

Abstract
The autoinducer-2 (AI-2) quorum-sensing system has been linked to diverse phenotypes and regulatory changes in pathogenic bacteria. In the present study, we performed a molecular and biochemical characterization of the AI-2 system in Yersinia pestis, the causative agent of plague. In strain CO92, the AI-2 signal is produced in a luxS-dependent manner, reaching maximal levels of 2.5 μM in the late logarithmic growth phase, and both wild-type and pigmentation (pgm) mutant strains made equivalent levels of AI-2. Strain CO92 possesses a chromosomal lsr locus encoding factors involved in the binding and import of AI-2, and confirming this assignment, an lsr deletion mutant increased extracellular pools of AI-2. To assess the functional role of AI-2 sensing in Y. pestis, microarray studies were conducted by comparing Δpgm strain R88 to a Δpgm ΔluxS mutant or a quorum-sensing-null Δpgm ΔypeIR ΔyspIR ΔluxS mutant at 37°C. Our data suggest that AI-2 quorum sensing is associated with metabolic activities and oxidative stress genes that may help Y. pestis survive at the host temperature. This was confirmed by observing that the luxS mutant was more sensitive to killing by hydrogen peroxide, suggesting a potential requirement for AI-2 in evasion of oxidative damage. We also show that a large number of membrane protein genes are controlled by LuxS, suggesting a role for quorum sensing in membrane modeling. Altogether, this study provides the first global analysis of AI-2 signaling in Y. pestis and identifies potential roles for the system in controlling genes important to disease.
INTRODUCTION
Yersinia pestis, the etiologic agent of plague, also called Black Death, has killed over 200 million people throughout recorded history (1). It has two transmission routes: the most common is through flea bites, and the other is via airborne droplets. The lifestyle of Y. pestis is complex and involves two distinct environments, the flea midgut and mammalian hosts (2). The distinct environments in which Y. pestis must survive require complex gene regulatory mechanisms to conserve energy. Gene regulation in Y. pestis is also responsive to temperature shifts between the ambient temperature of the flea vector and the temperature of the human host (3), complicating the regulatory circuits and data analysis.
Quorum sensing (QS) is a process of cell-to-cell communication that relies upon secretion and detection of chemical signals called autoinducers (AIs). Yersinia pestis possess two conserved QS systems, the AI-1 pathway, which utilizes acyl homoserine lactones (AHLs) as signals, and the LuxS pathway, which responds to AI-2 (4). The AI-2 signaling pathway is common to many bacteria through homologues of the luxS gene. LuxS generally has two functions: one is to generate the major methyl donor S-adenosylmethionine, and the other is to detoxify S-adenosyl-l-homocysteine to homocysteine and 4,5-dihydroxy-2,3-pentandione (DPD). DPD undergoes further cyclization rearrangements to yield AI-2 (5). AI-2 is derived from the ribose moiety S-ribosylhomocysteine (SRH). It has been proposed to serve as a universal signaling molecule for interspecies communication (5). In Escherichia coli and Salmonella, the AI-2 signal is the ligand for a periplasmic protein LuxP homologue (LsrB) and is detected by the ATP binding cassette (ABC) transporter/receptor on the cell membrane. AI-2 internalization is dependent on the Lsr transporter (luxS regulated). The lsr operon contains seven genes in the organization lsrACDBEFGR, and its transcription is induced by the presence of AI-2. The four promoter-proximal genes lsrACDB encode the components of the AI-2 importer (6). The lsrR gene encodes a repressor of the lsr operon but is a pseudogene in Y. pestis, suggesting that AI-2 synthesis is constitutive in this species.
Whether or not AI-2 quorum sensing is crucial for Y. pestis pathogenesis is unclear. While there is some evidence that mice challenged with a luxS::kan mutant did not show a loss of virulence, as determined by the 50% lethal dose (LD50)(4), an in-depth analysis of the role that QS, via AI-2, plays in controlling the expression of virulence genes has not been performed. Specifically, the number and types of genes controlled by QS have not been studied systematically. To obtain a better understanding of the role of AI-2 quorum sensing in Y. pestis, we performed transcriptional profiling experiments to identify AI-2 QS-regulated genes and networks at 37°C, representing the mammalian host temperature. Our results indicated that AI-2/LuxS quorum sensing is important for Y. pestis adaptation and survival in diverse environments, including oxidative stress.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The strains and plasmids used in this study are described in Table 1. Yersinia pestis strains were derived from a pgm deletion mutant of strain CO92 (7) and maintained in brain heart infusion (BHI) broth. For all microarray experiments, Y. pestis strains were grown overnight in BHI broth at 30°C. These cultures grown overnight were diluted 1:100 into fresh BHI culture medium supplemented with 2.5 mM calcium chloride and incubated at 37°C until the cell density reached an optical density at 600 nm (OD600) of 1.0, about 9 to 10 h. This was an optical density at which the culture demonstrated maximum induction of AI-2 under the conditions tested (Fig. 1). Cell growth was monitored on a Bausch and Lomb Spectronic 20 spectrophotometer at a wavelength of 600 nm. For experiments measuring AI-2 production, Y. pestis cells from glycerol stocks stored at −80°C were streaked onto BHI slants containing appropriate antibiotics and grown at 30°C for approximately 24 h. Cells were washed off slants by using 5 ml BHI broth containing appropriate antibiotics, and these cultures were grown overnight (∼12 h) at 30°C with shaking. The cultures grown overnight were then used to inoculate fresh BHI broth at a 1:500 dilution. Escherichia coli strains were maintained in Luria-Bertani (LB) broth, and Vibrio harveyi cells were maintained in autoinducer bioassay (AB) medium (8). Antibiotic concentrations (in μg/ml) used in bacterial cultures were as follows: 50 μg/ml kanamycin (Kan), 10 μg/ml chloramphenicol (Cam), and 100 μg/ml ampicillin (Amp). All reagents were purchased from Fisher Scientific (Pittsburg, PA) or Sigma (St. Louis, MO), unless otherwise indicated.
Table 1.
Bacterial strains used in this study
| Strain or plasmid | Genotype or description | Reference or source |
|---|---|---|
| Strains | ||
| Escherichia coli | ||
| BW25141 | Cloning strain | 9 |
| ER2566 | Overexpression strain | New England Biolabs |
| AH-pfs | ER2566/pET28a-pfs (Kanr) | This work |
| AH-luxS | ER2566/pET28a-luxS (Kanr) | This work |
| AH1070 | ER2566/pET28a-lsrB (Kanr) | This work |
| Yersinia pestis | ||
| CO92 | Yersinia pestis Lcr+ Pgm+ | 7 |
| R88 | CO92 Lcr+ Δpgm | R. D. Perry |
| R115 | CO92 Lcr+ Δpgm ΔypeIR ΔyspIR ΔluxS::kan | R. D. Perry |
| ISM1980 | R88 ΔluxS::kan | This work |
| YP21 | R88 ΔluxS::kan | This work |
| YP23 | R88 ΔluxS::frt/pJET1.2 (Ampr) | This work |
| YP21 | R88 ΔluxS::frt/pJET1.2-luxS | This work |
| YP24 | R88 Δlsr::frt | This work |
| Vibrio harveyi MM32 | luxN::cat luxS::Tn5kan | 12 |
| Plasmids | ||
| pET28a | Overexpression vector (Kanr) | Novagen |
| pET28a-pfs | Pfs expression vector (Kanr) | This work |
| pET28a-luxS | LuxS expression vector (Kanr) | This work |
| pET28a-lsrB | LsrB(Δ2–25) expression vector (Kanr) | This work |
| pJET1.2 | Cloning vector (Ampr) | Fermentas |
| pJET1.2-luxS | luxS complementation vector (Ampr) | This work |
Fig 1.
