

Abstract
The structures of membrane proteins are generally solved using samples dissolved in micelles, bicelles, or occasionally phospholipid bilayers using X-ray diffraction or magnetic resonance. Because these are less than perfect mimics of true biological membranes, the structures are often confirmed by evaluating the effects of mutations on the properties of the protein in their native cellular environments. Low-resolution structures are also sometimes generated from the results of site-directed mutagenesis when other structural data are incomplete or not available. Here, we describe a rapid and automated approach to determine structures from data on site-directed mutants for the special case of homo-oligomeric helical bundles. The method uses as input an experimental profile of the effects of mutations on some property of the protein. This profile is then interpreted by assuming that positions that have large effects on structure/function when mutated project toward the center of the oligomeric bundle. Model bundles are generated, and correlation analysis is used to score which structures have inter-subunit Cβ distances between adjoining monomers that best correlate with the experimental profile. These structures are then clustered and refined using energy-based minimization methods. For a set of 10 homo-oligomeric TM protein structures ranging from dimers to pentamers, we show that our method predicts structures to within 1–2 Å backbone RMSD relative to X-ray and NMR structures. This level of agreement approaches the precision of NMR structures solved in different membrane mimetics.
Keywords: correlation analysis, cross-linking, TOXCAT, M2, molecular modeling
Introduction
Helical transmembrane (TM) protein structure determination represents a significant challenge. Fewer than 2% of all experimentally determined structures deposited in the Protein Data Bank1 (PDB) are membrane proteins, yet 20–25% of open reading frames from recently sequenced genomes encode for proteins that embed in the membrane.2,3 Even with advances in conventional methods for protein structure determination such as X-ray crystallography and NMR spectroscopy, the fundamental problems of obtaining diffraction-quality crystals, protein expression and purification, and protein-size limitations still remain. Computational methods for modeling TM protein structure are becoming increasingly more important if we hope to decrease the discrepancy in structural information between globular and membrane proteins.
Depending on the scientific question being asked, the laborious (and sometimes insurmountable) task of experimentally determining the structure of a membrane protein using conventional methods may not be necessary. For example, Zhu et al. recently used disulfide cross-linking information to build models for the helical TM dimers glycophorin A (GpA) and integrin αIIβ3.4 The resulting models for GpA had a root-mean-square deviation (RMSD) over the backbone atoms of 1–1.5 Å with the NMR structures. Metcalf et al. used mutagenesis data and protein sequence variation to build models for the TM homo-dimers GpA and BNIP3 apoptosis factor.5 The RMSD for the GpA model was 1.3 Å. We hypothesize that other forms of low-resolution experimental data can potentially provide sufficient information to accurately model other TM protein structures. Experimental data from a variety of mutagenesis experiments are ideal for studying this possibility.
The earliest structural models by Treutlein et al. and Adams et al. for the TM region of GpA were based solely on the energetics of interaction between helices.6,7 The resulting models were compared against mutagenesis data showing the disruptive effects that nonpolar mutations had on GpA’s ability to dimerize.8 The structural models agreed with the mutagenesis data and showed that key residues oriented toward the helical interface were sensitive to nonpolar mutations.
The approach of modeling helical TM regions using the energetics of interaction between helices has been extended to larger homo-oligomers. Phospholamban is a TM homo-pentamer that is important in calcium storage and release in cardiocytes. Mutagenesis studies7,9 showed that mutations of key hydrophobic residues disrupted pentamer oligomerization. A global search of conformational space revealed five low-energy helical bundles,7 only one of which was found to be in agreement with an extensive set of mutagenesis data,7,9 and ultimately the experimentally determined structures.10,11 This 5-fold symmetrical structure has a left-handed twist; most critical residues lie at the helix/helix interface and show large interaction energies. Interestingly, the lowest-energy conformer did not agree with the experimental results, indicating that energy is a necessary—but insufficient—criterion for assessing models.
Herzyk and Hubbard used a different approach to model helical TM homo-oligomers.12 Using a combination of Monte Carlo/simulated annealing (MCSA) and molecular dynamics/simulated annealing along with a set of orientational restraints derived from published mutagenesis data,7,9 Heryzk and Hubbard constructed models for GpA and phospholamban. Unlike the modeling approach of Treutlin et al. and Adams et al. which made use of mutagenesis data after the model was constructed, Herzyk and Hubbard used restraints derived from mutagenesis data in their modeling procedure. The resulting model for GpA had an RMSD to native of 0.9 Å over the backbone atoms. A comparison of the profile between interaction energy and mutagenesis data revealed an excellent level of agreement for phospholamban.
More recent approaches for modeling helix TM homo-oligomers fall into one of these two categories: modeling methods based purely on energetics13–16 and those that use some combination of energetics and low-resolution experimental data.4,5,17,18 The incorporation of experimental data directly into the modeling process provides two obvious benefits. First, the experimental data correct for inaccuracies in the force field and for approximations regarding the environmental conditions. Second, through the use of experimental data directly in the modeling process, the conformational space that needs to be sampled can be greatly reduced.
We have developed a novel approach for modeling helical TM homo-oligomers that incorporates a variety of low-resolution mutagenesis data directly into the modeling process. Our modeling approach consists of two phases. In the first phase, we use a symmetric rigid-body search to generate an ensemble of models that is consistent with a given set of low-resolution data. In the second phase, we cluster and then refine only the centroid models using the CHARMM22 force field. At the heart of our rigid-body search is a simple scoring function that restrains the conformational search by maximizing the correlation between inter-subunit Cβ distance and experimental data while minimizing steric clashes between helices. Our correlation term allows us to use a variety of low-resolution mutagenesis data without the need for scaling the data or converting the data into distance restraints4 or angular restraints.12 We demonstrate the accuracy of our modeling approach by using a variety of low-resolution experimental data such as mutagenesis, ToxR, TOXCAT, ion channel, and cross-linking data to model the TM regions of GpA, phospholamban, M2, BM2, BNIP3, and the ephrin receptor tyrosine kinase (EphA1). The final models ranged in RMSD from 0.6 Å to 2.1 Å when compared to the native structures. This approach to modeling helical TM protein structure can be of enormous benefit when conventional methods of protein structure determination fall short.
