

Abstract
In colorectal cancer (CRC) the vitamin D catabolizing enzyme 1,25-dihydroxyvitamin D 24-hydroxylase (CYP24A1) is overexpressed with a potentially significant, positive impact on the catabolism of 1,25-dihydroxyvitamin D3 (1,25-D3). However, the underlying mechanism of CYP24A1 overexpression is poorly understood. In the present study, we investigated possible causes including hypomethylation of the CYP24A1 promoter, amplification of the CYP24A1 gene locus (20q13.2), and altered expression of CYP24A1-specific transcription factors. We quantified CYP24A1 gene copy-number, performed bisulfite sequencing of the CYP24A1 promoter to assess DNA methylation, and measured mRNA expression of CYP24A1, 25-hydroxyvitamin D 1α-hydroxylase (CYP27B1), vitamin D receptor (VDR) and retinoid X receptor (RXR). We found that 77 (60%) out of 127 colorectal tumors showed increased CYP24A1 gene copy-number and that more than 6 copies of CYP24A1 correlated positively with CYP24A1 mRNA expression suggestive of a causal relationship. No differences in CYP24A1 promoter methylation were found between tumor tissue and adjacent mucosa from the same patient or between tissues with high or low mRNA expression, thus excluding DNA hypomethylation as a possible cause of CYP24A1 overexpression in CRC. Furthermore, mRNA expression of several factors involved in replication licensing positively correlated with CYP24A1 mRNA expression, raising the possibility that CYP24A1 overexpression might favor increased proliferation in tumors by suppressing local 1,25-D3 levels. We conclude that high copy-number gain is a key determinant of CYP24A1 overexpression in CRC. Other postulated causes of CYP24A1 overexpression including promoter hypomethylation and enhanced VDR and/or RXR expression do not appear to be involved.
What’s new?
Recently, it has been suggested that the association between colorectal cancer and reduced levels of circulating vitamin D may be related to overexpression of the vitamin D-catabolizing enzyme, CYP24A1 in the tumor. In this search for a mechanistic explanation, increased CYP24A1 gene copy number was associated with the enzyme’s overexpression in 60 percent of colorectal tumors, and expression was correlated strongly with proliferation markers. The findings suggest that CYP24A1 overexpression is likely to deplete tumor calcitriol (1,25-dihydroxyvitamin D3) levels, possibly increasing the proliferative potential of the tumors.
Keywords: CYP24A1, colorectal cancer, 20q13.2, methylation, proliferation
Colorectal cancer is the third most common cancer in the world with over 1.2 million new cases in 2008. Colorectal cancer risk is inversely associated with the vitamin D status of the patient and epidemiological studies suggest that vitamin D supplementation decreases colorectal cancer incidence.1,2 25-hydroxyvitamin D3 (25-D3, calcidiol) is the circulating form of vitamin D. It is hydroxylated at position C-1 by 25-hydroxyvitamin D3 1α-hydroxylase (CYP27B1) to form the most active vitamin D metabolite 1,25-dihydroxyvitamin D3 (1,25-D3, calcitriol). Systemic levels of 1,25-D3 are controlled by the synthesizing enzyme CYP27B1 and the catabolizing enzyme 1,25-dihydroxyvitamin D 24-hydroxylase (CYP24A1) in the kidneys.3 To avoid hypercalcemia induced by excessively high blood levels of 1,25-D3, 1,25-D3 provides feedback inhibition of CYP27B1 expression and coordinately upregulates the expression of CYP24A1.4 The main endocrine function of 1,25-D3 is the modulation of calcium and phosphate homeostasis and bone mineralization.5
Expression of CYP27B1 and CYP24A1 is not limited to the kidneys, but is found in various tissues including breast and prostate, as well as colon.6 Extra-renally produced 1,25-D3 acts in an autocrine/paracrine manner5 and is considered to be anti-tumorigenic by regulating cell proliferation, apoptosis, angiogenesis and differentiation.7,8 The 1,25-D3 catabolizing enzyme CYP24A1 is overexpressed in several malignancies including tumor samples from patients with colorectal cancer.9 This abnormally high expression promotes the catabolism of local 1,25-D3 and appears to suppress its anti-tumorigenic effects. Thus, CYP24A1 has been identified as a proto-oncogene in colorectal and other cancers.10–12 The mechanisms responsible for upregulation of CYP24A1 in colorectal carcinogenesis are not well understood. Changes in promoter methylation and gene amplification have been identified as possible causes of aberrant CYP24A1 expression in cancer.13,14
DNA methylation is an epigenetic alteration and refers to the addition of a methyl group to cytosine bases in CpG dinucleotides. CpGs can either occur in isolation or in so-called CpG islands (CGIs) that are typically ≥500 bp, with GC contents that exceed 55%. CGIs are found in around 60% of gene promoter regions including the CYP24A1 promoter (bases −467 to +1273).15 During carcinogenesis, global DNA hypomethylation occurs, causing genomic instability and activation of proto-oncogenes.16,17 The CYP24A1 promoter contains regulatory elements including specificity protein 1 (SP1) binding sites and two vitamin D responsive elements (VDRE 1+2) controlling both basal and 1,25-D3 induced transcription,18 while distal enhancer elements19 are not located within CGIs. Reporter assays indicated that CYP24A1 promoter methylation lowers basal transcription and reduces responsiveness of CYP24A1 to 1,25-D3 dependent transcription,20,21 whereas demethylation promotes transcription.22 In prostate cancer, increased promoter methylation coincided with CYP24A1 downregulation.21,23 Besides osteoblastic ROS cells and prostate cancer cell lines, the CYP24A1 promoter is methylated also in healthy human placenta.20–22,24 Hypermethylation of other components of the vitamin D system were reported in breast cancer (VDR and CYP27B1),25,26 thus indicating an epigenetic control of the vitamin D system. A possible epigenetic control of CYP24A1 has been discussed in various recent reviews but has not been tested in the colon so far.14,27 Our study is the first to assess the methylation status of the CYP24A1 promoter in normal colon and colon cancer tissue, testing the hypothesis that the low expression of CYP24A1 in the normal colon mucosa could be due to promoter hypermethylation and that hypomethylation results in CYP24A1 overexpression in tumor tissue.
