importantly, 7f was not toxic to normal human cells. These results suggest that 7f is a novel compound as a potential anti-breast cancer agent without harming normal cells.
Introduction
Phenotypic screening played a fundamental role in the history of pharmacology by providing new molecular entities (NME) for drug discovery and chemical probes to study biological pathways.1-4 A recent analysis of FDA-approved drugs between 1999 and 2008 showed that the phenotypic screening method bears a prominent position in discovering the first-in-class drugs in the era of modern molecular biology.5 This renaissance of phenotypic screening in drug discovery1, 6 requires our access to novel chemical space in order to sustain further discoveries.7 Privileged chemical scaffolds are potential ligands to a diverse array of receptors.8, 9 Therefore, novel chemical space derived from these scaffolds can provide the basis to discover novel bioactive compounds with drug-like properties, perhaps with a unique mechanism of action.9-13
7H-Pyrrolo[3,2-f]quinazoline-1,3-diamine (1, Figure 1) and its derivatives, originally synthesized as antifolates in the 1970s,14 have been shown to possess a variety of biological activities including antibacterial, anticancer and antiparasitic activity.14-17 Antiviral activity against herpes simplex virus (HSV) has also been reported.18 The biochemical targets for these compounds include dihydrofolate reductase (DHFR) from various species,16, 19 thrombin receptors,20, 21 and protein tyrosine phosphatase 1B (PTP1B).22, 23 The wide spectrum bioactivity of 1 is not due to the so-called “frequent hitter”24 phenotype. Instead, these bioactivities are specific because a survey of the target-based and phenotypic screening assays involving 1 in PubChem showed it was only active in 35/528 or 6.6% of the assays (Table S1),25 suggesting that this particular chemotype is a privileged scaffold being intrinsically useful for different biological targets.8, 9
Figure 1.

The chemical structures (top) of 1 and regioselectively N-acetylated 2a, 4a, 7a and their corresponding molecular electrostatic potential (MEP) surfaces (bottom). The MEP surfaces were calculated at HF/6-31G** level of theory and mapped onto their electron densities. The Mulliken atomic charges, also calculated at HF/6-31G** level of theory, on N1, N3 and N7 of 1 are indicated in the parentheses. All the surfaces were normalized from −50 kcal/mol to +50 kcal/mol.
The significant biological activities associated with 1 spurred a great deal of interest in the synthesis and biological evaluation of its derivatives.16, 20, 23, 26 However, most of the modifications were based on N7-alkylation. To the best of our knowledge, nothing is known about the chemical space related to the regioselectively mono-N-acylated pyrrolo[3,2-f]quinazoline-1,3-diamines. The regioselectively mono-N-acylated compounds are expected to significantly modulate the electrostatics of the structural core as evidenced by the dramatically different molecular electrostatic potential (MEP) surfaces, calculated at HF/6-31G** level of theory,27 of 2a, 4a and 7a compared with that of 1 (Figure 1). Since electrostatic complementarity between ligands and receptors contributes a critical component to the overall binding interaction,28-30 these differences in electrostatics may provide leads for novel biological activities with a distinct mechanism of action. The significant challenge to rapidly synthesize these compounds could be the underlying reason for this unexplored chemical space because of the presence of multiple competing reactive sites in 1. Herein, we describe our strategies to achieve regioselective mono-N-acylation of 1 at N1, N3 and N7 (Figure 1), which significantly expand the accessible chemical space derived from 1. From this focused library of new compounds, a potent and nontoxic anti-breast cancer compound 7f was discovered by a phenotypic anti-cancer screen. These results illustrate the significant biological utility of this expanded chemical space.
Results and discussion
It is predicted that the proton attached to N7 is most acidic, however, the pKas of the protons attached to N1 and N3 are probably comparable. To further investigate this point, the structure of 1 was optimized at HF/6-31G** level of theory and the Mulliken atomic charges31 were calculated.28 Consistent with the prediction, N7 is least negatively charged among the three ionizable nitrogen atoms (Figure 1). N1 is slightly less charged than N3, suggesting that the order of pKa is N7< N1≤ N3. Therefore, we speculated that N7-H could be selectively deprotonated and acylated. Indeed, when 1, prepared from a reported procedure with slight modifications in 82% yield from 5-aminoindole (see Supporting Information),32 was deprotonated by NaH followed by treatment with acetic anhydride (3a), compound 2a was obtained in 78% yield (entry 1, Table 1). The diagnostic loss of N7-H at 11.55 ppm and loss of a triplet at 7.43 ppm attributed to C8-H in 1 clearly supported that the acetyl group was attached to N7. A few different anhydrides (3b-3d) were used and the corresponding N7-acylated products were obtained in comparable yields (entries 2-4, Table 1). Due to the limited commercial availability of anhydrides and loss of an acyl equivalent during reactions with anhydrides, we investigated the utility of N-hydroxysuccinimide (NHS) esters as the acylating reagents. Both aliphatic and aromatic carboxylic NHS esters were found to react readily to give the N7 acylated compounds in good to excellent yields (entries 5-6, Table 1). The discovery of NHS esters as efficient acylating agents substantially expands the variety of N7 acylated compounds that can be prepared through this route because of easy access to carboxylic acids. In general, the N7 acylated compounds 2 are poorly soluble in common organic solvents or water. Therefore, most of the products were not purified by column chromatography, but they were all found to be >95% pure based on 1H NMR analyses (see Supporting Information). In the case of 2a-2c, the solubility was so limited that high-quality 13C NMR spectra could not be obtained.
