

Abstract
Dendritic cells (DCs) represent a functionally diverse and flexible population of rare cells with the unique capability of binding, internalizing and detecting various microorganisms and their components. However, the response of DCs to innocuous or pathogenic microbes is highly dependent on the type of microbe-associated molecular patterns (MAMPs) recognized by pattern recognition receptors (PRRs) that interact with phylogenetically conserved and functionally indispensable microbial targets that involve both self and foreign structures such as lipids, carbohydrates, proteins, and nucleic acids. Recently, special attention has been drawn to nucleic acid receptors that are able to evoke robust innate immune responses mediated by type I interferons and inflammatory cytokine production against intracellular pathogens. Both conventional and plasmacytoid dendritic cells (cDCs and pDCs) express specific nucleic acid recognizing receptors, such as members of the membrane Toll-like receptor (TLR) and the cytosolic RIG-I-like receptor (RLR) families. TLR3, TLR7/TLR8 and TLR9 are localized in the endosomal membrane and are specialized for the recognition of viral double-stranded RNA, single-stranded RNA, and nonmethylated DNA, respectively whereas RLRs (RIG-I, MDA5, and LGP2) are cytosolic proteins that sense various viral RNA species. In this review we discuss the significance of detecting the genomic content of viruses by DC subsets capable of linking innate and adaptive immunity, and several viral evasion mechanisms that may allow us to better understand these responses. A particular attention is paid to the possible collaboration of TLR and RLR sensors in anti-viral protection.
Keywords: Pattern recognition receptors, cross-talk, dendritic cell subsets, interferon, inflammation
Introduction
Dendritic cells (DCs) play a pivotal role in bridging innate and adaptive immunity and in orchestrating strictly controlled immune responses, which ensure restoration of the resting state and the maintenance of self tolerance, or support the generation of effector and memory cells. Tissue resident DCs are able to engulf extracellular particles and soluble material and by continuous sampling of their environment they collect the actual molecular composition of a given tissue. Stress, inflammation or pathogenic evasion may alter the amount and content of the engulfed material and trigger the local stimulation of DCs [1]. The first response of tissue resident DCs to activation signals is the expression of cytokines, chemokines and their receptors followed by the rapid migration to secondary lymphoid organs to transport and present the stored material for naive circulating T-lymphocytes [2-4]. The nature, combination and duration of tissue-derived molecular signals determine the functional activities of DCs and have an impact on the polarization, magnitude, regulatory or stimulatory nature and duration of T-lymphocyte responses.
Under steady state conditions DCs are present throughout the body at low numbers representing ~1-2 % of white blood cells. They are characterized by high versatility, flexibility and multiple functional activities combined with their dual capacity to induce self tolerance or trigger immune responses. They also act as the most efficient antigen presenting cells (APC) to activate and instruct the differentiation of inntate, CD4+ and CD8+ T-lymphocytes and have been shown to be indispensible for inducing CD8+ cytotoxic T-cell priming [5]. Epithelial surfaces, such as the bronchial and intestinal tracts are continuously exposed to high doses of environmental antigens and pathogens, consequently they are considered more tolerogenic than the skin or other tissues, which also can be attacked by infectious agents, traumatic or toxic shock. These environmental changes are monitored preferentially by mechanisms of the innate immune system through epithelial, endothelial and stromal cells in collaboration with tissue resident macrophages, DCs and mast cells [6]. To our present view, the major function of DCs is to alarm the immune system against foreign and dangerous interventions, and to protect self tissues from damage to maintain self-tolerance. Discovering the coordination of these seemingly counteracting tasks may open up new avenues for stimulating or regulating immune responses and to develop preventive or therapeutic interventions for treating inflammatory and autoimmune diseases or cancer, as well as designing new types of vaccines based on DCs biology [7-10].
Development and specialization of human dendritic cell subtypes and subsets
DCs arise from bone marrow-derived CD34+ hematopoietic stem cells (HSC), which maintain their functional flexibility and are able to generate various DCs subsets. The two major subsets of DCs involve bone marrow derived plasmacytoid DCs (pDCs) and conventional DCs (cDC), which exhibit distinct phenotypic and functional attributes [11,12]. Based on their origin DCs can be further classified as conventional circulating CD1c+ and CD1c- blood DCs and monocyte-derived DCs [13]. Further separation identified the rare but highly specialized CD141+ blood DC population with the unique capacity to cross-present viral antigens, the monocyte derived CX3CR1+ non-migrating DC subset associated to the gut epithelium, and CD103+ DC present in the gut lamina propria and in mediastinal lymph nodes [14]. This heterogeneity reflects the functional specialization of defined DC subsets and suggests a rational distribution of labor at the level of DCs.
By analyzing more than 200 healthy donors we have previously described that the ratio of the CD1a+ and CD1a- monocyte-derived DCs (moDCs) vary among individuals. Our results also indicated that CD1a+ and CD1a- moDCs differ in the expression of some phenotypic markers, in their internalizing capacity, and migratory potential to lymph node-derived chemokines. Furthermore, a marked difference was found in the production of cytokines upon stimulation by CD40 ligand (CD40L) or various Pattern Recognition Receptor (PRR) ligands [15,16]. It was also shown that the ligand induced activation of the peroxisome proliferator-activated receptor-gamma (PPARγ), a member of the nuclear hormone receptors, can skew monocyte-derived DCs differentiation towards the CD1a- subset. As the expression of CD1 molecules (CD1a, b, c) could be down regulated by PPARγ, while CD1d expression was increased suggested opposing regulation of the expression of these lipid presenting molecules. In this context the pathways involved in this counter regulation were identified as serum lipids and lipoproteins, known modulators of PPARγ activity and consequently the dichotomy of CD1a- and CD1a+ cells [17]. A recent study confirmed our previous findings showing individual differences in CD1a expression by demonstrating CD1a deficiency as a common and genetically regulated phenomenon in the human population indicating a biologically relevant regulation [18]. Our findings and these new results identified the CD1a membrane protein as a marker of the phenotypically and functionally distinct CD1a+ DC subset.
Pattern recognition receptors of human dendritic cells
An important biological function of DCs relies on the continuous sampling of their tissue environment, responding to stress and danger signals and transducing the collected molecular information to other cell types of the immune system. DCs are equipped with unique sets of phylogenetically conserved PRRs, which are specialized to recognize Microbe Associated Molecular Patterns (MAMPs) and Danger Associated Molecular Patterns (DAMPs) [19,20]. The response of DCs to MAMPs and DAMPs is executed by the activation of resting DCs by microbial components, noxious or toxic insults. Activation of DCs results in the expression of costimulatory molecules, the production of cytokines, chemokines and other soluble mediators. Both resting and stimulated DCs are able to change their tissue location and migrate through peripheral and lymphoid tissues. Activation of DCs by MAMPs and DAMPS results in the rapid, chemokine-mediated translocation of DCs to draining lymph nodes where they have the chance to contact antigen-specific T-lymphocytes to initiate adaptive immune responses [21,22]. This process also ensures the transfer of molecular information collected in the periphery towards other cell types of both innate and adaptive immunity such as neutrophil granulocytes, NK and NKT cells, T- and B-lymphocytes.
