

Abstract
Vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) receptors are implicated in development and tumorigenesis and dual inhibitors like sunitinib are prescribed for cancer treatment. While mammalian VEGF and PDGF receptors are present in multiple isoforms and heterodimers, Drosophila encodes one ancestral PDGF/VEGF receptor, PVR. We identified PVR in an unbiased cell-based RNA interference (RNAi) screen of all Drosophila kinases and phosphatases for novel regulators of TORC1. PVR is essential to sustain target of rapamycin complex 1 (TORC1) and extracellular signal-regulated kinase (ERK) activity in cultured insect cells and for maximal stimulation by insulin. CG32406 (henceforth, PVRAP, for PVR adaptor protein), an Src homology 2 (SH2) domain-containing protein, binds PVR and is required for TORC1 activation. TORC1 activation by PVR involves Tsc1/Tsc2 and, in a cell-type-dependent manner, Lobe (ortholog of PRAS40). PVR is required for cell survival in vitro, and both PVR and TORC1 are necessary for hemocyte expansion in vivo. Constitutive PVR activation induces tumor-like structures that exhibit high TORC1 activity. Like its mammalian orthologs, PVR is inhibited by sunitinib, and sunitinib treatment phenocopies PVR loss in hemocytes. Sunitinib inhibits TORC1 in insect cells, and sunitinib-mediated TORC1 inhibition requires an intact Tsc1/Tsc2 complex. Sunitinib similarly inhibited TORC1 in human endothelial cells in a Tsc1/Tsc2-dependent manner. Our findings provide insight into the mechanism of action of PVR and may have implications for understanding sunitinib sensitivity and resistance in tumors.
INTRODUCTION
Vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) receptors are receptor tyrosine kinases (RTKs) broadly implicated in development and cancer pathogenesis. PDGF receptors (PDGFRs) are primarily expressed in fibroblasts and vascular mural cells, where they are important for cell survival, proliferation, and migration (1, 2). VEGF receptors (VEGFRs) are expressed in endothelial cells and are important for embryonic vasculogenesis and angiogenesis (3, 4). In addition, VEGFRs are also expressed in hematopoietic cells, where they support cell survival and regulate cell migration (5, 6). Recent evidence suggests that hematopoietic stem cells and endothelial cells are derived from the same precursors (7–9).
PDGF and VEGF receptors share common structural features, including an extracellular ligand binding domain with multiple immunoglobulin repeats, an intracellular juxtamembrane region (JMR), and an intracellular split kinase domain (3, 10). These receptors are in the same RTK subfamily and share a similar structure with KIT protein, the stem cell growth factor receptor (3, 11). In humans, there are two PDGF receptors, α and β, and three VEGF receptors, VEGFR1 to VEGFR3. Members of the PDGF and VEGF receptor families form both homo- and heterodimers. There are four genes encoding PDGFR ligands, PDGFA to PDGFD (2), and five genes encoding ligands for VEGFRs, VEGFA to VEGFD and placenta growth factor (PlGF) (3, 5, 12). VEGFRs and PDGFRs relay overlapping but not identical signals. Recruitment and activation of phospholipase C gamma (PLCγ) have been observed for all receptors except VEGFR3, and activation of phosphatidylinositol 3-kinase (PI3K) and Akt is observed downstream of all receptors except VEGFR1 (3, 10). Both PDGFRs activate protein kinase C (PKC) and c-Jun N-terminal kinase (JNK) (10, 13). In addition, activation of extracellular signal-regulated kinase (ERK) is observed upon stimulation of VEGFR2, VEGFR3, PDGFRα, and PDGFRβ (3, 10, 13, 14). Importantly, PDGF receptors and VEGF receptors have been found to be mutated in cancer, such as in gastrointestinal stromal tumors (GISTs) (11), various myeloproliferative syndromes (11, 15), and angiosarcomas (16).
An important regulator of cell growth and proliferation is target of rapamycin complex 1 (TORC1). TORC1 phosphorylates S6 kinase (S6K) and initiation factor 4E-binding protein 1 (4E-BP1), both regulators of protein translation (17, 18). TORC1 activation is observed upon VEGF stimulation in endothelial cells (19) and cardiac myocytes (20). Importantly, TORC1 inhibition with rapamycin blocks VEGF-induced endothelial cell proliferation (19). Similarly, PDGF stimulation leads to the activation of S6K (21, 22). Multiple mechanisms are implicated in TORC1 regulation by mitogens. The kinases ERK (23), ribosomal S6 kinase (RSK) (24), and Akt (25–28) have all been shown to directly phosphorylate the protein tuberous sclerosis complex 2 (TSC2). TSC2 binds tuberous sclerosis complex 1 (TSC1) to form a complex, TSC1/TSC2, with tumor suppressor function that acts as a GTPase activating protein toward Ras homolog enriched in brain (Rheb), a small GTPase required for TORC1 activation (18). While the molecular mechanism remains unclear, phosphorylation of TSC2 by ERK, RSK, and Akt leads to TORC1 activation. Akt also phosphorylates the proline-rich Akt substrate of 40 kDa (PRAS40) (29, 30), which functions as an inhibitory TORC1 substrate (31, 32). The importance of TORC1 in cancer biology is illustrated by the development and FDA approval of rapamycin analogs for cancer treatment, temsirolimus and everolimus (33).
