

Abstract
The glycocalyx of the cell is composed of highly hydrated saccharidic groups conjugated to protein and lipid cores. Although components of the glycocalyx are important in cell-cell interactions and other specific biological recognition events, a fundamental role of the glycocalyx is the inhibition of nonspecific interactions at the cell surface. Inspired by glycoproteins present in the glycocalyx, we describe a new class of synthetic antifouling polymer composed of saccharide containing N-substituted poly-peptide (glycopeptoid). Grafting of glycopeptoids to a solid surface resulted in a biomimetic shielding layer that dramatically reduced nonspecific protein, fibroblast and bacterial cell attachment. All-atom molecular dynamics simulation of grafted glycopeptoids revealed an aqueous interface enriched in highly hydrated saccharide residues. In comparison to saccharide-free peptoids, the interfacial saccharide residues of glycopeptoids formed a higher number of hydrogen bonds with water molecules. Moreover, these hydrogen bonds displayed a longer persistence time, which we believe contributed to fouling resistance by impeding interactions with biomolecules. Our findings suggest that the fouling resistance of glycopeptoids can be explained by the presence of both a ‘water barrier’ effect associated with the hydrated saccharide residues, as well as steric hindrance from the polymer backbone.
INTRODUCTION
Poly-N-substituted glycines, or peptoids, are peptide-mimetic macromolecules with a polyglycine backbone and side chain derivatization at the amide nitrogen instead of the α-carbon.1 In contrast to polypeptides, this structural substitution eliminates backbone chirality, prevents formation of β-sheet stabilizing hydrogen bonds, and increases protease resistance and stability in a wide range of salt, pH and solvent conditions.2,3 An important characteristic of peptoids is that they can be synthesized on automated peptide synthesizer in a sequence-specific manner with precisely controlled length and diverse natural or non-natural side chains. This feature permits the design of sequence-specific molecules useful in fundamental studies of protein folding and in a variety of biomedical applications.4,5 Biologically active peptoids have been investigated as mimics of HIV-Tat proteins, lung surfactant protein, and antibacterial peptides.6 Peptoids also have been explored for gene therapy, genetic analysis,7 bioseparations,8 and as ligands for cancer targeting.9
Biofouling involves the accumulation of biomolecules, cells, micro- and macroorganisms on surfaces, and is often an undesired and irreversible event of great practical importance in industrial, military, consumer and medical settings. In the healthcare area, biofouling of medical implants, surgical devices, and biosensors can hinder biological performance, affect device longevity, and increase healthcare costs. Common strategies to prevent biofouling include surface grafting of organic thin films including self-assembled monolayers (SAMs)10 and antifouling polymer brushes such as poly(ethylene glycol) (PEG),11 poly(2-oxazone),12 block copolymers containing PEG chains,13 polysaccharides,14 and zwitterionic polymers.15 We as well as others have investigated peptoids for prevention of surface biofouling, exploring a number of alkyl, ether and hydroxyalkyl side chains.16–18
The glycocalyx is the outermost surface of the cell membrane composed of a dense layer of highly glycosylated species (e.g., glycoproteins, glycolipids, and other glycol-conjugates). The idea of mimicking the carbohydrate-rich glycocalyx of plasma cell membranes is an attractive one due to the high natural resistance of the glycocalyx toward nonspecific interactions.19 Although carbohydrates have many characteristics suitable for antifouling applications (e.g., high hydrophilicity, neutral charge, flexible backbone), only a limited number of antifouling carbohydrates, glycopolymers and carbohydrate containing monolayers have been investigated. Dextran, which is a maltose polymer, exhibits very low non-specific protein interactions, and is widely used as an alternative to poly(ethylene glycol).20–22 Dextran-oligomer modified poly(vinyl amine) also showed good resistance against protein and platelet adhesion on hydrophobic surfaces (graphite and polycarbonate).23,24 It is also known that mannose, and other oligosaccharide containing self-assembled monolayers (SAMs) exhibit good short-term protein resistance.25–28 Glycocalyx-mimetic peptoids, i.e. peptoids containing saccharide or oligosaccharide side chains, have not been explored in an antifouling context.
Here we introduce a novel antifouling glycopeptide-mimetic polymer composed of three distinct functional domains (Figure 1): a peptide anchor, a polypeptoid backbone, and oligosaccharide side chain. Glucose and β-D-maltose were chosen as the oligosaccharide side chains because they provide physicochemical properties that are known to be important in fouling resistance, namely neutral charge and high hydrophilicity.29 2′-azidoethyl-β-D maltopyranose (Mal(OH)-N3) was prepared from β-D-maltose-monohydrate as shown in Scheme 1. Then, the disaccharide was coupled to the peptoid backbone via click chemistry as guided by previous reports of glycopeptoid30 and glycomimetic polymer synthesis (Figure 1 and Figure 2).31,32 The resulting polymer was used for surface modification of TiO2 substrates and the antifouling property was evaluated by testing resistance against protein adsorption, cell adhesion, and bacterial attachment. Insight into the role of terminally linked saccharides in mediating antifouling performance was provided by molecular dynamic simulations of surface-grafted peptoids, revealing new features of the interaction of water with glycopeptoids.