In addition to changes in methylation status, genomic instability in cancer results in chromosomal rearrangements and gene amplification. Although, copy-number gains may conceivably occur on all chromosomes, in colorectal cancer they are found with the highest frequencies on chromosomes 13q, 8q, and 20q.28 These regions are less frequently amplified in adenomas, suggesting that they might provide proliferative and/or metastatic advantage for the tumor and might be involved in the transition from adenoma to adenocarcinoma.28,29 CYP24A1 is located on 20q13.2, a region that is amplified in various malignancies including breast and pancreas, as well as colon.12,30 In previous studies, a broad range of colorectal tumors (9–92%) have been reported to exhibit 20q13 copy-number gain.31–33
The interest in using vitamin D as a potential cancer preventive factor or even as an adjuvant chemotherapeutic substance has increased tremendously in the last few years. However, the beneficial effects of 1,25-D3 are blunted by the upregulation of its degrading enzyme CYP24A1. Therefore, understanding the mechanisms behind CYP24A1 overexpression in tumors is of utmost importance, in order to take advantage of the entire anti-tumorigenic potential of vitamin D.
In this study, we investigated possible mechanisms underlying the upregulation of CYP24A1 in colorectal cancer. The current findings indicate that the DNA methylation state of the CYP24A1 promoter does not play a role in the pathogenesis of colorectal cancer. We focused on determining CYP24A1 copy-number and expression levels in the same patient cohort, demonstrating that high copy-number gains are closely associated with increased CYP24A1 mRNA and protein expression. Furthermore, our data suggest that one of the consequences of high CYP24A1 expression in colorectal tumors is increased proliferative potential.
Material and Methods
Tissue samples
Tissue samples (fresh frozen) were collected at the General Hospital of Vienna and Rudolfsstiftung Hospital after approval by the ethics committee of the Medical University of Vienna. Formal, written consent was obtained from all patients. 138 colorectal cancer tissue and adjacent mucosa from the same patient were collected, graded, and classified according to the TNM system by a pathologist. Because of the limited quantity of tissue samples we were unable to extract both RNA and DNA from some of the patients.
RNA isolation and reverse transcription
RNA was extracted with Trizol (LifeTechnologies (Invitrogen), Vienna, Austria) according to the manufacturer’s instructions and measured with a NanoDrop ND-1000 (Peqlab, Erlangen, Germany). RNA integrity was assessed by ethidium bromide staining on agarose gels and 2 µg of total RNA were reverse transcribed with RevertAid H Minus Reverse Transcriptase (Fermentas, St. Leon-Rot, Germany) using random hexamer primers according to the manufacturer’s instructions. cDNA was diluted 1:8 in water before qRT-PCR.
Quantitative reverse transcription PCR
We screened various housekeeping genes and found beta-actin to be stably expressed in colorectal cancer and normal mucosa. Quantitative reverse transcription PCR (qRT-PCR) was performed as described before.9 Samples were run in duplicates with POWER SYBR GREEN Mastermix (LifeTechnologies, Applied Biosystems), Vienna, Austria) on a Step One Plus qRT-PCR machine (LifeTechnologies). All values were set relative to total RNA calibrator and ΔΔCT was calculated relative to the housekeeping gene. Primer sequences of CYP24A1, VDR, CYP27B1, β-actin have been described before.9 Primer sequences for CYP3A4 (fwd: CAGGAGGAAATTGAGGCAGTTTT, rev: GTCAAGATACTCCATCTGTAGCAGAGT), RXRα (fwd: GGACATGCAGATGGACAAGAC, rev: CCTTGGAGTCAGGGTTAAAGAG), MCM2 (fwd: GCCAAGATGTACAGTGACCTGA, rev: GATGTGCCGCACCGTAAT), MCM4 (fwd: TTGAAGCCATTGATGTGGAA, rev: GGCACTCATCCCCGTAGTAA), MCM7 (fwd: CGGTGCTGGTAGAAGGAGAG, rev: AAACCCTGTACCACCTGTCG), CDC6 (fwd: CCTGTTCTCCTCGTGTAAAAGC, rev: GTGTTGCATAGGTTGTCATCG).
Immunofluorescence staining
Sections (5 μm) of paraffin-embedded tissue were incubated for 25 min at 60°C, deparaffinized, and rehydrated. After washing in phosphate-buffered saline (PBS, pH 7.2), we performed antigen-retrieval by boiling sections in 0.05% citraconic anhydride. Sections were washed in PBS, permeabilized in PBS, 0.2% Tween-20 (Sigma-Aldrich, Munich, Germany) for 15 min and blocked with 10% goat serum in PBS (Jackson ImmunoResearch, Suffolk, UK) for 45 min. CYP24A1 primary antibody was diluted 1:1,000 in 10% goat serum. Rabbit IgG (Abcam, Cambridge, UK) was used as negative control. After extensive washing, sections were incubated with Dylight 549 goat-anti-rabbit IgG (1:500, Vector Laboratories, UK). Nuclei were stained with Dapi (Roche, Vienna, Austria) for 10 min and coverslips were mounted using Fluoromount-G (Southern Biotech, Birmingham, AL). Whole tissue slide images were acquired using TissueFAXS 2.04 (TissueGnostics, Vienna, Austria). Staining intensities were classified as weak staining (i), weak positive (ii), moderate positive (iii), strong positive (iv) and highly positive (v).