Table 1.
Selective N7-acylation of 1.a

| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | acylating reagent | product | R | yield | |
| 1 | (CH3CO)2O | 3a | 2a | Me | 78 |
| 2 | (CH3CH2CO)2O | 3b | 2b | Et | 78 |
| 3 | (CH3CH2CH2CO)2O | 3c | 2c | Pr | 73 |
| 4 | [(CH3)2CHCO]2O | 3d | 2d | iPr | 78 |
| 5 | TBSO(CH2)4COOSu | 3e | 2e | TBSO(CH2)4 | 68 |
| 6 |
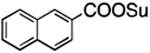
|
3f | 2f |
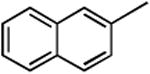
|
91 |
Compound 1 was treated with NaH (1.1 equiv) in DMF for 1 h, then an acylating reagent (1.1 equiv) was added. The yields refer to isolated yields.
N1 and N3 were predicted to be more nucleophilic than N7, however, all attempts to direct acetylation of either N1 or N3 in compound 1 with Ac2O failed to provide selectively mono-N-acetylated products. After considerable experimentation, we discovered that treatment of 2a with NaH gave N1-acetylated product 4a in 24% yield (Table 2). The regioselectivity of this reaction was confirmed by the positive nuclear Overhauser effect (NOE) between Ha and Hb, Ha and Hc observed in 4a. The mechanism for this transformation presumably involves an intermolecular acetyl transfer from N7 of one molecule to N1 of another molecule followed by cleavage of N7-acetyl group from the latter molecule. The major byproduct generated from this reaction was the deacetylated compound 1, which was isolated in 74% yield. The combined yields of 1 and 4a accounted for nearly quantitative recovery of 2a. The absence of N3-acylated product from this reaction supported the prediction of pKa order of N1< N3 (Figure 1) and illustrated that subtle differences in pKa can be synthetically exploited. All the aliphatic acylated substrates 2a-2e were successfully converted into N1-acylated products 4a-4e in 24-38% isolated yields (Table 2). In the case of aromatic acylated compound 4f, it was obtained in 29% yield existing as a 1:1 mixture of two inseparable yet clearly NMR-distinguishable tautomers 4f and 4f′ in DMSO-d6.33 Different bases exerted a great effect on the yield of this acyl transfer reaction. For example, the use of LDA gave 0% yield of 4a while 45% yield of 4a was obtained if LiHMDS was employed as a base (see Table S2). Similarly, 50% yield of 4f and 4f′ was obtained when LiHMDS was used as the base.
Table 2.
Synthesis of N1-acylated 4 from 2.a
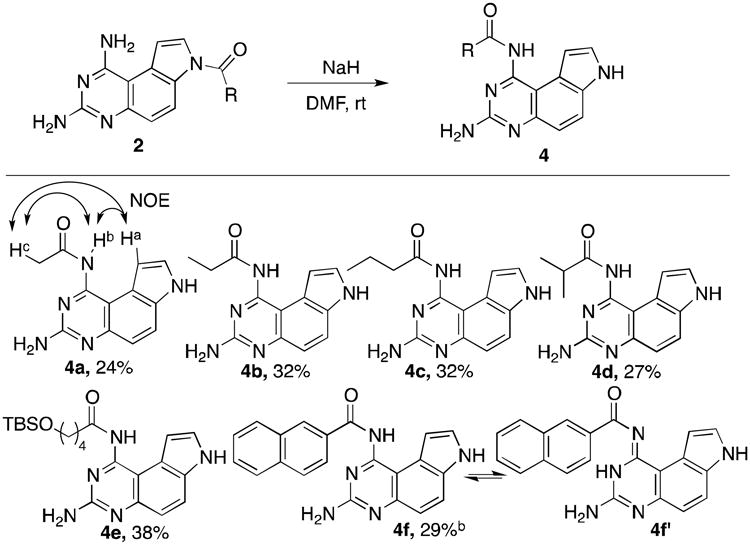
|
The reactions were carried out with compounds 2 (1.0 equiv) and NaH (1.1 equiv) in DMF. The yields refer to isolated yields.
Obtained as a 1:1 mixture of two tautomers 4f and 4f′ in DMSO-d6.
To achieve selective N3 acylation, a more elaborate and indirect scheme was designed (Table 3). The N1 amine was temporarily converted into a less nucleophilic hydroxyl group to give 5 in quantitative yield through acid hydrolysis.26, 34 Then the nucleophilic N3 in 5 was selectively acylated by treating with either an anhydride or NHS ester to generate compounds 6 in good to excellent yields (Table 3). With the acylated intermediates 6 in hand, the N1 amine was regenerated using an SNAr displacement reaction between ammonia (NH3/MeOH) and activated benzotriazole adducts generated between 6 and BOP35 to provide 7a-7f in moderate to good yields (Table 3). Therefore, the hydroxyl group in 5 served as a temporary protecting group for N1 amine. With this set of methods (Tables 1-3), we successfully completed regioselective monoacylation of N1, N3 and N7 of 1.
Table 3.
Synthesis of N3-acylated 7.