Figure 1.

Solid phase synthesis of M20Glu(OH) and M20Mal(OH). First, the peptide anchor was synthesized by traditional solid phase peptide synthesis (Step 1), followed by peptoid backbone synthesis by the submonomer approach (Step 2), after which oligosaccharide was conjugated by CuAAC (Step 3).
Scheme 1.
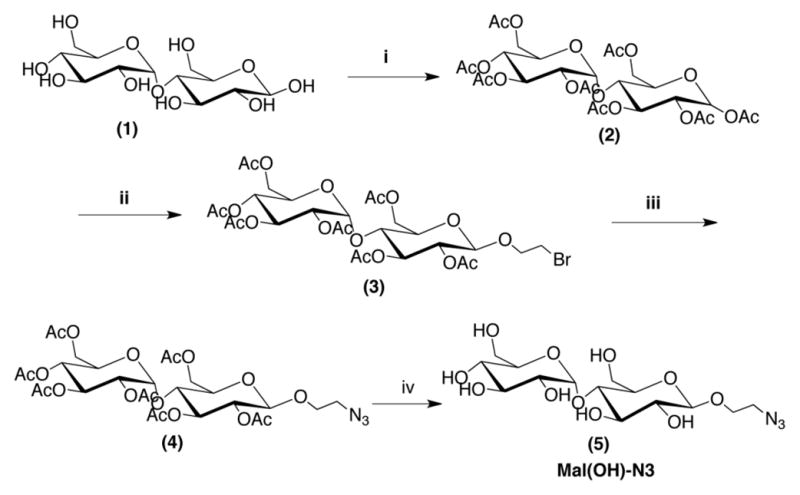
Synthesis of 2′azidoethyl-β-D-maltopyranose. i) excess Ac2O. ii) 2-bromoethanol and BF3-OEt2 in CH2Cl2. iii) NaN3 in DMF:DCM (6:1). iv) CH3ONa in CH3OH.
Figure 2.

Chemical structures of 20mer Nme peptoid (top), M20Glu(OH) (middle), and M20Mal(OH) (bottom) (a). Analytical RP-HPLC spectra (214 nm, 2–50% acetonitrile in H2O with 0.1% v/v TFA 1.0 ml/min) (b), and MALDI-TOF mass spectra of Na+ adducts (c) of purified M20Glu(OH) and M20Mal(OH).
RESULTS AND DISCUSSIONS
Characterization and evaluation of antifouling performance of glycopeptoid-coated surfaces
TiO2 was used in this study as a prototypical biomaterial that is representative of the surface composition present on titanium-based alloys used extensively in orthopedic, cardiovascular and other medical devices. The DOPA-Lys-DOPA-Lys-DOPA pentapeptide anchor is particularly well suited for grafting peptoids to TiO2 surfaces because of the strong affinity of the catechol OH groups of DOPA (pKa1 = 9.2; pKa2 = 13.0) to TiO2 (isoelectric point of TiO2 is ~ 6.2), which according to Redfern et al.33 is expected to occur through bidentate bridging bonds at pH 6.0.
The dry thickness of the M20Glu(OH) and M20Mal(OH) peptoid coatings adsorbed onto the TiO2 surfaces were measured by spectroscopic ellipsometry and the average thickness values are reported in Table 1. Although the average thickness values for M20Mal(OH)-modified surfaces appear to increase slightly compared to M20Glu(OH)-modified surfaces, the difference was not statistically significant, indicating that grafted films of M20Glu(OH) and M20Mal(OH) were of similar thicknesses.
Table 1.
Polymer thicknesses measured by spectroscopic ellipsometry, and average advancing (θa) and receding (θr) contact angles measured for bare TiO2 and polymer-modified substrates.