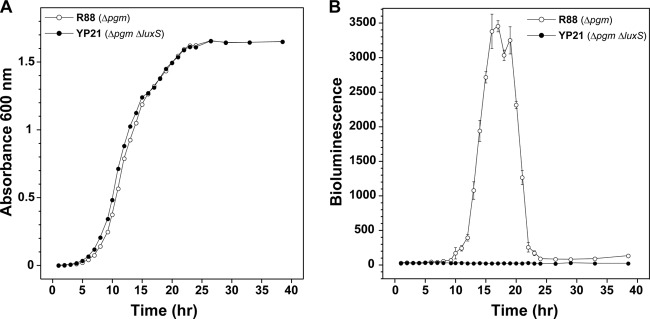
Growth and AI-2 production in Y. pestis strain R88 (Δpgm) and the ΔluxS mutant. (A) Growth of strains with shaking in BHI broth at 30°C. (B) At each time point during growth, cell supernatants were collected, filtered, and assayed for AI-2 by using the V. harveyi MM32 reporter strain (see Materials and Methods). Data represent the means of two Y. pestis cultures per strain (A) and means ± standard deviations of four wells of MM32 medium per time point for two experiments (n = 8) (B).
Strain construction.
The Y. pestis ΔluxS::frt mutant (strain YP21) was constructed by bacteriophage lambda Red-mediated recombination. Briefly, a Camr antibiotic resistance cassette was amplified from pKD3 (9) by using oligonucleotides YpluxS-S and YpluxS-AS (Tables 2 and 3). Purified PCR products were electroporated into strain R88 carrying plasmid pRedET (10). Individual Camr colonies were selected at room temperature and cultured at 37°C to screen for the loss of pRedET. The Camr cassette was subsequently removed by site-specific recombination by transformation with pCP20, as described previously (9). A similar luxS knockout mutation in R88 was also constructed with a Kanr marker by using pKD4 (9) as the template with the same primers, creating strain ISM1980.
Table 2.
Primers used for cloning

Underlined sequences are restriction sites included for cloning purposes.
Table 3.
Primers used for quantitative RT-PCR
| Gene | Primer sequence |
|
|---|---|---|
| Forward | Reverse | |
| aceA | AGATGTCTTGGGTGTACCAACGCT | AGCAGTACGGTCGCCGGTAATAAA |
| agaZ | GTGCAGCCTGGTGTTGAGTTTGAT | AATCGGTGGAGTGGGCTTCGAATA |
| caf1 | AACTTTACAGATGCCGCGGGTGAT | AGCGAACAAAGAAATCCTGGCTGC |
| deoB | ATCTGCCGGGCTATTTGGGTAACT | GGGCAATTTCGCACAGCTCATACA |
| fruK | GATCAACGTTGCCAAGGTGCTGAA | AGTCACTTCGCCATCTTTCTCGGT |
| galT | CAATCGCCCATGCTGCTGGATTAT | TAAGGCACTACCGCTAGCCAATGT |
| gltP | ATGCGAAGAAACTTGGCCGGATTG | TCAACGACGGTTAGCGCAGACATA |
| katY | ACCGATGTCAGTTCTTTCGCGGTA | TCAGGCACGGTCAGTTCCAGTTTA |
A luxS complementation plasmid was constructed by amplifying the luxS gene using primers Yp-luxS.S and Yp-luxS.AS. The PCR product (∼0.8 kbp) was gel purified, blunt ended, and cloned into the vector pJET by using the CloneJET PCR cloning kit (Fermentas). A luxS overexpression plasmid was constructed by amplifying the luxS gene by PCR using oligonucleotides forluxS and revluxS (Tables 2 and 3). The PCR product was digested with NheI and HindIII and ligated into pET28a (Novagen) cut with the same enzymes. A pfs overexpression plasmid was constructed by amplifying the pfs gene by PCR using oligonucleotides forpfs and revpfs (Tables 2 and 3). The PCR product was digested with NdeI and BamHI and cloned into pET28a cut with the same enzymes.
In a similar fashion, a Y. pestis Δlsr::kan operon deletion (strain YP26) was constructed. Oligonucleotides YplsrA2-ko.S and Yplsr-ko.AS were used to amplify a Kanr cassette from pKD4 (9). Purified PCR products were electroporated into R88 carrying plasmid pRedET, and Δlsr::kan mutants were isolated as described above.
Pfs and LuxS purification.
Plasmid pET28a-pfs or pET28a-luxS was then transformed into E. coli expression strain ER2566 by electroporation. For purification of either protein, the expression strain was grown overnight and inoculated at a dilution of 1:200 into 2 liters of LB broth supplemented with Kan. Cultures were grown at 37°C to an OD600 of ∼0.6, and gene expression of the protein of interest was induced with 1 mM isopropyl-β-d-thiogalactopyranoside. After 2 h under inducing conditions, cells were harvested by centrifugation at 6,000 × g for 10 min and suspended in 25 ml of ice-cold equilibration buffer (0.5 M NaCl, 5 mM imidazole, and 20 mM Tris [pH 7.9]). Cells were lysed with 3 ml of 10× BugBuster (Novagen) on a nutating mixer at 4°C for 2 h. Insoluble debris was removed by centrifugation at 30,000 × g for 35 min, and the cleared cell lysates were loaded onto 5-ml columns containing His-Select resin (Sigma) equilibrated at room temperature with equilibration buffer. The columns were washed with 30 ml of equilibration buffer followed by 50 ml of wash buffer (0.5 M NaCl, 60 mM imidazole, and 20 mM Tris [pH 7.9]), and the Pfs and LuxS proteins were then eluted with 50 ml of elution buffer (0.5 M NaCl, 1 M imidazole, 20 mM Tris [pH 7.9]). Fractions containing Pfs and LuxS proteins were identified by SDS-PAGE. Pooled fractions were concentrated by using 10-kDa-cutoff Amicon Ultra centrifugal filters (Millipore) and dialyzed against 10 mM Tris (pH 7.5) by using 10-kDa-cutoff Slide-A-Lyzer cassettes (Pierce). The protein concentration of the purified enzymes was determined by a Bradford assay (Bio-Rad), and glycerol was added to a final concentration of 33% before the Pfs and LuxS proteins were flash frozen at concentrations of 23 mg/ml and 28 mg/ml, respectively, by dripping the protein solutions into liquid nitrogen. Frozen pellets of Pfs and LuxS were stored at −80°C until they were used for biosynthesis of AI-2.
AI-2 biosynthesis.
AI-2 was biosynthesized from S-adenosylhomocysteine (SAH) in a two-step enzymatic process that used purified Y. pestis Pfs and LuxS enzymes, which was analogous to that described previously for the in vitro synthesis of AI-2 using purified Vibrio harveyi enzymes (11). In the first reaction, SAH was converted to S-ribosylhomocysteine (SRH) and adenine by the enzymatic action of Pfs. Specifically, 10 mM SAH in 50 μl of 50 mM Tris-HCl (pH 7.5) was incubated at 25°C with 4.2 μM purified Y. pestis Pfs (see the description of Pfs purification above). The reaction progress was monitored by measuring the time-dependent decrease in absorbance at 276 nm, which is due to adenine having a lower extinction coefficient than adenosine, and was found to be complete within 1 h. After 4 h of incubation, the Pfs reaction mixture was passed through an Amicon Ultra centrifugal filter device with a 5-kDa cutoff (Millipore) in order to remove the Pfs enzyme. In the second step of biosynthesis, S-ribosylhomocysteine produced by the Pfs reaction was enzymatically converted by LuxS to homocysteine and 4,5-dihydroxy-2,3-pentanedione (DPD), which spontaneously cyclizes to form bioactive AI-2. This was accomplished by adding purified Y. pestis LuxS (see the description of LuxS purification above) to the filtered Pfs reaction mixture at a concentration of 2.5 mg/ml and allowing the mixture to incubate at room temperature for up to 24 h. Production of DPD was monitored indirectly by periodically determining the concentration of homocysteine by using Ellman's reagent [5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB)], since the LuxS reaction produces equimolar quantities of DPD and homocysteine. Specifically, 15 μl of the LuxS reaction mixture was removed periodically and mixed with 985 μl of 100 μM DTNB in 100 mM Tris (pH 8.0), and the absorption at 412 nm was measured and converted to a molar concentration by using an extinction coefficient of 14,150 M−1 cm−1. LuxS was removed from the reaction mixture by passage through an Amicon Ultra centrifugal filter device with a 5-kDa cutoff, and aliquots were frozen and stored at −20°C until they were used for bioluminescent or microarray assays.