DNA isolation and copy-number assay
DNA was isolated by standard proteinase K (Sigma-Aldrich) digestion and subsequent phenol/chloroform extraction. DNA copy-number of CYP24A1 (fwd: GCTAACATCATATCCAACTCAG, rev: TGAAGTGTAAACCAGCAGTG) and RNaseP (fwd: CAGCGAAGTGAGTTCAATGG, rev: GGAGGAGAGTAGTCTGAATTGG) was assessed in duplicates by POWER SYBR GREEN (LifeTechnologies) qPCR on Step One Plus Real Time Machine (LifeTechnologies). The assay was adapted for CYP24A1 (20q13.2) and Ribonuclease P RNA component H1 (14q11.2) after the method of Ponchel et al.34 Primers were used at a final concentration of 500 nM (CYP24A1) and 300 nM (RNAseP) to achieve a ratio of 1:1 for both genes in samples with no gene amplifications. For this, ΔCt was calculated to achieve a value of 0 by the formula: ΔCT = (CT of the target CYP24A1) − (CT of the reference RNAseP). Human placenta was used as calibrator and the assay was validated with normal human colon and kidney as controls for normal copy-number. The melanoma cell line VM-17 was used as a positive control for gene amplification of CYP24A1 but not RNaseP, as previously determined by CGH.35,36 ΔΔCT was calculated, values below 2 were considered as normal copy-number (less than four copies), values above 2 as increased copy-number gain (more than four copies). For assay validation, we performed Fluorescence In Situ Hybridization (FISH) as described before (data not shown).”37
Bisulfite genomic sequencing
Bisulfite Genomic Sequencing Primers were designed using Methyl Primer Express v1.0 (LifeTechnologies). Region 1 contains 45 CpGs and represents the proximal promoter region (−494 to +3, primers fwd: ATTTTAGTTTAGGTTGGGGGTATTT, rev: CCATATTCCTATACCCAAAAACCAT), Region 2 is localized at the transcription start site and contains 39 CpGs (−18 to +609, primers fwd: TTTTTGGGTATAGGAATATGGAGAG, rev: CCCAACAATAACCAACTAATAAAAC). DNA was bisulfite-converted with the EpiTect Bisulfite Kit (Qiagen, Hilden, Germany). PCR amplification was performed using HotStarTaq DNA Polymerase (Qiagen), run on a 2% low-melt agarose gel and gel-purified with PureLink Quick Gel Extraction Kit (LifeTechnologies). Cloning was performed with the Topo TA Cloning Kit for Subcloning with either chemically competent or electrocompetent bacteria (LifeTechnologies) according to the manufacturer’s instructions. Miniprep and DNA-Sequencing of at least three clones was performed by Microsynth AG (Balgach, Switzerland). Sequencing results were analyzed with the BiQAnalyzer software.38
Statistical analysis
Data were log transformed to achieve normal distribution, paired t-tests were calculated on mRNA expression data for tumor samples and respective adjacent mucosa samples from the same patient. Number of patients and two-tailed significance levels are stated. The nonparametric Spearman Correlation was calculated without log transformation and the Spearman Correlation Coefficient and the two-tailed significance levels are given. All calculations were performed with SPSS Version 18, graphs were drawn using GraphPad Prism Version 5.
Results
Genes that control the metabolism of calcidiol are deregulated in colorectal tumor samples
Expression of CYP24A1 was significantly higher in colorectal tumor tissue compared with the respective adjacent mucosa (4.8-fold change; n = 69; p < 0.001; Table1 and Fig. 1a). This overexpression was confirmed on protein level by immunofluorescence staining of paraffin sections (Supporting Information Fig. S1). In the samples analyzed, CYP24A1 mRNA expression and protein expression strongly correlated (n = 29, Spearman Correlation Coefficient SCC = 0.615, p < 0.001, Fig. 1d). CYP24A1 overexpression did not correlate with any of the pathophysiological characteristics shown in Table1, such as lymph node involvement or tumor location. As CYP24A1 transcription is highly induced by 1,25-D3, we also determined expression of the 1,25-D3 synthesizing enzyme CYP27B1, as well as of VDR and RXR, the transcription factors that mediate 1,25-D3-dependent CYP24A1 transcription. CYP27B1 mRNA was significantly upregulated in the tumor tissue compared with the adjacent normal mucosa (twofold change; p < 0.001; Fig. 1a), whereas VDR and RXR were both significantly downregulated (p < 0.001, Fig. 1b). Specificity protein 1 (SP1) involved in both basal and vitamin D-dependent transcription of CYP24A1 was upregulated in adenocarcinomas suggestive of an 1,25-D3 independent upregulation (p < 0.001, Supporting Information Fig. S2). As an indicator of local 1,25-D3 activity, we measured the expression of one of its target genes CYP3A4. CYP3A4 mRNA expression was strongly decreased in colorectal tumor tissue compared with the adjacent normal mucosa (p < 0.001, Fig. 1c) consistent with the hypothesis that 1,25-D3 activity is suppressed in the context of CYP24A1 overexpression.
Table 1.
Patient cohort
| Tissue | Copy number analysis | mRNA expression | Overlap |
|---|---|---|---|
| n | 127 | 69 | 59 |
| Gender | |||
| Female | 66 (52%) | 34 (49%) | 31 (53%) |
| Male | 61 (48%) | 34 (49%) | 28 (47%) |
| Unknown | 0 | 1 | 0 |
| Age | |||
| Mean ± SD | 69.5 ± 12.6 | 68.8 ± 11.1 | 68.7 ± 8.0 |
| Tumor grading | |||
| Grade 1 | 1 | 1 | 1 |
| Grade 2 | 106 | 53 | 46 |
| Grade 3 | 18 | 12 | 10 |
| Unknown | 2 | 3 | 2 |
| Lymph node infiltration | |||
| 0 | 58 | 30 | 25 |
| 1 | 30 | 17 | 15 |
| 2 | 35 | 17 | 15 |
| 3 | 1 | 1 | 1 |
| Unknown | 3 | 4 | 3 |
| Site of the primary tumor | |||
| Cecum/ascending/transverse colon | 48 | 26 | 23 |
| Descending/sigmoid colon | 46 | 24 | 24 |
| Rectum | 30 | 17 | 11 |
| Unknown | 3 | 2 | 1 |
Figure 1.
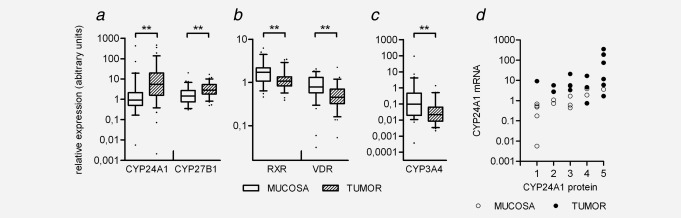
mRNA expression of vitamin D pathway genes differs between colorectal tumors and respective adjacent mucosa. mRNA expression was assessed in colorectal tumors and adjacent mucosa by qRT-PCR. 2-tailed paired t-tests on log transformed data were computed (*p < 0.05, **p < 0.001). Relative expression of CYP24A1 (n = 69) and CYP27B1 (n = 68) are shown in (a), RXR (n = 63) and VDR (n = 68) in (b), CYP3A4 (n = 70) in (c). Median, interquartile range and whiskers representing 5th to 95th percentile are shown. CYP24A1 protein expression in tumors and adjacent mucosa samples was determined by immunofluorescence stainings and was graded from 1–5 (low-high, x-axis), respective mRNA expression of CYP24A1 is shown on the y-axis (d).