| Substrate | Polymer Thickness (Å) | Contact angle (°±SD)
|
|
|---|---|---|---|
| Adv. (θa) | Rec. (θa) | ||
| Bare TiO2 | n/a | 7.8 ± 1.9 | 7.2 ± 2.8 |
| M20Glu(OH) | 29.2 ± 1.0 | 34.3 ± 0.1 | 33.7 ± 0.1 |
| M20Mal(OH) | 31.4 ± 1.1 | 31.5 ± 0.2 | 29.5 ± 0.4 |
Advancing and receding contact angles for unmodified TiO2, M20Glu(OH)-, and M20Mal(OH)-modified substrates were measured and the results are summarized in Table 1. Bare TiO2 immediately after oxygen plasma cleaning was shown to be extremely hydrophilic (<10°) while both M20Glu(OH)- and M20Mal(OH)- modified substrates demonstrated advancing contact angles of 30–35°. M20Mal(OH) was slightly more hydrophilic than M20Glu(OH), presumably due to the influence of the additional hydroxyl groups present in maltose versus glucose. The contact angle values for both glycopeptoid-modified surfaces fall near or between the reported values in the literature for self-assembled monolayers with hydroxyl ter al groups on gold (~10°)34 and the Nme peptoid backbone with the same length but without oligosaccharide (~39°).35
XPS survey scans reveal the adsorption of M20Glu(OH) and M20Mal(OH) onto TiO2 surfaces after immersion of the substrate into the polymer solution. Glycopeptoid modified substrates exhibited Ti signal from the underlying TiO2 as well as C, N and O signals from the chemical species found in glycopeptoids (see Figure S1). Compared to unmodified TiO2, the decrease in Ti content and significant increase in C and N signal intensity for glycopeptoid modified substrates indicate successful modification. To calculate the atomic composition of the polymer layer, detailed high-resolution spectra were acquired at 455–467 eV for Ti(2p), 280–292 eV for C(1s), 394–406 eV for N(1s), and 524–536 eV for O(1s); the results are shown in Table 2 and Figure 3. Of particular note in the high-resolution spectra were changes in the C1s and O1s spectra upon modification with glycopeptoid. The peak at 284.6 eV that emerges after peptoid modification is due to the aliphatic and aromatic carbons in the peptoid backbone and the DOPA-Lys-DOPA-Lys-DOPA anchoring groups, as well as some hydrocarbon contamination. Dramatic changes after polymer coating occurred for binding energies in the range 286–287 eV which correspond to the ether carbons (286.6 eV) of the Nme side chains, to the carbons adjacent to amino groups (286.0 eV [-(C)H2-NH2], 286.3 eV [-NH-(C)H-C=O]) of the peptoid backbone36, the hydroxyl carbons (286.7 eV), and carbons adjacent to the amine of the triazole linker (286.0 eV) and saccharide. The peak at 288.0 eV represents the amide groups of the peptoid backbone. The nitrogen signals originate from the nitrogens in the triazole linkage, peptide/peptoid backbone, and lysine side chains. Finally, the oxygen peaks are contributed by bulk TiO2 (529.9 eV), TiOH (531.1 eV), C-OH (532.5 eV), C-O (532.7 eV), and C=O (533.6 eV).36
Table 2.
Atomic compositions of unmodified, M20Glu(OH)-, and M20Mal(OH)- modified TiO2 substrates as determined from high-resolution XPS spectra.
| Substrate | Atomic Composition (%)
|
|||
|---|---|---|---|---|
| Ti | C | N | O | |
| Bare TiO2 | 24.8 | 26.9 | n/a | 48.3 |
| M20Glu(OH) | 7.4 | 53.8 | 9.2 | 29.5 |
| M20Mal(OH) | 8.7 | 50.3 | 10.0 | 31.0 |
Figure 3.

High-resolution XPS spectra of bare and glycopeptoid-modified TiO2, substrates. Shown in each panel are the spectral regions corresponding to Ti(2p) (a), C(1s) (b), N(1s) (c), and O(1s) (d).
As an initial antifouling performance test, resistance of the modified surfaces against fibrinogen adsorption was investigated. Both unmodified and modified TiO2 substrates were treated with buffered fibrinogen solution for 20 minutes, 24 hours, or 72 hours and the amount of adsorbed fibrinogen measured by ellipsometry. As shown in Figure 4, the fibrinogen adsorption values on both glycopeptoid-modified substrates were significantly lower than unmodified substrates for all time points. Whereas fibrinogen adsorption reached saturation (with a mass of > 500 ng/cm2) on unmodified TiO2 within 20 minutes exposure time, fibrinogen adsorption onto M20Glu(OH)- and M20Mal(OH)- modified surfaces under identical conditions was less than 1–10 % of the controls at all time points.
Figure 4.

Glycopeptoid-modified surfaces exhibit resistance to fibrinogen adsorption. Shown is the mass plot of fibrinogen adsorption at 20 min, 24 h and 72 h on unmodified and glycopeptoid-modified surfaces as measured by ellipsometry.
To further test the antifouling properties of the glycopeptoid-modified surfaces, cell adhesion studies were conducted with fibroblasts. Substrates were modified following the same procedure outlined above for protein adsorption, after which the substrates were seeded with fresh 3T3 fibroblasts suspended in serum containing media. Cell attachment was quantified from 4 hours to 7 days by live cell staining, fluorescent microscopy, and image analysis. At the initial 4 hour time point, fibroblasts attached readily to the unmodified TiO2 substrates while glycopeptoid modified substrates were all highly resistant to adhesion. A plot of % surface area covered by cells is shown in Figure 5. All glycopeptoid modified substrates remained highly resistant to fibroblast adhesion throughout the 7-day experiment, despite being challenged with fresh cells at each intermediate time point (4, 72 and 120 h). In contrast, fibroblast adhesion on unmodified TiO2 reached nearly a confluent monolayer by day 7. There was no significant difference in cell fouling resistance between M20Glu(OH) and M20Mal(OH) within the experimental time period. Low fibroblast adhesion suggests that serum protein adsorption on the glycopeptoid modified substrates remained low throughout the course of the in vitro experiment, thus indirectly confirming the results of the protein adsorption experiments, but over a longer time scale.
Figure 5.

Fibroblast adhesion on bare and glycopeptoid-modified TiO2 substrates. Representative fluorescent images from each time point measured (a) and quantified % surface area covered by fibroblasts (b).