Results
Overview of modeling protocol
Our modeling protocol can be broken down into two phases. The first phase involves rigid-body sampling (RBS) using an ideal or experimentally determined helix. The second phase involves side-chain placement, clustering, and refinement of the models with a molecular mechanics force field. We briefly describe the first phase here. A detailed description of the second phase can be found in Materials and Methods. RBS begins with a helix that is transformed to the global frame of reference so that the axis of the helix is coincident with the global z-axis and its geometric center is at the origin. Four degrees of freedom are required to define the relationship between monomers in a structure with exact rotational symmetry. Here, we apply two rotations and two translations to define the location of the helix in the unit cell. The individual steps in our modeling protocol are illustrated in Fig. 1.
Fig. 1.

An illustration of the steps used to model a TM oligomer with exact rotational symmetry. Step 1 starts with an ideal helix transformed to the global frame of reference such that the geometric center of the helix is positioned at the origin. Step 2 involves rotation about the global z-axis and determines which residues will form the interface of the dimer (denoted by the variable Φ). Step 3 is a translation along the global z-axis and will affect the point of closest approach (denoted by the variable Tz). Step 4 is a translation along the global x- axis and will affect the radius of the bundle (denoted by the variable Tx). Step 5 is a rotation about the global x-axis and will affect the tilt of the bundle with respect to the global z-axis (denoted by the variable θ). Step 6 is a rotation about the global z-axis used to generate the symmetry mate followed by optimization between experimental data and inter-subunit Cβ distance (see Materials and Methods for a description of the two-term scoring function used in the optimization step). Once an ensemble of 1000 poses has been generated, the ensemble is clustered, and side chains are added to the centroid models. The centroid models are then refined using Xplor-NIH. The spheres on the end of the helices denote the N-terminus (blue) and C-terminus (red). The individual axes on the global frame are color coded as follows: red denotes the positive x-axis, green denotes the positive y-axis, and blue denotes the positive z-axis.
At the heart of our RBS method is the use of the correlation coefficient (r) to evaluate the degree to which experimental data correlate with the projection of the side chains in the oligomer, as defined by the inter-subunit contact distance for each residue in the structure. The inter-subunit contact distance is defined here as the distance between Cβ atoms on identical residues of a homo-dimer, and this provides a quantitative measure that can be correlated with the extent of perturbation or cross-linking associated at the same position in the sequence. The correlation coefficient is a measure of the linear relationship between two variables and ranges from a value of 1 for two perfectly correlated variables to a value of −1 for two perfectly anti-correlated variables. The correlation coefficient is used to restrain the RBS protocol by incorporating it directly into a scoring function that is used to optimize each pose (see Materials and Methods). In this study, we correlate inter-subunit Cβ distance with the degree of experimental perturbation associated with mutations or the extent of cross-linking in a Cys-scanning experiment to determine how well a given hypothetical model agrees with experimental data. The extent of Cys cross-linking and the perturbational effects of mutations generally increase with decreasing inter-subunit distance (negative correlation). However, for simplicity, we refer to all correlations as positive for structures that are in agreement with the expected experimental outcome.
We demonstrate the utility of our RBS protocol by using it in three tests. In the first test, we use it to search for a set of idealized helical conformations using native inter-subunit Cβ distances as “experimental data.” The second test is similar to the first test but uses a set of nine symmetric helical TM structures obtained from the PDB instead of idealized helical arrangements. It should be noted that the first and second tests are used to determine how well our search strategy works under the most ideal conditions (i.e., where experimental data correlate perfectly with inter-subunit Cβ distances). In the third and final test, we model these same nine structures using low-resolution experimental data to restrain the search. The resulting ensembles of models from this test are clustered using a k-medoid clustering algorithm.19 Side chains are then added to each of the centroid models using SCAP20 followed by all-atom refinement using the CHARMM22 force field implemented in the Xplor-NIH package.21
Idealized hide-and-seek test
To test the RBS protocol, we constructed a set of 10 helix dimer conformations by randomly choosing values for the four search parameters (Tx, Tz, θ, and ϕ). Each set of four parameters is then used to position a 16-residue ideal poly-alanine helix in space. The symmetry mate is generated by rotating a copy of the helix 180° about the global z-axis. After construction of the 10 dimers, we determined the inter-subunit Cβ distances along the length of the helices. These distances were used as simulated experimental data to restrain the rigid-body search with the goal of recapitulating the original dimer conformation. In all 10 cases, the simple scoring function selects a model with an RMSD of 0.6 Å or less to the starting conformation (see Supplementary Table 1).
Hide-and-seek test using TM structures from the PDB
The RBS protocol can generate the native pose with high accuracy for idealized cases. A more challenging test would entail modeling actual helical structures from the PDB that may not contain idealized geometry. We repeated the hide-and-seek test on a set of nine symmetric helical TM structures from the PDB. Three of these structures are dimers, five are tetramers, and one is a pentamer. For each test case, we determined the inter-subunit Cβ distances from the first two chains of the native structure. If a glycine is present along the protein sequence, we computed the distance between Cα atoms. For structures solved using NMR, we use the average structure (see Materials and Methods) to obtain the native distances.
We use three separate measures of RMSD in assessing the performance of the RBS protocol on experimentally determined structures. The first measure, RMSDScore, denotes the RMSD between the best-scoring model in the ensemble and the native structure. The second measure, RMSDMin, denotes the smallest RMSD in the ensemble. The third measure, RMSDNative, denotes the RMSD of the best-scoring model when the native helix is used in place of the ideal helix in the rigid-body search. As shown in Table 1, all nine cases have an RMSDScore of 2.9 Å or less. The dimer BNIP3 gives the best results with an RMSDScore of 0.9 Å. The worst-performing case, phospholamban, gives an RMSDScore of 2.9 Å. The remaining cases yield RMSDScore values between 1.2 and 2.0 Å. While our scoring function does not select the lowest RMSD model in the ensemble, it does perform reasonably well at generating low-RMSD models (Fig. 2). With the exception of the BM2 case, a sizable population of models with RMSDs below 1.5 Å is always generated. Producing an ensemble of models with relatively low RMSD to native is critical for two reasons. First, models that are near native will generally yield more favorable scores in the refinement stage. Second, clustering will be more effective at assigning near-native models as centroids.
Table 1.