The importance of VEGF and PDGF signaling in cancer biology led to the development of VEGF and PDGF receptor inhibitors such as sunitinib (34). Sunitinib is a small molecule that mimics ATP and has direct antitumor effect on cells that depend on RTKs such as VEGFR2, PDGFRβ, or c-KIT for their survival or proliferation. Sunitinib also acts on cells of the tumor microenvironment, such as endothelial and vascular mural cells, where inhibition of VEGFR2 and PDGFRβ, respectively, has antiangiogenic effects. This has been proposed as the underlying mechanism of sunitinib action against renal cell carcinoma (35).
A challenge in the study of VEGF and PDGF signaling results from the existence of multiple ligands and receptors. Interestingly, however, there is a single receptor in Drosophila, the PDGF/VEGF receptor, or PVR (36, 37). Like VEGF and PDGF receptors, PVR consists of an extracellular domain composed largely of immunoglobulin repeats and a split tyrosine kinase domain (37). PVR is expressed in cells from the hematopoietic lineage (37, 38) and has been implicated in survival (38), proliferation (39, 40), and migration (36, 37). Like VEGF and PDGF receptors, PVR activates PI3K (41) and Ras (36–38, 42). In addition, PVR has also been implicated in the regulation of c-Jun N-terminal kinase (JNK) (43, 44), and this involves the adaptor protein Crk (43). However, whether PVR regulates TORC1 is unknown.
MATERIALS AND METHODS
Cell culture and drugs.
Kc and S2 Drosophila cells were maintained in 1× Schneider medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Sigma) and 1% penicillin-streptomycin (P-S) (Sigma) in a humidified 25°C incubator.
Human umbilical vein endothelial cells (HUVEC) and human dermal microvascular endothelial cells (HDMEC) were purchased from ScienCells Research Laboratories (Carlsbad, CA) and were maintained in endothelial cell medium (ECM) (catalog number 1001; ScienCells Research Laboratories). Plates were coated with 0.5% gelatin (Sigma) for 30 min and then 20% FBS in phosphate-buffered saline (PBS; Sigma) for 30 min at 37°C.
Rapamycin and sunitinib were purchased from LC Laboratories.
Fly reagents.
The following plasmids were generous gifts: pGAL4 (laboratory database reference, p489), pUAST (p490), pMT-V5hisA (p540), pHA-UAST (where HA is hemagglutinin) (p667) (J. Jiang, University of Texas [UT] Southwestern Medical Center), pCoPuromycin (p544) (T. Megraw, UT Southwestern Medical Center), pUAST-3HA-d4E-BP (p639) (45), pUAST-PVR (p575), pUAST-λ-PVR (p641) (36), and pUAST-HA-myr-dAKT (p638) (46). The following fly stocks were used in this study: He-Gal4, MS1096 Gal4, and an upstream activation sequence (UAS)-TOR RNA interference (RNAi) construct (Bloomington Drosophila Stock Center); UAS-λ-PVR and UAS-PVR (Pernile Röth) (36); UAS-PVR DN (where DN is dominant negative) (Denise Montell) (36); UAS-PVR RNAi A (Vienna Drosophila RNAi Center); UAS-PVR RNAi B (8222R-3; Fly Stocks of the National Institute of Genetics, Japan); UAS-Charybdis and UAS-Scylla (Ernst Hafen) (47).
Cloning and site-directed mutagenesis.
The coding sequence of the C-terminal 240 amino acids of PVR (PVR tail) was amplified from pUAST-PVR using the PVR-pet primer pair (see Table S1 in the supplemental material) and cloned into pET-11a (p492) (Novagen) using NdeI (New England BioLabs) and BamHI (New England BioLabs) to generate pET-PVR-his (p640).
To generate pUAST-PVR-N1159K (p683), pUAST-PVR-G1166P (p682), and pUAST-PVR-Y1160D (p684), site-directed mutagenesis was performed using pUAST-PVR as the template with primer pairs shown in Table S1 in the supplemental material and Pfu. Briefly, PCR products were purified using a PCR purification kit (Qiagen) and ligated with T4 DNA ligase (New England BioLabs); the parental plasmid was digested with DpnI, and the reaction product was transformed into XL1-Blue. cDNAs with mutations were confirmed by capillary sequencing.
The coding sequence for Drosophila Ras85D was amplified from a Drosophila Ras85D cDNA using a Ras85DV12 primer pair that included the G12V mutation (see Table S1) inserted into pUAST using EcoRI and NotI restriction sites to make pUAST-Ras85DV12 (p668).