Bacterial colonization of medical devices and implants can severely impair their performance and increase infection rates and health risks for patients.37 Three gram-positive and gram-negative bacterial strains commonly associated with medical device related infections were chosen for the static bacterial adhesion test on unmodified and glycopeptoid modified TiO2 substrates. S. epidermidis is a gram positive bacterium that is well known for its ability to colonize surfaces rapidly and is closely linked to device-associated clinical infections of the hip and urinary tract.38 E. coli is a gram negative strain of bacteria often found in surgically related infections;39 P. aeruginosa is gram negative and often associated with infections in patients with immune systems compromised by disease or trauma. The release of powerful exotoxins and endotoxins by P. aeruginosa can cause chronic and life-threatening conditions which persist even after the bacteria have been killed off by antibiotics.40
Unmodified, M20Glu(OH)-, and M20Mal(OH)-modified TiO2 substrates were exposed to static suspensions of S. epidermidis, P. aeruginosa, and E. coli for either 1 day or 4 days. Adherent bacterial cells were stained with Syto 9 and propidium iodide, and % surface area covered by bacterial cells was calculated and normalized to control substrates. Glycopeptoid modified substrates showed a 2–3 log reduction in bacterial adhesion (> 99.8%) compared to unmodified TiO2 substrates at both time points and against all three bacterial species as shown in Figure 6. For S. epidermidis in particular, it is notable that both M20Glu(OH) and M20Mal(OH) glycopeptoids demonstrated improved antifouling performance at 1 day and 4 days compared to previous findings with methoxyethyl (Nme) peptoids,18 a finding with potentially important practical implications and which suggests a fundamental difference in fouling resistance between glycopeptoids and 20mer Nme peptoids.
Figure 6.

S. epidermidis, P. aeruginosa, and E. coli adhesion to unmodified and glycopeptoid-modified TiO2 substrates after 1 day (top) and 4 day (bottom) exposure.
Molecular Dynamics (MD) of Water Interacting with Glycopeptoid Brush
It is well established that steric repulsions are responsible for the ability of grafted polymer layers to reduce protein adsorption.41–43 The term steric repulsion should be understood to include two main contributions. They are the higher osmotic pressure within the polymer layer due to the presence of the polymers and the reduction in conformational entropy of the polymer chains. Therefore increasing the polymer surface coverage reduces protein adsorption and in general working in the regime of stretched chains, so-called brush regime, provides optimal protein rejection. This concept should be applied to relatively long chains and in the regime where the conformational degrees of freedom of the polymers are accessible. In the limit of short chains and very high surface density, as those obtained in SAMs with short ethylene oxide chains or sugars, the prevention of protein adsorption has been suggested to be the result of the hydration layer around the terminal groups, the so called “water barrier”. Examples of this class of coatings include SAMs with a variety of functional groups25 including oligo(ethylene glycol),10 and charged or zwitterionic molecules.44,45 Saccharide-based SAMs also fall into this class and exhibit excellent antifouling performance.26,27,46,47 It has been speculated that the tightly bound hydration layer on saccharide containing SAMs creates a repulsive force on proteins, reducing the interaction between the protein and the surface.46,48
In our experiments, glycopeptoids were grafted on the TiO2 surface at surface chain densities that stretch the chains but are not nearly the close packed densities achieved in SAMs. Therefore, it may be expected that the ability of the peptoids to prevent protein adsorption is associated with a combination of both mechanisms. Namely, steric repulsion and hydration will combine to provide the non-fouling properties observed experimentally. Therefore understanding the role of oligosaccharide residues in suppressing protein, cell and bacterial adsorption was considered critical in understanding the antifouling mechanism of glycopeptoids.
In order to test whether the “water barrier” mechanism might play a significant role in the protein resistance of the grafted glycopeptoids, we performed extensive atomistic molecular dynamics (MD) simulations of the peptoid layer. It should be noted that experimental approaches to selectively probe the hydration layers around local functional groups are non-trivial. Therefore, all atom MD simulations on a system of 16 glycopeptoid (M20Glu(OH) or M20-Mal(OH)) chains grafted onto TiO2 at a surface chain density of 0.65 nm−2 were carried out in explicit water for 100ns. We focus on probing the physical characteristics of the first hydration layer surrounding the oligosaccharide residues in the surface-grafted glycopeptoid chains.
The proximal radial distribution functions, pG(r)49,50 of water oxygen atoms obtained from the simulations show that the thickness of the first hydration layer surrounding maltose and glucose residues is 4.4 and 4.7 Å, respectively (Figure 7a). For comparison, we also determined the pG(r) of Nme residue, for which the first hydration layer was found to be 4.7 Å thick. Using the thickness information we can calculate, for the chains grafted on TiO2 surface, the average distribution of the number of water molecules in the first hydration layer around the oligosaccharide functional groups. The results are summarized in Figure 7b. The first hydration layer contains on average 32.7 water molecules on each maltose residue, and 26.1 and 15.7 water molecules around each of the glucose and Nme residue, respectively. As the thickness of the first hydration layer was more or less the same among the residues, it is reasonable that the number of water molecules in the first hydration layer of each residue increases from Nme to glucose to maltose as the van der Waals volume of the residue increases in the same order.
Figure 7.

(a) The proximal radial distribution, pG(r), of water oxygen atoms surrounding maltose, glucose, and Nme residues, obtained from MD simulations. (b) The distribution of the average number of water oxygen atoms within the first hydration layer of maltose, glucose, and Nme, from MD simulations.
