

Abstract
The Dot/Icm type IV secretion system (T4SS) of Legionella pneumophila is crucial for the pathogen to survive in protozoa and cause human disease. Although more than 275 effector proteins are delivered into the host cell by the T4SS, the function of the majority is unknown. Here we have characterized the Dot/Icm effector LtpD. During infection, LtpD localized to the cytoplasmic face of the membrane of the Legionella-containing vacuole (LCV). In A549 lung epithelial cells, ectopically expressed LtpD localized to large vesicular structures that contained markers of endosomal compartments. Systematic analysis of LtpD fragments identified an internal 17-kDa fragment, LtpD471-626, which was essential for targeting ectopically expressed LtpD to vesicular structures and for the association of translocated LtpD with the LCV. LtpD471-626 bound directly to phosphatidylinositol 3-phosphate [PtdIns(3)P] in vitro and colocalized with the PtdIns(3)P markers FYVE and SetA in cotransfected cells. LtpD was also found to bind the host cell enzyme inositol (myo)-1 (or 4)-monophosphatase 1, an important phosphatase involved in phosphoinositide production. Analysis of the role of LtpD in infection showed that LtpD is involved in bacterial replication in THP-1 macrophages, the larvae of Galleria mellonella, and mouse lungs. Together, these data suggest that LtpD is a novel phosphoinositide-binding L. pneumophila effector that has a role in intracellular bacterial replication.
INTRODUCTION
Legionella pneumophila is found ubiquitously in freshwater reservoirs, where it replicates within free-living protozoa (1). Following the inhalation of contaminated aerosols, L. pneumophila can infect alveolar macrophages, resulting in a severe life-threatening pneumonia named Legionnaires' disease (2). L. pneumophila replicates in professional phagocytes by subverting the phagolysosomal pathway and avoiding killing (3). Instead, it establishes the specialized Legionella-containing vacuole (LCV), which acquires components of the early secretory pathway (4). Gradual fusion of the LCV with vesicles derived from the endoplasmic reticulum (ER) generates an intracellular niche that ultrastructurally resembles the rough ER and supports bacterial replication (5).
L. pneumophila encodes a Dot (defective in organelle trafficking)/Icm (intracellular multiplication) type IV secretion system (T4SS) which is indispensable for the establishment of the LCV (6, 7). The T4SS is a multiprotein complex able to translocate at least 300 effector proteins directly into host cells (8). T4SS effectors have been shown to modulate many aspects of cellular signaling, including apoptosis, host cell stress responses, immune signaling, and endosomal and secretory vesicular traffic (9). For example, the effector SidK inhibits the vacuolar H+-ATPase to prevent acidification of the LCV (10), while several effectors, including RalF, SidM, SidD, AnkX, Lem3, and LidA, target small GTPases involved in vesicle recruitment and fusion (11). In addition, it is becoming increasingly apparent that L. pneumophila effectors exploit host cell lipids, in particular phosphatidylinositol (PtdIns)-phosphates (PtdInsPs), during the formation of the LCV. PtdIns consists of a phosphatidic acid membrane anchor that is connected via a phosphodiester bond to myo-d-inositol. Positions 3, 4, and 5 of the inositol ring can be phosphorylated, resulting in PtdInsPs with different biophysical properties and protein interaction profiles. This diversity makes them versatile signaling molecules and, by preferential enrichment of a PtdInsP species, gives identity to organelle membranes, e.g., phosphatidylinositol 3-phosphate [PtdIns(3)P] is localized to early endosomes, whereas phosphatidylinositol 4-phosphate [PtdIns(4)P] is found on the Golgi apparatus (12). A limiting step in the formation of PtdInsPs is the production of inositol via dephosphorylation of inositol monophosphate by the conserved enzyme inositol (myo)-1 (or 4)-monophosphatase 1 (IMPA1) (13, 14). Inhibition of IMPA1 has pleiotropic effects on cellular function, including modulation of second messenger signaling (14), autophagy (15), and cell death (16).
L. pneumophila actively regulates PtdInsP levels on the LCV by recruiting host cell PtdInsP-converting enzymes or by direct conversion of PtdIns through the enzymatic activity of bacterial effectors. For example, SidF is a phosphatidylinositol polyphosphate 3-phosphatase that removes the D3 phosphate from phosphatidylinositol 3,4-phosphate [PtdIns(3,4)P2] and PtdIns(3,4,5)P3 (17). L. pneumophila also recruits the inositol polyphosphate-5-phosphatase OCRL1 to the LCV, which dephosphorylates PtdIns(4,5)P2, yielding PtdIns(4)P (18). The accumulation of PtdIns(4)P on the LCV serves as a membrane anchor for SidC and SidM (19, 20), whereas LidA, SetA, and LpnE are localized to the LCV membrane and bind PtdIns(3)P in vitro (18, 21, 22), suggesting that the LCV membrane also contains PtdIns(3)P. The aim of the present study was to investigate the effector LtpD (23), which we found to bind both PtdIns(3)P and IMPA1.
MATERIALS AND METHODS
Bacterial and yeast strains and growth conditions.
Bacterial strains are listed in Table 1. Legionella pneumophila strains were grown in ACES [N-(2-acetamido)-2-aminoethanesulfonic acid]-buffered yeast extract broth (AYE) or on charcoal-buffered yeast extract agar plates (CYE) as previously described (24). Escherichia coli strains were cultured in Luria-Bertani broth or on agar. Antibiotics were added to media at the following final concentrations for L. pneumophila (or E. coli): kanamycin, 25 μg/ml (100 μg/ml); chloramphenicol, 6 μg/ml (30 μg/ml); and ampicillin, 100 μg/ml (100 μg/ml). Saccharomyces cerevisiae strains AH109 and Gold were cultured at 30°C according to the manufacturer's instructions (Clontech laboratories, Inc.).
Table 1.
Bacterial strains used in this study
| Strain | Serogroup or genotypea | Source or reference |
|---|---|---|
| L. pneumophila | ||
| 130b (ATCC BAA-74) | O1; clinical isolate | 29 |
| 130b ΔdotA mutant | dotA gene disrupted with a Kanr cassette | 23 |
| 130b ΔltpD (ICC1064) mutant | lptD gene replaced with a Kanr cassette | This study |
| E. coli | ||
| BL21(DE3) | F− ompT hsdSB (rB− mB−) gal dcm | Novagen |
| Top10 | F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 nupG recA1 araD139 Δ(ara-leu)7697 rpsL (Strr) endA1 λ− | Invitrogen |
Strr, streptomycin resistance; Kanr, kanamycin resistance.
Cell culture.
The alveolar epithelial cell line A549 and the cervical epithelial HeLa cell line were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and GlutaMAX (Invitrogen). The human monocytic cell line THP-1 was maintained in RPMI 1640 medium, supplemented with 10% FBS and GlutaMAX (Invitrogen). All cell lines were obtained from the American Tissue Culture Collection and subjected to minimal passaging. THP-1 cells were seeded into 24-well tissue culture plates at a density of 5 × 105 cells/well and differentiated into macrophage-like adherent cells with 10 ng of phorbol 12-myristate 13-acetate/ml for 48 to 72 h. Transfection of HeLa or A549 cells with mammalian expression plasmids was performed using GeneJuice (Novagen) according to the manufacturer's instructions.