The action of DCs can be divided into the recognition phase followed by phases of signal transduction pathways assisted by adaptors and mediated by posttranslational modifications such as phosphorylation and ubiquitination events leading to the activation of transcription factors, and gene transcription accompanied by the production of soluble factors [23,24]. In this cascade few receptor complexes ligated by their specific ligands allow enormous signal amplification. It has also been shown that the generation of fully active and stable DCs requires the parallel activation of multiple signaling pathways [25]. This suggests that signals through a single receptor may result in partial activation only, which may be reverted by signals which favor the differentiation of regulatory DCs. Signals generated by Toll-like receptors, cytokines, chemokines, eicosanoids, free oxygen radicals, and various inflammatory mediators all contribute to a “signaling matrix” and influence the phenotype and functional responses of DCs.
TLRs and RLRs: sensors of viral nucleic acids
The immune system acts as an evolutionally conserved and advanced host defense mechanism against invading pathogens. Innate immune responses are triggered by phylogenetically conserved microbial components that are essential for the survival of a given type of organism. Upon pathogenic infection, these pathogen-associated molecular patterns (PAMPs) are recognized by specific PRRs that are germline encoded and are usually expressed constitutively in the host [26-28]. The overall picture however, is far more complex as successful microbial moieties are also found in non-pathogenic microbes, and thus the presence of different PAMPs per se is not sufficient to discriminate “pathogenic” and “non-pathogenic” life forms. Furthermore, certain PRRs also sense host-derived/“self” components that become available as a result of cellular/tissue injury. The list of endogenous DAMPs is continuously growing but their impact on immune homeostasis are yet to be clarified. A recent review focuses on the role of these endogenous molecules in eliciting inflammation and cell death by activating innate PRRs [29]. Growing body of evidence also suggests an evolutionary link between innate immunity and cell death signaling. For example, several studies discuss the emerging role of mitochondria in the activation of innate signaling, and the connection between apoptotic cell death and innate immunity [30,31]. According to the symbiotic theory, the mitochondrion is an organelle derived from Gram-negative bacteria and thus the development of cellular machineries involved in cell death and innate defense against microbial pathogens have developed from ancestral mechanisms associated with bacteria. Thus far, five classes of PRRs have been identified: i) Transmembrane TLRs, which are integrated to cell surface or endosomal membranes of various cell types; ii) Membrane C-type lectin receptors (CLRs) characterized by the presence of a carbohydrate-binding domain; iii) Three additional families of intracellular sensors, which are localized to the cytosol of various cell types and involve NOD-like receptors (NLRs), RLRs, and the recently described AIM2-like receptors (ALRs), all with nucleotide recognition capabilities [32-34].
Upon binding of their specific ligands TLRs activate the NF-κB/AP-1 and the interferon-regulatory factor 3/7 (IRF-3/7) pathways to coordinate innate and initiate adaptive immunity [35,36]. RLRs are essential viral sensors in the cytoplasm and comprise Retinoic acid inducible gene-I (RIG-I), Melanoma differentiation-associated gene-5 (MDA5), and Laboratory of genetics and physiology 2 (LGP2), respectively [37-39]. RIG-I and MDA5 have been identified as receptors for double-stranded RNA [40], Nucleotide-binding oligomerization domain (NOD)-like receptors mediate primarily antibacterial immunity through the activation of NF-kappaB or inflammasomes [41], whereas RIG-I-like helicases have a fundamental role in the induction of antiviral immune responses [27]. Both RIG-I and MDA5 contain a C-terminal DExD/H box RNA helicase domain and two N-terminal caspase-recruitment domains (CARDs) required for eliciting downstream signaling pathways, while LGP2 lacks the CARD-domain and acts as a primary regulator of the RIG-I/MDA5-inititated signaling pathway (Figure 1) [42]. RIG-I and MDA5 have different ligand specificity but both of them are able to induce the production of type I interferons (IFNs) and pro-inflammatory cytokines in a tightly regulated and balanced manner [43,44].
Figure 1.
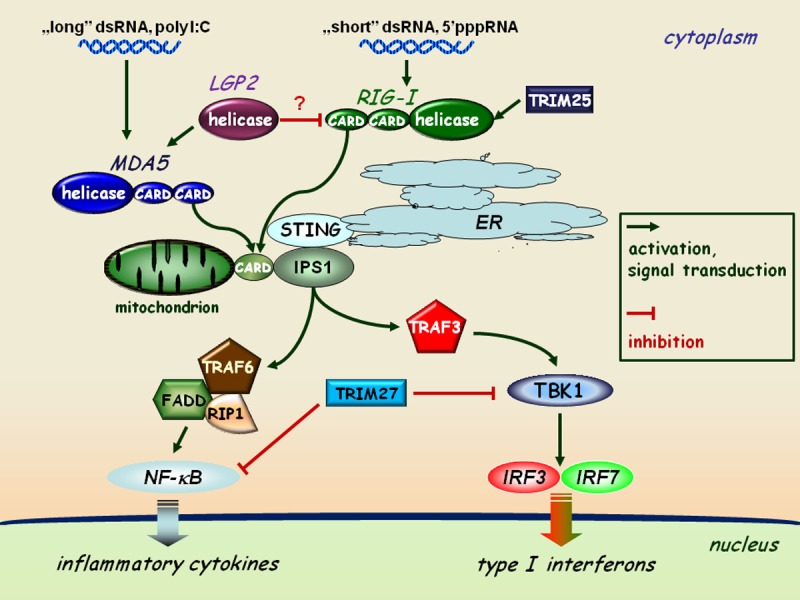
RLR-mediated pathways of type I interferon and inflammatory responses. The interaction of dsRNA as a viral genome or as a replication intermediate of RNA viruses with the helicase domain of RLRs (RIG-I or MDA5) induces association of the CARD domains of RIG-I/MDA5 and the adaptor protein IPS1 localized to the mitochondrial membrane. This receptor-adaptor interaction results in the activation of TBK1 and the subsequent phosphorylation of IRF3 and IRF7 on specific serine residues, resulting in their homodimerization. These dimers can translocate to the cell nucleus and activate the transcription of type I IFN genes. The expression of IRF3, IRF7, RIG-I and MDA5 is coordinately upregulated by type I IFN-mediated signaling acting as an amplification circuit. This pathway together with IPS1 is coupled to the NF-κB signaling pathway through the interaction of FADD (FAS-associated via death domain), RIP1, and TRAF6 resulting in the induction of inflammatory cytokine genes, such as IL-1β, IL-6 and TNFα. The TRIM proteins shown act as specific regulators of this pathway.