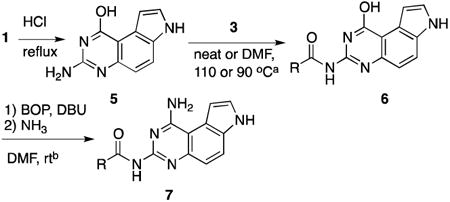
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| entry | R | acylating reagent | 6 | yieldc | 7 | yieldc |
| 1 | Me | 3a | 6a | 83 | 7a | 48 |
| 2 | Et | 3b | 6b | 84 | 7b | 44 |
| 3 | Pr | 3c | 6c | 85 | 7c | 37 |
| 4 | iPr | 3d | 6d | 67 | 7d | 40 |
| 5 | TBSO(CH2)4 | 3e | 6e | 62 | 7e | 50 |
| 6 |
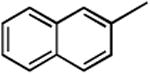
|
3f | 6f | 56 | 7f | 25 |
This step was carried out with compound 5 (1.0 equiv) and an anhydride in neat or an NHS ester (1.5 equiv) in DMF.
This operation was executed with 6 (1.0 equiv), BOP (1.3 equiv) and DBU (1.5 equiv) for 4 h. Then NH3 (7 N in MeOH) was added.
Isolated yields.
To further demonstrate the versatility of the synthetic methods for selective acylation, we investigated the possibility of selective bisacylation to prepare compounds 4a′, 8a and 8b, which are N1, N3-, N3, N7-, and N1, N7-bisacetyled, respectively (Scheme 1). We found that selective deprotonation of N7-H of 7a by NaH followed by treatment with Ac2O gave 8a in 81% yield. Similarly, compound 8b was obtained from 4a′ by NaH/Ac2O in 53% yield. As was the case for compounds 2, treatment of 8a with LiHMDS resulted in selective acetyl transfer from N7 to N1 to afford 4a, which existed as a 10:1 mixture of tautomers 4a′ and 4a″ in DMSO-d6. Finally, a synthesis of N1, N3, N7-triacetylated compound 9 was accomplished in 71% yield by heating a mixture of 1 and Ac2O at 100 °C for 2 h (Scheme 1). Compound 9 was presented as a 3.4:1 mixture of two tautomers 9 and 9 in DMSO-d6.
Scheme 1.

Synthesis of bis- and tri-acetylated compounds 4a′, 8a-8b and 9.
To illustrate the biological utility of the expanded chemical space derived from 1, we carried out a phenotypic antiproliferative assay with this focused library of new compounds in breast cancer cell lines. The N7-acylated compounds 2, 8a-8b and 9 were found to be inappropriate for biological testing because of poor solubility in DMSO or aqueous buffers. Two triple negative breast cancer (TNBC) cell lines, MDA-MB-231 and MDA-MB-468, were selected to evaluate potential anticancer activity of 4 and 7 by an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay.36, 37 TNBC represents a unique subtype of breast cancer clinically characterized by the lack of expression of estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2) and carries poor prognosis.38, 39 Current treatment options for TNBC are limited to conventional cytotoxic chemotherapeutics without much success and novel nontoxic agents are in dire need.40 The antiproliferative activities of 4 and 7 in MDA-MB-231 and MDA-MB-468 cells are presented in Table 4. The biological activities of compound 1 and an FDA-approved breast cancer drug doxorubicin (Dox) were also included for comparison. In general, compounds 7 are more potent than 4(4d-4f vs 7d-7f). In the series of N1-acylated compounds 4, the potency followed the order of 1 > 4f > 4e > 4b > 4c > 4a > 4d, while an order of 7f > 7e > 1 > 7b > 7c > 7d ≈7a was observed in the series of N3-acylated compounds 7. These potency differences indicate the complex interplay of regiochemistry, electronics and sterics in contributing to their anticancer activity. While the bulky groups in 4e-f and 7e-f rendered them more potent, the sterics per se did not predict this potency improvement because the bulky isopropyl group in 4d and 7d did not provide more benefit than the corresponding less bulky 4a-4c and 7a-7c. Instead, the opposite was observed. Compound 4a′ was the only bisacetylated compound tested for antiproliferative activity in MDA-MB-231 and MDA-MB-468 cells. However, no significant activity was observed (Table 1).
Table 4.
Antiproliferative activities of 4 and 7 in MDA-MB-231 and MDA-MB-468 cells.a
| compd | GI50 (μM) | compd | GI50 (μM) | ||
|---|---|---|---|---|---|
|
|
|
||||
| MDA-MB-231 | MDA-MB-468 | MDA-MB-231 | MDA-MB-468 | ||
| 4a | 32.62 ± 13.97 | 56.46 ± 17.97 | 7a | 27.17 ± 11.40 | 53.54 ± 29.54 |
| 4b | 18.66 ± 2.24 | 20.28 ± 7.40 | 7b | 21.43 ± 9.86 | 24.41 ± 3.33 |
| 4c | 26.86 ± 12.90 | 26.27 ± 4.83 | 7c | 25.52 ± 9.93 | 27.37 ± 4.52 |
| 4d | >100 | >100 | 7d | 39.66 ± 22.46 | 29.80 ± 8.41 |
| 4e | 15.64 ± 7.66 | 11.11 ± 1.73 | 7e | 2.43 ± 0.13 | 2.24 ± 0.40 |
| 4f | 8.46 ± 2.56 | 8.91 ± 1.28 | 7f | 1.60 ± 0.51 | 0.44 ± 0.14 |
| 4a′ | >100 | >100 | 1 | 4.13 ± 0.54 | 3.34 ± 0.93 |
| Dox | 0.15 ± 0.57 | 0.085 ± 0.029 | |||
The GI50 values, which represent the concentrations needed to inhibit the growth of the cancer cells by 50% during a 72 h incubation period, are presented as mean ± SD of at least two independent experiments performed in duplicates. If the GI50 did not reach at the highest concentration tested at 100 μM, it is designated as >100.