Thus, CYP24A1 overexpression does not correlate with VDR or RXR expression. Since both VDR and RXR were significantly downregulated in the tumors, it is unlikely that the physiological 1,25-D3-VDR-induced transcription accounts for CYP24A1 overexpression.
CYP24A1 promoter methylation is comparable between tumor and normal mucosa
To test the hypothesis that the expression of CYP24A1 is low due to dense promoter methylation in normal colon and that hypomethylation upregulates CYP24A1 during carcinogenesis, we determined the methylation status of two distinct regions of the CYP24A1 promoter (Fig. 2a) in samples of colorectal tumors and their respective adjacent mucosa (n = 20). We found very low levels of methylation (<3% mean methylation) in promoter region 1 in both adenocarcinomas and the adjacent normal tissue, indicating the absence of methylation-dependent transcriptional silencing (Fig. 2b). A similar low level of methylation (<4%) was observed in the tissue obtained from a healthy control subject (data not shown). Interestingly, methylation of promoter region 2 was higher than that in region 1 but, as for promoter region 1, there was no significant difference between the levels of methylation in tumor tissue and adjacent mucosal tissue as calculated by paired t-tests on log-transformed data, respectively (median region 1: mucosa: 4%, tumor 2%; region 2: mucosa 10%, tumor 7%). In addition, promoter methylation of CYP24A1 did not correlate with mRNA expression (Supporting Information Table S1). Figure 2c shows the average CpG methylation frequency in tumor and mucosa. The transcription start site (TSS) and TATA box are located between region 1 and 2 and do not contain CpGs. There appeared to be no difference in the methylation patterns of responsive elements between tumor and mucosa. A methylation hot spot was observed in region 2 approximately 220bp downstream of the TSS (Fig. 2c). In summary, the similar methylation levels found in adjacent mucosa and tumor samples for both CYP24A1 promoter regions 1 and 2 do not suggest involvement of DNA methylation in regulation of CYP24A1 expression.
Figure 2.

Promoter methylation of CYP24A1 in adjacent mucosa and tumor tissue. Schematic overview of the CYP24A1 promoter region including position of regulatory elements (vitamin D responsive elements (VDRE1 and VDRE2), specificity protein 1 (SP1), vitamin D stimulatory element (VSE) and TATA box) as well as position of bisulfite genomic sequencing primers (black arrows) are shown (a). Bisulfite genomic sequencing of the CYP24A1 promoter was performed in 20 human adenocarcinoma and 20 adjacent mucosa samples. For each region, the mean methylation per sample is blotted, median is indicated (b). Incidence of CpG nucleotide methylation is shown for tumor and adjacent mucosa (c).
60% of colorectal tumors carry increased number of copies of the CYP24A1 gene
The long arm of chromosome 20 and specifically the region 20q13 are amplified in various cancers, including colorectal cancer. To assess the genomic copy-number of CYP24A1, located at 20q13.2, we developed a quantitative real-time PCR assay based on the methods of Ponchel et al.34 and Muhamad et al.39 We determined CYP24A1 copy-number in colorectal tumors and respective adjacent mucosa of 127 patients (Table1). As the assay does not give absolute values, we considered fewer than four copies as normal, four to six copies of CYP24A1 as moderately amplified, and greater than six copies of CYP24A1 as highly amplified. Of the adjacent normal mucosa samples studied, 103 (81%) exhibited normal CYP24A1 copy-numbers and 24 (19%) samples carried more than four copies of CYP24A1 (Fig. 3a). However, of the adenocarcinoma samples studied, only 51 (40%) exhibited normal CYP24A1 copy-number and 76 (60%) had elevated copy-number. Of these 76 adenocarcinomas, 51 (67%) had four to six copies and 25 (33%) had more than six copies. Figure 3a shows the distribution of patients in the above mentioned groups. CYP24A1 copy-number did not correlate with patient age, sex, or anatomical location of the tumor. To validate our results of the PCR assay, we performed fluorescence in situ hybridization (FISH) analysis on two cell lines and five patients. The FISH analysis of the melanoma cell line VM-17 used as a positive control in our qPCR assay, showed clear CYP24A1 gene amplification. At the same time in the colon cancer cell line Caco-2/15 we found both CYP24A1 gene amplification as well as chromosome 20 polysomy. The FISH analysis of the patient samples also confirmed increases in copy-numbers, however, it suggested that the higher copy-numbers are primarily due to chromosome 20 polysomy and only partially to 20q13.2 gene locus amplification.
Figure 3.
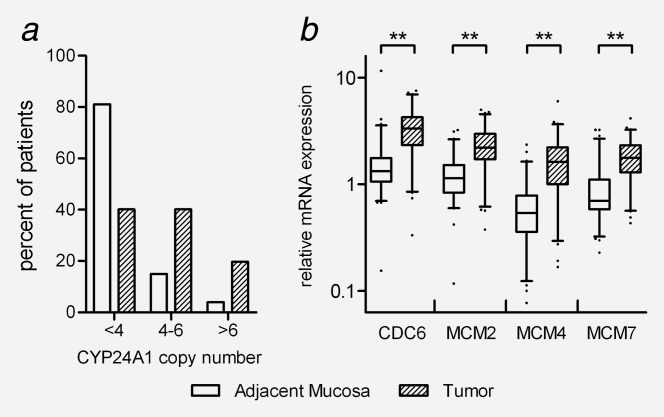
CYP24A1 gene copy-number and proliferation marker expression in colorectal tumors. Genomic copy-number of CYP24A1 was determined in tumor tissue and adjacent mucosa (n = 127). Patients were grouped in three categories, less than four copies (normal), four to six copies (amplified) and more than six copies (highly amplified), data are presented as % of total patients (a). mRNA expression of the proliferation markers CDC6, MCM2, MCM4 and MCM7 were assessed in tumor and adjacent mucosa. Paired samples t-test was computed on log transformed data, 2-tailed p-values (** p < 0.001) are indicated (b). Median, interquartile range and whiskers representing 5th to 95th percentile are shown.