Plasmid and strain construction.
Plasmids were constructed according to standard molecular biology techniques using the primers and restriction enzymes described in Table S1 in the supplemental material. An ltpD deletion mutant of L. pneumophila was constructed by replacing the ltpD gene with the kanamycin resistance cassette from pSB315 (25). Deletion mutants were selected on CYE agar with kanamycin and confirmed by PCR and sequencing. For the construction of LtpDΔ472-626 and LtpDΔ472-556, the 5′ and 3′ fragments were amplified separately, ligated together, and inserted into the plasmids. Plasmids were transformed into L. pneumophila by electroporation as described previously (26).
Infection of eukaryotic cells.
Infections with L. pneumophila were done at multiplicity of infection (MOI) of 5 for THP-1 cells or 100 for A549 cells as described previously (23). To synchronize infection, plates were centrifuged for 10 min at 500 × g. To analyze the role of IMPA1 in infection, the cells were treated with the IMPA1 inhibitors L-690,330 (10 mM) or the more cell-permeable analogue L-690,488 (10 to 100 μM) (Tocris Biosciences).
Immunofluorescence microscopy.
Cells on coverslips were washed, fixed with 4% paraformaldehyde for 20 min, and quenched with 50 mM ammonium chloride for 10 min. The cells were permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (PBS) and then blocked with blocking buffer (2% [wt/vol] bovine serum albumin [BSA] in PBS). Samples were then incubated for 1 h with the primary antibody in blocking buffer, washed three times in PBS, and incubated for a further 1 h with secondary antibodies before mounting using ProLong Gold antifade reagent (Invitrogen). The following primary antibodies were used: mouse anti-hemagglutinin (anti-HA) conjugated to TRITC (tetramethyl rhodamine isothiocyanate; catalog no. H9037; Sigma), mouse anti-Myc conjugated to fluorescein isothiocyanate (F2047-100; Sigma), mouse anti-Myc (05-724; Millipore), rabbit anti-Legionella lipopolysaccharide (LPS; PA1-7227; Affinity Bioreagents), mouse anti-EEA1 (catalog no. 610457; BD Transduction Laboratories), mouse anti-LAMP2 (catalog no. 555803; BD Pharmingen), mouse anti-FLAG (F1804; Sigma). The secondary antibodies used were donkey anti-rabbit IgG-Alexa Fluor 488 (Jackson Immunoresearch), donkey anti-rabbit IgG-Rhodamine Red-X (RRX; Jackson Immunoresearch), rabbit anti-mouse IgG-RRX (Jackson Immunoresearch), and rabbit anti-mouse IgG-Alexa Fluor 488 (Jackson Immunoresearch). To visualize DNA, 5 μg of DAPI (4′,6′-diamidino-2-phenylindole)/ml was used. Samples were analyzed using either an Axio M1 Imager or an Axio Z1 Imager microscope, and images were processed using AxioVision software (Carl Zeiss) and Photoshop (Adobe).
Protein purification.
Production of GST fusion proteins in E. coli BL21(DE3) was induced at a cell optical density at 600 nm of 0.6 by adding 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 5 to 6 h at 37°C. Bacterial lysates were prepared using an EmulsiFlex B15 cell disruptor (Avestin). Fusion proteins were purified from the soluble fraction on glutathione-Sepharose beads (Amersham Biosciences) according to the manufacturer's instructions and eluted using 10 mM reduced glutathione (in PBS). His-IMPA1 was purified as previously described (27). The purity of the proteins was analyzed by SDS-PAGE, and all proteins were stored in 50 mM Tris-HCl (pH 8).
Protein-lipid overlay assay.
To test the direct binding of glutathione S-transferase (GST)–LtpD472-626 fusion protein to lipids, experiments were carried out with commercially available membrane lipid strips and PIP arrays (Echelon Biosciences), using GST-tagged PH domains of PLC-δ1 (PIP2 Grip; Echelon Biosciences) and GST alone as controls. After blocking, the strips were incubated with purified GST-LtpD472-626 or controls overnight in 2% fatty-acid-free BSA (Millipore) in Tris-buffered saline with 0.1% Tween 20, washed, and incubated with the primary anti-GST antibody (Abcam) and the secondary peroxidase-conjugated goat anti-mouse antibody (Jackson Immunoresearch). Membranes were then revealed using EZ-ECL (Geneflow) and a Fuji Las3000 imager.
Intracellular replication assays.
THP-1 cells were infected with L. pneumophila wild type (WT) or the ΔdotA, ΔltpD, or ΔltpD(p4HA-LtpD) strain. A549 cells were seeded 24 h before infection in 24-well plates at 5 × 105 cells/well and were infected at an MOI of 40. The infection was allowed to proceed for 1 h, media were removed, and the cells were incubated with gentamicin (100 μg/ml) for 1 h. Cells were then washed twice and incubated in RPMI 1640 or DMEM supplemented with 6 μg of chloramphenicol/ml and 1 mM IPTG. At 0, 5, 18, 24, 48, and 72 h postinfection (p.i.), the cells were lysed with digitonin (10 μg/ml), and the intra- and extracellular fractions were pooled and plated onto CYE with 6 μg of chloramphenicol/ml to determine the total CFU. The results are the mean ± the standard deviation of at least three separate experiments.
Y2H assay.
All yeast work was performed according to the Clontech protocol handbook. A yeast two-hybrid (Y2H) screen was performed using LtpD as a bait in the S. cerevisiae Gold strain with the Matchmaker HeLa cDNA library (Clontech Laboratories, Inc.), and clones containing interaction partners were selected on high-stringency quadruple-dropout media (QDO) lacking leucine, tryptophan, histidine, and adenine in the presence of Aureobasidin A and X-α-Gal (Clontech Laboratories, Inc.). Single clones from QDO screening plates were restreaked onto fresh QDO for a second round of selection. After repeated QDO selection, the cDNA fragments contained in the prey plasmids were identified by sequencing. For direct Y2H assays, the impa1 gene was amplified from IMPA1 cDNA (Open Biosystems), inserted into pGADT7, and directly transformed into the yeast strain AH109 with pGBT9-LtpD or pGBT9 empty vector as a negative control. Successful transformation with bait and prey plasmids was selected by plating on double-dropout media (DDO) lacking leucine and tryptophan. Bait-prey interactions were assessed by streaking the transformed clones from DDO onto QDO selection medium.
Coimmunoprecipitation.
HeLa cells were transfected using GeneJuice (Novagen) according to the manufacturer's instructions, incubated for 48 h, lysed in radioimmunoprecipitation assay buffer, and then centrifuged at 13,000 rpm for 1 h at 4°C. Cleared lysate was incubated with agarose beads directly conjugated to anti-Myc antibody (Sigma) for 1 h at 4°C on a rotor, washed once with 0.1% Tween-PBS (PBST), and then washed three times with PBS. Proteins bound to the beads were eluted, separated by SDS-PAGE, transferred to polyvinylidene difluoride membrane (GE Healthcare), blocked for 1 h in 2% fat-free milk-PBST, and incubated overnight with anti-Myc (1:2,500) or anti-FLAG (1:1,000) antibodies. Membranes were washed three times with PBST and incubated with peroxidase-conjugated goat anti-mouse secondary antibody (Jackson Immunoresearch) for 1 h. Blots were revealed using EZ-ECL (Geneflow) and a Fuji Las3000 imager.