Application of the sampling method using inter-helical Cβ distances from experimentally determined helical TM structures
| Name | Oligomer | PDB ID | Residues | RMSDScore | RMSDMin | RMSDNative |
|---|---|---|---|---|---|---|
| GpA | Dimer | 1AFO | 73–96 | 1.7 | 1.4 | 0.1 |
| EphA1 | Dimer | 2K1L | 549–560 | 1.3 | 1.0 | 0.2 |
| BNIP3 | Dimer | 2KA2 | 170–184 | 0.9 | 0.9 | 0.3 |
| M2(xtal) | Tetramer | 3LBW | 26–43 | 1.4 | 1.4 | 1.4 |
| M2(NMR) | Tetramer | 2RLF | 26–43 | 1.5 | 1.2 | 0.1 |
| M2(ssNMR1) | Tetramer | 2KQT | 26–43 | 1.3 | 1.2 | 0.0 |
| M2(ssNMR2) | Tetramer | 2LOJ | 26–43 | 1.5 | 1.2 | 0.2 |
| BM2 | Tetramer | 2KIX | 7–25 | 2.0 | 1.6 | 0.6 |
| Phospholamban | Pentamer | 1ZLL | 37–52 | 2.9 | 0.9 | 0.3 |
Fig. 2.
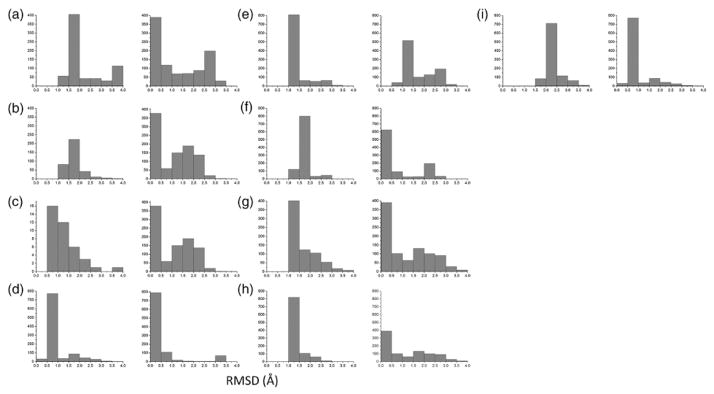
RMSD distributions for models generated using native inter-subunit Cβ distances. Each panel consists of two distributions. The distribution on the left was generated using an ideal helix. The distribution on the right was generated using the native helix. The RMSD value is between each model in the generated ensemble and the native structure. The distributions are: (a) GpA, (b) EphA1, (c) BNIP3, (d) phospholamban, (e) M2(xtal), (f) M2(NMR), (g) M2(ssNMR1), (h) M2(ssNMR2), and (i) BM2.
We suspected that our sampling algorithm could generate a larger population of near-native models if we introduced natural curvature into the starting helix. Superimposing an ideal helix onto the corresponding native helix gives an RMSD that is larger than 1.0 Å for GpA, BM2, and all of the M2 structures. To better assess how this deviation from ideality influences the final result, we carried out the same search using the native helix in place of the ideal helix. The resulting RMSDNative values are 0.6 Å or less for all cases with the exception of the M2(xtal) case (Table 1). However, we note the existence of models with RMSDNative values of 0.6 Å or less for all of the ensembles generated using a native helix (Fig. 2).
Restrained sampling using low-resolution experimental data
The first two tests show that when sufficient information between monomers is given in the form of native distances, our RBS protocol can generate models with RMSDMin values between 0.9 and 1.6 Å. However, in a practical situation, exact distance information will likely be unavailable. Therefore, to assess the ability of the sampling protocol to perform similarly in a practical situation, we used low-resolution experimental data to restrain the search. Besides being the most stringent test thus far, given the inherent noise present in low-resolution experimental data, this test will provide a meaningful benchmark in terms of the practicality of our method. A description of the low-resolution experimental data is provided in Materials and Methods.
Before carrying out the search, we wanted to test our hypothesis that inter-subunit Cβ distance correlates with low-resolution experimental data. To do this, we determined the correlation coefficient and the associated p values between the inter-subunit Cβ distance data obtained from each native structure and the corresponding set of experimental data (see Supplementary Tables 2–8 for the experimental data). Phospholamban has the strongest correlation with |r|=0.91 (p=4.6E-7). The dimer GpA has roughly the same |r| value of 0.78 (with an approximate p value of 5.0-E-6) for both the cross-linking and mutagenesis data. The dimer EphA1 has |r|=0.76 (p= 3.2E-3). The M2 cases have roughly the same |r|=0.72 (with a p value of about 3.7E-4). BNIP3 and BM2 have the weakest correlations with |r|=0.44 (p= 5.7E-2) and |r|=0.58 (p= 4.7E-3), respectively. For all but one of the cases, the p value for the correlation between experimental data and inter-subunit Cβ distance is less than 0.05, indicating that the correlation is unlikely due to chance. Based on the |r| values and associated p values obtained for the native structures, it would seem that correlating inter-subunit Cβ distance with low-resolution experimental data can provide a useful filter when modeling TM homo-oligomers (Fig. 3).
Fig. 3.

PI versus inter-subunit Cβ distance profiles for (a) native M2(NMR) structure and (b) our best-scoring model after refinement with Xplor-NIH. A superimposition of our best-scoring model and the native M2(NMR) structure is shown on the right.
Using the low-resolution experimental data to generate TM bundles, we obtained an RMSDScore of 2.1 Å or less for 8 out of 10 cases (Table 2). BNIP3 and phospholamban are the largest outliers with RMSDScore values of 3.0 Å. For BNIP3, it is not surprising that the RMSDScore is so large given the weak correlation between inter-subunit Cβ distance and the experimental data. For phospholamban, we noticed that the bundle radius for the top-scoring model is about 1.0 Å smaller than in the native structure. A more important measure of performance of the sampling protocol is how close to native conformation our sampling can reach. Clearly, if the sampling protocol cannot generate a sufficient number of models that are close to native, it is likely that all-atom refinement will be of little value in generating good models. The RMSDMin value is 1.6 Å or less for 9 out of 10 cases (Table 2). With the exception of BM2, the sampling protocol generates ensembles with a significant fraction of models with less than 2.0 Å RMSD to native (Fig. 4). Based on these results, it appears that when inter-subunit Cβ distance data correlate strongly with mutational data, RBS alone can be used to generate reasonable starting conformations that can be further refined. However, since our scoring function is designed as a filter, it may not select the most energetically favorable conformation in the ensemble of models. For this, we use a more detailed all-atom scoring function.
Table 2.