Among the tested compounds, 7f displayed superior activity in both MDA-MB-231 (GI50 = 1.60 μM) and MDA-MB-468 (GI50 = 0.44 μM) cells and was ∼3-8-fold more potent than 1 (Table 4). One of the limitations of current therapeutics for TNBC is their toxicity to normal cells. Therefore, we investigated if 7f was toxic to normal human mammary epithelial cells (HMEC). We found that no toxicity in HMEC was observed up to 5 μM after continuous 72 h incubation (Figure 2) although some toxicity was seen at 10 μM. If the drug treatment was reduced to 24 h, no toxicity was observed even at 10 μM (Figure 2). Reduction of drug exposure time in cancer cells did not significantly decrease activity, further demonstrating 7f's selective sensitivity in cancer cells (Figure S1). In contrast, the cytotoxic chemotherapeutic agent Dox started to present toxic effect in HMEC even at 0.1 μM (Figure 2). These results suggest that 7f is a potential novel nontoxic anti-TNBC agent.
Figure 2.

Compound 7f was not toxic to normal human mammary epithelial cells (HMEC). HMEC cells were treated with different concentrations of Dox or 7f for either 24 h or 72 h. For those cells treated with drugs for 24 h, the cells were further incubated in drug-free media for 48 h. At the end of the 72 h incubation period, the number of viable cells was quantified by an MTT assay. The data are presented as mean ± SD of a representative experiment performed in triplicates.
Compound 1 was known to be a human DHFR inhibitor, which is a potential mechanism contributing to its antiproliferative activity in TNBC cells.16, 19 Based on the orientation of an N7-alkylated 1 bound to DHFR from Candida albicans and its structural similarity to human DHFR,41 it was predicted that the bulky naphthyl group in 7f would not be accommodated in the human DHFR binding pocket (Figure S2). Indeed, we found that 7f did not inhibit human DHFR up to 10 μM concentration (Figure S2), indicating that the potent antiproliferative activity of 7f in TNBC cells is independent of DHFR inhibition. We recently showed that small molecule inhibitors of CREB (cyclic-AMP response element binding protein) are potential novel anticancer agents.36, 42 Therefore, we also tested if 7f was able to inhibit CREB-mediated gene transcription using a CREB reporter assay in HEK 293T cells.43 However, no inhibition was observed up to 10 μM concentration (Figure S3), excluding the CREB pathway as a potential target of 7f.
Conclusions
In conclusion, we have developed efficient methods to selectively monoacylate N1, N3 and N7 of compound 1. The distinct differences in the nucleophilicity and pKa of the nitrogen atoms were exploited to achieve regioselectivity. These methods significantly expand the accessible chemical space of this privileged chemical scaffold for phenotypic screening. Similar strategies should be applicable to other heterocycles with multiple potentially competing reactive sites. To demonstrate the biological utility of the newly developed chemical space, a phenotypic anti-cancer screen identified 7f as a potent and nontoxic anti-breast cancer agent independent of DHFR or CREB inhibition.
Experimental section
General
The solvents used for each reaction were purified from the Glass Contout solvent purification system. All the other chemicals were used as provided without further purification unless otherwise stated. Melting points were determined in capillary tubes using Mel-Temp and are uncorrected. NMR spectra were recorded at 400 MHz (1H NMR) and 100 MHz (13C NMR). Chemical shifts (δ) are reported in ppm relative to the residual CHCl3 (1H, 7.26 ppm, 13C, 77.0 ppm) or DMSO (1H, 2.50 ppm, 13C, 39.5 ppm). The following abbreviations were used to describe the splitting pattern of individual peaks if applicable: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. The coupling constants (J) were reported in Hertz (Hz). Silica gel flash chromatography was performed using 230– 400 mesh silica gel (EMD). The mass spectra were obtained from an LTQ Orbitrap Discovery mass spectrometer (Thermo Scientific, West Palm Beach, FL) with electrospray operated either in positive or negative mode. All final compounds were confirmed to be of >95% purity based on HPLC (Waters) analysis using an XBridge C18 column (4.6 × 150 mm) and detected at 254 nm (due to the poor solubility, compound 2a-2d were not evaluated by HPLC). The mobile phases for HPLC are water and acetonitrile, both of which contain 0.1% TFA (for compounds 2e, 4e, 7e which contain a TBS group, 0.01% TFA was used due to instability of these compounds in 0.1% TFA).
Representative procedure for N7-acylation: synthesis of (1,3-Diamino-7H-pyrrolo[3,2-f]quinazolin-7-yl)ethanone (2a)
NaH (20.0 mg, 60% in mineral oil, 0.50 mmol) was added to a stirred solution of 1 (90.0 mg, 0.45 mmol) in dry DMF (5 mL) at 25 °C under Ar atmosphere. The reaction mixture was stirred for 1 h, when Ac2O (47.3 L, 0.50 mmol) was added and the mixture was stirred for another 3 h. The solvent was removed and the residue was treated with water. The solid was collected by filtration and dried in vacuum. Then it was treated with DCM (2 mL) and collected by filtration to give the desired product 2a as a yellowish solid (85.0 mg, 78%): mp 202-204 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.56 (d, J = 8.8 Hz, 1 H), 7.99 (d, J = 3.6 Hz, 1 H), 7.39 (d, J = 4.0 Hz, 1 H), 7.18 (d, J = 9.2 Hz, 1 H), 6.90 (s, 2 H), 5.92 (s, 2 H), 2.69 (s, 3 H); HRMS (ESI) Calcd for C12H12N5O+ (M + H)+ 242.10364; Found 242.10313.
