
Figure 4. Unbiased and targeted quantitative proteomics reveals abundance changes in a small subset of proteins.
(A) Example mass spectra demonstrating both m/z peaks used for peptide sequencing and iTRAQ reporter ion signal to measure relative abundance across time points. (B) Hierarchical clustering heat map of paired mRNA and ribosome footprint relative read density vs 0 hr with relative iTRAQ protein abundance for 2572 proteins. (C) Inset of heat map for proteins increased in relative abundance by >50% at ≥2 time points shows few proteins are measurably produced during bortezomib-induced apoptosis. (D) Targeted selected reaction monitoring (SRM) assays orthogonally validate iTRAQ data for 152 proteins. In this representative data, each colored trace monitors the intensity of a given parent and fragment ion pair, as demarcated in the peptide sequence; multiple co-eluting peaks positively identify a targeted peptide. (E) Protein abundance measured by both SRM and iTRAQ demonstrate strong correlation across time points (r = 0.80).
DOI: http://dx.doi.org/10.7554/eLife.01236.016

Figure 4—figure supplement 1. Protein abundance comparison.

Figure 4—figure supplement 2. Comparison of baseline (0 hr) read density of mRNA transcripts vs number of identified peptides mapping to each protein in iTRAQ proteomics.

Figure 4—figure supplement 3. Relative 5′ UTR translation across the time course for upregulated genes.
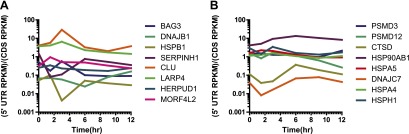
Figure 4—figure supplement 4. Unbiased and targeted proteomics comparison to deep sequencing data.