Application of the sampling method using low-resolution experimental data
| Name | Type of data | RMSDScore | RMSDMin |
|---|---|---|---|
| GpA | Mutagenesis | 1.4 | 1.4 |
| GpA(Cross-linking) | Cross-linking | 1.6 | 1.4 |
| EphA1 | TOXCAT | 1.6 | 1.4 |
| BNIP3 | TOXCAT/mutagenesis | 3.0 | 1.6 |
| M2(xtal) | Ion conductance | 1.9 | 1.5 |
| M2(NMR) | Ion conductance | 1.4 | 1.3 |
| M2(ssNMR1) | Ion conductance | 1.6 | 1.5 |
| M2(ssNMR2) | Ion conductance | 1.7 | 1.3 |
| BM2 | Ion conductance | 2.1 | 1.8 |
| Phospholamban | Mutagenesis | 3.0 | 1.0 |
Fig. 4.

RMSD distributions for models generated using low-resolution experimental data. Each panel consists of a single distribution that was generated using an ideal helix. The RMSD value is obtained between each model in the ensemble and the native structure. The distributions are as follows: (a) GpA, (b) GpA(Cross-linking), (c) BNIP3, (d) EphA1, (e) BM2, (f) M2(xtal), (g) M2(NMR), (h) M2(ssNMR1), (i) M2 (ssNMR2), and (j) phospholamban.
Refinement using Xplor-NIH
The resolution of our simple scoring function does not capture detailed energetic interactions such as van der Waals packing and Coulombic interactions. These interactions are important for obtaining optimal packing between helices. To capture these important interactions, we first cluster the ensemble of models generated using our RBS protocol, add side chains to all centroids, and then subject them to all-atom refinement using the CHARMM22 force field in Xplor-NIH.21 The most favorable scoring model according to XPLOR is deemed our best prediction.
Refinement of the centroids gives an RMSDScore of 2.1 Å or better for all 10 cases (Fig. 5). For the dimers GpA, GpA(Cross-linking), EphA1, and BNIP3, the RMSDScore is 1.4 Å or less. Results for larger homo-oligomeric states are equally as impressive with RMSDScore ranging in value from 1.1 to 2.1 Å. Given the spread in RMSD values between individual models in the native NMR ensembles, which can be as large as 0.9 Å for some of the structures considered here, our results would indicate that the RBS protocol coupled to clustering and refinement with Xplor-NIH has the potential to generate models comparable in accuracy to those obtained using medium-resolution NMR. The importance of using a detailed all-atom scoring function is clearly illustrated for the case of BNIP3. Using our simple scoring function to select a model from the ensemble will give an RMSD to native of 3.0 Å. If we refine all of the models in the ensemble and then select the most favorable scoring model according to CHARMM22, we obtain an RMSD to native of 0.6 Å. Clearly, refining the entire ensemble of 1000 models would be a time-consuming task, and so we cluster the ensemble of models first and then refine only the centroids. Using this approach, we also obtain a model with an RMSD to native of 0.6 Å but do so in a fraction of the time it would take to refine the entire ensemble of models. The r value between the experimental data and the inter-subunit Cβ distance for the refined models either remained the same or improved when compared with the corresponding value for native.
Fig. 5.
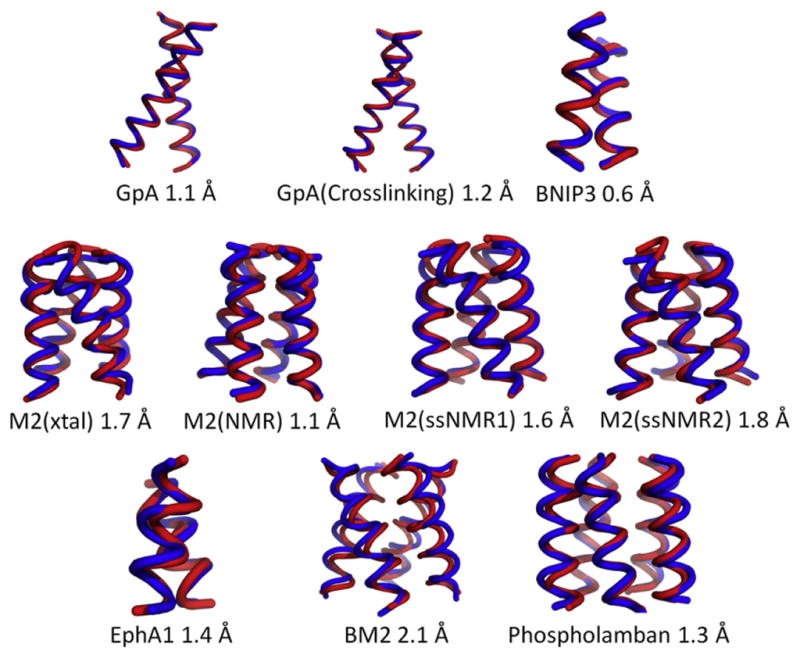
A comparison between the backbone of the native structure (shown in blue) and best-scoring model (shown in red) after refinement.
As a control, we applied the same Xplor-NIH refinement protocol to all the native structures. This involved refinement of all the individual models in each NMR ensemble and not the average model. We expect the experimentally determined structures after refinement to have scores that are similar to or more favorable than the scores of our centroid models. We observe this trend for all cases with the exception of BM2 (Fig. 6). We find that the refined native models for BM2 are about 100 XPLOR energy units less favorable than our best-scoring model. This seems to imply that the native BM2 bundle may not be tightly packed, which ultimately leads to a less favorable van der Waals score. For most cases, refinement with Xplor-NIH does not significantly perturb the native structure. The RMSD between the unrefined and refined native models is on average less than 1.0 Å (represented as blue circles in Fig. 6). For phospholamban and BM2, refinement perturbs the native conformation to a larger extent. In particular, the RMSD after refinement of the native BM2 ensemble resulted in two models having RMSDs larger than 1.7 Å.
Fig. 6.

Energy profiles versus RMSD to native after refinement with Xplor-NIH. Blue circles show the RMSD between the starting native model (from the NMR ensemble) and the native model after refinement with Xplor-NIH. Red circles show the RMSD between each of the centroids and the corresponding native structure. The filled green circles represent the most favorable scoring model among the 20 centroid models. Only models with RMSD values below 2.5 Å are displayed on the graph. The panels are labeled as follows: (a) GpA, (b) GpA(Cross-linking), (c) EphA1, (d) BNIP3, (e) M2(xtal), (f) M2(NMR), (g) BM2, (h) phospholamban, (i) M2(ssNMR1), and (j) M2(ssNMR2).