The TLR family is an important class of PRRs through which the innate immune system detects the major types of invasive microorganisms. TLRs are also important in the non-immediate phase of the immune response, such as the recruitment of phagocytes to infected tissue areas. Recent studies revealed that TLRs are able to recognize several microorganisms, such as bacteria, fungi, protozoa, and viruses [45]. Once TLRs have been activated by their specific ligands, they trigger signal transduction cascades that mount immune responses through the activation of the transcription factors NF-κB, IRFs and the mitogen-activated protein kinases (MAPKs) p38, ERK1/2, and c-Jun N-terminal kinase (JNK). This process altogether results in the expression of a common set of genes whose products, such as cytokines, chemokines, and co-stimulatory molecules are essential for the orchestration of both innate and adaptive immunity.
Apart from the TLR “master adaptor” Myeloid differentiation primary response gene 88 (MyD88), other important adaptor molecules also take part in downstream TLR signaling events: i) the MyD88-adapter-like or TIR domain-containing adapter (TIRAP/Mal); ii) the TIR domain-protein TRIF/TICAM-1; iii) the TRIF-related adapter molecule (TRAM) also known as TICAM-2; iv) the protein that contains sterile α and HEAT-Armadillo motifs (SARM). As not all members of the TLR family bind the same adapter(s), their differential contribution ensures the initiation of separate signaling cascades triggered by different TLRs (Figure 2). A good example is the regulation of IRF3 via the adapter TRIF, that can be induced only by TLR3 or TLR4 to initiate the TRIF-dependent activation of IRF3 and thereby the production of type I IFNs. TLR3 activation is linked to both IRF3 and IRF7 however, the baseline expression level of IRF3 in DCs and in most cells is far higher than that of IRF7, and thus the primary target of TLR3-mediated activation is IRF3 [36,46,47], whereas the ligation of TLR7, TLR8, and TLR9 triggers IRF7 activation (Figure 2). Signaling through TLRs in DCs leads to pro-inflammatory cytokine and IFN responses and results in the recruitment of other inflammatory cell types such as granulocytes and natural killer (NK) cells. Thus, the coordinated activation and interaction of different cell types involved in the mobilization of innate immune cells are able to create a local microenvironment that allows the regulated activation of adaptive immunity [48]. As various TLRs are expressed in a cell type specific manner and a given cells may express at least one or a defined combination of TLRs, these receptors and their adaptors may have evolved to act as regulators of physiological functions upon responding to hazardous signals [49].
Figure 2.
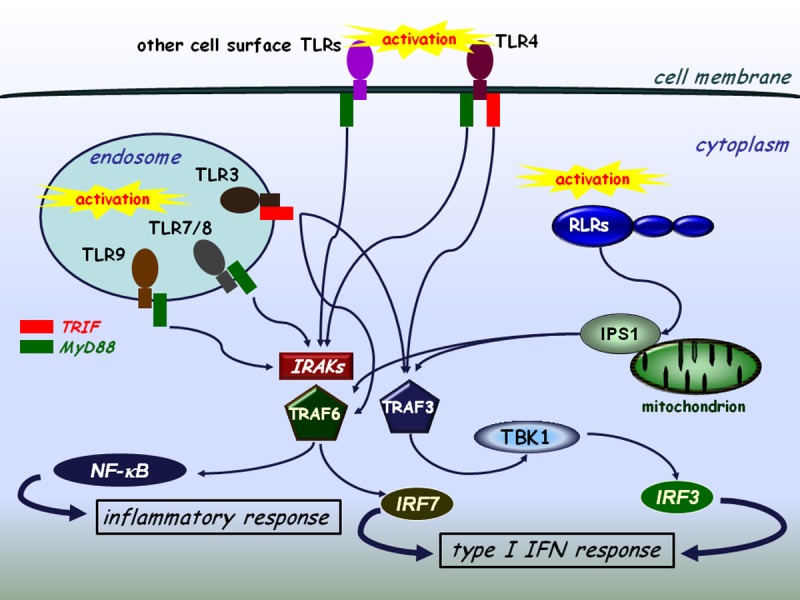
The interplay of TLR and RLR signaling. TLRs expressed on the cell surface or localized on intracellular membranes recognize various sets of pathogenic structures among them viral genomes or replication intermediates and transduce signals through the NF-κB/type I IFN pathways. The TLR3 and TLR4 mediated signaling pathways are independent of MyD88 and IRAKs (IRAK-1/2/4), whereas the other TLRs use the MyD88 pathway. TLR4 is capable of using both MyD88-dependent and independent signaling. TRAF3 and TRAF6 have a cardinal role in both the collaboration and the essential signaling processes of TLRs and RLRs.
Several studies have revealed the importance of TLR-independent virus sensing mechanisms occurring in the cytoplasm. It is known that TLR7 and TLR9 are crucial endosomal detectors of viral nucleic acids in plasmacytoid dendritic cells (pDCs), but several cell types recognize viral RNA through the RLR system [50]. Initially, both RIG-I and MDA5 were thought to recognize viral dsRNA of the same type. Recent studies however, have shown differential recognition of viral PAMPs by RIG-I and MDA5. According to Hornung et al., the main ligand for RIG-I is ssRNA with a 5’triphosphate motif [51], but this helicase can also be activated by short dsRNA fragments [52]. 5’pppRNA is a general genome constituent or the product of in vitro transcripts of most RNA viruses. Endogenous 5’ppp moieties are removed by adding a 7-methyl-guanosine cap in host cells, therefore these self-patterns are refractory to detection by RIG-I. In addition, the recognition of 5’ppp-RNA is strictly dependent on the presence of a short double-stranded section of the molecule, because short dsRNAs trigger RIG-I only weakly and a single 5’ppp strand by itself is unable to do so. It was also demonstrated that the transfection of AT-rich dsDNA (synthetic polyd-AdT:polydAdT) leads to the production of type I IFNs. This phenomenon was shown to be RIG-I-dependent and mediated by a host DNA-dependent RNA polymerase III that transcribes polydAdT:polydAdT to 5’pppRNA [53,54]. This latter mechanism may explain IFN induction by some DNA viruses and intracellular bacteria. A recent study reported that besides RIG-I, PKR can also recognize the 5’ppp signature however, this interaction does not lead to IFN production and hence the biological significance of this simultaneous activation is still obscure [55].