IMPA1 activity assay.
IMPA1 activity in vitro was measured using a malachite green assay kit (Cambridge Biosciences). The assay was performed at 37°C for 30 min in assay buffer (50 mM Tris-HCl [pH 8.0]) with 0.7 mM myo-inositol monophosphate, 1 mM EGTA, 2 mM MgCl2, and 0.5 μg of purified His-IMPA1/ml with or without 0.5 μg of purified GST-LtpD/ml and a 1 mM concentration of IMPA1 inhibitor L-690,330 (Tocris Bioscience). The reaction was quenched by the addition of 5 μl of MG acidic solution (Cambridge Biosciences) and incubated for 10 min at room temperature, and then 25 μl of MG blue solution added to visualize released phosphate, followed by incubation for a further 20 min at room temperature. Quantification was carried out by measuring the absorbance at 690 nm and comparing to a phosphate standard curve. The assay was performed in triplicate, and results are means of three separate experiments ± the standard deviations.
Infection of G. mellonella.
Infection of larvae and bacterial growth assays in G. mellonella were performed as described previously (28). G. mellonella larvae were infected with 107 L. pneumophila in a 1:1 WT/ΔltpD mutant or WT/ΔltpD(p4HA-LtpD) mutant ratio. At 24 h p.i., hemolymph was extracted as described above, and serial dilutions were plated onto CYE agar with spectinomycin and with or without kanamycin. Colonies were counted to determine the numbers of viable bacteria and the ratios of WT to mutant bacteria colonizing the larvae.
Infection of A/J mice.
Mouse procedures were approved by the University of Melbourne Animals Ethics Committee. The comparative virulence of L. pneumophila 130b and the lptD mutant and complemented strain were examined as described previously (29).
RESULTS
LtpD localizes to the cytoplasmic side of the LCV membrane during infection.
In order to determine the localization of LtpD (Lpw03701) during infection, A549 lung epithelial cells were infected with WT and ΔdotA mutant L. pneumophila 130b strains expressing LtpD fused to four N-terminal HA tags. At 5 h p.i., the cells were fixed and stained using anti-HA and anti-Legionella antibodies. LtpD was exclusively observed surrounding the WT, but not ΔdotA bacteria (Fig. 1A). The same localization was observed during infection of the human macrophage cell line THP-1 (data not shown).
Fig 1.
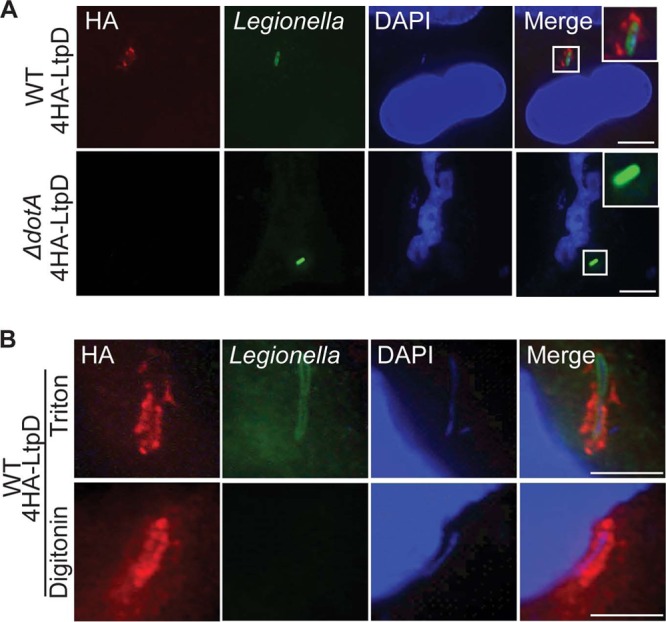
4HA-LtpD localizes to the cytoplasmic surfaces of the LCVs. A549 cells were infected for 5 h with the L. pneumophila 130b WT or ΔdotA mutant expressing 4HA-LtpD, fixed, permeabilized, and immunostained with anti-L. pneumophila (green) and anti-HA (red) antibodies. DNA was visualized with DAPI (blue). (A) After permeabilization with Triton X-100, 4HA-LtpD staining was visible around the WT but not around the ΔdotA bacteria in a manner that suggests the association of 4HA-LtpD with the LCV. (B) After selective permeabilization of membranes with digitonin, 4HA-LtpD staining was detected surrounding the intracellular bacteria, whereas these bacteria were not accessible for anti-L. pneumophila immunostaining. This indicates that 4HA-LtpD is localized on the cytoplasmic surfaces of the LCVs. Images are representative of at least three independent experiments. Scale bars, 10 μm.
The LtpD staining is reminiscent of proteins localized on the membrane of the LCV. In order to confirm this and to determine whether LtpD (which is not predicted to be a transmembrane protein) was localized to the outer or inner leaflet of the LCV, infected cells were fixed and subjected to permeabilization with either digitonin, which does not disrupt the endoplasmic reticulum (ER), and thus presumably the ER-like LCV, or Triton X-100, which permeabilizes both plasma and ER membranes. As expected, antibodies raised against Legionella LPS did not stain bacteria in LCVs, which were visualized with the DNA stain DAPI, when the cells were permeabilized with digitonin (Fig. 1B). In contrast, LtpD staining surrounding the bacteria was visible in cells permeabilized by both methods, suggesting that 4HA-LtpD is localized on the outer leaflet of the LCV.
LtpD associates with vesicles.
Bioinformatic sequence analysis revealed that LtpD might have evolved by fusion of two L. pneumophila proteins; the N terminus of LtpD, LtpD1-469, shares 33% sequence identity with LtpK (Lpw27671), a conserved, uncharacterized effector among L. pneumophila strains, whereas the C terminus, LtpD472-679, shares 64% sequence identity with the C terminus of Lpp0356, an uncharacterized ankyrin repeat-containing protein found only in the L. pneumophila strain Paris (30).
In order to examine the subcellular localization of Myc-tagged LtpD and to determine the role of the two domains (Fig. 2A), LtpD or both domains of LtpD individually were ectopically expressed in HeLa cells, which yielded higher transfection efficiencies compared to A549 cells. Myc-LtpD mostly localized to large perinuclear vesicular structures and to some smaller vesicles throughout the cell (Fig. 2B). Although Myc-LtpD1-469 displayed a diffuse cytosolic localization, Myc-LtpD472-679 showed a vesicular localization. Notably, the Myc-LtpD472-679 vesicular structures seemed distinct from the full-length protein with smaller, more diffusely distributed vesicles, suggesting that the N-terminal region may contribute to formation of the large vesicular structures.
Fig 2.