V. harveyi AI-2 bioassay.
For all experiments measuring growth-dependent production of AI-2 by Y. pestis, the cultures were grown overnight, as described above, by using the following procedure. Cells were diluted 1:500 in 5 ml of fresh BHI broth and grown at 30°C with shaking for ∼20 h. This culture was diluted 1:1,000 in 50 ml of fresh BHI broth, and AI-2 production was measured. At the indicated time points, 1 ml of culture was removed, the OD600 was measured by using a Tremo Spectronic Genesys 20 spectrophotometer, and 600 μl (of the 1 ml) was sterilized by using 0.22-μm Costar Spin-X centrifuge tube filters (Corning). The sterile conditioned medium was diluted 1:10 and 1:100 with BHI broth (1 ml was prepared for each dilution), and all tubes were frozen and stored at −80°C until the AI-2 concentration was measured.
AI-2 concentrations were measured by using V. harveyi reporter strain MM32 (12). This strain will produce light only when AI-2 is supplied exogenously, since it contains deletions of luxN and luxS (Table 1) that render it incapable of sensing homoserine lactones or synthesizing AI-2. For measurement of AI-2 concentrations, a culture of MM32 grown overnight in AB medium was used to inoculate fresh AB medium at a 1:500 dilution. After ∼1 h of growth at 30°C with shaking, 180-μl aliquots of the MM32 culture were transferred onto 96-well microtiter plates (Costar 3603), to which 20 μl of Y. pestis conditioned medium (4 wells per dilution per time point) had already been added. After mixing, the final dilutions of the conditioned medium relative to the Y. pestis culture were 1:10, 1:100, and 1:1,000. The microtiter plates were incubated at 30°C with shaking, and at 2-h intervals (for 10 h), the OD600 and luminescence were measured by using either a Tecan GENios or a Tecan Infinite 200 M microtiter plate reader. The luminescence values and AI-2 concentrations reported are those measured after 6 h of MM32 growth.
AI-2-induced luminescence by MM32 was converted to a molar concentration by developing a standard curve using known signal concentrations of biosynthesized AI-2. The standard curve was made by using the same MM32 culture used to measure AI-2 in conditioned medium, and OD and luminescence measurements were taken at the same time points in order to minimize differences. Luminescence values, measured at 6 h of MM32 growth, were normalized to OD595 values and plotted versus the AI-2 concentration. The resulting plot of OD-normalized luminescence versus AI-2 concentration was fit to a 5-parameter logistic fit by using KaleidaGraph software (Synergy Software). The resulting standard curve was used to convert OD-normalized luminescence values measured for Y. pestis conditioned medium to molar AI-2 concentrations.
AI-2 levels in virulent Y. pestis CO92 cells.
Virulent strain Y. pestis CO92 was obtained from the Biodefense and Emerging Infections Research Resources Repository. The strain was maintained and grown in the biosafety level 3 (BSL-3) facility at the Department of Veterinary Microbiology and Preventive Medicine, Iowa State University, according to established protocols. For the AI-2 time course, strain CO92 was grown in BHI in a similar manner as described above. Spent medium was collected at the indicated time points, sterilized by using a 0.22-μm filter, and confirmed as being culture negative. Samples were saved and tested for AI-2 as described above.
Microarray experimental design.
For each microarray experiment, the transcriptomes of the luxS mutant (ISM1980) or the luxS yspIR ypeIR triple mutant (R115) were compared with that of the CO92 Δpgm isogenic parent strain R88. In these experiments, the cells were harvested at mid- to late log phase (OD600 = 1.0), which was the time when cultures exhibited the greatest amount of AI-2 production (Fig. 1). Two additional experiments were performed with the Δpgm background strain, in which cells were treated with purified QS signals at an early stage of growth prior to the cells achieving quorum sensing (Fig. 1) (13). In one experiment, AI-2 signal, prepared as described above, was added to cultures. In the second experiment, AI-2 plus the two acyl homoserine lactones (AHLs) produced and used by Y. pestis [N-hexanoyl-dl-homoserine lactone and N-(3-oxooctanoyl)-l-homoserine lactone] (Sigma) were added simultaneously. Cultures grown overnight were washed three times to remove any quorum-sensing signals and diluted 1:100 in BHI broth. After 2 h of incubation at 37°C to establish early-log-phase growth, either single AI-2 (500 nM final concentration) or three-signal AI-2 (500 nM final concentration) and AHLs (both at 5 μM final concentrations) were added. The cells were incubated for 4 h at 37°C and then isolated by centrifugation, RNA Protect reagent (Invitrogen) was added, and the cells were stored at −70°C until the RNA was isolated. The control was an identical aliquot of cells from the same culture without added QS signals.
For each experiment, labeled cDNAs generated from RNA from six mutant cultures (biological replicates) were paired with controls for hybridization on a two-color microarray slide. Cy3 and Cy5 dyes were used as labels, and dye assignments for control and mutant samples were reversed for three of the arrays to account for variation in labeling efficiencies (dye bias). Table 4 describes the microarray experiments performed in this study.
Table 4.
Numbers of significantly regulated genes by different transcriptional studies
| Microarray design | No. of significantly regulated genes at a P value of <0.05a | GEO accession no. |
|---|---|---|
| R88 vs luxS::kan | 615 | GSE22849 |
| R88 vs ΔypeIR ΔyspIR luxS::kan | 194 | GSE21911 |
| Δpgm untreated vs AI-2 treated | 32 | GSE22848 |
| Δpgm untreated vs AI-2 plus AHL treated | 30 | GSE22846 |
All with a fold change of >1.5.
RNA extraction, cDNA synthesis, and labeling.
The cells from each culture were collected and were treated with RNA Protect bacterial reagent (Qiagen) and stored at −70°C. RNA was extracted from frozen cell pellets by using the RNeasy minikit (Qiagen) according to the manufacturer's protocol. The RNAs were treated with DNase I (Ambion) for 30 min at 37°C to remove contaminating genomic DNA. PCR was used to confirm DNA removal. The RNAs were then centrifuged through Microcon Ultracel/YM30 filters (Millipore). Ten micrograms of total RNA from each sample was used to synthesize aminoallyl-labeled cDNA by using SuperScript III reverse transcriptase (Invitrogen) and 10 μg of random hexamers (Integrated DNA Technologies). Fluorescently labeled targets were prepared as described previously (14). The frequency of dye incorporation and yield were determined by spectrophotometry (ND-1000; Nanodrop).
Hybridization.
Microarray slides were prewashed in water for 5 min and rinsed twice with water for 1 min. Corresponding equal amounts of dye-labeled cDNA targets (1.5 μg) were mixed together, dried, resuspended in 225 μl Long Oligo hybridization solution (Corning), incubated at 95°C for 5 min, and centrifuged for 4 min at 10,000 rpm. This solution was then injected into a Lucidea Slidepro (Amersham Biosciences, Piscataway, NJ) hybridization station containing the prewashed microarray slides. The hybridization lasted for 16 h at 42°C, and the slides were then washed in a series of wash buffers (2× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 0.1% SDS, 1× SSC, and 0.1× SSC) by the hybridization station and dried by centrifugation at 1,500 × g for 30 s.
To enhance the discovery of differentially expressed genes, the hybridized arrays were scanned three times with various laser power and photomultiplier tube values by using a ScanArray HT scanner (PerkinElmer) to detect excited wavelengths, as described previously (15). Image processing and normalization were performed as described previously (15). The normalized values for triplicate spots were averaged within each array to produce one normalized measure of expression of fluorescence for each probe sequence. A mixed-model analysis was conducted as previously described (14).
Real-time RT-PCR.