MDA5 is turned out to be the key intracellular sensor for longer viral dsRNAs capped by di- or mono-5’phosphate moieties and for the nucleotide-analogue poly I:C [56]. RIG-I and MDA5 respond differently to viral infections by various strains: RIG-I mainly senses viruses with short ssRNA genome such as those exhibited by paramyxoviruses or orthomyxoviruses, while MDA5 can be activated for example by picornaviruses, which produce large amounts of dsRNA during their replication (Table 1) [39,57]. Since host RNAs are single stranded (ssRNAs) capped by a methylguanosine or are protected by monophosphate at the 5’end, they do not activate RIG-I/MDA5 ensuring the discriminative recognition of self and foreign RNA. The third member of the RLR family, LGP2 lacks the CARD domain and is unable to activate IFN and pro-inflammatory cytokine responses upon viral stimuli, so it was originally considered acting as a negative regulator of RIG-I/MDA5 [58,59]. A recent study by Satoh and colleagues showed that LGP-/- mice produce decreased amounts of IFN upon infection by Vesicular stomatitis virus (VSV) or Encephalomyocarditis virus (EMCV) suggesting that LGP2 may cooperate with RIG-I and MDA5 to sense viral nucleic acids in the cytoplasm [60]. The Janus-faced activity of LGP2 through inhibiting and/or facilitating RLR activity is still a subject of controversy. Furthermore, very recently LGP2 was shown to regulate CD8+ cytotoxic T lymphocyte (CTL) survival and fitness during the clonal expansion of lymphocytes provoked by viral infection. This effect was mediated via T-cell receptor signaling inducing the expression of LGP2 in CTLs [61].
Table 1.
Examples of viruses detected by RLRs
| RIG-I | MDA5 | |
|---|---|---|
| Taxonomy | Paramyxoviruses: | Picornaviruses: |
| Measles virus, Mumps virus, Respiratory syncytial virus | Encephalomyocarditis virus, Mengo virus, Theiler’s virus | |
| Orthomyxoviruses: | Reoviruses: | |
| Influenza virus (type A, B and C), Thogotovirus | Reovirus, Rotavirus | |
| Rhabdoviridae: | ||
| Rabies virus, Vesicular stomatitis virus | ||
| Flaviviruses: | ||
| Japanese encephaltis virus | ||
| Type of nucleic acid | ssRNA | dsRNA |
Interaction of the specific ligand with the helicase domain of RIG-I or MDA5 induces the ubiquitin-dependent association of CARD of RLR with the CARD domain of the CARD-adaptor protein inducing interferon-β (CARDIF, also known as IPS-1, MAVS or VISA), which is localized in the mitochondrial membrane [62,63].This receptor-adaptor interaction results in the activation of TANK-binding kinase 1 (TBK1) through TRAF-family-member-associated NF-κB activator (TANK) binding. Activated TBK1 induces the phosphorylation of IRF3/IRF7 on specific serine residues, resulting in their homodimerization [64]. These dimers then translocate to the nucleus and activate the transcription of type I IFN genes (Figure 1). The expression of IRF3, IRF7, RIG-I and MDA5 is coordinately upregulated by type I IFN-mediated signaling acting as an amplification process. This pathway is implicated to be connected to the NF-κB activation pathway through the interaction of FADD (FAS-associated via death domain), Receptor-interacting protein (RIP1) and TNFR-associated factor 6 (TRAF6) together with CARDIF, which results in the induction of proinflammatory cytokine genes and proteins such as IL-1β, IL-6 and TNF-α (Figure 1) [65].
Lately, another protein, known as Stimulator of IFN genes (STING) has been described, which is located in the endoplasmic reticulum (ER) membrane [66]. STING acts as a co-factor of RIG-I-CARDIF, but not in MDA5-CARDIF signaling. Another group of proteins, called the tripartite motif-containing (TRIM) superfamily also play a significant role in RIG-I/MDA5 regulated induction of type I IFNs and of pro-inflammatory cytokines. TRIM proteins are expressed in response to IFNs and take part in many biological processes connected to innate immunity [67]. For instance, TRIM27 interacts with inhibitor of NF-κB (IκB) kinases (IKKs) and are able to block the activation of NF-κB, IRF3 and IRF7. Another member of the TRIM family, TRIM25 binds to RIG-I and conjugates ubiquitin to the CARD domain of the helicase thus facilitating the activation of downstream signaling pathways (Figure 1) [68,69].
Viral evasion mechanisms of TLR and RLR recognition and signaling
Taken the diversity of receptors specialized for nucleic acid recognition the genetic material seems to be a phylogenetically validated and important target of recognition. Nucleic acids derived from microbial genomes generated during viral or bacterial life-cycles are potent ligands for PRRs. To ensure escape from immediate recognition, various pathogens developed numerous alternatives of evasion mechanisms [27]. RNA viruses, such as poxviruses are able to integrate into the mRNA processing apparatus of host cells in order to cap their viral mRNAs or like picornaviruses can protect their own genomic RNA with a covalently linked protein at the 5’end [70]. The genomic RNA of ssRNA viruses (like influenza A virus) possess a 5’end triphosphate motif and therefore efficiently activate the RIG-I pathway. Viruses with a dsRNA genome or with dsRNA as a replicative intermediate (positive-sense RNA viruses) are main targets of MDA5. Moreover, DNA viruses can produce high amounts of dsRNA and consequently trigger an RLR-related response. In order to avoid recognition, some viruses encode protective dsRNA-binding proteins, like the HIV-1 Tat or VACV E3L, which can defend dsRNA species from detection by cytoplasmic receptors [71].
RLR signaling can also be the target of viral evasion mechanisms mediated either by direct blocking of helicases or by inhibition of other members of the signaling cascade [72]. According to Mibayashi et al. the influenza A virus NS1 protein can block downstream signaling upon binding to the RIG-I-IPS1 complex [73], while poliovirus can induce the degradation of MDA5 by caspase-related enzymatic cleavage [74]. The elimination of IPS1 is a „common” target of viral proteins, e.g. the hepatitis A 3ABC protein triggers the degradation of IPS1, thus decoupling the signaling cascade from RLRs and IRF3 [75]. The downstream branches of the RLR and TLR signaling pathways can also be blocked and the transcriptional control of the ifnb promoter by IRFs can also be inhibited by several ways. The V proteins of paramyxoviruses behave as a mimic of IRF3 and act as a competitve antagonist for phosphorylation by TBK1 [76]. Another example is the Kaposi’s sarcoma-associated herpesvirus (KSHV) viral mimic IRF7 protein, which can dimerize with cellular IRF7 consequently inhibiting its DNA-binding ability [77], whereas the KSHV protein K-bZIP is known as a strong inhibitor of IFNβ expression by competing the binding of host IRF3 to the promoter site [78]. Other type of viral control of IRFs is mediated by e.g. the rotavirus non-structural protein 1 (NSP1), which targets IRF3, IRF5, and IRF7 for degradation [79]. Thus the cytoplasmic level of IRFs can be dynamically regulated by various viral factors. A putative function of the IFN-stimulated protein of 15kDa (ISG15) is to prevent the virus-mediated degradation of IRFs when it binds to and stabilizes of IRF3 in Newcastle disease virus infections [80].