High CYP24A1 gene copy-number correlates with increased mRNA expression
To determine the impact of CYP24A1 copy-number gain on mRNA expression, we performed correlation analysis between number of CYP24A1 copies and mRNA expression in the same patient cohort (n = 58, Table1). We used the nonparametric Spearman Correlation Coefficient to test for statistical dependence between CYP24A1 mRNA expression and CYP24A1 copy-numbers. A Spearman Correlation Coefficient (SCC) of “1” indicates almost linear correlation, “0” absence of a correlation. We found a significant correlation between copy-number status and mRNA expression in the overall patient cohort (SCC = 0.379). However, in the subgroup of patients with high CYP24A1 copy-number (6–11), the correlation increased considerably (SCC = 0.580; Table2, dot-blots shown in Supporting Information Fig. S3) indicating that high copy-numbers are associated with increased mRNA expression, consistent with the idea that copy-number gain results in enhanced CYP24A1 transcription.
Table 2.
CYP24A1 gene copy-number correlates with increased mRNA expression
| Correlation CYP24A1 copy number—mRNA | |||
|---|---|---|---|
| CYP24A1 copy number | n | SCC | Sig. (two-tailed) |
| Total | 118 | 0.379 | 0.001 |
| >6 | 13 | 0.580 | 0.038 |
CYP24A1 mRNA expression correlates with proliferation
Increased expression of CYP24A1 lowers the half-life of 1,25-D3 and thus suppresses local 1,25-D3 concentrations. Such an outcome is predicted to promote proliferation and impair differentiation. As more than 75% of tumors were classified as moderately differentiated (i.e., grade 2; Table1), we were unable to assess the relationship between tumor grade and CYP24A1 expression. Therefore, to assess the possible impact of CYP24A1 mRNA expression on the proliferative potential of the tumors, we quantified mRNA expression of several genes responsible for initiating cell cycle turnover, including cell division cycle 6 homolog (CDC6) and mini-chromosome maintenance complex components 2, 4 and 7 (MCM2, MCM4, MCM7). All licensing factors were expressed at elevated levels in the colorectal tumor samples compared with the adjacent mucosa (log transformed data; p < 0.001; Fig. 3b). Furthermore, CYP24A1 mRNA expression correlated with CDC6 (SCC = 0.596), MCM2 (SCC = 0.566), MCM4 (SCC = 0.569) and MCM7 (SCC = 0.568) mRNA expression in the overall cohort (Table3, Supporting Information Fig. S4). Interestingly, in the subgroup of patient samples with more than six copies of CYP24A1, these correlations were significantly strengthened (CDC6: SCC = 0.607; MCM2: SCC = 0.829; MCM4: SCC = 0.789; MCM7: SCC = 0.768, Table3, Supporting Information Fig. S5). Therefore, high CYP24A1 copy-number was associated with upregulated expression of key activators of cell cycle turnover raising the possibility that enhanced 1,25-D3 degradation and suppression of local 1,25-D3 levels removes an important inhibitory restraint on cell proliferation.
Table 3.
CYP24A1 mRNA expression correlates with expression of markers of proliferation
| >6 genomic copies of CYP24A1 | ||||||
|---|---|---|---|---|---|---|
| Gene | n | SCC | Sig. (two-tailed) | n | SCC | Sig. (two-tailed) |
| CDC6 | 120 | 0.596 | 0.001 | 15 | 0.607 | 0.016 |
| MCM2 | 120 | 0.566 | 0.001 | 15 | 0.829 | 0.001 |
| MCM4 | 120 | 0.569 | 0.001 | 15 | 0.789 | 0.001 |
| MCM7 | 120 | 0.568 | 0.001 | 15 | 0.768 | 0.001 |
Discussion
In this study, we show that high CYP24A1 copy-number leads to increased mRNA expression of CYP24A1. Furthermore, CYP24A1 expression correlated with several proliferation markers, suggesting that high CYP24A1 expression might result in stronger proliferating tumors. In addition, we provide evidence that overexpression of CYP24A1 in colorectal cancer is not caused by promoter hypomethylation.
Changes in 1,25-D3 levels are dependent on the relative expressions of the 1,25-D3 synthesizing enzyme CYP27B1 and the degrading enzyme CYP24A1. Although CYP24A1 is overexpressed in most cancer types,9,40–43 reports on changes in CYP27B1 expression are inconsistent.44 In our colorectal cancer patient cohort the expression of mRNAs encoding both enzymes was elevated. Previously, coordinate upregulation of CYP24A1 and CYP27B1 was observed in breast cancer with an apparent net outcome of enhanced 1,25-D3 catabolism.41 As both, the absolute expression levels as well as the fold-increases of CYP24A1 were higher than for CYP27B1 in tumor samples, the results suggest a shift to enhanced 1,25-D3 breakdown in our patient cohort. Consistent with a local reduction in 1,25-D3 levels, we observed reduced expression of the 1,25-D3 target gene CYP3A4 in the tumor samples. Comparing CYP3A4 expression levels between tumor and normal mucosa in the same patient controls for impact of possible drug treatment on CYP3A4 level. Our study is the first to show that in colorectal tumors not only VDR9,10 but also RXR, two key mediators of 1,25-D3-dependent transcription of CYP24A1 were both downregulated, while SP1 was elevated in the tumors. This suggests an 1,25-D3-independent upregulation of CYP24A1, possibly via a noncanonical control mechanism such as promoter hypomethylation or increased CYP24A1 copy-number.
As the methylation status of CYP24A1 in normal colon and colonic tumor tissue has been previously unknown, we hypothesized that CYP24A1 expression may be limited by high levels of promoter methylation in normal colon as observed in placenta and that hypomethylation during carcinogenesis may drive strong increases in transcription. However, we observed only low levels of CYP24A1 promoter methylation both in a sample of healthy colon and in the apparently normal mucosa adjacent to tumors in patients with colorectal cancer. Previously, we have demonstrated that in colon cancer cell lines the methylation status of the CGI of the CYP24A1 promoter was not involved in regulation of CYP24A1 expression.45 Taken together, our results indicate that hypomethylation of the CYP24A1 promoter is not responsible for the upregulation of CYP24A1 expression in colorectal tumors.