Representative procedure for N1-acylation: synthesis of N-(3-Amino-7H-pyrrolo[3,2-f]quinazolin-1-yl)acetamide (4a)
Method A: NaH (6.0 mg, 0.15 mmol) was added to a stirred suspension of 2a (33 mg, 0.137 mmol) in dry DMF (3 mL) under argon atmosphere at 25 °C. The reaction mixture was stirred at 25 °C for 2 h, when a few drops of water was added to quench the reaction. Then the solvents were removed and the residue was purified by column chromatography on silica gel, eluting with 15:1 DCM:MeOH containing 1% DIPEA to give a sticky solid, which was treated with water (1 mL) at room temperature for 1 h. Then the solid was collected by filtration to give the desired product 4a (8.0 mg, 23%) as a yellowish solid. Method B: A suspension of 2a (40 mg, 0.166 mmol) in dry DMF under argon atmosphere was cooled to 0 °C. LiHMDS (182 μL, 1 M, 0.182 mmol) was added dropwise to the suspension. The reaction mixture was stirred for 40 min at 0 °C and then a few drops of water was added to quench the reaction. Then the solvents were removed in vacuo and the residue was purified by column chromatography on silica gel, eluting with 15:1 DCM:MeOH containing 1% DIPEA to give compound 4a (18 mg, 45%) as a yellowish solid: mp 236-238 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.61 (s, 1 H), 10.22 (s, 1 H), 7.80 (d, J = 8.8 Hz, 1 H), 7.43 (brs, 1 H), 7.17 (d, J = 8.8 Hz, 1 H), 6.70 (brs, 1 H), 6.38 (s, 2 H), 2.20 (s, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 169.7, 159.0, 157.0, 152.1, 130.6, 124.8, 121.2, 119.9, 118.8, 109.2, 103.5, 23.5; HRMS (ESI) Calcd for C12H12N5O+ (M + H)+ 242.10364; Found 242.10361.
3-Amino-7H-pyrrolo[3,2-f]quinazolin-1-ol (5)
A suspension of 1 (200 mg, 1.0 mmol) in 6 N aqueous HCl (10.0 mL) was heated under reflux for overnight. The reaction mixture was adjusted to pH = 10-11 with 10.0 N NaOH and the resulting black solution was stirred for 1 h. Then the pH was adjusted to 6-7 and the precipitate was collected by filtration to give the desired product 5 (200 mg, 99%) as a brown solid: mp 284-286 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.34 (s, 1 H), 10.95 (brs, 1 H), 7.64 (d, J = 8.8 Hz, 1 H), 7.41 (t, J = 2.4 Hz, 1 H), 7.10 (s, 1 H), 6.97 (d, J = 8.4 Hz, 1 H), 6.17 (s, 2 H); 13C NMR (100 MHz, DMSO-d6) δ 162.7, 150.9, 142.9, 131.8, 127.0, 123.9, 119.2, 115.2, 107.6, 102.5; HRMS (ESI) Calcd for C10H9N4O+ (M + H)+ 201.07709; Found 201.07708.
Representative procedure for N3-acylation: synthesis of N-(1-Hydroxy-7H-pyrrolo[3,2-f]quinazolin-3-yl)acetamide (6a)
A suspension of 5 (50 mg, 0.25 mmol) in Ac2O (3 mL) was stirred at 110 °C for 1.5 h. The reaction mixture was cooled to room temperature. The excess of acetic anhydride was removed and the residue was treated with DCM (3 mL). The solid was collected by filtration to give the desired product 6a (50 mg, 83%) as a brown solid, which was used for the next step without further purification: mp 266-268 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.95 (s, 1 H), 11.61 (s, 1 H), 11.49 (s, 1 H), 7.83 (d, J = 8.4 Hz, 1 H), 7.55 (t, J = 2.8 Hz, 1 H), 7.23-7.20 (m, 2 H), 2.17 (s, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 173.4, 160.4, 144.8, 144.7, 132.8, 127.3, 123.7, 119.6, 119.0, 111.1, 103.0, 23.8;HRMS (ESI) Calcd for C12H9N4O2− (M - H)− 241.07310; Found 241.07294.
Representative procedure for NN3-acylation: synthesis of N-(1-Amino-7H-pyrrolo[3,2-f]quinazolin-3-yl)acetamide (7a)
BOP (83.1 mg, 0.188 mmol) and DBU (32.3 L, 0.216 mmol) were added to a stirred solution of 6a (35.0 mg, 0.144 mmol) in dry DMF (3 mL). The resulting reaction mixture was stirred for 4 h, when NH3 (7 N in MeOH, 0.82 mL, 5.7 mmol) was added. The reaction mixture was stirred at room temperature for 16 h. The solvents were removed and the residue was purified by column chromatography on silica gel, eluting with 1.5:1 EtOAc:THF containing 1% DIPEA to give a yellow solid, which was further treated with DCM (2 mL) and collected by filtration to give the desired compound 7a (17.0 mg, 48%) as a yellowish solid: mp 260-262 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.79 (s, 1 H), 9.76 (s, 1 H), 7.84 (d, J = 8.4 Hz, 1 H), 7.56 (brs, 1 H), 7.26 (d, J = 8.8 Hz, 1 H), 7.23 (brs, 1 H), 7.11 (brs, 2 H), 2.25 (s, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 169.7, 161.9, 152.5, 148.4, 131.9, 125.6, 120.0, 119.6, 119.4, 102.5, 24.6; HRMS (ESI) Calcd for C12H12N5O+ (M + H)+ 242.10364; Found 242.10359.