Discussion
We have presented a method for modeling helical TM homo-oligomers that uses a rotationally symmetric rigid-body search followed by clustering and energy refinement using the CHARMM22 force field in Xplor-NIH. At the heart of our modeling procedure is a simple scoring function composed of a VDW clash term and a correlation coefficient between mutational data and inter-subunit Cβ distance. The simple scoring function is optimized to obtain maximal agreement with experimental data while avoiding clashes between helices. The novelty of our method is in its ability to directly restrain the search using low-resolution experimental data. This prevents the search from needlessly meandering through space and focuses the sampling to give the best agreement with experimental data.
Our method performs best when the experimental data correlate with |r|>0.5 with the native inter-subunit Cβ distance. In these cases, the rigid-body search does a reasonable job at generating near-native backbone conformations. As the correlation becomes weaker, so does the structural similarity between the native structure and the best-scoring model. The combination of clustering, all-atom refinement, and ranking with the Xplor-NIH scoring function improves the RMSD value to native. In 7 of the 10 cases, the RMSD to native is 1.6 Å or less.
Modeling TM homo-dimers
As a prerequisite for addressing the general TM homo-oligomer problem, we first applied our modeling approach to the homo-dimer GpA using two sets of mutational data. One set of data is from a fairly recent study and is composed of cross-linking efficiency.4 Another data set consists of dimer disruption data and has been used extensively by others to propose different methods for modeling the TM region of GpA.5,12–14 Through the use of either a combination of energetics and restraints derived from mutational data or energetics alone, all of these methods generate models for the TM region of GpA with RMSDs to native in the range of 0.7–1.5 Å. Through the use of either set of low-resolution experimental data, our modeling approach achieves a similar level of accuracy for GpA.
Earlier work in our group made use of an MCSA protocol to propose a model for the TM region of BNIP3.5 The MCSA method used two energy terms that would penalize both neutral and disruptive mutations. The method we propose here is different in two ways. First, we do not use a stochastic approach for sampling conformational space. Second, the present method does not rely solely on the energy to decide on the plausibility of a model but instead also relies on how well the inter-subunit Cβ distance correlates with mutational data. While both methods manage to accurately model the backbone of BNIP3, only the MCSA protocol correctly models the hydrogen bond between Nε2 of His173 and Oγ from Ser172 reported by Sulistijo and Mackenzie.22 Since our refinement protocol in Xplor-NIH does not incorporate side-chain rotamer sampling, we could not optimize hydrogen bond interactions between side chains. This prompted us to see if we could model this hydrogen bond by simply changing the rotameric state of His173 and Ser172 in our best-scoring model. Changing the rotameric states results in a new model that scores better than our original model. This suggests that the hydrogen bond may not be absolutely necessary for dimerization of the helices (our best-refined model did not have this hydrogen bond) but, if formed, produces a slightly more stable complex conferring specificity to the dimer as pointed out in the recent work of Lawrie et al.23
Modeling larger TM homo-oligomeric complexes
Our modeling protocol performed well on larger TM homo-oligomeric complexes. The largest complex we considered is the pentamer phospholam-ban. Similar to the case for GpA, the mutagenesis data for phospholamban have been used extensively in proposing a model for the TM region.7,12,14 It is difficult to compare our results directly with earlier studies since they were carried out before publication of the NMR structure for phospholamban. However, a plot of the inter-helical van der Waals energy per residue for phospholamban reveals a similar periodic pattern observed in plots from earlier studies (Supplementary Fig. 1). A salient feature of using inter-subunit Cβ distance over interaction energy when constructing a profile is that the former descriptor is less sensitive to force field effects. We note that our model for phospholamban has a smaller radius than what is seen in the NMR structure. However, since our modeling protocol does not account for the membrane environment or make use of experimentally derived distance information (i.e., intermonomer nuclear Overhauser enhancements), the effect of the non-bonded forces from the molecular mechanics force field dominates, resulting in tightly packed helices.
We also applied our modeling approach to the influenza proton transporters M2 and BM2. Our modeling protocol generates models for M2 with an RMSD of 1.7 Å to the high-resolution X-ray structure. When compared to the NMR model of M2, our protocol achieves an RMSD of about 1.0 Å. Our automated method provides predictions for M2 that are better than earlier predictions that relied heavily on the expertise and intuition of the investigators.24 We also applied our modeling protocol to the recent solid-state NMR structures of Sharma et al.25 and Cady et al.26 Our best-scoring models have RMSDs of 1.8 Å and 1.6 Å, respectively, to these solid-state structures. As a point of comparison, the NMR (solution and solid state) and the high-resolution X-ray structures show a spread in RMSD between 0.8 and 1.6 Å.
In an earlier study, we also made use of correlation analysis in modeling the BM2 proton transporter.18 In our previous approach, we adopted a less efficient method that included the generation of a large ensemble of sterically feasible helical bundles (both ideal and coiled helices). The ensemble was scored using the correlation coefficient between the pertubility index (PI) and an estimate of the phase angle for the helix. The surviving models were subjected to refinement and then clustered. Two out of eight proposed models from our earlier study are within 1.0 Å of our current best-scoring model. It should be noted that all of the models from our previous study exhibit a weaker correlation with the experimental data than the model we propose here. The current study along with our earlier study shows the generality of the use of the correlation approach in modeling TM homo-oligomers; different geometric descriptors between helices can be used in modeling TM homo-oligomers.
Two clear strengths with our modeling protocol are speed (~8 min on a single 2.40-GHz processor) and the ability to use data directly from experiments conducted in native cellular membranes. This is in contrast to previous methods that often require the conversion of experimental data into distance restraints,4,27 angular restraints,12 or pseudo-energy terms.5 A potential downside of these approaches is their reliance on setting thresholds a priori. In contrast, we use experimental data directly to correlate against geometric descriptors between helices. We feel that this makes for a simpler protocol and allows us to avoid choosing “optimal” values through an intermediate training step. Moreover, since our method relies heavily on the correlation value, data from a variety of different experimental contexts can be used without the need for scaling. Other approaches need to determine different thresholds and penalty functions for different sets of experimental data, which can make them difficult to apply.
One potential drawback of our modeling protocol is the requirement of both neutral and destabilizing mutations. If a mutagenesis experiment is carried out only on residues at the dimer interface, our correlation approach will fail due to its reliance upon detectable differences between residues close to the dimer interface and those farther away. Put another way, if all the values for a particular mutagenesis experiment are identical, the correlation value would be undefined since the difference between each experimental value would be identical with the mean value. Alternative approaches do not have the same constraint. However, we anticipate that most mutagenesis experiments would involve mutations at a number of consecutive residues to determine which residues are located at the interface.