During carcinogenesis, genomic rearrangements and chromosome aneuploidy are caused by increased genomic instability. Comparative genome hybridization (CGH) studies have demonstrated that there is a nonrandom distribution of copy-number gains in colorectal cancer, with a hotspot on chromosome 20q12,28,32. Although the frequency and level of 20q13 gain corresponding to the location of CYP24A1 and neighboring genes varied significantly in colorectal tumors31,33, this suggests a selection advantage for tumors carrying these amplifications. Recently, it was suggested that this advantage is not caused by a single gene but rather a cluster of genes, including aurora kinase A (AURKA, 20q13.2), which is involved in mitotic spindle assembly and chromosome segregation. In a recent study, gene amplification of AURKA was linked to increased expression of AURKA mRNA29, however, no increase in CYP24A1 expression was observed. In this study, we found that enhanced CYP24A1 expression correlated with gene copy-number and this was more significant if the CYP24A1 gene copy-number was greater than 6. One of the transcription factors that drives basal transcription (SP1) was overexpressed in colorectal tumors. This raises the possibility that high copy-number together with increased basal transcription drives CYP24A1 overexpression.
The copy-number gains detected in 19% of adjacent mucosa samples suggest that, as reported previously, this tissue cannot be considered healthy normal tissue as it already harbors alterations46,47. Inclusion of a healthy control group in future studies would assist the evaluation of adjacent tissue for genetic and phenotypic abnormalities. The approach taken in the present study to compare samples of tumor tissue with the adjacent mucosa from the same patient has yielded clear evidence for increased number of CYP24A1 copies in tumors and has the advantage of controlling for differences in nutrition, lifestyle, environment, and genetic predisposition.
The high frequency of CYP24A1 copy-number gains (60% of all tumor samples) in our patient cohort suggested a selection advantage for tumors carrying this modification. Therefore, it is of great importance to determine the consequences of CYP24A1 copy-number gain. Increased CYP24A1 activity limits the biological half-life of 1,25-D3 and thereby counteracts the anti-tumorigenic effects of 1,25-D3, possibly resulting in increased proliferation. Previously, we demonstrated a correlation between CYP24A1 protein expression and the expression of the proliferation marker Ki-67 protein in colorectal tumors9. In this study, we examined whether CYP24A1 mRNA expression correlates with very early phases of DNA replication, namely with expression of replication licensing factors. CDC6 is involved in the loading of the MCM helicase complex onto DNA. MCM2 expression was shown previously to correlate with the proliferative potential of colorectal cancers and MCM2 overexpression was also detected in gastric carcinomas48,49. We found significant correlation between CYP24A1 expression and the expression of replication licensing factors. Interestingly, the correlation was considerably strengthened in the high copy-number group, expressing extremely high levels of CYP24A1 protein, suggesting that colorectal tumors harboring more than six copies of CYP24A1 are highly proliferative. Whether CYP24A1 overexpression is sufficient to drive tumor growth is not yet clear. However, we previously showed that in cells with high basal CYP24A1 expression, the anti-proliferative effects of 1,25-D3 can only be restored by inhibition of CYP24A1 activity50.
In conclusion, upregulation of CYP24A1 gene expression in colorectal cancer is not caused by DNA hypomethylation. Instead, we found high CYP24A1 gene copy-number in 60% of colorectal tumors and the copy-number gain was independent of sex, anatomical location and age. Furthermore, high CYP24A1 copy-number correlated with increased mRNA and protein expression, suggesting that increased CYP24A1 copy-number contributes to the overexpression of CYP24A1. In addition, CYP24A1 expression correlated with recognized proliferation markers including CDC6, MCM2, MCM4 and MCM7. Indeed, the correlation was strongest in samples from patients with more than six genomic copies of CYP24A1. Assuming that 24-hydroxylase activity changes in concert with CYP24A1 mRNA and protein expression it seems reasonable to hypothesize that the role of CYP24A1 in malignant transformation arises from downregulation of 1,25-D3 leading to loss of its anti-proliferative, pro-differentiating effects. Consistent with this idea, CYP3A4 a gene whose expression is positively modulated by 1,25-D3, was downregulated in colorectal tumor samples when compared with adjacent mucosa tissue. Mechanistic studies are required to prove causality of the observed relationship between CYP24A1 overexpression and proliferation. If overexpression of CYP24A1 directly affects tumor proliferation, tumor-targeted treatment with CYP24A1-specific inhibitors may be effective in slowing tumor growth.
Acknowledgments
The authors thank Christine Pirker for providing the VM-17 DNA sample and CGH results and Teresa Manhardt and Rita Lang for their support. They thank Dr. Arthur Conigrave for critically revising the manuscript and for his suggestions for improvement. The CYP24A1 antibody was a kind gift from Dr. Pamela A. Hershberger.
Glossary
- 1,25-D3, calcitriol
1,25-dihydroxyvitamin D3
- AURKA
aurora kinase A
- CDC6
cell division cycle 6 homolog
- CGH
comparative genome hybridization
- CGIs
CpG islands
- CRC
colorectal cancer
- CYP24A1
1,25-dihydroxyvitamin D 24-hydroxylase
- CYP27B1
25-hydroxyvitamin D3 1α-hydroxylase
- MCM
mini-chromosome maintenance complex
- RXR
retinoid X receptor
- SCC
spearman correlation coefficient
- SP1
specificity protein 1
- TSS
transcription start site
- VDR
vitamin D receptor
- FISH
fluorescence in situ hybridization
Additional Supporting Information may be found in the online version of this article.