The SensiMix SYBR No-Rox One-Step kit (Bioline) was used to perform the real-time reverse transcription-PCR (RT-PCR) assay on the Mx3005P QPCR system (Stratagene). The quantification cycle (Cq) values were exported from MxPro 4.1 software. Three biological Y. pestis replicates were used, and all original RNAs were equally diluted to 4 ng/μl. Each RNA template was then diluted 3 times, as described previously for the PREXCEL-Q software program (16). The leuB gene was used as the internal reference gene because it showed no transcriptional changes by microarray under any of the experimental conditions. The final reaction mixture volume for each well was 15 μl and contained 7.5 μl SensiMix SYBR No-Rox One-Step, 0.6 μl of each primer at 2.5 μM, 0. 3 μl of 10 U/μl RiboSafe RNase inhibitor, 5 μl RNA, and 1 μl diethyl pyrocarbonate (DEPC)-water. The thermal cycling conditions included a reverse transcription step at 50°C for 30 min; a polymerase activation step at 95°C for 10 min; 40 PCR cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s; and a melting-curve step of 95°C for 1 min, 55°C for 30 s, and 95°C for 30 s. The data were analyzed according to methods described previously by Gallup and Ackermann (16).
Hydrogen peroxide killing assays.
Strains R88 and YP21 (ΔluxS::Cm) were grown as 5-ml cultures overnight in BHI broth at 37°C with 4 mM CaCl2 supplementation to an OD600 of 3 to 4. Cells were pelleted by centrifugation, the growth medium was decanted, and the cell pellet was suspended in an equal volume of 10 mM phosphate-buffered saline. Strains were diluted to an OD600 of 0.25 in a 1-ml total volume in 15-ml culture tubes. After treatment with different concentrations of hydrogen peroxide for 1 h, 50 μl of the cell suspension was diluted into 1 ml in PBS with 1 mg/ml catalase (Sigma) to quench the reaction. The samples were further serially diluted, spotted onto BHI agar plates, and grown at 30°C to determine the CFU per ml.
Microarray data accession numbers.
The microarrays used in these studies can be found in the National Center for Biological Informatics Gene Expression Omnibus (GEO) under platform accession numbers GPL9009 and GPL10017. All microarray data are available under the GEO accession numbers given Table 4.
RESULTS
LuxS-dependent production of AI-2.
To initiate studies on AI-2 signaling, the luxS gene was deleted from the Δpgm mutant of CO92 (strain R88) by using lambda Red recombinase, resulting in strain YP21 (Table 1). No growth defects in YP21 were observed compared to the parent during growth at 30°C in rich medium (Fig. 1A). For monitoring AI-2 production, Vibrio harveyi reporter strain MM32 was utilized throughout this report. MM32 is unable to synthesize AI-2 due to a luxS mutation, and additionally, the presence of a luxN mutation strain blocks the bioluminescent response to AI-1 (12). Thus, MM32 will bioluminesce only in the presence of exogenously added AI-2, making it a robust and more sensitive AI-2 reporter than the commonly used strain BB170. The bioluminescent response of MM32 was confirmed through control experiments using culture supernatants from known producers of AI-2 (data not shown).
To generate a profile of AI-2 production in Y. pestis, the levels of AI-2 were monitored over time in R88 and its isogenic ΔluxS knockout. Cultures were grown at 30°C in rich medium, and cell-free supernatants were assayed for AI-2 by using the MM32 reporter strain. The R88 strain produced robust levels of AI-2 during an approximately 10-h window in late logarithmic phase (Fig. 1B). As anticipated, deletion of luxS in YP21 eliminated AI-2 production. As the cells entered stationary phase, the extracellular AI-2 concentration in R88 dropped off dramatically to almost undetectable levels. To confirm the role of LuxS, a plasmid containing the luxS gene and its native promoter were transformed into YP21 to create strain YP24. Compared to the empty vector control (strain YP23), the luxS plasmid complemented AI-2 production to levels similar to those of strain R88 (Fig. 2), indicating that the lack of AI-2 production was not due to secondary mutations. These findings demonstrate that the LuxS enzyme is essential for Y. pestis AI-2 production during late logarithmic growth.
Fig 2.
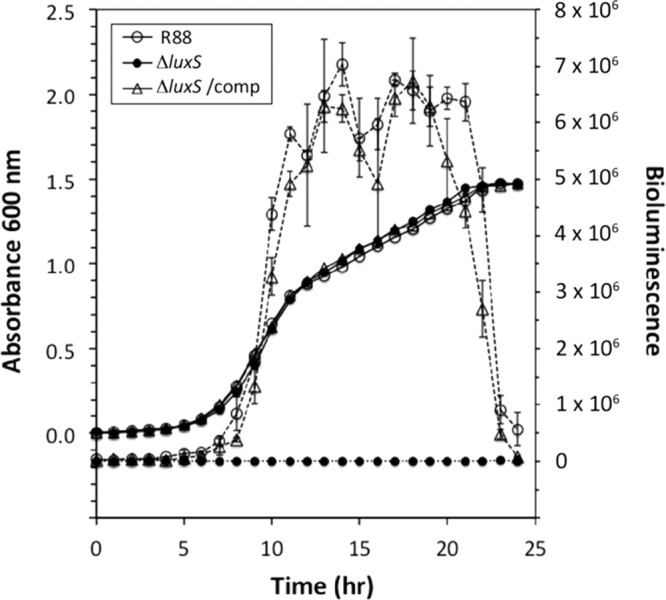
Complementation of the ΔluxS mutant. Yersinia pestis strain R88, the ΔluxS mutant, and the complemented strain were grown with shaking in BHI broth at 30°C. At each time point during growth, cell supernatants were collected, filtered, and assayed for AI-2 by using the V. harveyi MM32 reporter strain. Data represent the means ± standard deviations of triplicate wells per time point and strain.
Δpgm mutation does not affect AI-2 production.
The Δpgm mutation in Y. pestis strain R88 removes 102 kb of chromosomal DNA, resulting in attenuation (17). Since this deletion is large and encompasses many genes, it was important to assess the contribution of this mutation to AI-2 production. For this comparison, a time course experiment was performed to measure the expression of extracellular levels of AI-2 produced by wild-type CO92 and strain R88. Overall, the Δpgm mutation had no observable effect on AI-2 production, as near-identical AI-2 production was observed between the two strains (Fig. 3). Throughout the rest of this report, all studies were performed with Δpgm mutant strain R88.
Fig 3.
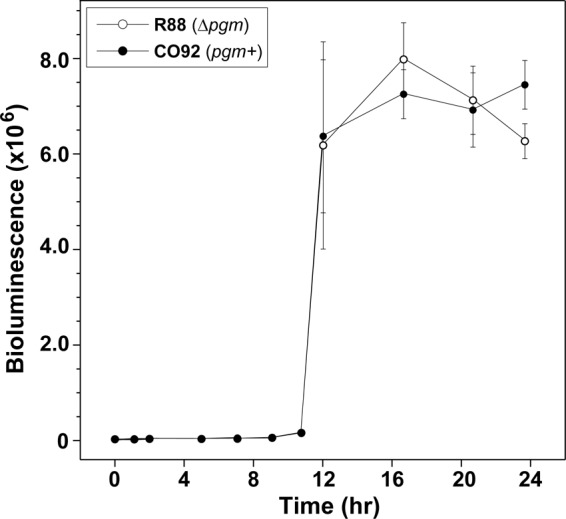
Comparison of AI-2 production in Y. pestis wild-type strain CO92 and strain R88 (Δpgm). Yersinia pestis strain R88 and wild-type strain CO92 were grown with shaking in BHI broth at 30°C in a BSL-3 facility. At each time point during growth, cell supernatants were collected, filtered, and assayed for AI-2 by using the V. harveyi MM32 reporter strain. Data represent the means ± standard deviations of triplicate wells per time point.
The lsr chromosomal region.