A second potential drawback of our method is the use of an “ideal” helix during the rigid-body search. This drawback has been discussed by Kim et al. who note that experimentally determined helices can contain large deviations from ideal geometry that result in significant kinks or curvature.14 The importance of accounting for curvature as seen in experimentally determined helices was demonstrated by performing a hide-and-seek test using the low-resolution data; carrying out the search using the native helix yielded an RMSDMin in the range 0.3–1.1 Å, while using an ideal helix yields an RMSDMin in the range 1.1–1.8 Å. One way of incorporating experimentally determined helices into our modeling protocol is through the use of helical protein structures deposited in the PDB. Initial tests using experimentally derived helices extracted from the PDB reveal significant improvements in RMSDMin when compared to the case of using an ideal helix. Using the native helix from each target in Table 2, we searched the PDB using a rapid distance-matrix structure search method28 and extracted all helices below an RMSD of 0.5 Å to the native helix. The helix with the smallest RMSD to the native helix was used to carry out a rigid-body search using the low-resolution experimental data. The RMSDMin value when using the PDB-derived helices range from 0.5 to 1.3 Å, which is not significantly different from the case of using the native helix. However, these PDB-derived helices were obtained using the native helix, which will likely be unavailable in a practical modeling situation. It is clear that devising a way to incorporate structural information from the PDB into our modeling protocol would provide substantial enrichment of near-native conformations.
The ultimate utility of our method to experimental biologists would be to avoid performing exhaustive mutagenesis experiments when attempting to model homo-oligomeric helical TM structure. Earlier work in our group used phylogeny information along with lattice models to determine how much experimental information is needed to make reasonable predictions.29 For the method developed in our current study, we find that the more experimental data points provided as input, the more accurate the final predictions will be. However, a judicious choice of sequence region to target for carrying out the mutagenesis experiments can yield accurate results with far fewer experimental data points. We find that, for GpA, eight contiguous experimental data points are sufficient to generate predictions that are within 1 Å RMSD to native (Supplementary Table 9). Selecting 8 contiguous experimental data points from the N-terminal, center, or C-terminal regions of the TM sequence produces results that are similar to what is obtained using the full set of 23 experimental data points. Splitting the eight contiguous experimental data points into four contiguous experimental data points at both N-terminal and C-terminal ends of the TM sequence for GpA yields a prediction that is also near 1 Å RMSD to native. We find a similar result for the phospholamban case when using four contiguous experimental data points at both the N-terminal and C-terminal ends.
Future improvements to our method will include adding additional terms to our simple two-term scoring function. One possibility would be to include knowledge-based terms to improve the packing between helices. Work by Harrington and Ben-Tal.30 shows that five types of chemical interactions common to TM helices could be used to essentially generate sub-angstrom predictions. It would be interesting to see if these five types of chemical interactions could be used to complement our current scoring function to filter out conformations that do not exhibit structural determinants common to TM helices. Such an approach would incur minimal computational cost while enriching the ensemble with more native-like models.
While the manuscript was in review, a refined structure for phospholamban was published by Verardi et al.11 Comparing our prediction for phospholamban to this new structure (PDB ID: 2KYV) gives a final prediction of 0.8 Å. Using four contiguous experimental data points at both the N-terminal and C-terminal ends also gives a prediction around 0.8 Å.
Conclusion
In summary, we have developed a tool for rapidly modeling helical TM homo-oligomers that uses low-resolution experimental data directly in the modeling process. At the heart of our modeling protocol is the use of a correlation term that restrains the RBS and avoids costly searches in regions of conformational space that do not correlate with experimental information. We show that correlating mutagenesis, cross-linking, and ion channel data with inter-subunit Cβ distance data followed by refinement provides accurate models for helical TM proteins exhibiting exact rotational symmetry. One area where our modeling approach is likely to have a significant impact is in situations where it is either too difficult or time consuming to obtain a complete set of NMR data.
Materials and Methods
Details regarding the low-resolution experimental data
At the core of our sampling methodology is the use of experimental information in the form of mutagenesis data, cross-linking data, and TOXCAT and TOXR data. We provide a short description of the data below. All of the experimental data used in this study can be found in Supplementary Tables 2–8.
Phospholamban
Phospholamban is a homo-pentameric bundle located in the sarcoplasmic reticulum of cardiocytes and is responsible for calcium transport. The mutagenesis data for phospholamban were taken from Table 2 of Simmer-man et al.9 The data from this table show the extent of pentamer formation following mutation to either an alanine or a phenylalanine along the TM region. For this study, we used the alanine mutational data only.
M2 and BM2
Both M2 (A/M2) and BM2 are homo-tetrameric TM proton transporters belonging to different types of influenza viruses. These proton transporters are responsible for acidifying the interior of the virus, which ultimately leads to virion uncoating in the endosomes. For both M2 and BM2, we used PI, which is a combination of reversal potential, current and specific activity data (see Pinto et al.24 for details). PI data for M2 from were obtained from Fig. 1 of Pinto et al.24 PI data for BM2 were obtained from Fig. 3 of Ma et al.18
GpA
GpA is homo-dimeric sialoglycoprotein from erythrocyte cells. Two sets of data for the TM region of of GpA were used. The first set of data was obtained from Fig. 5 of Lemmon et al. and shows the relative degree of disruption of the GpA dimer by mutation of the native sequence with a nonpolar residue.8 The degree of disruption of the dimer interface uses a scale of 0 (no effect on dimer formation) to 3 (no dimer formation). The second set of data was taken from Supplementary Tables of Zhu et al. and shows the percentage of cross-linking between residues in the TM region of the αIIbβ3/GpA chimera.4 For this study, we considered only symmetric cross-linking data between residues.
BNIP3
BNIP3 is a homo-dimer and a member of the Bcl-2 homology domain-3 subfamily of proapoptotic Bcl-2 proteins. BNIP3 is associated with apoptotic response in the myocardium. Mutational data for the TM region of BNIP3 dimer come from Fig. 7 of Lawrie et al. and represent a combination of TOXCAT and SDS-PAGE page phenotype scores based on percentage dimer disruption.23 The “unified score” gives the average disruptive effect of different amino acid substitution along the protein sequence of the TM helix. The unified scale ranges in value from 0 (no dimer formation) to 10 (strong dimerization). We used all the phenotype scores from the alanine mutations.