References
- 1.Giovannucci E. Epidemiology of vitamin D and colorectal cancer. Anticancer Agents Med Chem. 2013;13:11–9. [PubMed] [Google Scholar]
- 2.Lazzeroni M, Serrano D, Pilz S, et al. Vitamin D supplementation and cancer: review of randomized controlled trials. Anticancer Agents Med Chem. 2013;13:118–25. [PubMed] [Google Scholar]
- 3.Deeb KK, Trump DL, Johnson CS. Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer. 2007;7:684–700. doi: 10.1038/nrc2196. [DOI] [PubMed] [Google Scholar]
- 4.Jones G, Strugnell SA, DeLuca HF. Current understanding of the molecular actions of vitamin D. Physiol Rev. 1998;78:1193–231. doi: 10.1152/physrev.1998.78.4.1193. [DOI] [PubMed] [Google Scholar]
- 5.Dusso AS, Brown AJ, Slatopolsky E. Vitamin D. Am J Physiol Renal Physiol. 2005;289:F8–F28. doi: 10.1152/ajprenal.00336.2004. [DOI] [PubMed] [Google Scholar]
- 6.Zehnder D, Bland R, Williams MC, et al. Extrarenal expression of 25-hydroxyvitamin d(3)-1 alpha-hydroxylase. J Clin Endocrinol Metab. 2001;86:888–94. doi: 10.1210/jcem.86.2.7220. [DOI] [PubMed] [Google Scholar]
- 7.González-Sancho JM, Larriba MJ, Ordóñez-Morán P, et al. Effects of 1alpha,25-dihydroxyvitamin D3 in human colon cancer cells. Anticancer Res. 2006;26:2669–81. [PubMed] [Google Scholar]
- 8.Welsh J. Cellular and molecular effects of vitamin D on carcinogenesis. Arch Biochem Biophys. 2012;523:107–14. doi: 10.1016/j.abb.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horváth HC, Lakatos P, Kósa JP, et al. The candidate oncogene CYP24A1: A potential biomarker for colorectal tumorigenesis. J Histochem Cytochem. 2010;58:277–85. doi: 10.1369/jhc.2009.954339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson MG, Nakane M, Ruan X, et al. Expression of VDR and CYP24A1 mRNA in human tumors. Cancer Chemother Pharmacol. 2006;57:234–40. doi: 10.1007/s00280-005-0059-7. [DOI] [PubMed] [Google Scholar]
- 11.Parise RA, Egorin MJ, Kanterewicz B, et al. CYP24, the enzyme that catabolizes the antiproliferative agent vitamin D, is increased in lung cancer. Int J Cancer. 2006;119:1819–28. doi: 10.1002/ijc.22058. [DOI] [PubMed] [Google Scholar]
- 12.Albertson DG, Ylstra B, Segraves R, et al. Quantitative mapping of amplicon structure by array CGH identifies CYP24 as a candidate oncogene. Nat Genet. 2000;25:144–6. doi: 10.1038/75985. [DOI] [PubMed] [Google Scholar]
- 13.Komagata S, Nakajima M, Takagi S, et al. Human CYP24 catalyzing the inactivation of calcitriol is post-transcriptionally regulated by miR-125b. Mol Pharmacol. 2009;76:702–9. doi: 10.1124/mol.109.056986. [DOI] [PubMed] [Google Scholar]
- 14.King AN, Beer DG, Christensen PJ, et al. The vitamin D/CYP24A1 story in cancer. Anticancer Agents Med Chem. 2010;10:213–24. doi: 10.2174/1871520611009030213. [DOI] [PubMed] [Google Scholar]
- 15.Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Leung FC. An evaluation of new criteria for CpG islands in the human genome as gene markers. Bioinformatics. 2004;20:1170–7. doi: 10.1093/bioinformatics/bth059. [DOI] [PubMed] [Google Scholar]
- 17.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tashiro K, Ishii C, Ryoji M. Role of distal upstream sequence in vitamin D-induced expression of human CYP24 gene. Biochem Biophys Res Commun. 2007;358:259–65. doi: 10.1016/j.bbrc.2007.04.103. [DOI] [PubMed] [Google Scholar]
- 19.Meyer MB, Goetsch PD, Pike JW. A downstream intergenic cluster of regulatory enhancers contributes to the induction of CYP24A1 expression by 1alpha,25-dihydroxyvitamin D3. J Biol Chem. 2010;285:15599–610. doi: 10.1074/jbc.M110.119958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Novakovic B, Sibson M, Ng HK, et al. Placenta-specific methylation of the vitamin D 24-hydroxylase gene: implications for feedback autoregulation of active vitamin D levels at the fetomaternal interface. J Biol Chem. 2009;284:14838–48. doi: 10.1074/jbc.M809542200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo W, Karpf AR, Deeb KK, et al. Epigenetic regulation of vitamin D 24-hydroxylase/CYP24A1 in human prostate cancer. Cancer Res. 2010;70:5953–62. doi: 10.1158/0008-5472.CAN-10-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khorchide M, Lechner D, Cross HS. Epigenetic regulation of vitamin D hydroxylase expression and activity in normal and malignant human prostate cells. J Steroid Biochem Mol Biol. 2005;93:167–72. doi: 10.1016/j.jsbmb.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 23.Deeb KK, Luo W, Karpf AR, et al. Differential vitamin D 24-hydroxylase/CYP24A1 gene promoter methylation in endothelium from benign and malignant human prostate. Epigenetics. 2011;6:994–1000. doi: 10.4161/epi.6.8.16536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohyama Y, Kusada T, Yamasaki T, et al. Extensive methylation of CpG island of CYP24 gene in osteoblastic ROS17/2.8 cells. Nucleic Acids Res. 2002;(Suppl):249–50. doi: 10.1093/nass/2.1.249. [DOI] [PubMed] [Google Scholar]
- 25.Marik R, Fackler M, Gabrielson E, et al. DNA methylation-related vitamin D receptor insensitivity in breast cancer. Cancer Biol Ther. 2010;10:44–53. doi: 10.4161/cbt.10.1.11994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi H, Yan PS, Chen CM, et al. Expressed CpG island sequence tag microarray for dual screening of DNA hypermethylation and gene silencing in cancer cells. Cancer Res. 2002;62:3214–20. [PubMed] [Google Scholar]
- 27.Luo W, Hershberger PA, Trump DL, et al. 24-Hydroxylase in cancer: impact on vitamin D-based anticancer therapeutics. J Steroid Biochem Mol Biol. in press. [DOI] [PMC free article] [PubMed]