Synthesis of N-(7-Acetyl-1-amino-7H-pyrrolo[3,2-f]quinazolin-3-yl)acetamide (8a)
NaH (40 mg, 60% in mineral oil, 1.0 mmol) was added to a stirred solution of 7a (220 mg, 0.91 mmol) in dry DMF (25 mL) at 25 °C under Ar atmosphere. The reaction mixture was stirred for 1 h, when Ac2O (95 L, 1.0 mmol) was added and the mixture was stirred for another 1 h. The white solid was collected by filtration and washed with DCM (2 mL). Then the solid was treated with H2O (6 mL). The solid was collected by filtration to give 8a as a gray solid (210 mg, 81%): mp 261-263 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.84 (s, 1 H), 8.73 (d, J = 9.2 Hz, 1 H), 8.12 (d, J = 4.0 Hz, 1 H), 7.56 (d, J = 4.0 Hz, 1 H), 7.46 (d, J = 9.2 Hz, 1 H), 7.38 (brs, 2 H), 2.73 (s, 3 H), 2.28 (s, 3 H); HRMS (ESI) Calcd for C14H14N5O2+ (M + H)+ 284.11420; Found 284.11410.
Synthesis of N,N′-(7H-Pyrrolo[3,2-f]quinazoline-1,3-diyl)diacetamide (4a) and N-(3-acetamido-2,7-dihydro-1H-pyrrolo[3,2-f]quinazolin-1-ylidene)acetamide (4a″)
A suspension of 8a (94 mg, 0.332 mmol) in dry DMF (9.4 mL) under Ar atmosphere was cooled to 0 °C. LiHMDS (365 μL, 1 M in THF/ethylbenzene, 0.365 mmol) was added dropwise to the suspension. The reaction mixture was stirred at 0 °C for 40 min, when a few drops of water was added to the reaction mixture to quench the reaction. The solvents were removed in vacuo and the residue was purified by column chromatography on silica gel, eluting with 2:1-1:1 DCM:THF containing 1% DIPEA to give a yellowish compound (40 mg, 43%), which existed as a 10:1 tautomeric mixture of 4a′ and 4a″ in DMSO-d6: mp 220-222 °C. 1H NMR (400 MHz, DMSO-d6) δ 15.60 (s, 0.1 H), 11.90 (s, 1 H), 11.74 (s, 0.1 H), 11.49 (s, 0.1 H), 10.49 (s, 1 H), 10.46 (s, 1 H), 8.03 (d, J = 8.8 Hz, 1 H), 7.93 (d, J = 8.8 Hz, 0.1 H), 7.68 (t, J = 2.0 Hz, 0.1 H), 7.59 (t, J = 2.4 Hz, 1.1 H), 7.47 (d, J = 8.8 Hz, 1 H), 7.26 (d, J = 8.4 Hz, 0.1 H), 6.88 (brs, 1 H), 2.30 (s, 3 H), 2.29 (s, 0.3 H), 2.24 (s, 3 H), 2.16 (s, 0.3 H); 13C NMR (100 MHz, DMSO-d6) δ 184.5, 171.8, 170.2, 169.2, 156.9, 154.9, 151.8, 150.5, 145.7, 142.6, 133.0, 132.3, 126.8, 125.8, 123.1, 122.1, 120.9, 119.8, 119.3, 118.9, 111.6, 110.3, 105.9, 104.3, 28.8, 24.5, 23.8; HRMS (ESI) Calcd for C14H14N5O2+ (M + H)+ 284.11420; Found 284.11412.
Synthesis of N-(7-Acetyl-3-amino-7H-pyrrolo[3,2-f]quinazolin-1-yl)acetamide (8b)
NaH (4.4 mg, 60% in mineral oil, 0.11 mmol) was added to a stirred solution of 4a (24.1 mg, 0.1 mmol) in dry DMF (1 mL) at 25 °C under Ar atmosphere. The reaction mixture was stirred for 1 h, when Ac2O (11 L, 0.11 mmol) was added and the mixture was stirred for another 3 h. The solvent was removed and the residue was treated with THF (1.5 mL). The solid was collected by filtration and then treated with H2O (1.5 mL). The solid was collected by filtration to give 8b as a yellowish solid (15.0 mg, 53%): mp 232-234 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1 H), 8.69 (d, J = 9.2 Hz, 1 H), 7.94 (d, J = 3.6 Hz, 1 H), 7.36 (d, J = 9.2 Hz, 1 H), 6.90 (d, J = 2.8 Hz, 1 H), 6.68 (s, 2 H), 2.70 (s, 3 H), 2.22 (s, 3 H); 13C NMR (100 MHz, DMSO-d6) δ 170.1, 159.8, 158.0, 152.7, 129.7, 127.2, 124.4, 123.4, 121.9, 108.8, 108.7, 23.9, 23.7; HRMS (ESI) Calcd for C14H14N5O2+ (M + H)+ 284.11420; Found 284.11403.