LtpD consists of two domains and associates via a 17-kDa internal region, LtpD472-626, with vesicles. LtpD or selected fragments of LtpD were ectopically expressed in HeLa cells. Cells were then stained with anti-Myc antibody (green) and DAPI DNA stain (blue). (A) Schematic overview of the LtpD fragments investigated in the present study. (B) Myc-LtpD localizes to large, vesicular structures around the nucleus. Analysis of fragments corresponding to the N- and C-terminal domains of LtpD shows that LtpD472-679 is sufficient to mediate association with vesicles. (C) Systematic analysis of fragments of the C-terminal domain of LtpD revealed that an internal domain, LtpD472-626, is required and sufficient for localization of LtpD to vesicles. Scale bars, 10 μm.
In order to map the vesicular-binding domain further, additional truncations of LtpD (Fig. 2A) were made according to the predicted secondary structure. Visualization of these LtpD fragments (Fig. 2C) resulted in the identification of an internal 17-kDa fragment, LtpD472-626, which showed vesicular localization following transfection. The smaller fragments Myc-LtpD472-556 and Myc-LtpD556-679, which contained only parts of the 17-kDa region, showed diffuse cytosolic distribution. This defines Myc-LtpD472-626 as the minimal fragment of LtpD able to associate with vesicles. To validate this conclusion, an internal deletion of this domain in LtpD (Myc-LtpDΔ472-626) was created. Myc-LtpDΔ472-626 did not localize to membranes upon transfection (Fig. 2C), demonstrating that LtpD472-626 is both necessary and sufficient for membrane localization during ectopic expression.
LtpD472-626 is sufficient for LtpD recruitment to the LCV during infection.
We next analyzed whether LtpD472-626 is also important in the localization of LtpD to the LCV. To achieve this, A549 cells were infected for 5 or 24 h with WT L. pneumophila expressing 4HA-LtpD, 4HA-LtpD472-679 or 4HA-LtpDΔ472-626. LtpD and LtpD472-679 localized to the LCV at both time points (see Fig. S1 in the supplemental material and data not shown). LtpDΔ472-626 could not be detected at 5 h p.i. (data not shown), which could be due to less efficient translocation or an insufficient local concentration for detection. However, at 24 h p.i. when cells contained large numbers of bacteria, a diffuse cytosolic and nuclear staining of LtpDΔ472-626 was observed (see Fig. S1 in the supplemental material). This demonstrates that LtpD472-626 is required for the association of LtpD with the LCV.
In order to determine whether LtpD472-626 was sufficient for the recruitment of LtpD to the LCV, A549 cells were transfected with selected Myc-LtpD truncation or deletion derivatives; 24 h later, the cells were infected with WT or ΔdotA bacteria for 5 h. Cells transfected with full-length Myc-LtpD and infected with WT, but not the ΔdotA mutant, showed recruitment of Myc-LtpD to the bacteria. Similarly, recruitment to WT L. pneumophila was also observed when cells were transfected with LtpD472-679 and LtpD472-626 but not in cells expressing LtpDΔ472-626 (see Fig. S2 in the supplemental material). Taken together, these data suggest that LtpD472-626 is necessary and sufficient for the localization of LtpD to the LCV containing WT L. pneumophila. However, selective recruitment to L. pneumophila WT, but not the ΔdotA mutant, suggests that Dot/Icm T4SS-dependent LCV remodeling is required for LtpD localization.
GST-LtpD472-626 binds phosphatidylinositol 3-phosphate [PtdIns(3)P].
To determine whether LtpD associates with the LCV by binding a membrane lipid anchor, we purified LtpD472-626 fused to GST and performed a protein-lipid overlay assay using a PIP array, including serial dilutions of all seven phosphoinositides spotted on a membrane. Under these conditions, GST-LtpD472-626 was primarily seen bound to PtdIns(3)P and, with lower affinity, PtdIns(4)P (Fig. 3A). The positive control PLC-δ1 PH domain fused to GST bound strongly to PtdIns(4,5)P2, whereas GST alone did not bind to any lipid.
Fig 3.

GST-LtpD472-626 binds to PtdIns(3)P in vitro and localizes to PtdIns(3)P-containing membranes. (A) Protein-lipid overlay. Purified GST-LtpD472-626, GST, or a GST fusion of the PH domain of PLC-δ1 was incubated with membranes on which serial dilutions of various PtdIns species were spotted, as indicated. Protein binding to lipids was detected using an anti-GST antibody. LtpD472-626-GST bound preferentially to PtdIns(3)P and, to a smaller extent, to PtdIns(4)P. The PH domain of PLC-δ1 was used as a positive control and bound strongly to PtdIns(3,4)P2. The results are representative of three separate experiments. (B) Upon cotransfection of HeLa cells, Myc-LtpD colocalized with 2×FYVE-GFP and 4HA-SetAPIP3, reporters for PtdIns(3)P, but not HA-SidCPIP4, a marker for PtdIns(4)P. Scale bars, 10 μm. (C) Upon infection of A549 cells with either L. pneumophila 4HA-4LtpD or 4HA-SetAPIP3, 4HA-SetAPIP3 localizes to the LCV, indicating the presence of PtdIns(3)P in the LCV membrane. Scale bars, 5 μm.
To analyze if LtpD472–626 and LtpD bind to PtdIns(3)P on cellular membranes, HeLa cells were cotransfected with full-length LtpD or LtpD472-679 and the PtdIns(3)P-reporter proteins 2×FYVE-green fluorescent protein (GFP) and HA-SetAPIP3. FYVE is a eukaryotic PtdIns(3)P-binding domain and a well-established marker of PtdIns(3)P in cellular membranes (31). SetAPIP3 corresponds to the PtdIns(3)P-binding domain of the L. pneumophila effector SetA (22). We used the PtdIns(4)P-binding domain of the effector SidC, SidCPIP4, as an additional control (19, 20). Myc-LtpD or Myc-LtpD472-626 (data not shown) colocalized with FYVE and SetAPIP3, but not SidCPIP4, in vesicular structures (Fig. 3B), supporting the conclusion that LtpD binds to PtdIns(3)P in cellular membranes.
Although the presence of PtdIns(4)P on the LCV membrane is well established (19, 20), it is less clear whether PtdIns(3)P is also present. In order to address this, we expressed the SetAPIP3 reporter fused to 4HA tags in L. pneumophila. 4HA-SetAPIP3 was translocated into A549 cells and found surrounding the bacteria similarly to 4HA-LtpD (Fig. 3C), suggesting that that PtdIns(3)P is also present in the LCV membrane.
Ectopically expressed LtpD colocalizes with markers of the endocytic pathway.
Since LtpD is associated with vesicular structures, we analyzed whether it colocalized with markers of various cellular compartments by cotransfection (Fig. 4A) or by antibody straining (Fig. 4B) of HeLa cells. Myc-LtpD did not (or only to very small extent) coincidentally colocalize with or redistribute markers of the ER (calnexin), Golgi (GM130), ER/Golgi (Rab1-GFP), peroxisomes (catalase), or mitochondria (MitoTracker) (Fig. 4A and see Fig. S3 in the supplemental material). In contrast, Myc-LtpD redistributed and colocalized, in large perinuclear vesicular structures, with markers (arrows) of the endocytic pathway including early endosomes (Rab5-GFP and EEA1), early/recycling endosomes (Rab4-GFP), late endosomes (Rab7-GFP), and lysosomes (LAMP2). Despite the strong phenotype upon ectopic expression, we observed no differential recruitment or localization of Rab5, Rab7, or LAMP2 to the LCV of L. pneumophila overexpressing LtpD (data not shown). These results suggest that LtpD can modulate endocytic vesicle traffic; however, this effect may be masked by the action of other effectors during infection.