Genetic and molecular studies of Salmonella enterica have revealed that AI-2 signaling occurs through a different mechanism than that in the V. harveyi paradigm model. Central to the difference is the presence of the lsr chromosomal locus, encoding factors necessary for AI-2 binding, import, and intracellular regulation (18, 19). Our bioinformatic analysis of the CO92 chromosome indicates the presence of a gene cluster with striking similarity to the S. enterica and E. coli lsr loci. Other research groups have made similar claims about the presence of a potential lsr locus in Y. pestis through sequence analysis (20, 21).
The cluster stretches from Y. pestis CO92 open reading frames (ORFs) YPO415 through YPO407 (Fig. 4). ORFs YPO414 and YPO415 encode proteins with similarity to the LsrR transcriptional regulator and LsrK AI-2 kinase, respectively. In the intergenic region, there is a short gene, called YPO413, that does not exist in other enteric lsr regions. Divergently transcribed from the predicted LsrR regulator, ORFs YPO412 through YPO410 share similarity with the ABC transporter LsrACD, while YPO409 is similar to the periplasmic receptor called LsrB. Finally, the YPO408 and YPO407 ORFs share similarity to the LsrF and LsrG enzymes, respectively.
Fig 4.
The lsr region of Y. pestis CO92 compared to that of E. coli. The CO92 open reading frame designations (YPO407 to YPO415) are shown on the bottom of the schematic. The percent identity of the CO92-encoded proteins with the corresponding E. coli Lsr proteins is indicated. An additional open reading frame, YPO413, is present in the intergenic region of the CO92 genome.
We hypothesized that mutations in the lsr locus would alter the extracellular level of AI-2 over time. To investigate this question, a complete deletion of the locus was constructed by lambda Red recombination. The time course to monitor AI-2 production was performed similarly to that for luxS mutant strain YP26. Similar levels of AI-2 were detected in the spent media of YP26 and the parent strain R88 during logarithmic growth (Fig. 5A). Interestingly, the extracellular level of AI-2 remained high in YO26 as it entered stationary phase, while the level of AI-2 in R88 dropped off dramatically.
Fig 5.
AI-2 production profile of the Y. pestis Δlsr mutant and quantification of signal levels. (A) Yersinia pestis strain R88 and the Δlsr mutant were grown with shaking in BHI broth at 30°C. At each time point during growth, cell supernatants were collected, filtered, and assayed for AI-2 by using the V. harveyi MM32 reporter strain. (B) AI-2 was biosynthesized by using purified Y. pestis CO92 Pfs and LuxS enzymes (see Materials and Methods). A standard curve was prepared by using synthesized AI-2 in the V. harveyi bioassay. (C) Conversion of the bioluminescence output into AI-2 concentrations by using the developed standard curve. Data represent the means of two cultures per Y. pestis strain (A), means ± standard deviations of two cultures with 4 wells per time point (B), and the same data as in panel A with bioluminescence converted to μM by using the curve in panel B (C).
To quantify the difference between YP26 and R88, a bioassay standard curve was generated by using biosynthesized AI-2 (Fig. 5B). AI-2 was obtained by overexpressing and affinity purifying the Y. pestis CO92 Pfs and LuxS enzymes and performing a two-step conversion of S-adenosylhomocysteine to DPD, as described in Materials and Methods. By extrapolation to the standard curve, the AI-2 concentration was determined throughout the time course (Fig. 5C). In both strains R88 and YP26, peak production of AI-2 reached levels of ∼2.5 μM in the late logarithmic growth phase. In stationary phase, the AI-2 levels dropped to 0.2 μM by 25 h of growth and remained fairly constant in the 0.2 to 0.3 μM range. In contrast, the AI-2 levels in YP26 ranged from 1.1 to 1.4 μM in stationary phase but eventually dropped to under 1 μM at later time points (Fig. 5C). Altogether, YP26 showed no defect in AI-2 production, but the mutant displayed an altered phenotype in the stationary-phase levels of the signal. Thus, these findings implicate the Y. pestis lsr chromosomal region in stationary-phase degradation or transport of the AI-2 signal.
Effect of QS pathways on gene expression.
To obtain a better understanding of the role of LuxS, Ype, and Ysp pathways at the mammalian host temperature, we measured transcript levels in R88 and compared them to the levels in triple mutant strain R115, as described above. In this experiment, 194 genes were found to be differentially expressed at least 1.5-fold (P < 0.05), and 92 genes were differentially expressed with a P value of <0.01 (see Table S1 in the supplemental material). Genes upregulated in response to all three QS systems with the highest fold changes included lsrK (YPO0415) (kinase that phosphorylates carbon-5 of the open form of DPD), lamB and malMKEFG (maltose transport and metabolism), agaYZ (tagatose metabolism), gltA and icdA (Krebs cycle), tcaA (insecticidal toxin), yapM (YPO0823) (autotransporter), and metA (homoserine O-succinyltransferase). Genes downregulated in response to all three QS systems included yopEPT (effector proteins), sycEN (Yop chaperones), and yscABCDEFGHIJKLMNOPQRSTXY and lcrDF (low-calcium response).
luxS-regulated genes.
To clarify the role of the luxS gene at the host temperature, we measured transcript levels in ISM1980 and compared them to the transcript levels in R88 at 37°C, as described above. In this experiment, 615 genes were found to be differentially expressed at least 1.5-fold at a P value of <0.05, and 220 genes were differentially expressed at a P value of P <0.01 (see Table S2 in the supplemental material). Genes upregulated in response to AI-2 included the lsr operon (lsrABCDGFKR) (AI-2 regulation), mglBC and galEKT (monosaccharide galactose uptake and metabolism), lamB and malMKEFGSZ (maltose metabolism), aceABK (glyoxylate bypass), acs (acetyl coenzyme A synthetase), araFGH (arabinose operon), deoB (phosphopentomutase), gcsH and gcvPT (glycine degradation), yfiA (putative σ54 modulation protein), ail (YPO2905) (attachment and invasion of mammalian cells), katA (catalase), katY (catalase), fadABDH (fatty acid degradation), treBC (trehalose metabolism), terZ (tellurium resistance protein), metK (S-adenosylmethionine synthetase), ureABCDEFG, and ompW (putative exported protein), and genes involved in the Krebs cycle. Genes downregulated in response to AI-2 included rpl (ribosomal proteins), yopMP and yscBCDEFGHIJKLMS (type III secretion system), fabABFJ (fatty acid biosynthesis), hmuSTU (hemin transport), waaL (putative O-antigen biosynthesis protein), and lolCDE (lipoprotein-releasing system).
Differentiated genes in response to purified AI-2.
To decipher the role of AI-2 signaling at mammalian host temperatures, an AI-2 add-in study was performed as described above. The results showed a total of 32 differentially regulated genes at a P value of <0.05 and 9 genes at a P value of <0.01 (see Table S3 in the supplemental material). Upregulated genes in AI-2-treated cells with a fold change of >1.5 included only two genes, YPO4086 (lipoprotein) and fliH (flagellar assembly protein). Downregulated genes in AI-2-treated cells included psaAB (synthesis of the pH 6 antigen precursor, antigen 4), mgtB (Mg2+ transport ATPase protein B), terA (tellurite resistance protein), ompC (outer membrane porin protein C), YPO0276, and yfiA (σ54 modulation protein involved in broad regulatory functions).
Differentiated genes in response to AI-2 and both AHLs.
A three-signal add-in study was performed, and the array results showed a total of 30 differentially regulated genes at a P value of <0.05 and 21 genes at a P value <0.01 (see Table S4 in the supplemental material). Upregulated genes in the 3-signal-treated cells with the highest fold change included cspA1, cspA2, and cspB (cold shock proteins); ibpB (heat shock protein); asnA (aspartate-ammonia ligase); gntT (gluconate permease); psaE (regulatory protein); ompAC (cell envelope proteins); and gntT and fruAB (carbohydrate transport). Downregulated genes in the three-signal-treated cells included two membrane proteins, YPO0800 and YPO1499; iucC (siderophore biosynthesis protein); and metE (5-methyltetrahydropteroyltriglutamate–homocysteine S-methyltransferase).