Synthesis of N,N′-(7-Acetyl-7H-pyrrolo[3,2-f]quinazoline-1,3-diyl)diacetamide (9) and N-(3-acetamido-7-acetyl-2,7-dihydro-1H-pyrrolo[3,2-f]quinazolin-1-ylidene)acetamide (9′)
A mixture of 1 (100 mg, 0.5 mmol) and Ac2O (189 μL, 2.0 mmol) in DMF (6 mL) was stirred at 100 °C for 2 h. Then the reaction mixture was cooled to room temperature and the solid was collected by filtration, washed with DCM (2 mL) to give a yellowish compound (115 mg, 71%), which existed as a 3.4:1 tautomeric mixture of 9 and 9′ in DMSO-d6: mp > 300 °C. 1H NMR (400 MHz, DMSO-d6) δ 15.52 (s, 0.3 H), 11.60 (s, 0.3 H), 10.80 (s, 1 H), 10.61 (s, 1 H), 8.91 (d, J = 9.2 Hz, 1 H), 8.80 (d, J = 8.8 Hz, 0.3 H), 8.09 (d, J = 4.0 Hz, 1.3 H), 7.95 (d, J = 4.0 Hz, 0.3 H), 7.70 (d, J = 9.2 Hz, 1 H), 7.45 (d, J = 9.2 Hz, 0.3 H), 7.05 (d, J = 3.6 Hz, 1 H), 2.74 (s, 3 H), 2.73 (s, 1 H), 2.32 (s, 3 H), 2.31 (s, 1 H), 2.26 (s, 3 H), 2.17 (s, 1 H); HRMS (ESI) Calcd for C16H16N5O3+ (M + H)+ 326.12477; Found 326.12494.
Supplementary Material
Acknowledgments
This work was partially supported by NIH (R01GM087305) and Susan G. Komen for the Cure. We thank Dr. Andrea DeBarber (OHSU) for expert mass spectroscopic analyses.
Footnotes
Electronic supplementary information. Additional experimental procedures, supporting Figures, 1H NMR and 13C NMR spectra.
References
- 1.Lee JA, Uhlik MT, Moxham CM, Tomandl D, Sall DJ. J Med Chem. 2012;55:4527–4538. doi: 10.1021/jm201649s. [DOI] [PubMed] [Google Scholar]
- 2.Burdine L, Kodadek T. Chem Biol. 2004;11:593–597. doi: 10.1016/j.chembiol.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Cisar JS, Cravatt BF. J Am Chem Soc. 2012;134:10385–10388. doi: 10.1021/ja304213w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang D, Wang J, Bonamy GM, Meeusen S, Brusch RG, Turk C, Yang P, Schultz PG. Angew Chem Int Ed Engl. 2012;51:9302–9305. doi: 10.1002/anie.201204589. [DOI] [PubMed] [Google Scholar]
- 5.Swinney DC, Anthony J. Nat Rev Drug Discov. 2011;10:507–519. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- 6.Kotz J. SciBX. 2012;5 doi: 10.1038/scibx.2012.1380. [DOI] [Google Scholar]
- 7.Ziegler S, Pries V, Hedberg C, Waldmann H. Angew Chem Int Ed Engl. 2013;52:2744–2792. doi: 10.1002/anie.201208749. [DOI] [PubMed] [Google Scholar]
- 8.Evans BE, Rittle KE, Bock MG, DiPardo RM, Freidinger RM, Whitter WL, Lundell GF, Veber DF, Anderson PS, Chang RSL, Lotti VJ, Cerino DJ, Chen TB, Kling PJ, Kunkel KA, Springer JP, Hirshfield J. J Med Chem. 1988;31:2235–2246. doi: 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- 9.Welsch ME, Snyder SA, Stockwell BR. Curr Opin Chem Biol. 2010;14:347–361. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serrao E, Debnath B, Otake H, Kuang Y, Christ F, Debyser Z, Neamati N. J Med Chem. 2013;56:2311–2322. doi: 10.1021/jm301632e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mei ZW, Wang L, Lu WJ, Pang CQ, Maeda T, Peng W, Kaiser M, El Sayed I, Inokuchi T. J Med Chem. 2013;56:1431–1442. doi: 10.1021/jm300887b. [DOI] [PubMed] [Google Scholar]
- 12.Schimer J, Cigler P, Vesely J, Grantz Saskova K, Lepsik M, Brynda J, Rezacova P, Kozisek M, Cisarova I, Oberwinkler H, Kraeusslich HG, Konvalinka J. J Med Chem. 2012;55:10130–10135. doi: 10.1021/jm301249q. [DOI] [PubMed] [Google Scholar]
- 13.Ding K, Lu Y, Nikolovska-Coleska Z, Qiu S, Ding Y, Gao W, Stuckey J, Krajewski K, Roller PP, Tomita Y, Parrish DA, Deschamps JR, Wang S. J Am Chem Soc. 2005;127:10130–10131. doi: 10.1021/ja051147z. [DOI] [PubMed] [Google Scholar]
- 14.Ledig KW. US4118561. U S Pat. 1978
- 15.Gamo FJ, Sanz LM, Vidal J, de Cozar C, Alvarez E, Lavandera JL, Vanderwall DE, Green DV, Kumar V, Hasan S, Brown JR, Peishoff CE, Cardon LR, Garcia-Bustos JF. Nature. 2010;465:305–310. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 16.Kuyper LF, Baccanari DP, Jones ML, Hunter RN, Tansik RL, Joyner SS, Boytos CM, Rudolph SK, Knick V, Wilson HR, Caddell JM, Friedman HS, Comley JC, Stables JN. J Med Chem. 1996;39:892–903. doi: 10.1021/jm9505122. [DOI] [PubMed] [Google Scholar]