Fig 4.

Myc-LtpD colocalizes with and redistributes marker proteins of the endocytic pathway. HeLa cells were transfected with Myc-LtpD and either cotransfected (A) or specifically immunostained (B) for markers of the endocytic pathway. (A) Myc-LtpD colocalized with (arrows) and redistributed the endocytic markers Rab5-GFP, Rab4-GFP, and Rab7-GFP but not the ER/Golgi marker Rab1-GFP. (B) Myc-LtpD also colocalized with EEA1 and LAMP2. Images are representative of at least three separate experiments. Scale bars, 10 μm.
LtpD binds the host protein inositol (myo)-1 (or 4)-monophosphatase-1.
To gain insight into the function of LtpD we set out to identify its host cell binding partners. A yeast two-hybrid (Y2H) screen using LtpD fused to the GAL4 DNA-binding domain as bait and a HeLa cDNA library of prey proteins fused to the GAL4 activation domain was performed. After repeated screening of the isolate clone on a selective QDO medium, DNA sequencing of the contained prey plasmids revealed 8 different cDNA inserts (see Table S2 in the supplemental material), including a fragment (residues 218 to 278) of the enzyme IMPA1 as a potential binding partner. Because of its importance in eukaryotic inositol signaling and because IMPA1 has previously been found in a proteomic screen of the LCV (32), we focused on this hit for further investigation. Direct Y2H experiments confirmed the interaction between LtpD and full-length IMPA1 (Fig. 5A) since only yeast containing both proteins could grow on QDO selective media. This revealed that LtpD472-679 was sufficient to reconstitute growth on selective media, showing that the interaction with IMPA1 occurs through the C-terminal domain of LtpD (Fig. 5B).
Fig 5.

LtpD interacts with the host cell enzyme inositol (myo)-1 (or 4)-monophosphatase 1 (IMPA1). (A) AH109 yeast cotransformed with pGBT9_LtpD and pGADT7_IMPA1 grew on both double-dropout (DDO) and quadruple-dropout (QDO) selective media, whereas cotransformation of either pGBT9_LtpD or pGADT7_IMPA1 with the empty pGADT7 or pGBT9 vector, respectively, was unable to allow yeast growth on QDO medium. This demonstrates interaction of LtpD with IMPA1. (B) Cotransformation of AH109 with pGADT7_IMPA1 and pGBT9_LtpD1-471 or pGBT9_LtpD472-679 showed that LtpD472-679 is necessary and sufficient for the interaction between LtpD and IMPA1. (C and D) Coimmunoprecipitation experiments indicate LtpD/IMPA1 interaction through LtpD472-679. HeLa cells were cotransfected with Myc-LtpD (full length), Myc-LtpD1-469, or Myc-LtpD472-679 and IMPA1-FLAG. IMPA1-FLAG could be coimmunoprecipitated with Myc-LtpD and Myc-LtpD472-679 but not with Myc-LtpD1-469.
To confirm the interaction of IMPA1 with LtpD, we tested whether Myc-LtpD and FLAG-IMPA1 coimmunoprecipitated from cotransfected HeLa cells. FLAG-IMPA1 was specifically coimmunoprecipitated with Myc-LtpD (Fig. 5C). Furthermore, confirming the results from the Y2H, Myc-LtpD471-679 was sufficient to coimmunoprecipitate FLAG-IMPA1 (Fig. 5D). Analysis of the localization of IMPA1 in infected A549 cells by immunostaining showed strong IMPA1 staining throughout the cytoplasm but no enrichment of IMPA1 on the LCVs of WT and ΔltpD strains or on L. pneumophila overexpressing LtpD (data not shown), suggesting the IMPA1 is recruited transiently or at a low level to the LCV, an effect that could be masked by the strong cytoplasmic staining.
IMPA1 is an enzyme which dephosphorylates myo-inositol monophosphate to generate inositol. We analyzed the impact of LtpD binding on its phosphatase activity by performing a malachite green phosphate release assay with recombinant proteins (see Fig. S4 in the supplemental material and the supplemental methods); however, the addition of GST-LtpD did not affect the activity of IMPA1, indicating that LtpD does not modulate the activity under these conditions or that additional factors might be required. The use of pharmacological inhibitors (L-690,488) or small interfering RNA (siRNA) to abolish IMPA1 activity resulted in significant death of macrophage-like THP-1 cells (data not shown), which made it impossible to investigate the specific role of IMPA1 for L. pneumophila intracellular replication.
Taken together, our data suggest that LtpD binds IMPA1. This interaction does not seem to directly modulate IMPA1 activity but may facilitate the interaction of IMPA1 with other effectors or host cell proteins.
LtpD enhances the replication of L. pneumophila in THP-1 cells.
In order to determine the role of LtpD during infection, we constructed a gene deletion mutant (ΔltpD) in the L. pneumophila 130b strain and compared the intracellular growth of the WT and mutant strains after infection of THP-1 cells. No difference was recorded between the WT and ΔltpD strains in terms of bacterial internalization (data not shown) or replication during the first 18 h p.i. However, after 24 h of infection, the ΔltpD strain had a subtle but significant growth defect compared to the WT strain (Fig. 6, P = 0.0021 at 48 h p.i., unpaired Student t test). This growth defect could be complemented by the addition of 4HA-LtpD on a plasmid, confirming that the defect was due to the absence of LtpD.
Fig 6.
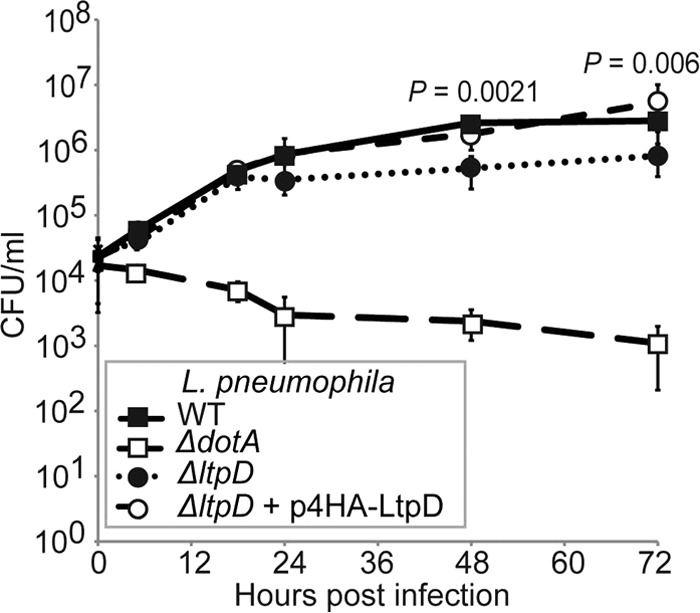
LtpD enhances the replication of L. pneumophila 130b in THP-1 macrophages. (A) THP-1 macrophages were infected with the L. pneumophila WT, ΔdotA strain, ΔltpD strain, or ΔltpD strain expressing 4HA-LtpD (ΔltpD/p4HA-LtpD), and bacterial CFU at set time points were quantified by plating. The ΔdotA strain did not replicate. From 24 h p.i., the ΔltpD strain showed a small but significant growth defect compared to the WT strain. This growth defect could be complemented by the addition of p4HA-LtpD. The results are means of at least three separate experiments ± the standard deviations. Significance was calculated by using an unpaired Student t test.