The role and control of the NF-κB pathway during viral infection is far more complex than that of IRF3/IRF7 [27]. NF-κB is an important factor not only in the production of several cytokines and anti-viral IFNs but also able to inhibit apoptosis and foster cell proliferation, which effects are apparently beneficial to virus replication. In fact, some viruses activate NF-κB signaling in order to avoid apoptotic death of the host cell [81]. An example of the unique viral regulation of NF-κB pathway is the African swine fever virus (ASFV) protein A238L that inhibits NF-κB activity at the early stage of infection to delay provocation of the innate immune response [82]. However, at the late stage of viral infection another ASFV protein, called A224L activates NF-κB and deactivates caspases [83]. Similarly, the KSHV protein K13 can interact with the IKK-complex and accordingly is able to activate the NF-κB pathway [84]. This biphasic mode of action allows the virus to propagate in a more efficient manner. By the early inhibition of NF-κB the virus gains time to establish a supportive environment for infection. Later, the virus has an impact on the regulation of PRR-induced signaling-related factors and promotes the activation of NF-κB, thereby promoting fast multiplication. Referring to Bowie et al., one of the key challenges in future research is to harness the information learned from viral evasion studies for the benefit of human health [27].
Interaction of TLR- and RLR-mediated pathways: to impel or to labefy?
The collaboration of PRRs and the consequent secretion of type I interferons and inflammatory cytokines can be highly efficient against pathogens. After viral infections, innate defense mechanisms are activated promptly and allow the development of adaptive immune responses. DCs play an essential role in the orchestration of humoral and cellular immunity and the induction and maintenance of long-term immunological memory [36,85]. Interaction of microbes with the innate immune system involves the induction of multiple PRR pathways triggered simultaneously by various PAMPs of the whole pathogen [86].
The possible interaction of two or more signaling pathways in biochemical systems can either be potentiating (synergistic) or weakening (competitive, hampering). Multiple evidences have been accumulated in the past few years that reflect the enormous complexity of these processes. For instance in moDCs and monocyte-derived Langerhans cells (moLCs) co-ligation of TLR3/TLR7 and TLR3/Dectin-1 led to increased Th1/Th17 responses, in contrast to TLR3 and Langerin ligation, which had an opposite effect [87]. Similarly, another group found that RLR/TLR co-activation caused decreased Th1/Th17 responses upon bacterial infection [88]. This cross-interference of RLR and TLR signaling might have important implications in the design of future vaccination strategies, and the possible spectrum may be expanded to other non-immune cell types as well [89,90]. miRNAs also possess the capability of fine tuning TLR and RLR signaling [91]. Common regulatory members of both pathways such as miR-146 can have a strong impact on the counter-regulation of activation mediated by TLRs and RLRs.
The interplay between TLRs and RLRs also has an important role during in vivo viral infections. Infection by RNA viruses is detected by RLRs and TLR7/8 resulting in the production of type I IFNs (Figure 2). Kumagai and colleagues found that in Newcastle disease virus (NDV) infected mice the major source of type I IFN was not pDCs [92], even though they were able to produce vast amounts of IFNs in the absence of alveolar macrophages (AM) suggesting that pDCs play an important role when the first line of AM-mediated defense is disrupted. Since many viruses evolved RLR evasion mechanisms, pDCs may function as a backup for antiviral immunity when RLR signaling is shut down [45]. Detection of Hepatitis C virus (HCV) is carried out by both TLR3 and RIG-I but the virus evades type I IFN responses by expressing a viral protein called NS3-NS4A. This HCV protease cleaves TRIF and IPS1 and renders hepatocytes incapable of producing IFNβ [93]. On the other hand the presence of HCV induces a robust type I IFN response by pDCs, which infiltrate the liver during infection. HCV RNA is delivered to pDCs by a direct cell-to-cell contact between infected hepatocytes and pDCs. This process leads to type I IFN production via TLR7. Moreover, stimulation of TLR7 or TLR9 by selective ligands can also upregulate the cytoplasmic expression of RIG-I protein in pDCs in a type I IFN-independent manner showing the importance of collaborative signaling between these two PRR families (Szabo et al. unpublished results).
While these sensors were shown to be crucial for innate and adaptive host defense, their inappropriate activation has been associated with autoimmunity and inflammatory diseases. Hence, a more complete appreciation of TLRs, RLRs, and their complex signaling processes will provide important insights into new therapeutic modalities that can either enhance immune responses or inhibit functions to diminish the deleterious effects of uncontrolled inflammation [94].
An interesting contemporary approach to the topic has been carried out by using systems biology, bioinformatics, and biophysics, as tools of better understanding. This approach proposed that instead of single cell analyses one should move towards a more holistic understanding of signaling systems. The meta-network of biological entities is considered to possess both microscopic and macroscopic dynamics as observed in physical sciences. The origin of averaging effects from stochastic responses of a single cell when collected to form a population should also be taken into account [95,96]. It is very likely that the emergence of an average cell deterministic response (e.g. following a TLR and/or RLR stimulus) from single cell stochastic responses complement each other [97,98]. Thus the stochastic fluctuations in the IFN response of a single dendritic cell or a single signaling pathway are necessary to induce probabilistic differentiation from identical cells or interacting pathways of the same PRR family. This might allow multicellular organisms or complex, interacting PRR signaling networks to switch cell fates or states to yield diversity, fine-tuning and reach the proper response that cannot be achieved by a purely deterministic system. Recent studies of multi-component, non-linear modeling of different TLR pathways verified the success of this approach [98,99] by identifying cross-talk mechanisms between the MyD88- and TRAM-dependent pathways and led to the concept of signaling flux redistribution (SFR). This proposal is based on the law of conservation where the removal of MyD88 leads to increased activation of the entire alternative TRAM-pathway. Thus, total signaling flux information from a receptor through final downstream gene activation in the network is conserved. The group experimentally validated the SFR theory by using MyD88-/- and TRAF6-/- KO mice and their data generated interesting interpretations [99], which may open up new aspects towards the deeper understanding of cellular signaling processes.