- 28.Meijer GA, Hermsen MA, Baak JP, et al. Progression from colorectal adenoma to carcinoma is associated with non-random chromosomal gains as detected by comparative genomic hybridisation. J Clin Pathol. 1998;51:901–9. doi: 10.1136/jcp.51.12.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sillars-Hardebol AH, Carvalho B, Tijssen M, et al. TPX2 and AURKA promote 20q amplicon-driven colorectal adenoma to carcinoma progression. Gut. 2011;61:1568–75. doi: 10.1136/gutjnl-2011-301153. [DOI] [PubMed] [Google Scholar]
- 30.Fukushige S, Waldman FM, Kimura M, et al. Frequent gain of copy number on the long arm of chromosome 20 in human pancreatic adenocarcinoma. Genes Chromosomes Cancer. 1997;19:161–9. doi: 10.1002/(sici)1098-2264(199707)19:3<161::aid-gcc5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 31.Lassmann S, Weis R, Makowiec F, et al. Array CGH identifies distinct DNA copy number profiles of oncogenes and tumor suppressor genes in chromosomal-and microsatellite-unstable sporadic colorectal carcinomas. J Mol Med (Berl) 2007;85:293–304. doi: 10.1007/s00109-006-0126-5. [DOI] [PubMed] [Google Scholar]
- 32.Postma C, Hermsen MA, Coffa J, et al. Chromosomal instability in flat adenomas and carcinomas of the colon. J Pathol. 2005;205:514–21. doi: 10.1002/path.1733. [DOI] [PubMed] [Google Scholar]
- 33.Hidaka S, Yasutake T, Takeshita H, et al. Differences in 20q13.2 copy number between colorectal cancers with and without liver metastasis. Clin Cancer Res. 2000;6:2712–7. [PubMed] [Google Scholar]
- 34.Ponchel F, Toomes C, Bransfield K, et al. Real-time PCR based on SYBR-Green I fluorescence: an alternative to the TaqMan assay for a relative quantification of gene rearrangements, gene amplifications and micro gene deletions. BMC Biotechnol. 2003;3:18. doi: 10.1186/1472-6750-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mathieu V, Pirker C, Schmidt WM, et al. Aggressiveness of human melanoma xenograft models is promoted by aneuploidy-driven gene expression deregulation. Oncotarget. 2012;3:399–413. doi: 10.18632/oncotarget.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pirker C, Lötsch D, Spiegl-Kreinecker S, et al. Response of experimental malignant melanoma models to the pan-Aurora kinase inhibitor VE-465. Exp Dermatol. 2010;19:1040–7. doi: 10.1111/j.1600-0625.2010.01182.x. [DOI] [PubMed] [Google Scholar]
- 37.Wiesner T, Streubel B, Huber D, et al. Genetic aberrations in primary cutaneous large B-cell lymphoma: a fluorescence in situ hybridization study of 25 cases. Am J Surg Pathol. 2005;29:666–73. doi: 10.1097/01.pas.0000155163.40668.e7. [DOI] [PubMed] [Google Scholar]
- 38.Bock C, Reither S, Mikeska T, et al. BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics. 2005;21:4067–8. doi: 10.1093/bioinformatics/bti652. [DOI] [PubMed] [Google Scholar]
- 39.Muhamad P, Chaijaroenkul W, Congpuong K, et al. SYBR Green I and TaqMan quantitative real-time polymerase chain reaction methods for the determination of amplification of Plasmodium falciparum multidrug resistance-1 gene (pfmdr1) J Parasitol. 2011;97:939–42. doi: 10.1645/GE-2792.1. [DOI] [PubMed] [Google Scholar]
- 40.Bareis P, Bises G, Bischof MG, et al. 25-hydroxy-vitamin d metabolism in human colon cancer cells during tumor progression. Biochem Biophys Res Commun. 2001;285:1012–7. doi: 10.1006/bbrc.2001.5289. [DOI] [PubMed] [Google Scholar]
- 41.Townsend K, Banwell CM, Guy M, et al. Autocrine metabolism of vitamin D in normal and malignant breast tissue. Clin Cancer Res. 2005;11:3579–86. doi: 10.1158/1078-0432.CCR-04-2359. [DOI] [PubMed] [Google Scholar]
- 42.Friedrich M, Rafi L, Mitschele T, et al. Analysis of the vitamin D system in cervical carcinomas, breast cancer and ovarian cancer. Recent Results Cancer Res. 2003;164:239–46. doi: 10.1007/978-3-642-55580-0_17. [DOI] [PubMed] [Google Scholar]
- 43.Chen G, Kim SH, King AN, et al. CYP24A1 is an independent prognostic marker of survival in patients with lung adenocarcinoma. Clin Cancer Res. 2011;17:817–26. doi: 10.1158/1078-0432.CCR-10-1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hobaus J, Thiem U, Hummel DM, et al. Role of calcium, vitamin d, and the extrarenal vitamin d hydroxylases in carcinogenesis. Anticancer Agents Med Chem. 2013;13:20–35. [PMC free article] [PubMed] [Google Scholar]
- 45.Höbaus J, Fetahu IS, Khorchide M, et al. Epigenetic regulation of the 1,25-dihydroxyvitamin D(3) 24-hydroxylase (CYP24A1) in colon cancer cells. J Steroid Biochem Mol Biol. 2012 doi: 10.1016/j.jsbmb.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edelstein PS, Thompson SM, Davies RJ. Altered intracellular calcium regulation in human colorectal cancers and in “normal” adjacent mucosa. Cancer Res. 1991;51:4492–4. [PubMed] [Google Scholar]
- 47.Pretlow TP, Barrow BJ, Ashton WS, et al. Aberrant crypts: putative preneoplastic foci in human colonic mucosa. Cancer Res. 1991;51:1564–7. [PubMed] [Google Scholar]
- 48.Yang C, Wen Y, Li H, et al. Overexpression of minichromosome maintenance 2 predicts poor prognosis in patients with gastric cancer. Oncol Rep. 2012;27:135–42. doi: 10.3892/or.2011.1473. [DOI] [PubMed] [Google Scholar]
- 49.Giaginis C, Georgiadou M, Dimakopoulou K, et al. Clinical significance of MCM-2 and MCM-5 expression in colon cancer: association with clinicopathological parameters and tumor proliferative capacity. Dig Dis Sci. 2009;54:282–91. doi: 10.1007/s10620-008-0305-z. [DOI] [PubMed] [Google Scholar]
- 50.Lechner D, Manhardt T, Bajna E, et al. A 24-phenylsulfone analog of vitamin D inhibits 1alpha,25-dihydroxyvitamin D(3) degradation in vitamin D metabolism-competent cells. J Pharmacol Exp Ther. 2007;320:1119–26. doi: 10.1124/jpet.106.115451. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.