LtpD enhances bacterial survival in in vivo infection models.
We next investigated the role of LtpD in two in vivo infection models. We recently showed that L. pneumophila virulence in the Galleria mellonella model is Dot/Icm T4SS-dependent (28, 33, 34). Ten G. mellonella larvae were infected with 107 CFU of bacteria, and larval survival was monitored over 72 h p.i. There was no significant difference in larval mortality between the WT and ΔltpD strains over the course of the infection (Fig. 7A).
Fig 7.

LtpD has a role in in vivo infection models. Ten G. mellonella larvae were infected with 107 L. pneumophila 130b (the WT, ΔdotA strain, ΔltpD strain, or ΔltpD strain expressing 4HA-LtpD). (A) The ΔltpD strain did not affect larval mortality under these conditions. (B) The hemolymph of infected larvae was extracted at 0, 5, 18, and 24 h p.i., and the CFU were quantified. The ΔltpD strain replicated at the same level as the WT over the first 18 h but displayed at 24 h p.i. a significant drop in CFU (P = 0.004, unpaired Student t test), which could be complemented by the addition of 4HA-LtpD on a plasmid. The results in panels A and B are means of at least three separate experiments ± the standard deviations. (C) Larvae were injected with the WT and either the ΔltpD or ΔltpD (p4HA-LtpD) strain in a 1:1 ratio, and the ratio of WT to each mutant was calculated at 24 h p.i. The WT significantly (P = 0.01, unpaired Student t test) outcompeted the ΔltpD strain. Each point represents a separate experiment (from the pooled hemolymph of three larvae), while the bars show the means ± the standard deviations. (D) Pulmonary infections of A/J mice with L. pneumophila 130b WT and either the mutant ΔltpD strain or the ΔltpD(p4HA-LtpD) strain in a 1:1 ratio were introduced into the lungs of A/J mice by intratracheal inoculation. At 72 h p.i., the ratio of the WT to the ΔltpD strain in infected lungs was determined. The ΔltpD strain was outcompeted (CI = 0.372) by the WT in the mouse lung, and this effect could be complemented. The data are representative of actual values obtained per mouse, and a solid bar represents the mean value (P = 0.017, unpaired Student t test).
We then quantified bacterial replication over time by plating the hemolymph extracted at 0, 5, 18, and 24 h p.i. Although the ΔdotA strain was cleared, no difference in bacterial CFU was detected between WT and ΔltpD strains until 18 h p.i. (Fig. 7B). Importantly, a small but significant (P < 0.005) difference between the WT and ΔltpD strains was observed at 24 h p.i, which could be complemented by expressing 4HA-LtpD from a plasmid.
We next performed mixed infections aimed at establishing the competitive index (CI) of the mutant. After infecting larvae with a 1:1 ratio of WT to ΔltpD or ΔltpD(p4HA-LtpD) bacteria, hemolymph was extracted at 24 h p.i., and the CFU of both strains were determined by plating. At 24 h p.i., WT L. pneumophila significantly (P = 0.01) outcompeted the ΔltpD strain, whereas complementation with the plasmid p4HA-LtpD restored the fitness of the ΔltpD strain (Fig. 7C).
Finally, we examined the competitive fitness of the ΔltpD mutant in a mixed intranasal infection of susceptible A/J mice. Determination of bacterial CFU after 72 h showed a mean CI of 0.372 for the ΔltpD mutant versus the WT and a mean CI of 1.219 for the ΔltpD(p4HA-LtpD) strain versus a WT strain infection (Fig. 7D), mirroring the results found in G. mellonella and indicating that LtpD is required for optimal survival of L. pneumophila in the mouse lung.
DISCUSSION
In this study we report that LtpD localizes to the cytoplasmic face of the LCV membrane, making it accessible for interaction with cytosolic proteins or incoming vesicles. Localization studies of ectopically expressed LtpD enabled us to map the region required for vesicle association to a 17-kDa internal region, LtpD472-626. We further demonstrated the requirement of this region for localization on the LCV during infection; LtpD lacking the 17-kDa region was not retained on the LCV, but found diffuse in the cytosol. Protein-lipid overlay assays showed that LtpD472-626 bound PtdIns(3)P and, with lower affinity, PtdIns(4)P.
Exploitation of host cell lipids by pathogens is emerging as a process critical for microbial infection (35). LtpD is the fourth L. pneumophila effector shown to bind preferentially to PtdIns(3)P; SetA and LpnE show high binding specificity for this phosphoinositide, whereas LidA also binds PtdIns(4)P with slightly lower affinity (18, 21, 22). SidM, which interacts preferentially with PtdIns(4)P and also binds to PtdIns(3)P in vitro (20). Since LtpD472-679, which contains the PtdIns(3)P-binding domain, has 64% identity to the C terminus of the L. pneumophila Paris protein Lpp0356, this protein might also bind to PtdIns(3)P. It is noteworthy that although LtpD is not conserved across L. pneumophila isolates, the strain-to-strain variation of the T4SS effector pool is high. Alternatively, SetA, LpnE, and LidA are conserved among sequenced isolates (36), indicating that exploitation of PtdIns(3)P provides an evolutionary advantage.
Since the PtdInsP-binding specificities of LtpD472-626 in vitro might deviate from the full-length protein inside the cell, we performed cotransfections with PtdInsP reporter proteins. LtpD colocalized with the PtdIns(3)P reporters 2×FYVE-GFP and SetAPIP3, but not the PtdIns(4)-binding domain of the effector SidC. This supports the conclusion that LtpD specifically binds and is targeted to PtdIns(3)P-containing membranes.
In eukaryotic cells, PtdIns(3)P is found exclusively on early endosomes (37). It has been previously shown that early endosomal proteins are excluded from the LCV (38), whereas L. pneumophila specifically upregulates the level of PtdIns(4)P (17, 18). We confirm here that the translocated SetAPIP3 reporter localizes to the LCV similarly to LtpD, suggesting that PtdIns(3)P is available as membrane anchor on the LCV.
Host or L. pneumophila PtdInsP-metabolizing enzymes that directly influence the PtdIns(3)P level on the LCV have not yet been identified. In preliminary experiments addressing this question, we observed that localization of SetAPIP3 and LtpD appears to be insensitive to treatment with the phosphoinositide 3-kinase inhibitor wortmannin (unpublished results). Our experiments demonstrated that ectopically expressed full-length LtpD could be selectively recruited to WT L. pneumophila. This indicates that other Dot/Icm T4SS effectors are required to remodel and possibly regulate PtdIns(3)P levels in the LCV membrane. Recently, the conserved effectors SidP, which is a phosphatidylinositol 3-phosphatase hydrolyzing PtdIns(3)P and PtdIns(3,5)P2 in vitro, and SidF, a phosphatidylinositol polyphosphate 3-phosphatase with specificity for PtdIns(3,4)P2 and PtdIns(3,4,5)P3,were described (17, 39). It is tempting to speculate that L. pneumophila also encodes phosphoinositide 3-kinases to optimally adjust PtdIns(3)P levels on the LCV when required during the infection cycle. Analysis of effector mutants and quantification of PtdIns(3)P levels of LCVs are needed to clarify the mechanisms underlying PtdIns(3)P exploitation by L. pneumophila.