EphA1
Ephrin receptor A1 is part of a receptor tyrosine kinase and is involved in animal development and certain cancers. ToxR data were taken from Table 3 of Volynsky et al.15 It should be noted that the ToxR data from Volynsky et al. do not consider every possible residue along the TM region. For our purposes, the mutations to glutamine and serine were not as informative, since mutation to a polar residue in the membrane could cause the dimer to be disrupted even when the residue is not along the dimer interface. Similarly, glycine mutations may cause a structural change in the helices despite not residing at the oligomer interface. Therefore, we concentrated on only the hydrophobic mutations (i.e., the isoleucine, alanine, and valine mutations).
Generation of the homo-oligomeric models
Generation of the oligomer begins with the construction of an ideal helix using CHARMM22 internal geometry with ϕ and Ψ dihedral angles set to –60° and –40°, respectively. Modeling was carried out only for the TM region that had experimental data. As such, the sequence length of the helix was dependent on the available experimental data.
The sampling procedure begins with an ideal helix containing the native sequence. Side chains were not considered at this stage, but all residues (with the exception of glycine) contained a Cβ atom. The individual steps used to position the helix in space are depicted in cartoon form in Fig. 1. Each search for the best dimer configuration begins with a set of initial parameters for the four variables Tx, Tz, θ, and ϕ that were applied to an ideal helix centered at the origin of the global frame of reference. To maximize the correlation of experimental data with inter-subunit Cβ distance while maintaining a sterically feasible distance between helices, we optimized the scoring function below using the Nelder–Mead simplex algorithm from the Gnu Scientific Library31
The weighting parameters C1 (kcal/mol Å2) and C2 (kcal/mol) were set to 75 and 100, respectively. The scale variable S is used to soften the van der Waal’s radii. For the study carried out here, S was set to 0.80. is the sum of the van der Waal’s radii of two atoms ij (the radii for different atoms were taken directly from the XPLOR manual32). The distance between two atoms is denoted as “R(Tx, Tz,θ,φ)ij” and is a function of the four search parameters Tx, Tz, θ, and Φ. The “corr” term is the correlation between the inter-subunit Cβ distances and the corresponding experimental data. The experimental data (denoted “y” in the above equation) denote the same residue on symmetric helices and are correlated with the distance between these residues. The variable M represents all the atoms from each helix, while the variable N represents only the Cβ atoms.
Initial values for the sampling parameters were obtained by coarse enumeration between a suitable set of numerical boundaries and were subject to the three following conditions: (1) the bundle radius Tx for a dimer must lie between 2 and 4 Å. For oligomeric states larger than 2, the bundle radius should be restrained between 6 and 9 Å. (2) The tilt angle θ must lie in the range of –30° to 30°. (3) The translation along the z-axis, Tz, measured from the geometric center of the helix lies in the range of −10 to 10 Å. To avoid a combinatorial explosion of values for the parameters, we capped the maximum number of initial values to 1000. Only models with a Score less than 50 were retained for the refinement phase.
Clustering
Models were clustered using a k-medoid algorithm from the C clustering library.19 The number of initial clustering attempts was set to 100. The model with smallest RMSD to all other models in the cluster was selected as the centroid model.
Side-chain placement
Side chains were added to all the centroid models using the side-chain prediction program SCAP.20 Default options were used with SCAP.
Refinement with Xplor-NIH
After side-chain addition, all of the centroid models from the first phase were subjected to 100 steps of rigid-body minimization using Xplor-NIH with the CHARMM22 force field. The goal of this step in the refinement procedure is to enforce proper packing between helices while removing any steric clashes that arise as a result of having used a reduced representation for the side chain during the generation of the oligomer in the first phase. The rigid-body minimization step is then followed by two thousand steps of Powell minimization. The dielectric constant was set to 4, and the nonbonded cutoff distance was set to 12.0 Å. All other options were left at their default values.
Correlation versus anti-correlation
It should be noted that while some experimental data will correlate positively with inter-subunit Cβ distance, some data will correlate negatively with inter-subunit Cβ distance. This can be understood by considering the case for cross-linked residues. If two corresponding residues in a homo-dimer are close in space, the distance between the Cβ atoms will be small. In this scenario, the Cβ atoms should cross-link strongly. The data would then be anti-correlated with a maximal value of −1 since a small distance yields a stronger (large magnitude) cross-linking signal. The statistical significance attributed to r is the probability of arriving at the current value if the correlation coefficient were in fact actually 0 (the null hypothesis). For the purposes of this study, r is considered statistically significant if the associated p value is less than 5% (p<0.05). Correlation coefficients and their respective p values were calculated in Matlab. The p values are one sided and represent the probability that two uncorrelated sequences of the given length would have a correlation value as good as the calculated correlation by chance.
RMSD calculations
The RMSD is computed by optimally superimposing N, Cα, C, and O atoms from a model onto the native structure. For the purposes of comparing our best prediction with an NMR models, we used the average model computed from all the individual models in the NMR ensemble. For the results involving refinement of the native models, we superimposed the refined native model onto the unrefined native model to determine the RMSD.
Supplementary Material
Acknowledgments
We acknowledge support from grants 54616 and 60610 from the National Institutes of Health to W.F.D. and support from the Materials Research Science and Engineering Center program of the National Science Foundation. B.T.H. would like to thank the United States Department of Defense for support through a National Defense Science and Engineering Graduate Fellowship. We would like to acknowledge Dr. Gevorg Grigoryan for carefully reading the manuscript and for allowing us to use his rapid distance-matrix-based method for scanning the PDB. We also would like to thank Gerald Cadena for help with Fig. 1. A web interface is available at http://www.degradolab.org/OSTRICh.