The stability of the interfacial water layer was probed by measuring the average number and the lifetime of the hydrogen bonds between the oligosaccharide residues and water molecules. The hydrogen bonds were identified by a widely-accepted geometric criterion: a pair is considered to be hydrogen bonded if the oxygen-oxygen distance is no greater than 3.5 Å, and simultaneously, the O-H…O angle is less than 30°.51 The distributions of the number of oligosaccharide-water and Nme-water hydrogen bonds are summarized in Figure 8a. The average number of hydrogen bonds is 11.8, 6.5 and 1.4 for the maltose-water, glucose-water, and Nme-water pair, respectively. The results show that the maltose residues in the surface-grafted glycopeptoids typically form the largest number of hydrogen bonds with water, the glucose-water pair about half as many, and each Nme residue in the surface-grafted Nme-20mer chains forms less than 2 hydrogen bonds with water on average. The results are consistent with the number of hydrogen bond-eligible sites available in each residue.
Figure 8.

(a) The distribution of the average number of hydrogen bonds of maltose, glucose, and Nme with water, obtained from MD simulations. (b) The autocorrelation functions of hydrogen bonds for maltose-water, glucose-water, Nme-water, acetylated glucose (Ac. Glucose)-water, and water-water pairs, calculated from MD simulations.
Perhaps the most interesting results involve the lifetime of the hydrogen bonds, which is a crucial indicator of the hydrogen bond stability. The lifetime of the hydrogen bonds was calculated from the autocorrelation function C(t) of the hydrogen bond operator h(t). The hydrogen bond autocorrelation has been used to characterize the kinetic relaxation behavior of hydrogen bonds52,53 and is defined as follows:54
Here the hydrogen bond operator h(t) = 1 if a tagged pair is hydrogen bonded at time t, and h(t) = 0 otherwise. The bracket represents the ensemble average.
Figure 8b shows the autocorrelation functions for the maltose-water and glucose-water hydrogen bonds from the glycopeptoid chains grafted on the TiO2 substrate, and for the water-water hydrogen bonds from a pure water system, calculated from the MD simulations. The autocorrelations of the hydrogen bonds between maltose residues and water as well as between the glucose residues and water for the surface-grafted glycopeptoid chains are found to decay much more slowly than the autocorrelation of water-water hydrogen bonds. The relaxation time of the hydrogen bonds, defined as the time when the autocorrelation drops to e−1, was found to be 14.1 ps for maltose-water, 14.4 ps for glucose-water, and 2.2 ps for water-water pairs, showing the relaxation times being more than 6 times larger for the maltose-water and glucose-water hydrogen bonds than for the water-water hydrogen bonds. Moreover, the long tail of the autocorrelation at longer times for the oligosaccharide residues is reminiscent of the relaxation process of disordered quenched systems such as glasses. Therefore, the hydration layers around the oligosaccharide residues are very long lived and possess glass-like dynamics which can further enhance the protein resistance by causing incoming proteins to slip and make the replacement of water molecules by protein very difficult.
The autocorrelation function of the hydrogen bonds between Nme and water, calculated from the simulation system of a 20mer Nme peptoid grafted on TiO2 and also shown in Figure 8b, decays in a similar way to those for maltose and glucose, suggesting that the Nme -water hydrogen bond in Nme-20mer system is as persistent as the maltose-water and glucose-water hydrogen bonds in glycopeptoid systems. However, the number of hydrogen bonds between Nme and water is significantly less than for the maltose-water and glucose-water pairs as shown in Figure 8a resulting, as discussed below, in a less effective protection.
In Figure 8b, we also present the autocorrelation function of the hydrogen bonds between water and the acetylated glycopeptoid (abbreviated as “Ac. Glucose”) chains grafted on TiO2 at the same surface chain density. The molecular structure of the acetylated glycopeptoid chain is identical to the M20Glu(OH) chain, except that all five hydrogen atoms in the alcohol groups in the glucose residue are substituted with acetyl group (this is essentially the “protected” form of the glycopeptoid). With no alcohol functional groups available, the acetylated glucose residue no longer has the ability to act as a hydrogen bond donor and has only a limited capability to form hydrogen bonds with water molecules as a weak acceptor via the ester oxygens. As a result, the first hydration layer of the acetylated glucose residue should be very weakly bound as corroborated by the fast-decaying autocorrelation function with the relaxation time of 3.9 ps, shown in Figure 8b. The weakly bound hydration layer of the acetylated glucose residue suggests a poor resistance to protein adsorption for the acetylated glycopeptoid chains. Indeed, our experiments confirmed that the surfaces modified with acetylated glycopeptoid chains displayed a high degree of fibrinogen adsorption at short times (experimental data not shown). These results validate the importance of hydration layers around surface-grafted molecules in determining their effectiveness to protein resistance. Further, our analysis illustrates a possibility that the autocorrelation function of hydrogen bonds might be used as a simple and effective predictor to scan for potential protein resistance for systems where steric repulsions are suppressed and hence the interfacial hydration layer is likely to make a major contribution toward fouling resistance.