Acknowledgements
This work was supported by the TÁMOP 4.2.2.A-11/1/KONV-2012-0023 “VÉD-ELEM” project, and OTKA NK 101538 grants (both for ER).
Disclosure of conflict of interest
The authors have declared no competing interests.
References
- 1.Underhill DM, Goodridge HS. Information processing during phagocytosis. Nat Rev Immunol. 2012;12:492–502. doi: 10.1038/nri3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 3.Steinman RM. Dendritic cells: understanding immunogenicity. Eur J Immunol. 2007;37(Suppl 1):S53–60. doi: 10.1002/eji.200737400. [DOI] [PubMed] [Google Scholar]
- 4.Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- 5.Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12:557–569. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 6.O’Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol. 2013;13:453–460. doi: 10.1038/nri3446. [DOI] [PubMed] [Google Scholar]
- 7.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12:265–277. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palucka K, Banchereau J. Human dendritic cell subsets in vaccination. Curr Opin Immunol. 2013;25:396–402. doi: 10.1016/j.coi.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity. 2013;39:38–48. doi: 10.1016/j.immuni.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Szabo A, Gogolak P, Pazmandi K, Kis-Toth K, Riedl K, Wizel B, Lingnau K, Bacsi A, Rethi B, Rajnavolgyi E. The two-component adjuvant IC31(R) boosts type i interferon production of human monocyte-derived dendritic cells via ligation of endosomal TLRs. PLoS One. 2013;8:e55264. doi: 10.1371/journal.pone.0055264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol. 2011;29:163–183. doi: 10.1146/annurev-immunol-031210-101345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol. 2007;7:543–555. doi: 10.1038/nri2103. [DOI] [PubMed] [Google Scholar]
- 14.Haniffa M, Shin A, Bigley V, McGovern N, Teo P, See P, Wasan PS, Wang XN, Malinarich F, Malleret B, Larbi A, Tan P, Zhao H, Poidinger M, Pagan S, Cookson S, Dickinson R, Dimmick I, Jarrett RF, Renia L, Tam J, Song C, Connolly J, Chan JK, Gehring A, Bertoletti A, Collin M, Ginhoux F. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity. 2012;37:60–73. doi: 10.1016/j.immuni.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gogolak P, Rethi B, Szatmari I, Lanyi A, Dezso B, Nagy L, Rajnavolgyi E. Differentiation of CD1a- and CD1a+ monocyte-derived dendritic cells is biased by lipid environment and PPARgamma. Blood. 2007;109:643–652. doi: 10.1182/blood-2006-04-016840. [DOI] [PubMed] [Google Scholar]
- 16.Szabo A, Bene K, Gogolak P, Rethi B, Lanyi A, Jankovich I, Dezso B, Rajnavolgyi E. RLR-mediated production of interferon-beta by a human dendritic cell subset and its role in virus-specific immunity. J Leukoc Biol. 2012;92:159–169. doi: 10.1189/jlb.0711360. [DOI] [PubMed] [Google Scholar]
- 17.Szatmari I, Gogolak P, Im JS, Dezso B, Rajnavolgyi E, Nagy L. Activation of PPARgamma specifies a dendritic cell subtype capable of enhanced induction of iNKT cell expansion. Immunity. 2004;21:95–106. doi: 10.1016/j.immuni.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 18.Seshadri C, Shenoy M, Wells RD, Hensley-McBain T, Andersen-Nissen E, McElrath MJ, Cheng TY, Moody DB, Hawn TR. Human CD1a Deficiency Is Common and Genetically Regulated. J Immunol. 2013;191:1586–1593. doi: 10.4049/jimmunol.1300575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pisetsky DS. The origin and properties of extracellular DNA: from PAMP to DAMP. Clin Immunol. 2012;144:32–40. doi: 10.1016/j.clim.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimizu K, Fujii S. An adjuvant role of in situ dendritic cells (DCs) in linking innate and adaptive immunity. Front Biosci. 2008;13:6193–6201. doi: 10.2741/3147. [DOI] [PubMed] [Google Scholar]
- 21.Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol. 2005;5:617–628. doi: 10.1038/nri1670. [DOI] [PubMed] [Google Scholar]
- 22.Bachmann MF, Kopf M, Marsland BJ. Chemokines: more than just road signs. Nat Rev Immunol. 2006;6:159–164. doi: 10.1038/nri1776. [DOI] [PubMed] [Google Scholar]
- 23.Pao LI, Badour K, Siminovitch KA, Neel BG. Nonreceptor protein-tyrosine phosphatases in immune cell signaling. Annu Rev Immunol. 2007;25:473–523. doi: 10.1146/annurev.immunol.23.021704.115647. [DOI] [PubMed] [Google Scholar]
- 24.Hammer GE, Ma A. Molecular control of steady-state dendritic cell maturation and immune homeostasis. Annu Rev Immunol. 2013;31:743–791. doi: 10.1146/annurev-immunol-020711-074929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moresco EM, LaVine D, Beutler B. Toll-like receptors. Curr Biol. 2011;21:R488–493. doi: 10.1016/j.cub.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 26.Nunez G. Intracellular sensors of microbes and danger. Immunol Rev. 2011;243:5–8. doi: 10.1111/j.1600-065X.2011.01058.x. [DOI] [PubMed] [Google Scholar]
- 27.Bowie AG, Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol. 2008;8:911–922. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawai T, Akira S. Antiviral signaling through pattern recognition receptors. J Biochem. 2007;141:137–145. doi: 10.1093/jb/mvm032. [DOI] [PubMed] [Google Scholar]
- 29.Rock KL, Lai JJ, Kono H. Innate and adaptive immune responses to cell death. Immunol Rev. 2011;243:191–205. doi: 10.1111/j.1600-065X.2011.01040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tal MC, Iwasaki A. Mitoxosome: a mitochondrial platform for cross-talk between cellular stress and antiviral signaling. Immunol Rev. 2011;243:215–234. doi: 10.1111/j.1600-065X.2011.01038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11:389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barber GN. Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Curr Opin Immunol. 2011;23:10–20. doi: 10.1016/j.coi.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olive C. Pattern recognition receptors: sentinels in innate immunity and targets of new vaccine adjuvants. Expert Rev Vaccines. 2012;11:237–256. doi: 10.1586/erv.11.189. [DOI] [PubMed] [Google Scholar]
- 34.Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annu Rev Immunol. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- 35.Kawai T, Akira S. Toll-like receptor and RIG-I-like receptor signaling. Ann N Y Acad Sci. 2008;1143:1–20. doi: 10.1196/annals.1443.020. [DOI] [PubMed] [Google Scholar]