AI-2 is required for resistance to oxidative damage.
There are several reports that AI-2 may be important for resistance to oxidative stress in Gram-negative bacteria (22, 23). In addition, our microarray data indicated that a mutation in luxS (strain ISM1980) resulted in downregulation of katY (see Table S2 in the supplemental material). To assess this phenotype in Y. pestis, hydrogen peroxide was used in growth inhibition assays with strain R88 and ΔluxS strain YP26. In the presence of the oxidant, YP26 displayed a longer growth lag in culture medium and increased sensitivity on agar plates (data not shown), in accordance with our microarray results (see Table S2 in the supplemental material). To quantify this phenotype, a dose-response curve of peroxide exposure at 37°C (Fig. 6A) and 30°C (Fig. 6B) with calcium and at 30°C without supplementation (Fig. 6C) was performed, and viable cells were enumerated. Under every condition, the ΔluxS mutant displayed increased sensitivity to hydrogen peroxide exposure.
Fig 6.
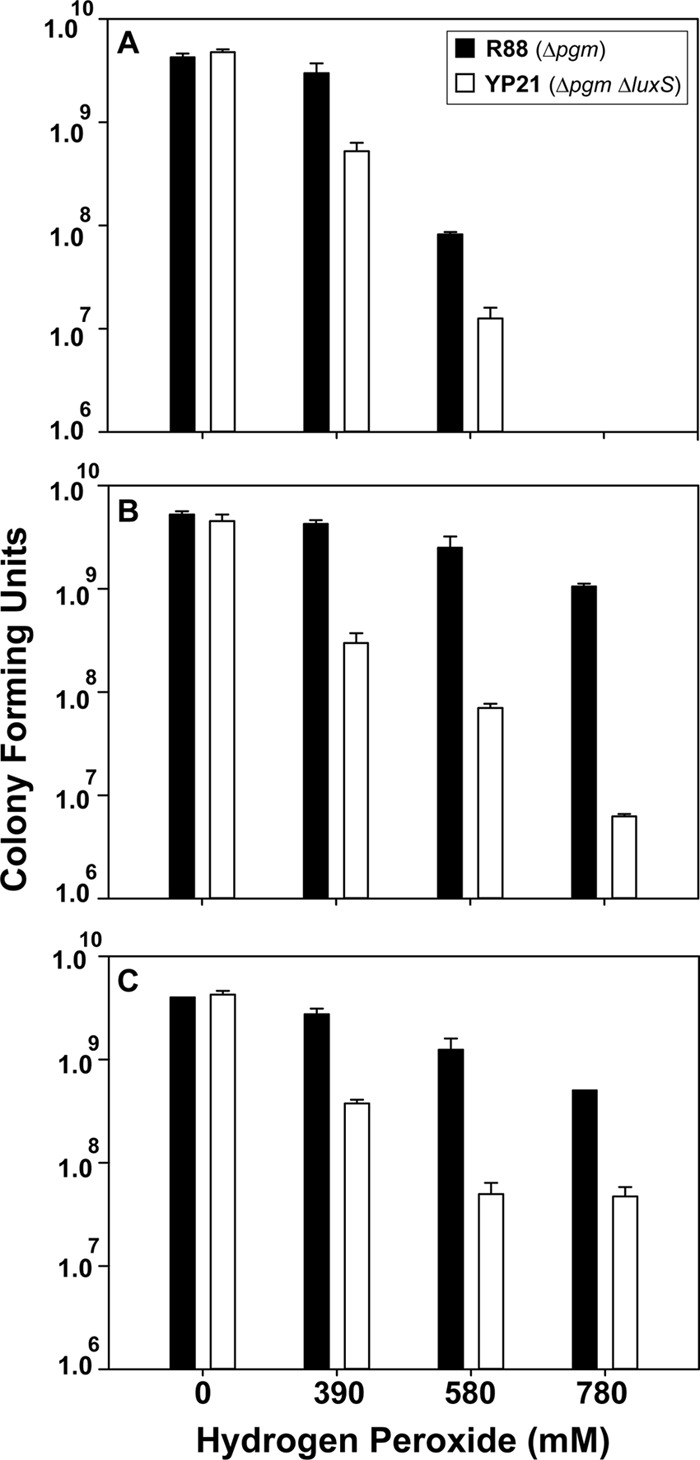
The Yersinia pestis ΔluxS strain is more sensitive to hydrogen peroxide killing. Strains R88 and YP21 (ΔluxS mutant) were grown in BHI broth and exposed to various concentrations of hydrogen peroxide for 1 h. The reactions were quenched, and colony counts were performed. (A) Cells grown at 37°C with supplemented calcium. (B) Cells grown at 30°C with supplemented calcium. (C) Cells grown at 30°C. Data represent the means ± standard deviations of biological triplicates per peroxide concentration tested.
DISCUSSION
In this report, we demonstrate that Y. pestis CO92 utilizes an AI-2 QS mechanism that is similar to those of other enteric pathogens. Yersinia pestis makes AI-2 in a LuxS-dependent manner during late logarithmic growth, and we identified an lsr chromosomal locus that modulates the stationary-phase level of extracellular AI-2. Finally, our findings indicate that AI-2 sensing may be important for resistance to oxidative stress.
A recent study of Y. pestis strain KIM demonstrated that luxS mutants were unable to make AI-2 and that this phenotype could be complemented (4). Our time course analysis has taken these observations a step further and demonstrated that AI-2 is produced only in a tight window during logarithmic growth (Fig. 1). In stationary phase, the AI-2 levels drop precipitously in an Lsr-dependent manner, similar to what has been observed for S. enterica and E. coli (6, 18). Whether the concentration decline is due to extracellular degradation of AI-2 or transport is not clear. Considering that the lsr locus encodes an LsrACD transporter that is functional in other enteric pathogens (19, 24), it seems likely that the disappearance is due to AI-2 transport.
The results of the AI-2 time course study with the Y. pestis Δlsr mutant (Fig. 5) mirror the findings for S. enterica and E. coli (18, 24). In all three pathogens, when the Lsr transporter is deactivated, the extracellular AI-2 levels are prolonged but eventually drop to lower levels. These observations prompted the proposal that another low-affinity or cryptic AI-2 transporter may exist. The lsrACDBFGE operon shows striking parallels to the rbs operon (19), which encodes functions required for ribose transport. In the oral pathogen Actinobacillus actinomycetemcomitans, the periplasmic ribose receptor RbsB binds AI-2 with strong affinity (12). However, a knockout of the rbs system in A. actinomycetemcomitans had only a minimal effect on AI-2 extracellular levels (25), paralleling observations made previously with E. coli (26). Taken together, the Lsr system is the primary mechanism of AI-2 transport in most bacteria, and the Rbs system functions at a secondary level. Both the lsr and rbs systems need to be deactivated for maintaining extracellular AI-2 levels (25). Similar to other bacteria, a chromosomal locus with similarity to the rbs operon is present in Y. pestis CO92 (ORFs YPO3633 to YPO3639), which could be functioning in this capacity. Other transporters, such as the TqsA protein (27), could also be contributing to the stationary-phase drop in AI-2 levels in the Δlsr mutant (Fig. 5), and a TqsA homologue is present on the Y. pestis CO92 chromosome (YPO2450). Further investigation is necessary to clarify possible roles of the putative rsb locus or TqsA in AI-2 binding and transport.
Our findings with the Y. pestis ΔluxS mutant demonstrating an increased sensitivity to hydrogen peroxide killing compared to the luxS+ control strain (R88) suggest a role for AI-2 in the oxidative stress response. Yersinia pestis possesses two functional catalases, KatA and KatY, that are known to contribute to hydrogen peroxide resistance (28). Reduced levels of one or both of these enzymes could explain the increased sensitivity to oxidative stress (Fig. 4). Although subtle temperature regulation of the katY gene has been reported (28), we did not observe significant temperature or calcium dependence in our killing assay.