Antifouling Mechanism and the Role of Water
The overall picture emerging from the above analysis of the interfacial hydration layer surrounding the terminal saccharide residues of the surface-grafted glycopeptoid chains is the following: the maltose and glucose residue carry a large number of interfacial water molecules due to their large sizes. Many of these interfacial water molecules are tightly bound to maltose and glucose residues via hydrogen bonds that have a long duration time and hence are likely to be much stronger than their water-water counterparts. The tightly bound interfacial water molecules surrounding the oligosaccharide residues mean that a protein would need a longer time to displace the interfacial water molecules surrounding the oligosaccharide residues before it can penetrate the hydration layer and interact with the grafted polymer chains, leading to a diminished possibility for protein to adsorb on the surface. This could serve as one of the major mechanisms that allow the glycopeptoid chains to effectively resist protein adsorption as observed in our experiments.
The importance of the bound water layer is clear, however, it should not be considered the only component in the non-fouling capabilities of polymer layers. The application of a molecular theory to study protein resistance for end-grafted PEG and polypetptoids has also shown the importance of the polymer conformational degrees of freedom, the deformation of the polymer layer, and the osmotic pressure repulsion to quantitatively predict protein adsorption. Therefore, we believe that it is the combination of the steric repulsions and the water layer that are responsible for protein resistance. Actually, the two effects complement each other since the long lived, bound, water molecules make the effective volume of the peptoids units large and highly hydrophilic. Therefore, the steric barrier is larger when water molecules are bound. This further explains the differences between the peptoids with sugars as compared to Nme. The latter has a smaller number of bound water and therefore, for the same surface coverage it produces a smaller steric barrier (recall that we refer to the steric barrier as the combination of conformational entropy loss and osmotic pressure).
The glycopeptoids used in this study have a 20mer N-methoxyethyl (Nme) peptoid linker that tethers the terminal oligosaccharide residue to the TiO2 surface. Nme peptoids have previously been shown to exhibit excellent antifouling properties,18,55 and predictions from the molecular theory mentioned above are in good agreement with the experimental observation,35,56 suggesting that steric repulsion is one of the main contributions to the antifouling mechanism provided by the Nme peptoid linker of the glycopeptoids.
Thus, we find that the non-fouling behavior of the glycopeptoids is the result of the combination of the steric repulsions from the 20mer Nme peptoid linker of the glycopeptoid, as well as the repulsion arising from the volume and bound water surrounding the terminal oligosaccharide residues. Although the results for the acetylated glycopeptoids indicate that the bound water may be the major mechanism of protein resistance for the glycopeptoid chains, it is not entirely clear whether the substitution of the hydroxyl groups by acetyl groups changes the bare interaction between the protein and the peptoid, resulting in strong attractions that lead to the observed adsorption.
Finally, it is important to emphasize the differences between our study and the previously proposed water barrier. The earlier simulation work describing the importance of the bound water on the non-fouling capabilities of sugars has been done on high density SAM’s and therefore the analysis is based on complete water layers parallel to the SAM.57 In our work we study the water molecules bound to the sugar functional group in the polymer layer at finite, experimentally relevant, surface coverage well below that of short oligomers SAMs. Therefore, our study shows how bound water molecules contribute to the non-fouling in addition to the steric repulsions arising from the conformational degrees of freedom of the polymers and the osmotic pressure contribution.
CONCLUSION
We reported the synthesis and characterization of a novel class of antifouling polymers that mimic the glycocalyx in both composition and ability to inhibit nonspecific fouling interactions. CuAAC and high-fidelity solid phase methods allowed the incorporation of saccharides as N-substituted side chains of a peptidomimetic polymer backbone, providing precise control of chemical composition, molecular weight, and architecture of the polymers. The glycopeptoid polymers readily adsorbed onto TiO2 substrates, conferring significant protein, cell and bacterial antifouling properties. All-atom molecular dynamics simulations were conducted to provide insight into the role of oligosaccharides in antifouling, demonstrating the existence of strongly bound water molecules at the terminal saccharide residues, which are likely to play an important role in fouling resistance through inhibition of protein-surface interactions in combination with polymer steric repulsions. In the future, more detailed explorations of chemical composition and architecture of glycopeptoids on fouling resistance may provide further insight into antifouling mechanism and inform the design of more effective glycomimetic antifouling polymers.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by NIH grant R01 EB005772.