- 36.Benko S, Magyarics Z, Szabo A, Rajnavolgyi E. Dendritic cell subtypes as primary targets of vaccines: the emerging role and cross-talk of pattern recognition receptors. Biol Chem. 2008;389:469–485. doi: 10.1515/bc.2008.054. [DOI] [PubMed] [Google Scholar]
- 37.Yoneyama M, Fujita T. Function of RIG-I-like receptors in antiviral innate immunity. J Biol Chem. 2007;282:15315–15318. doi: 10.1074/jbc.R700007200. [DOI] [PubMed] [Google Scholar]
- 38.Loo YM, Gale M Jr. Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato H, Takahasi K, Fujita T. RIG-I-like receptors: cytoplasmic sensors for non-self RNA. Immunol Rev. 2011;243:91–98. doi: 10.1111/j.1600-065X.2011.01052.x. [DOI] [PubMed] [Google Scholar]
- 40.Fujita T, Onoguchi K, Onomoto K, Hirai R, Yoneyama M. Triggering antiviral response by RIG-I-related RNA helicases. Biochimie. 2007;89:754–760. doi: 10.1016/j.biochi.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 41.Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- 42.Bruns AM, Horvath CM. Activation of RIG-I-like receptor signal transduction. Crit Rev Biochem Mol Biol. 2012;47:194–206. doi: 10.3109/10409238.2011.630974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumagai Y, Takeuchi O, Akira S. Pathogen recognition by innate receptors. J Infect Chemother. 2008;14:86–92. doi: 10.1007/s10156-008-0596-1. [DOI] [PubMed] [Google Scholar]
- 44.Eisenacher K, Krug A. Regulation of RLR-mediated innate immune signaling--it is all about keeping the balance. Eur J Cell Biol. 2012;91:36–47. doi: 10.1016/j.ejcb.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 45.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 46.Mamane Y, Heylbroeck C, Genin P, Algarte M, Servant MJ, LePage C, DeLuca C, Kwon H, Lin R, Hiscott J. Interferon regulatory factors: the next generation. Gene. 1999;237:1–14. doi: 10.1016/s0378-1119(99)00262-0. [DOI] [PubMed] [Google Scholar]
- 47.Hansen JD, Vojtech LN, Laing KJ. Sensing disease and danger: a survey of vertebrate PRRs and their origins. Dev Comp Immunol. 2011;35:886–897. doi: 10.1016/j.dci.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 48.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 49.Yamamoto M, Takeda K, Akira S. TIR domain-containing adaptors define the specificity of TLR signaling. Mol Immunol. 2004;40:861–868. doi: 10.1016/j.molimm.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 50.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 51.Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 5’-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 52.Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol. 2009;10:1065–1072. doi: 10.1038/ni.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138:576–591. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nallagatla SR, Hwang J, Toroney R, Zheng X, Cameron CE, Bevilacqua PC. 5’-triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science. 2007;318:1455–1458. doi: 10.1126/science.1147347. [DOI] [PubMed] [Google Scholar]
- 56.Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. 2008;20:17–22. doi: 10.1016/j.coi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 57.Yoneyama M, Fujita T. Structural mechanism of RNA recognition by the RIG-I-like receptors. Immunity. 2008;29:178–181. doi: 10.1016/j.immuni.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 58.Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale M Jr, Akira S, Yonehara S, Kato A, Fujita T. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175:2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- 59.Komuro A, Bamming D, Horvath CM. Negative regulation of cytoplasmic RNA-mediated antiviral signaling. Cytokine. 2008;43:350–358. doi: 10.1016/j.cyto.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Satoh T, Kato H, Kumagai Y, Yoneyama M, Sato S, Matsushita K, Tsujimura T, Fujita T, Akira S, Takeuchi O. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc Natl Acad Sci U S A. 2010;107:1512–1517. doi: 10.1073/pnas.0912986107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suthar MS, Ramos HJ, Brassil MM, Netland J, Chappell CP, Blahnik G, McMillan A, Diamond MS, Clark EA, Bevan MJ, Gale M Jr. The RIG-I-like receptor LGP2 controls CD8(+) T cell survival and fitness. Immunity. 2012;37:235–248. doi: 10.1016/j.immuni.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, Xu M, Chen ZJ. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315–330. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 64.Unterholzner L, Bowie AG. The interplay between viruses and innate immune signaling: recent insights and therapeutic opportunities. Biochem Pharmacol. 2008;75:589–602. doi: 10.1016/j.bcp.2007.07.043. [DOI] [PubMed] [Google Scholar]
- 65.Lee MS, Kim YJ. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu Rev Biochem. 2007;76:447–480. doi: 10.1146/annurev.biochem.76.060605.122847. [DOI] [PubMed] [Google Scholar]
- 66.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, Guffanti A, Minucci S, Pelicci PG, Ballabio A. The tripartite motif family identifies cell compartments. EMBO J. 2001;20:2140–2151. doi: 10.1093/emboj/20.9.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 69.Ozato K, Shin DM, Chang TH, Morse HC 3rd. TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol. 2008;8:849–860. doi: 10.1038/nri2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee YF, Nomoto A, Detjen BM, Wimmer E. A protein covalently linked to poliovirus genome RNA. Proc Natl Acad Sci U S A. 1977;74:59–63. doi: 10.1073/pnas.74.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weeks KM, Ampe C, Schultz SC, Steitz TA, Crothers DM. Fragments of the HIV-1 Tat protein specifically bind TAR RNA. Science. 1990;249:1281–1285. doi: 10.1126/science.2205002. [DOI] [PubMed] [Google Scholar]