- 17.Li Q, Kozar MP, Shearer TW, Xie LH, Lin AJ, Smith KS, Si Y, Anova L, Zhang J, Milhous WK, Skillman DR. Antimicrob Agents Chemother. 2007;51:2898–2904. doi: 10.1128/AAC.00932-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dicker IB, Blasecki JW, Seetharam S. Antiviral Res. 1995;28:213–224. doi: 10.1016/0166-3542(95)00049-r. [DOI] [PubMed] [Google Scholar]
- 19.McCormack JJ, Allen BA, Ledig KW, Hillcoat BL. Biochem Pharmacol. 1979;28:3227–3229. doi: 10.1016/0006-2952(79)90066-2. [DOI] [PubMed] [Google Scholar]
- 20.Ahn HS, Arik L, Boykow G, Burnett DA, Caplen MA, Czarniecki M, Domalski MS, Foster C, Manna M, Stamford AW, Wu Y. Bioorg Med Chem Lett. 1999;9:2073–2078. doi: 10.1016/s0960-894x(99)00339-x. [DOI] [PubMed] [Google Scholar]
- 21.Nadal-Wollbold F, Bocquet A, Bourbon T, Letienne R, Le Grand B. Eur J Pharmacol. 2010;644:188–194. doi: 10.1016/j.ejphar.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 22.Berthel SJ, Cheung AWH, Kim K, Thakkar KC, Yun W. WO 2004101568. PCT Int Appl. 2004
- 23.Cheung AW, Banner B, Bose J, Kim K, Li S, Marcopulos N, Orzechowski L, Sergi JA, Thakkar KC, Wang BB, Yun W, Zwingelstein C, Berthel S, Olivier AR. Bioorg Med Chem Lett. 2012;22:7518–7522. doi: 10.1016/j.bmcl.2012.10.035. [DOI] [PubMed] [Google Scholar]
- 24.Che J, King FJ, Zhou B, Zhou Y. J Chem Inf Model. 2012;52:913–926. doi: 10.1021/ci300005y. [DOI] [PubMed] [Google Scholar]
- 25.http://pubchem.ncbi.nlm.nih.gov/ accessed on Nov 26 2012.
- 26.Guan J, Zhang Q, O'Neil M, Obaldia N, 3rd, Ager A, Gerena L, Lin AJ. Antimicrob Agents Chemother. 2005;49:4928–4933. doi: 10.1128/AAC.49.12.4928-4933.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang M, Li BX, Xie F, Delaney F, Xiao X. J Med Chem. 2012;55:4020–4024. doi: 10.1021/jm300043c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiao X, Antony S, Pommier Y, Cushman M. J Med Chem. 2005;48:3231–3238. doi: 10.1021/jm050017y. [DOI] [PubMed] [Google Scholar]
- 29.Muzet N, Guillot B, Jelsch C, Howard E, Lecomte C. Proc Natl Acad Sci. 2003;100:8742–8747. doi: 10.1073/pnas.1432955100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kangas E, Tidor B. J Phys Chem B. 2001;105:880–888. [Google Scholar]
- 31.Mulliken RS. J Chem Phys. 1955;23:1833–1840. [Google Scholar]
- 32.Jones ML, Kuyper LF, Styles VL, Caddell JM. J Heterocycl Chem. 1994;31:1681–1683. [Google Scholar]
- 33.The tautomeric mixture became a single tautomer 4f upon treatment with an aqueous NaOH solution (see supporting information for its NMR spectra). In addition, all the active protons in 4f disappeared in its 1H NMR due to H-D exchange with HDO generated from reaction of NaOH with DMSO-d6. For the same reason, the signals from the residual solvents in both 1H NMR and 13C NMR spectra became very complicated.
- 34.Trattner RB, Elion GB, Hitchings GH, Sharefkin DM. J Org Chem. 1964;29:2674–2677. [Google Scholar]
- 35.Wan ZK, Wacharasindhu S, Levins CG, Lin M, Tabei K, Mansour TS. J Org Chem. 2007;72:10194–10210. doi: 10.1021/jo7020373. [DOI] [PubMed] [Google Scholar]
- 36.Li BX, Yamanaka K, Xiao X. Bioorg Med Chem. 2012;20:6811–6820. doi: 10.1016/j.bmc.2012.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mosmann T. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 38.Kang SP, Martel M, Harris LN. Curr Opin Obstet Gynecol. 2008;20:40–46. doi: 10.1097/GCO.0b013e3282f40de9. [DOI] [PubMed] [Google Scholar]
- 39.The Cancer Genome Atlas Network. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shastry M, Yardley DA. Curr Opin Obstet Gynecol. 2013;25:40–48. doi: 10.1097/GCO.0b013e32835c1633. [DOI] [PubMed] [Google Scholar]
- 41.Whitlow M, Howard AJ, Stewart D, Hardman KD, Kuyper LF, Baccanari DP, Fling ME, Tansik RL. J Biol Chem. 1997;272:30289–30298. doi: 10.1074/jbc.272.48.30289. [DOI] [PubMed] [Google Scholar]
- 42.Xiao X, Li BX, Mitton B, Ikeda A, Sakamoto KM. Curr Cancer Drug Targets. 2010;10:384–391. doi: 10.2174/156800910791208535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li BX, Xiao X. ChemBioChem. 2009;10:2721–2724. doi: 10.1002/cbic.200900552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.