We acknowledge Dr. Xinqi Chen for his assistance with XPS studies performed at the Keck-II/NIFTI facilities of NUANCE Center at Northwestern University. We thank Yufei Tian for technical support. NMR and Mass spectroscopy analysis were performed in the IMSERC facility at Northwestern University.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. / All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Experimental details for glycopeptoid synthesis, surface modification and characterization; additional NMR data including peak assignments, and XPS data before and after glycopeptoid modification. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Simon RJ, Kania RS, Zuckermann RN, Huebner VD, Jewell DA, Banville S, Ng S, Wang L, Rosenberg S, Marlowe CK. Proc Natl Acad Sci USA. 1992;89:9367. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kirshenbaum K, Barron AE, Goldsmith RA, Armand P, Bradley EK, Truong KT, Dill KA, Cohen FE, Zuckermann RN. Proc Natl Acad Sci USA. 1998;95:4303. doi: 10.1073/pnas.95.8.4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zuckermann RN, Kerr JM, Kent SBH, Moos WH. J Am Chem Soc. 1992;114:10646. [Google Scholar]
- 4.Nam KT, Shelby SA, Choi PH, Marciel AB, Chen R, Tan L, Chu TK, Mesch RA, Lee BC, Connolly MD, Kisielowski C, Zuckermann RN. Nat Mater. 2010;9:454. doi: 10.1038/nmat2742. [DOI] [PubMed] [Google Scholar]
- 5.Shin SB, Yoo B, Todaro LJ, Kirshenbaum K. J Am Chem Soc. 2007;129:3218. doi: 10.1021/ja066960o. [DOI] [PubMed] [Google Scholar]
- 6.Patch JA, Barron AE. Curr Opin Chem Biol. 2002;6:872. doi: 10.1016/s1367-5931(02)00385-x. [DOI] [PubMed] [Google Scholar]
- 7.Vreeland WN, Meagher RJ, Barron AE. Anal Chem. 2002;74:4328. doi: 10.1021/ac0258094. [DOI] [PubMed] [Google Scholar]
- 8.Vreeland WN, Slater GW, Barron AE. Bioconjugate Chem. 2002;13:663. doi: 10.1021/bc0155871. [DOI] [PubMed] [Google Scholar]
- 9.Hooks JC, Matharage JP, Udugamasooriya DG. Peptide Science. 2011;96:567. doi: 10.1002/bip.21596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harder P, Grunze M, Dahint R, Whitesides GM, Laibinis PE. J Phys Chem B. 1998;102:426. [Google Scholar]
- 11.Lu HB, Campbell CT, Castner DG. Langmuir. 2000;16:1711. [Google Scholar]
- 12.Konradi R, Pidhatika B, Muhlebach A, Textor M. Langmuir. 2008;24:613. doi: 10.1021/la702917z. [DOI] [PubMed] [Google Scholar]
- 13.Bearinger JP, Terrettaz S, Michel R, Tirelli N, Vogel H, Textor M, Hubbell JA. Nat Mater. 2003;2:259. doi: 10.1038/nmat851. [DOI] [PubMed] [Google Scholar]
- 14.Osterberg E, Bergstrom K, Holmberg K, Schuman TP, Riggs JA, Burns NL, Van Alstine JM, Harris JM. J Biomed Mater Res. 1995;29:741. doi: 10.1002/jbm.820290610. [DOI] [PubMed] [Google Scholar]
- 15.Huang CJ, Li Y, Jiang S. Anal Chem. 2012;84:3440. doi: 10.1021/ac3003769. [DOI] [PubMed] [Google Scholar]
- 16.Crowley MM, Zhang F, Koleng JJ, McGinity JW. Biomaterials. 2002;23:4241. doi: 10.1016/s0142-9612(02)00187-4. [DOI] [PubMed] [Google Scholar]
- 17.Lin S, Zhang B, Skoumal MJ, Ramunno B, Li X, Wesdemiotis C, Liu L, Jia L. Biomacromolecules. 2011;12:2573. doi: 10.1021/bm200368p. [DOI] [PubMed] [Google Scholar]
- 18.Statz AR, Barron AE, Messersmith PB. Soft Matter. 2008;4:131. doi: 10.1039/B711944E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reitsma S, Slaaf D, Vink H, van Zandvoort M, oude Egbrink M. Pflugers Arch. 2007;454:345. doi: 10.1007/s00424-007-0212-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Massia SP, Stark J, Letbetter DS. Biomaterials. 2000;21:2253. doi: 10.1016/s0142-9612(00)00151-4. [DOI] [PubMed] [Google Scholar]
- 21.Perrino C, Lee S, Choi SW, Maruyama A, Spencer ND. Langmuir. 2008;24:8850. doi: 10.1021/la800947z. [DOI] [PubMed] [Google Scholar]
- 22.Martwiset S, Koh AE, Chen W. Langmuir. 2006;22:8192. doi: 10.1021/la061064b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holland NB, Qiu Y, Ruegsegger M, Marchant RE. Nature. 1998;392:799. doi: 10.1038/33894. [DOI] [PubMed] [Google Scholar]
- 24.Sen Gupta A, Wang S, Link E, Anderson EH, Hofmann C, Lewandowski J, Kottke-Marchant K, Marchant RE. Biomaterials. 2006;27:3084. doi: 10.1016/j.biomaterials.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 25.Chapman RG, Ostuni E, Takayama S, Holmlin RE, Yan L, Whitesides GM. J Am Chem Soc. 2000;122:8303. [Google Scholar]