- 72.Leung DW, Basler CF, Amarasinghe GK. Molecular mechanisms of viral inhibitors of RIG-I-like receptors. Trends Microbiol. 2012;20:139–146. doi: 10.1016/j.tim.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mibayashi M, Martinez-Sobrido L, Loo YM, Cardenas WB, Gale M Jr, Garcia-Sastre A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol. 2007;81:514–524. doi: 10.1128/JVI.01265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barral PM, Morrison JM, Drahos J, Gupta P, Sarkar D, Fisher PB, Racaniello VR. MDA-5 is cleaved in poliovirus-infected cells. J Virol. 2007;81:3677–3684. doi: 10.1128/JVI.01360-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang Y, Liang Y, Qu L, Chen Z, Yi M, Li K, Lemon SM. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc Natl Acad Sci U S A. 2007;104:7253–7258. doi: 10.1073/pnas.0611506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lu LL, Puri M, Horvath CM, Sen GC. Select paramyxoviral V proteins inhibit IRF3 activation by acting as alternative substrates for inhibitor of kappaB kinase epsilon (IKKe)/TBK1. J Biol Chem. 2008;283:14269–14276. doi: 10.1074/jbc.M710089200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Joo CH, Shin YC, Gack M, Wu L, Levy D, Jung JU. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi’s sarcoma-associated herpesvirus viral IRF homolog vIRF3. J Virol. 2007;81:8282–8292. doi: 10.1128/JVI.00235-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lefort S, Soucy-Faulkner A, Grandvaux N, Flamand L. Binding of Kaposi’s sarcoma-associated herpesvirus K-bZIP to interferon-responsive factor 3 elements modulates antiviral gene expression. J Virol. 2007;81:10950–10960. doi: 10.1128/JVI.00183-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barro M, Patton JT. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J Virol. 2007;81:4473–4481. doi: 10.1128/JVI.02498-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lu G, Reinert JT, Pitha-Rowe I, Okumura A, Kellum M, Knobeloch KP, Hassel B, Pitha PM. ISG15 enhances the innate antiviral response by inhibition of IRF-3 degradation. Cell Mol Biol (Noisy-le-grand) 2006;52:29–41. [PubMed] [Google Scholar]
- 81.Hiscott J, Nguyen TL, Arguello M, Nakhaei P, Paz S. Manipulation of the nuclear factor-kappaB pathway and the innate immune response by viruses. Oncogene. 2006;25:6844–6867. doi: 10.1038/sj.onc.1209941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tait SW, Reid EB, Greaves DR, Wileman TE, Powell PP. Mechanism of inactivation of NF-kappa B by a viral homologue of I kappa b alpha. Signal-induced release of i kappa b alpha results in binding of the viral homologue to NF-kappa B. J Biol Chem. 2000;275:34656–34664. doi: 10.1074/jbc.M000320200. [DOI] [PubMed] [Google Scholar]
- 83.Rodriguez CI, Nogal ML, Carrascosa AL, Salas ML, Fresno M, Revilla Y. African swine fever virus IAP-like protein induces the activation of nuclear factor kappa B. J Virol. 2002;76:3936–3942. doi: 10.1128/JVI.76.8.3936-3942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Matta H, Mazzacurati L, Schamus S, Yang T, Sun Q, Chaudhary PM. Kaposi’s sarcoma-associated herpesvirus (KSHV) oncoprotein K13 bypasses TRAFs and directly interacts with the IkappaB kinase complex to selectively activate NF-kappaB without JNK activation. J Biol Chem. 2007;282:24858–24865. doi: 10.1074/jbc.M700118200. [DOI] [PubMed] [Google Scholar]
- 85.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 86.Ramos HJ, Gale M Jr. RIG-I like receptors and their signaling crosstalk in the regulation of antiviral immunity. Curr Opin Virol. 2011;1:167–176. doi: 10.1016/j.coviro.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dzopalic T, Rajkovic I, Dragicevic A, Colic M. The response of human dendritic cells to co-ligation of pattern-recognition receptors. Immunol Res. 2012;52:20–33. doi: 10.1007/s12026-012-8279-5. [DOI] [PubMed] [Google Scholar]
- 88.Negishi H, Yanai H, Nakajima A, Koshiba R, Atarashi K, Matsuda A, Matsuki K, Miki S, Doi T, Aderem A, Nishio J, Smale ST, Honda K, Taniguchi T. Cross-interference of RLR and TLR signaling pathways modulates antibacterial T cell responses. Nat Immunol. 2012;13:659–666. doi: 10.1038/ni.2307. [DOI] [PubMed] [Google Scholar]
- 89.Goutagny N, Estornes Y, Hasan U, Lebecque S, Caux C. Targeting pattern recognition receptors in cancer immunotherapy. Target Oncol. 2012;7:29–54. doi: 10.1007/s11523-012-0213-1. [DOI] [PubMed] [Google Scholar]
- 90.Szabo A, Osman RM, Bacskai I, Kumar BV, Agod Z, Lanyi A, Gogolak P, Rajnavolgyi E. Temporally designed treatment of melanoma cells by ATRA and polyI: C results in enhanced chemokine and IFNbeta secretion controlled differently by TLR3 and MDA5. Melanoma Res. 2012;22:351–361. doi: 10.1097/CMR.0b013e328357076c. [DOI] [PubMed] [Google Scholar]
- 91.Li Y, Shi X. MicroRNAs in the regulation of TLR and RIG-I pathways. Cell Mol Immunol. 2013;10:65–71. doi: 10.1038/cmi.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E, Aozasa K, Kawai T, Akira S. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity. 2007;27:240–252. doi: 10.1016/j.immuni.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 93.Lemon SM. Induction and evasion of innate antiviral responses by hepatitis C virus. J Biol Chem. 2010;285:22741–22747. doi: 10.1074/jbc.R109.099556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fekete T, Szabo A, Beltrame L, Vivar N, Pivarcsi A, Lanyi A, Cavalieri D, Rajnavolgyi E, Rethi B. Constraints for monocyte-derived dendritic cell functions under inflammatory conditions. Eur J Immunol. 2012;42:458–469. doi: 10.1002/eji.201141924. [DOI] [PubMed] [Google Scholar]
- 95.Zhao M, Zhang J, Phatnani H, Scheu S, Maniatis T. Stochastic expression of the interferon-beta gene. PLoS Biol. 2012;10:e1001249. doi: 10.1371/journal.pbio.1001249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hwang SY, Hur KY, Kim JR, Cho KH, Kim SH, Yoo JY. Biphasic RLR-IFN-beta response controls the balance between antiviral immunity and cell damage. J Immunol. 2013;190:1192–1200. doi: 10.4049/jimmunol.1202326. [DOI] [PubMed] [Google Scholar]
- 97.Gutierrez J, St Laurent G 3rd, Urcuqui-Inchima S. Propagation of kinetic uncertainties through a canonical topology of the TLR4 signaling network in different regions of biochemical reaction space. Theor Biol Med Model. 2010;7:7. doi: 10.1186/1742-4682-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Selvarajoo K. Macroscopic law of conservation revealed in the population dynamics of Toll-like receptor signaling. Cell Commun Signal. 2011;9:9. doi: 10.1186/1478-811X-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Selvarajoo K, Takada Y, Gohda J, Helmy M, Akira S, Tomita M, Tsuchiya M, Inoue J, Matsuo K. Signaling flux redistribution at toll-like receptor pathway junctions. PLoS One. 2008;3:e3430. doi: 10.1371/journal.pone.0003430. [DOI] [PMC free article] [PubMed] [Google Scholar]