Upon ectopic expression, we found that LtpD colocalizes and redistributes a number of endosomal proteins. EEA1 and Rab5 are usually found on early endosomes, which are enriched in PtdIns(3)P, possibly explaining the colocalization of LtpD with these markers. However, LtpD also seems to induce a redistribution of early endosomes to large perinuclear structures, which contain markers typically found in late endosomes or lysosomes. Endocytic vesicle traffic is a dynamic process, which is tightly regulated by a network of protein-protein and protein-lipid interactions (40). Our data suggest that LtpD could interfere with endosomal vesicle trafficking. L. pneumophila is commonly assumed to avoid the endocytic pathway; however, selective recruitment of markers such as Rab7 to the LCV shows that interactions may occur (32, 41). Since we could not detect differential recruitment of endosomal markers to LCVs of L. pneumophila overproducing or lacking LtpD (unpublished results), the effects of LtpD are most likely balanced by other T4SS effectors. The effector Lpw27671 and the N-terminal domains of two other uncharacterized proteins, Lpp0303 in L. pneumophila Paris and FdumT_02243 in L. dumoffii, show homology to LtpD1-469 but contain different C-terminal extension domains (42). This suggests that LtpD might belong to a family of hybrid effector proteins found in different Legionella species.
We found that LtpD binds IMPA1 and that this interaction is mediated via the C-terminal domain, LtpD472-679. The D. discoideum homologue of IMPA1 was previously found as a component of the proteome of the LCV (32). However, this study was carried out using L. pneumophila strain Philadelphia, which does not contain an LtpD homologue, indicating that the recruitment of IMPA1 in D. discoideum can occur independently of LtpD. In human cells, we observed strong IMPA1 staining throughout the host cytoplasm but did not detect enrichment of IMPA1 on LCVs (unpublished results), suggesting that the presence of IMPA1 on the LCV may be transient or below the detection level. Alternatively, other effectors might modulate IMPA1 and mask effects induced by LtpD.
The enzymatic activity of IMPA1 has been investigated thoroughly due to its proposed role in the pathogenesis of bipolar disorder (43), and yet it has not been previously identified as a target for bacterial virulence factors. IMPA1 occupies an essential role in the production of PtdIns, and its modulation therefore impacts cell fate. Indeed, inhibition and silencing of IMPA1 expression cause apoptosis in J774.1 macrophages (16). Similarly, we observed that the pharmacological inhibitors of IMPA1 or siRNA transfection were cytotoxic, which made it impossible to draw conclusions about the specific role of IMPA1 in intracellular bacterial replication. Biochemical activity assays did not reveal a direct modulation of IMPA1 activity by LtpD. Our data suggest that LtpD might facilitate the interaction of IMPA1 with a second host or effector protein to fine-tune IMPA1 activity and host lipid metabolism.
Taken together, our findings demonstrate that the novel Dot/Icm T4SS effector LtpD binds to the lipid PtdIns(3)P and the essential host cell protein IMPA1. Importantly, the presence of LtpD contributes to the fitness of L. pneumophila 130b in the infection of macrophages, G. mellonella, and the lungs of susceptible mice. Future studies will focus on elucidating the function of the N-terminal domain of LtpD and its homologous hybrid effectors in L. pneumophila pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jean Celli (Rocky Mountain Labs, National Institute of Allergy and Infectious Disease) for the 2×FYVE-GFP vector and Miguel Seabra (Imperial College London) for the Rab5-GFP, Rab7-GFP, Rab4-GFP, and Rab1-GFP plasmids.
This study was supported by grants from the Australian National Health and Medical Research Council, the German Academic Exchange Service, the Wellcome Trust, and the Medical Research Council.
Footnotes
Published ahead of print 3 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01054-13.
REFERENCES
- 1.Rowbotham TJ. 1980. Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae. J. Clin. Pathol. 33:1179–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Newton HJ, Ang DK, van Driel IR, Hartland EL. 2010. Molecular pathogenesis of infections caused by Legionella pneumophila. Clin. Microbiol. Rev. 23:274–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horwitz MA. 1983. The Legionnaires' disease bacterium (Legionella pneumophila) inhibits phagosome-lysosome fusion in human monocytes. J. Exp. Med. 158:2108–2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Derre I, Isberg RR. 2004. Legionella pneumophila replication vacuole formation involves rapid recruitment of proteins of the early secretory system. Infect. Immun. 72:3048–3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swanson MS, Isberg RR. 1995. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect. Immun. 63:3609–3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berger KH, Isberg RR. 1993. Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol. Microbiol. 7:7–19 [DOI] [PubMed] [Google Scholar]
- 7.Marra A, Blander SJ, Horwitz MA, Shuman HA. 1992. Identification of a Legionella pneumophila locus required for intracellular multiplication in human macrophages. Proc. Natl. Acad. Sci. U. S. A. 89:9607–9611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lifshitz Z, Burstein D, Peeri M, Zusman T, Schwartz K, Shuman HA, Pupko T, Segal G. 2013. Computational modeling and experimental validation of the Legionella and Coxiella virulence-related type-IVB secretion signal. Proc. Natl. Acad. Sci. U. S. A. 110:E707–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hubber AR, Roy CR. 2010. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 26:261–283 [DOI] [PubMed] [Google Scholar]
- 10.Xu L, Shen X, Bryan A, Banga S, Swanson MS, Luo ZQ. 2010. Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog. 6(3):e1000822. 10.1371/journal.ppat.1000822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardiman CA, McDonough JA, Newton HJ, Roy CR. 2012. The role of Rab GTPases in the transport of vacuoles containing Legionella pneumophila and Coxiella burnetii. Biochem. Soc. Trans. 40:1353–1359 [DOI] [PubMed] [Google Scholar]
- 12.Falkenburger BH, Jensen JB, Dickson EJ, Suh BC, Hille B. 2010. Phosphoinositides: lipid regulators of membrane proteins. J. Physiol. 588:3179–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atack JR, Broughton HB, Pollack SJ. 1995. Structure and mechanism of inositol monophosphatase. FEBS Lett. 361:1–7 [DOI] [PubMed] [Google Scholar]
- 14.King JS, Teo R, Ryves J, Reddy JV, Peters O, Orabi B, Hoeller O, Williams RS, Harwood AJ. 2009. The mood stabilizer lithium suppresses PIP3 signaling in Dictyostelium and human cells. Dis. Model Mech. 2:306–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarkar S, Rubinsztein DC. 2006. Inositol and IP3 levels regulate autophagy: biology and therapeutic speculations. Autophagy 2:132–134 [DOI] [PubMed] [Google Scholar]