- 26.Fyrner T, Lee HH, Mangone A, Ekblad T, Pettitt ME, Callow ME, Callow JA, Conlan SL, Mutton R, Clare AS, Konradsson P, Liedberg B, Ederth T. Langmuir. 2011;27:15034. doi: 10.1021/la202774e. [DOI] [PubMed] [Google Scholar]
- 27.Luk YY, Kato M, Mrksich M. Langmuir. 2000;16:9604. [Google Scholar]
- 28.Smith EA, Thomas WD, Kiessling LL, Corn RM. J Am Chem Soc. 2003;125:6140. doi: 10.1021/ja034165u. [DOI] [PubMed] [Google Scholar]
- 29.Ostuni E, Chapman RG, Liang MN, Meluleni G, Pier G, Ingber DE, Whitesides GM. Langmuir. 2001;17:6336. [Google Scholar]
- 30.Ryle AP. Nature. 1965;206:1256. doi: 10.1038/2061256a0. [DOI] [PubMed] [Google Scholar]
- 31.Geng J, Mantovani G, Tao L, Nicolas J, Chen G, Wallis R, Mitchell DA, Johnson BRG, Evans SD, Haddleton DM. J Am Chem Soc. 2007;129:15156. doi: 10.1021/ja072999x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Slavin S, Burns J, Haddleton DM, Becer CR. Eur Polym J. 2011;47:435. [Google Scholar]
- 33.Redfern PC, Zapol P, Curtiss LA, Rajh T, Thurnauer MC. J Phys Chem B. 2003;107:11419. [Google Scholar]
- 34.Lee SW, Laibinis PE. Biomaterials. 1998;19:1669. doi: 10.1016/s0142-9612(98)00044-1. [DOI] [PubMed] [Google Scholar]
- 35.Lau KHA, Ren C, Park SH, Szleifer I, Messersmith PB. Langmuir. 2011;28:2288. doi: 10.1021/la203905g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang NP, Michel R, Voros J, Textor M, Hofer R, Rossi A, Elbert DL, Hubbell JA, Spencer ND. Langmuir. 2000;17:489. [Google Scholar]
- 37.Hall-Stoodley L, Costerton JW, Stoodley P. Nat Rev Micro. 2004;2:95. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 38.Katsikogianni M, Missirlis YF. Eur Cell Mater. 2004;8:37. doi: 10.22203/ecm.v008a05. [DOI] [PubMed] [Google Scholar]
- 39.Ruggieri P, Angelini A, Pala E, Mercuri M. Spine. 2012;37:420. doi: 10.1097/BRS.0b013e3182213a44. [DOI] [PubMed] [Google Scholar]
- 40.Anwar H, Strap JL, Chen K, Costerton JW. Antimicrob Agents Chemother. 1992;36:1208. doi: 10.1128/aac.36.6.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Gennes PG. Macromolecules. 1981;14:1637. [Google Scholar]
- 42.Jeon SI, Lee JH, Andrade JD, De Gennes PG. J Colloid Interface Sci. 1991;142:149. [Google Scholar]
- 43.Szleifer I. Biophys J. 1997;72:595. doi: 10.1016/s0006-3495(97)78698-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen S, Yu F, Yu Q, He Y, Jiang S. Langmuir. 2006;22:8186. doi: 10.1021/la061012m. [DOI] [PubMed] [Google Scholar]
- 45.Cho WK, Kong B, Choi IS. Langmuir. 2007;23:5678. doi: 10.1021/la063737w. [DOI] [PubMed] [Google Scholar]
- 46.Ederth T, Ekblad T, Pettitt ME, Conlan SL, Du CX, Callow ME, Callow JA, Mutton R, Clare AS, D’Souza F, Donnelly G, Bruin A, Willemsen PR, Su XJ, Wang S, Zhao Q, Hederos M, Konradsson P, Liedberg B. ACS Appl Mater Interfaces. 2011;3:3890. doi: 10.1021/am200726a. [DOI] [PubMed] [Google Scholar]
- 47.Prime K, Whitesides G. Science. 1991;252:1164. doi: 10.1126/science.252.5009.1164. [DOI] [PubMed] [Google Scholar]
- 48.Szleifer I. Physica A. 1997;244:370. [Google Scholar]
- 49.Wei T, Carignano MA, Szleifer I. Langmuir. 2011;27:12074. doi: 10.1021/la202622s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Merzel F, Smith JC. Proc Natl Acad Sci USA. 2002;99:5378. doi: 10.1073/pnas.082335099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luzar A, Chandler D. J Chem Phys. 1993;98:8160. [Google Scholar]
- 52.Ebbinghaus S, Kim SJ, Heyden M, Yu X, Heugen U, Gruebele M, Leitner DM, Havenith M. Proc Natl Acad Sci USA. 2007;104:20749. doi: 10.1073/pnas.0709207104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Starr FW, Nielsen JK, Stanley HE. Phys Rev E: Stat, Nonlinear, Soft Matter Phys. 2000;62:579. doi: 10.1103/physreve.62.579. [DOI] [PubMed] [Google Scholar]
- 54.Luzar A, Chandler D. Nature. 1996;379:55. [Google Scholar]
- 55.Statz AR, Meagher RJ, Barron AE, Messersmith PB. J Am Chem Soc. 2005;127:7972. doi: 10.1021/ja0522534. [DOI] [PubMed] [Google Scholar]
- 56.Statz A, Kuang J, Ren C, Barron A, Szleifer I, Messersmith P. Biointerphases. 2009;4:FA22. doi: 10.1116/1.3115103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hower JC, He Y, Bernards MT, Jiang S. J Chem Phys. 2006;125:214704. doi: 10.1063/1.2397681. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.