Abbreviations used
- PDB
Protein Data Bank
- TM
transmembrane
- MCSA
Monte Carlo/simulated annealing
- RBS
rigid-body sampling
- PI
pertubility index
Footnotes
Supplementary data to this article can be found online at doi:10.1016/j.jmb.2011.10.016
References
- 1.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kyogoku Y, Fujiyoshi Y, Shimada I, Nakamura H, Tsukihara T, Akutsu H, et al. Structural genomics of membrane proteins. Acc Chem Res. 2003;36:199–206. doi: 10.1021/ar0101279. [DOI] [PubMed] [Google Scholar]
- 3.Elofsson A, von Heijne G. Membrane protein structure: prediction versus reality. Annu Rev Biochem. 2007;76:125–140. doi: 10.1146/annurev.biochem.76.052705.163539. [DOI] [PubMed] [Google Scholar]
- 4.Zhu J, Luo BH, Barth P, Schonbrun J, Baker D, Springer TA. The structure of a receptor with two associating transmembrane domains on the cell surface: integrin alphaIIbbeta3. Mol Cell. 2009;34:234–249. doi: 10.1016/j.molcel.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Metcalf DG, Law PB, DeGrado WF. Mutagenesis data in the automated prediction of transmembrane helix dimers. Proteins. 2007;67:375–384. doi: 10.1002/prot.21265. [DOI] [PubMed] [Google Scholar]
- 6.Treutlein HR, Lemmon MA, Engelman DM, Brunger AT. The glycophorin A transmembrane domain dimer: sequence-specific propensity for a right-handed supercoil of helices. Biochemistry. 1992;31:12726–12732. doi: 10.1021/bi00166a003. [DOI] [PubMed] [Google Scholar]
- 7.Adams PD, Arkin IT, Engelman DM, Brunger AT. Computational searching and mutagenesis suggest a structure for the pentameric transmembrane domain of phospholamban. Nat Struct Biol. 1995;2:154–162. doi: 10.1038/nsb0295-154. [DOI] [PubMed] [Google Scholar]
- 8.Lemmon MA, Flanagan JM, Treutlein HR, Zhang J, Engelman DM. Sequence specificity in the dimerization of transmembrane alpha-helices. Biochemistry. 1992;31:12719–12725. doi: 10.1021/bi00166a002. [DOI] [PubMed] [Google Scholar]
- 9.Simmerman HK, Kobayashi YM, Autry JM, Jones LR. A leucine zipper stabilizes the pentameric membrane domain of phospholamban and forms a coiled-coil pore structure. J Biol Chem. 1996;271:5941–5946. doi: 10.1074/jbc.271.10.5941. [DOI] [PubMed] [Google Scholar]
- 10.Oxenoid K, Chou JJ. The structure of phospholamban pentamer reveals a channel-like architecture in membranes. Proc Natl Acad Sci USA. 2005;102:10870–10875. doi: 10.1073/pnas.0504920102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verardi R, Shi L, Traaseth NJ, Walsh N, Veglia G. Structural topology of phospholamban pentamer in lipid bilayers by a hybrid solution and solid-state NMR method. Proc Natl Acad Sci USA. 2011;108:9101–9106. doi: 10.1073/pnas.1016535108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herzyk P, Hubbard RE. Using experimental information to produce a model of the transmembrane domain of the ion channel phospholamban. Biophys J. 1998;74:1203–1214. doi: 10.1016/S0006-3495(98)77835-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fleishman SJ, Ben-Tal N. A novel scoring function for predicting the conformations of tightly packed pairs of transmembrane alpha-helices. J Mol Biol. 2002;321:363–378. doi: 10.1016/s0022-2836(02)00590-9. [DOI] [PubMed] [Google Scholar]
- 14.Kim S, Chamberlain AK, Bowie JU. A simple method for modeling transmembrane helix oligomers. J Mol Biol. 2003;329:831–840. doi: 10.1016/s0022-2836(03)00521-7. [DOI] [PubMed] [Google Scholar]
- 15.Volynsky PE, Mineeva EA, Goncharuk MV, Ermolyuk YS, Arseniev AS, Efremov RG. Computer simulations and modeling-assisted ToxR screening in deciphering 3D structures of transmembrane alpha-helical dimers: ephrin receptor A1. Phys Biol. 2010;7:15. doi: 10.1088/1478-3975/7/1/016014. [DOI] [PubMed] [Google Scholar]
- 16.Rouse SL, Carpenter T, Stansfeld PJ, Sansom MS. Simulations of the BM2 proton channel transmembrane domain from influenza virus B. Biochemistry. 2009;48:9949–9951. doi: 10.1021/bi901166n. [DOI] [PubMed] [Google Scholar]
- 17.Dieckmann GR, DeGrado WF. Modeling transmembrane helical oligomers. Curr Opin Struct Biol. 1997;7:486–494. doi: 10.1016/s0959-440x(97)80111-x. [DOI] [PubMed] [Google Scholar]
- 18.Ma C, Soto CS, Ohigashi Y, Taylor A, Bournas V, Glawe B, et al. Identification of the pore-lining residues of the BM2 ion channel protein of influenza B virus. J Biol Chem. 2008;283:15921–15931. doi: 10.1074/jbc.M710302200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 20.Xiang Z, Steinbach PJ, Jacobson MP, Friesner RA, Honig B. Prediction of side-chain conformations on protein surfaces. Proteins. 2007;66:814–823. doi: 10.1002/prot.21099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 22.Sulistijo ES, Mackenzie KR. Structural basis for dimerization of the BNIP3 transmembrane domain. Biochemistry. 2009;48:5106–5120. doi: 10.1021/bi802245u. [DOI] [PubMed] [Google Scholar]
- 23.Lawrie CM, Sulistijo ES, MacKenzie KR. Intermonomer hydrogen bonds enhance GxxxG-driven dimerization of the BNIP3 transmembrane domain: roles for sequence context in helix-helix association in membranes. J Mol Biol. 2010;396:924–936. doi: 10.1016/j.jmb.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pinto LH, Dieckmann GR, Gandhi CS, Papworth CG, Braman J, Shaughnessy MA, et al. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc Natl Acad Sci USA. 1997;94:11301–11306. doi: 10.1073/pnas.94.21.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath DD, et al. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science. 2010;330:509–512. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cady SD, Schmidt-Rohr K, Wang J, Soto CS, Degrado WF, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–692. doi: 10.1038/nature08722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sale K, Faulon JL, Gray GA, Schoeniger JS, Young MM. Optimal bundling of transmembrane helices using sparse distance constraints. Protein Sci. 2004;13:2613–2627. doi: 10.1110/ps.04781504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grigoryan G, Kim YH, Acharya R, Axelrod K, Jain RM, Willis L, et al. Computational design of virus-like protein assemblies on carbon nanotube surfaces. Science. 2011;332:1071–1076. doi: 10.1126/science.1198841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nanda V, DeGrado WF. Automated use of mutagenesis data in structure prediction. Proteins. 2005;59:454–466. doi: 10.1002/prot.20382. [DOI] [PubMed] [Google Scholar]
- 30.Harrington SE, Ben-Tal N. Structural determinants of transmembrane helical proteins. Structure. 2009;17:1092–1103. doi: 10.1016/j.str.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 31.GNU Scientific Library. http://www.gnu.org/software/gsl/
- 32.Brunger A. X-PLOR Version 3.1 A System for X-ray Crystallography and NMR. Yale University Press; New Haven, CT: 1991. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.