- 16.De Meyer I, Martinet W, Van Hove CE, Schrijvers DM, Hoymans VY, Van Vaeck L, Fransen P, Bult H, De Meyer GR. 2011. Inhibition of inositol monophosphatase by lithium chloride induces selective macrophage apoptosis in atherosclerotic plaques. Br. J. Pharmacol. 162:1410–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu F, Zhu W, Brennan L, Tao L, Luo ZQ, Mao Y. 2012. Structural basis for substrate recognition by a unique Legionella phosphoinositide phosphatase. Proc. Natl. Acad. Sci. U. S. A. 109:13567–13572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weber SS, Ragaz C, Hilbi H. 2009. The inositol polyphosphate 5-phosphatase OCRL1 restricts intracellular growth of Legionella, localizes to the replicative vacuole and binds to the bacterial effector LpnE. Cell Microbiol. 11:442–460 [DOI] [PubMed] [Google Scholar]
- 19.Weber SS, Ragaz C, Reus K, Nyfeler Y, Hilbi H. 2006. Legionella pneumophila exploits PI(4)P to anchor secreted effector proteins to the replicative vacuole. PLoS Pathog. 2(5):e46. 10.1371/journal.ppat.0020046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brombacher E, Urwyler S, Ragaz C, Weber SS, Kami K, Overduin M, Hilbi H. 2009. Rab1 guanine nucleotide exchange factor SidM is a major phosphatidylinositol 4-phosphate-binding effector protein of Legionella pneumophila. J. Biol. Chem. 284:4846–4856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neunuebel MR, Mohammadi S, Jarnik M, Machner MP. 2011. Legionella pneumophila LidA affects nucleotide binding and activity of the host GTPase Rab1. J. Bacteriol. 194:1389–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jank T, Bohmer KE, Tzivelekidis T, Schwan C, Belyi Y, Aktories K. 2012. Domain organization of Legionella effector SetA. Cell Microbiol. 14:852–868 [DOI] [PubMed] [Google Scholar]
- 23.Schroeder GN, Petty NK, Mousnier A, Harding CR, Vogrin AJ, Wee B, Fry NK, Harrison TG, Newton HJ, Thomson NR, Beatson SA, Dougan G, Hartland EL, Frankel G. 2011. Legionella pneumophila strain 130b possesses a unique combination of type IV secretion systems and novel Dot/Icm secretion system effector proteins. J. Bacteriol. 192:6001–6016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edelstein PH. 1981. Improved semiselective medium for isolation of Legionella pneumophila from contaminated clinical and environmental specimens. J. Clin. Microbiol. 14:298–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galan JE, Ginocchio C, Costeas P. 1992. Molecular and functional characterization of the Salmonella invasion gene invA: homology of InvA to members of a new protein family. J. Bacteriol. 174:4338–4349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen DQ, Huang SS, Lu YJ. 2006. Efficient transformation of Legionella pneumophila by high-voltage electroporation. Microbiol. Res. 161:246–251 [DOI] [PubMed] [Google Scholar]
- 27.Ohnishi T, Ohba H, Seo KC, Im J, Sato Y, Iwayama Y, Furuichi T, Chung SK, Yoshikawa T. 2007. Spatial expression patterns and biochemical properties distinguish a second myo-inositol monophosphatase IMPA2 from IMPA1. J. Biol. Chem. 282:637–646 [DOI] [PubMed] [Google Scholar]
- 28.Harding CR, Schroeder GN, Reynolds S, Kosta A, Collins JW, Mousnier A, Frankel G. 2012. Legionella pneumophila pathogenesis in the Galleria mellonella infection model. Infect. Immun. 80:2780–2790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newton HJ, Sansom FM, Dao J, Cazalet C, Bruggemann H, Albert-Weissenberger C, Buchrieser C, Cianciotto NP, Hartland EL. 2008. Significant role for ladC in initiation of Legionella pneumophila infection. Infect. Immun. 76:3075–3085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cazalet C, Rusniok C, Bruggemann H, Zidane N, Magnier A, Ma L, Tichit M, Jarraud S, Bouchier C, Vandenesch F, Kunst F, Etienne J, Glaser P, Buchrieser C. 2004. Evidence in the Legionella pneumophila genome for exploitation of host cell functions and high genome plasticity. Nat. Genet. 36:1165–1173 [DOI] [PubMed] [Google Scholar]
- 31.Gillooly DJ, Morrow IC, Lindsay M, Gould R, Bryant NJ, Gaullier JM, Parton RG, Stenmark H. 2000. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 19:4577–4588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Urwyler S, Nyfeler Y, Ragaz C, Lee H, Mueller LN, Aebersold R, Hilbi H. 2009. Proteome analysis of Legionella vacuoles purified by magnetic immunoseparation reveals secretory and endosomal GTPases. Traffic 10:76–87 [DOI] [PubMed] [Google Scholar]
- 33.Aurass P, Schlegel M, Metwally O, Harding CR, Schroeder GN, Frankel G, Flieger A. 2013. The Legionella pneumophila Dot/Icm-secreted effector PlcC/CegC1, together with PlcA and PlcB, promotes virulence and belongs to a novel zinc metallophospholipase C family present in bacteria and fungi. J. Biol. Chem. 288:11080–11092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harding CR, Stoneham CA, Schuelein R, Newton HJ, Oats CV, Hartland E, Schroeder GN, Frankel G. 2013. The Dot/Icm effector SdhA is necessary for virulence of Legionella pneumophila in Galleria mellonella and A/J. mice. Infect. Immun. 81:2598–2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Meer-Janssen YP, van Galen J, Batenburg JJ, Helms JB. 2012. Lipids in host-pathogen interactions: pathogens exploit the complexity of the host cell lipidome. Prog. Lipid Res. 49:1–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomez-Valero L, Buchrieser C. 2013. Genome dynamics in Legionella: the basis of versatility and adaptation to intracellular replication. Cold Spring Harbor Perspect. Med. 1:3(6). 10.1101/cshperspect.a009993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Behnia R, Munro S. 2005. Organelle identity and the signposts for membrane traffic. Nature 438:597–604 [DOI] [PubMed] [Google Scholar]
- 38.Clemens DL, Lee BY, Horwitz MA. 2000. Deviant expression of Rab5 on phagosomes containing the intracellular pathogens Mycobacterium tuberculosis and Legionella pneumophila is associated with altered phagosomal fate. Infect. Immun. 68:2671–2684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Toulabi L, Wu X, Cheng Y, Mao Y. 2013. Identification and structural characterization of a Legionella phosphoinositide phosphatase. J. Biol. Chem. 10.1074/jbc.M113.474239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huotari J, Helenius A. 2011. Endosome maturation. EMBO J. 30:3481–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clemens DL, Lee BY, Horwitz MA. 2000. Mycobacterium tuberculosis and Legionella pneumophila phagosomes exhibit arrested maturation despite acquisition of Rab7. Infect. Immun. 68:5154–5166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin T, Cui Y, Cen Z, Liang T, Ren H, Yang X, Zhao X, Liu Z, Xu L, Li D, Song Y, Yang R, Shao Z. 2012. Draft genome sequences of two Legionella dumoffii strains, TEX-KL and NY-23. J. Bacteriol. 194:1251–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams RS, Cheng L, Mudge AW, Harwood AJ. 2002. A common mechanism of action for three mood-stabilizing drugs. Nature 417:292–295 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.