Knockouts of the luxS biosynthetic gene in many pathogenic bacteria have revealed broad-spectrum phenotypes. These phenotypic changes can range from biofilm defects to altered expression of virulence factors and have been summarized in review articles (29). While we have identified a putative role for AI-2 signaling during oxidative stress, other studies have linked the system to Y. pestis biofilm formation (4), an important factor during growth in the flea. However, the biofilm phenotype was observed in combination with acyl homoserine lactone knockouts, suggesting that the contribution of AI-2 to this phenotype is unclear.
Our studies show that the Y. pestis AI-2 quorum-sensing system functions in a manner similar to that in the S. enterica paradigm model. Both pathogens make AI-2 during logarithmic growth, modulate extracellular levels by using the Lsr system, and recognize the (2R,4S)-2-methyl-2,3,3,4-tetrahydroxytetrahydrofuran (R-THMF) form of the signal (30). We have gone further and uncovered a function for AI-2 in the Y. pestis oxidative stress response, shedding light on a potential role for the system in host interactions. Beyond these enteric pathogens, recent studies with the plant symbiont Sinorhizobium meliloti indicate that the Lsr system is also present in nonpathogenic bacteria (21). Interestingly, S. meliloti does not possess luxS but responds to the R-THMF form of exogenous AI-2 by using an Lsr system, providing a new twist on this signaling theme. As the list of bacteria with functional AI-2 systems continues to grow, we are gaining an improved understanding of the contribution of this communication mechanism to the lifestyles of diverse Gram-negative and Gram-positive bacteria.
More than 100 bacteria can produce and detect AI-2 quorum-sensing signals; therefore, AI-2/LuxS quorum sensing is thought to represent an important means of intergeneric communication (31). To clarify the function of AI-2-mediated QS in Y. pestis, we compared the transcriptomes of the Δpgm strain of CO92 with mutant QS strains using microarrays. This gave us insight into the role of AI-2 in gene expression on a global basis. According to the number of significantly regulated genes in the microarray experiments in this study (see Table S2 in the supplemental material), AI-2 quorum sensing was an important regulator of gene expression at 37°C, with an effect on 615 genes at a P value of <0.05 and an absolute-value fold change of >1.5 in the ΔluxS::kan mutant study (strain ISM1980).
Since this experiment involved full induction of both AHL pathways, the AI-2 addition study might better isolate only the AI-2 contribution to gene expression, with a more modest number of 32 significant genes at a P value of <0.05 (see Table S3 in the supplemental material). In this experiment, only AI-2 induction would have been operative since the growth phase was too early for an AHL contribution, as shown in previous studies (13).
In this study, we found numerous metabolism-associated genes, such as those involved with maltose, galactose, arabinose, and fatty acid metabolism, that were downregulated in the ΔluxS::kan mutant. In support of this, there is evidence that AI-2 QS can regulate carbohydrate metabolism in Streptococcus gordonii (32). Bacteria transport maltose and galactose from extracellular fluid into the cytosol. Why is it important? We know that maltose or galactose can be utilized for nutrition, although fatty acids are toxic to E. coli. The malQ gene, encoding a maltodextrin phosphorylase, is a pseudogene in Y. pestis. This mutation prevents glucose-1-phosphate from converting back to glucose to complete the O-antigen unit. We know that Y. pestis contains the core constituents of lipopolysaccharide but lacks the extended O-group side chains (33). The agaYZ operon is involved in the metabolism of N-acetylgalactosamine and N-acetylgalactosamine. Both sugars are concentrated in sensory nerve structures, are components of the interstitial tissues of both humans and animals, and are necessary for intercellular communication. Disruption of host tissues by degradation of these sugars might enhance bacterial spread. The agaYZ genes were downregulated in ΔluxS::kan mutant strain ISM1980 and triple QS mutant strain ISM1185, suggesting that agaYZ is under quorum-sensing control.
As a Gram-negative bacterium, Y. pestis requires that nutrients pass through outer and inner membranes. Around 84 inner and outer membrane proteins, including lipoproteins and porins, were up- or downregulated in the ΔluxS::kan mutant. This suggests that remodeling of the cell envelope might be one response when the luxS gene is mutated. yapM (YPO0823) encodes an autotransporter membrane protein that showed decreased expression levels in ΔluxS::kan mutant strain ISM1980. These results suggest that yapM is regulated by AI-2/LuxS. The function of yapM requires further investigation. The gene for the envelope protein OmpW, a putative exported protein with unknown function, was also downregulated in the ΔluxS::kan mutant.
Oxidative stress is encountered by Y. pestis under a variety of conditions by exposure to various highly reactive oxygen derivatives. In response to the luxS mutation, the catalase gene katA (YPO1207); katY (YPO3319), which encodes catalase-peroxidase; and ahpC (YPO3194) (alkyl hydroperoxide reductase subunit C) were upregulated in R88 to supposedly help Y. pestis combat oxidative stress inside host cells. The ahpC gene also has a role in resistance to peroxynitrite and stage-specific survival in macrophages. To support this hypothesis, we showed that a luxS mutant was more sensitive to hydrogen peroxide exposure than the control strain (Fig. 6).
The 70-kb virulence plasmid pCD in Y. pestis encodes a type III secretion system (TTSS) that injects cytotoxins, termed Yops, into the mammalian cytosol. The TTSS transcripts were upregulated in both strains containing a luxS mutation, ISM1980 and R115. Although there is some evidence that LuxS does not affect the expression of the TTSS in Salmonella (34), our data show that this does not appear to be the case in Y. pestis CO92. Two other virulence-related genes, psaA and mgtB, were also downregulated in AI-2-treated cells. These results indicate that luxS and three QS systems can decrease the expression levels of some virulence factors that may have important consequences for disease. Despite the fact that a KIM5 luxS mutant failed to reduce the LD50 in a mouse model of virulence (4), other disease parameters and models, i.e., pneumonic disease and tissue specificities, etc., should be studied in light of these new findings.
In this report, we demonstrated that Y. pestis CO92 utilizes an AI-2 quorum-sensing mechanism that is similar to those of other enteric pathogens. Yersinia pestis makes AI-2 in a LuxS-dependent manner during late logarithmic growth. To study this further, we performed a comparative analysis with a lsr chromosomal locus that modulates the stationary-phase level of extracellular AI-2. Our array data indicate that AI-2 QS affects the general physiology of the cell and may impact the expression of virulence factors as well. Finally, our findings indicate that AI-2 sensing may be important for resistance to oxidative stress and membrane modeling during quorum sensing. The consequences of such changes and their impact on disease are not understood and should be studied further.
Supplementary Material
ACKNOWLEDGMENTS
We thank R. Perry for Y. pestis strains R88 and R115 and Carrie Oster and Jenny Boonyakuakel for technical assistance.
This work was supported by Office of Naval Research grant N00014-06-1-1176 to F.C.M., A.R.H, and G.J.P.
Footnotes
Published ahead of print 19 August 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00880-13.
REFERENCES
- 1.Achtman M, Zurth K, Morelli G, Torrea G, Guiyoule A, Carniel E. 1999. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. U. S. A. 96:14043–14048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ayyadurai S, Houhamdi L, Lepidi H, Nappez C, Raoult D, Drancourt M. 2008. Long-term persistence of virulent Yersinia pestis in soil. Microbiology 154:2865–2871 [DOI] [PubMed] [Google Scholar]
- 3.Brubaker RR. 1983. The Vwa+ virulence factor of yersiniae: the molecular basis of the attendant nutritional requirement for Ca++. Rev. Infect. Dis. 5(Suppl 4):S748–S758 [DOI] [PubMed] [Google Scholar]
- 4.Bobrov AG, Bearden SW, Fetherston JD, Khweek AA, Parrish KD, Perry RD. 2007. Functional quorum sensing systems affect biofilm formation and protein expression in Yersinia pestis. Adv. Exp. Med. Biol. 603:178–191 [DOI] [PubMed] [Google Scholar]
- 5.Chen X, Schauder S, Potier N, Van Dorsselaer A, Pelczer I, Bassier BL, Hughson FM. 2002. Structural identification of a bacterial quorum-sensing signal containing boron. Nature 415:545–549 [DOI] [PubMed] [Google Scholar]
- 6.Xavier KB, Miller ST, Lu W, Kim JH, Rabinowitz J, Pelczer I, Semmelhack MF, Bassler BL. 2007. Phosphorylation and processing of the quorum-sensing molecule autoindiucer-2 in enteric bacteria. ACS Chem. Biol. 2:128–136 [DOI] [PubMed] [Google Scholar]
- 7.Doll JM, Zeitz PS, Ettestad P, Bucholtz AL, Davis T, Gage K. 1994. Cat-transmitted fatal pneumonic plague in a person who traveled from Colorado to Arizona. Am. J. Trop. Med. Hyg. 51:109–114 [DOI] [PubMed] [Google Scholar]
- 8.Greenberg EP, Hastings JW, Ulitzur S. 1979. Induction of luciferase synthesis in Beneckea harveyi by other marine bacteria. Arch. Microbiol. 120:87–91 [Google Scholar]
- 9.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, Sarov M, Rientjes J, Fu J, Hollak H, Kranz H, Xie W, Stewart AF, Zhang Y. 2006. An improved recombineering approach by adding RecA to lambda Red recombination. Mol. Biotechnol. 32:43–53 [DOI] [PubMed] [Google Scholar]
- 11.Schauder S, Shokat K, Surette MG, Bassler BL. 2001. The LuxS family of bacterial autoinducers: biosynthesis of a novel quorum-sensing signal molecule. Mol. Microbiol. 41:463–476 [DOI] [PubMed] [Google Scholar]
- 12.Miller ST, Xavier KB, Campagna SR, Taga ME, Semmelhack MF, Bassler BL, Hughson FM. 2004. Salmonella typhimurium recognizes a chemically distinct form of the bacterial quorum-sensing signal AI-2. Mol. Cell 15:677–687 [DOI] [PubMed] [Google Scholar]
- 13.LaRock C, Yu J, Horswill AR, Parsek MR, Minion FC. 2013. Transcriptome analysis of acyl-homoserine lactone-based quorum sensing regulation in Yersinia pestis. PLoS One 8:e62337. 10.1371/journal.pone.0062337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carruthers MD, Minion C. 2009. Transcriptome analysis of Escherichia coli O157:H7 EDL933 during heat shock. FEMS Microbiol. Lett. 295:96–102 [DOI] [PubMed] [Google Scholar]
- 15.Madsen ML, Nettleton D, Thacker EL, Edwards R, Minion FC. 2006. Transcriptional profiling of Mycoplasma hyopneumoniae during heat shock using microarrays. Infect. Immun. 74:160–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gallup JM, Ackermann MR. 2006. Addressing fluorogenic real-time qPCR inhibition using the novel custom Excel file system ‘Focusfield2-6GallupqPCRSet-upTool-001′ to attain consistently high fidelity qPCR reactions. Biol. Proced. Online 8:87–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fetherston JD, Schuetze P, Perry RD. 1992. Loss of the pigmentation phenotype in Yersinia pestis is due to the spontaneous deletion of 102 kb of chromosomal DNA which is flanked by a repetitive element. Mol. Microbiol. 6:2693–2704 [DOI] [PubMed] [Google Scholar]
- 18.Taga ME, Miller ST, Bassler BL. 2003. Lsr-mediated transport and processing of AI-2 in Salmonella typhimurium. Mol. Microbiol. 50:1411–1427 [DOI] [PubMed] [Google Scholar]
- 19.Taga ME, Semmelhack JL, Bassler BL. 2001. The LuxS-dependent autoinducer AI-2 controls the expression of an ABC transporter that functions in AI-2 uptake in Salmonella typhimurium. Mol. Microbiol. 42:777–793 [DOI] [PubMed] [Google Scholar]
- 20.Sun J, Daniel R, Wagner-Dèobler I, Zeng AP. 2004. Is autoinducer-2 a universal signal for interspecies communication: a comparative genomic and phylogenetic analysis of the synthesis and signal transduction pathways. BMC Evol. Biol. 4:36. 10.1186/1471-2148-4-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pereira CS, MacAuley JR, Taga ME, Xavier KB, Miller ST. 2008. Sinorhizobium meliloti, a bacterium lacking the autoinducer (AI-2) synthase, responds to AI-2 supplied by other bacteria. Mol. Microbiol. 70:1223–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He YW, Frye JG, Strobaugh TP, Chen CY. 2008. Analysis of AI-2/LuxS-dependent transcription in Campylobacter jejuni strain 81-176. Foodborne Pathog. Dis. 5:399–415 [DOI] [PubMed] [Google Scholar]
- 23.Krin E, Chakroun N, Turlin E, Givaudan A, Gaboriau F, Bonne I, Rousselle JC, Frangeul L, Lacroix C, Hullo MF, Marisa L, Danchin A, Derzelle S. 2006. Pleiotropic role of quorum-sensing autoinducer 2 in Photorhabdus luminescens. Appl. Environ. Microbiol. 72:6439–6451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xavier KB, Bassler BL. 2005. Regulation of uptake and processing of the quorum-sensing autoinducer AI-2 in Escherichia coli. J. Bacteriol. 187:238–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shao H, James D, Lamont RJ, Demuth DR. 2007. Differential interaction of Aggregatibacter (Actinobacillus) actinomycetemcomitans LsrB and RbsB proteins with autoinduer 2. J. Bacteriol. 189:5559–5565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hardie KR, Cooksley C, Green AD, Winzer K. 2003. Autoinducer 2 activity in Escherichia coli culture supernatants can be actively reduced despite maintenance of an active synthase, LuxS. Microbiology 149:715–728 [DOI] [PubMed] [Google Scholar]
- 27.Herzberg M, Kaye IK, Peti W, Wood TK. 2006. YdgG (TqsA) controls biofilm formation in Escherichia coli K-12 through autoinducer 2 transport. J. Bacteriol. 188:587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han Y, Geng J, Qui Y, Guo Z, Zhou D, Bi Y, Du Z, Song Y, Wang X, Tan Y, Zhu Z, Zhai J, Yang R. 2008. Physiological and regulatory characterization of KatA and KatY in Yersinia pestis. DNA Cell Biol. 27:453–462 [DOI] [PubMed] [Google Scholar]
- 29.Vendeville A, Winzer K, Heurlier K, Tang CM, Hardie KR. 2005. Making ‘sense' of metabolism: autoinducer-2, LuxS and pathogenic bacteria. Nat. Rev. Microbiol. 3:383–396 [DOI] [PubMed] [Google Scholar]
- 30.Kavanaugh JS, Gakhar L, Horswill AR. 2011. The structure of LsrB from Yersinia pestis complexed with autoinducer-2. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 67:1501–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xavier KB, Bassler BL. 2003. LuxS quorum sensing: more than just a numbers game. Curr. Opin. Microbiol. 6:191–197 [DOI] [PubMed] [Google Scholar]
- 32.McNab R, Ford SK, El-Sabaeny A, Barbieri B, Cook GS, Lamont RJ. 2003. LuxS-based signaling in Streptococcus gordonii: autoinducer 2 controls carbohydrate metabolism and biofilm formation with Porphyromonas gingivalis. J. Bacteriol. 185:274–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perry RD, Fetherston JD. 1997. Yersinia pestis—etiologic agent of plague. Clin. Microbiol. Rev. 10:35–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perrett CA, Karavolos MH, Humphrey S, Mastroeni P, Martinez-Argudo I, Spenser H, Bulmer DM, Winzer K, McGhie E, Koronakis V, Williams P, Khan CM, Jepson MA. 2009. LuxS-based quorum sensing does not affect the ability of Salmonella enterica serovar Typhimurium to express the SPI-1 type 3 secretion system, induce membrane ruffles, or invade epithelial cells. J. Bacteriol. 191:7253–7259 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.