The coding sequence of CG32406 was amplified using a CG32406 cloning primer pair (see Table S1 in the supplemental material) from Drosophila CG32406 cDNA. The PCR product was digested with BglII and NotI and ligated into pHA-UAST (previously cut with the same enzymes) to generate pUAST-HA-CG32406 (p678). pMT-HA-CG32406 (p843) was generated by cutting out the CG32406 coding sequence from pUAST-HA-CG32406 using EcoRI and NotI and then ligating it into the pMT-V5hisA digested with the same enzymes. The integrity of cDNAs or mutations generated by PCR or site-directed mutagenesis was confirmed by capillary sequencing.
Drosophila RNAi.
Double-stranded RNAs (dsRNAs) were generated as described previously (48) using the primers listed in Table S1 in the supplemental material. RNAi was performed as described previously (48). Three micrograms of each dsRNA was used per well of a 12-well plate containing 1 × 106 to 1.25 × 106 Kc or S2 cells. When two or more dsRNAs were used, total dsRNA amounts were kept constant using control dsRNA (either β-galactosidase [β-Gal] or luciferase [Luc]). β-Galactosidase and luciferase dsRNAs were generated using genomic DNA from Redd1β-geo/β-geo cells (49) or pcDNA3-Luc (p216) as templates.
Plasmid transfections and stable Kc cell line generation.
Approximately 1 × 106 Kc cells were plated per well (12-well plate), and the next day, cells were transfected with 0.45 and 0.15 μg/well of expression vector and pGAL4 driver, respectively, using Effectene (Qiagen) according to the manufacturer's instructions.
To generate Kc stable cell lines, cells were plated as described above. On the next day, cells were transfected with 270 ng of pMT-CG32406 and 30 ng of pCoPuromycin using Effectene as described above. Three days later, cells harboring plasmids were selected using 2 μg/ml puromycin (Sigma). Every 3 days, cells were split, or medium was changed until all cells in the negative-control plate were dead. To induce protein expression, CuSO4 (0.7 μM; Sigma) was added to cells and incubated for 20 h before samples were harvested for analysis.
Cell proliferation assays.
Approximately 5,000 cells (Kc or S2) were plated per well on a 96-well white clear-bottom plate (Fisher), and ∼24 h later, sunitinib or dimethyl sulfoxide (DMSO) was added to the indicated final concentrations (see figures and legends). Proliferation was assayed using a CellTiter-Glo kit (Promega) and PolarStar plate reader (BMG Labtech).
Proliferation assays of Kc cells treated with different dsRNAs was performed using 24-well plates, and RNAi was performed as described above with the following changes: Kc cells were plated at 4 × 105 to 5 × 105 cells/well, and 1.5 μg of dsRNA/gene to be knocked down was used.
PVR antibody generation.
pET-PVR-His was transformed into BL21(DE3) tonA::Tn10, and protein expression was induced with 100 μM isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 h. Bacteria were pelleted, resuspended in PBS, and lysed using Cell Lytic Express (Sigma) according to the manufacturer's instructions. Insoluble material was removed by centrifugation at 10,000 × g for 15 min at 4°C, and the supernatant was used as the input for purification of His-PVR tail using Talon resin (Clontech). Purified protein was used to raise PVR antibodies in rabbits by ProSci Incorporated (Poway, California).
Apoptosis assay.
Cells treated for 3 days with either Luc or PVR dsRNA were evaluated using a DeadEnd fluorometric TUNEL (terminal deoxynucleotidyltransferase-mediated fluorescein-12-dUTP nick end labeling) System (Promega) according to the manufacturer's instructions and assessed by fluorescence-assisted cell sorting (FACS) using a FACS ARIA I instrument (BD). In the case of Diap1 dsRNA, cells were harvested for FACS analysis after 24 h of treatment. When cells were treated with Z-VAD (N-benzyloxycarbonyl-Val-Ala-Asp; Sigma), 20 μM was added immediately after the RNAi procedure. To assess apoptosis of cells exposed to either rapamycin or sunitinib, cells were plated at 15 × 106 per 100-mm-diameter plate for 24 h and then exposed to vehicle (methanol for rapamycin or DMSO for sunitinib), rapamycin (25 nM), or sunitinib (10 μM) for 48 h.
BrdU incorporation assay.
Cells were plated in 60-mm-diameter discs at 6 × 106 cells/plate for 24 h and then exposed to vehicle or sunitinib for another day. On the third day, bromodeoxyuridine (BrdU) (50 μM; Sigma) was added and incubated for 20 h. Cells were pelleted and processed according to Crevel et al. (50), and 2 μl of 20 ng/μl of total DNA, undiluted or serially diluted, was spotted on Amersham Hybond 0.45-μm-pore-size nylon membrane. BrdU incorporation was detected using mouse monoclonal anti-BrdU antibody purchased from BD.
Antibodies, immunofluorescence, IP, and Western blot analysis.
PVR antibodies for Western blotting were a generous gift from Pernile Rörth (36). The PVR antibody generated was used for immunofluorescence and immunoprecipitation (IP). Drosophila phospho-p70-S6K (T398), phospho-p70-S6K (T389), phospho-4E-BP (T37/46), phospho-S6 (S235/236), and phospho-S6 (S240/244) antibodies were from Cell Signaling; anti-HA antibodies were from Covance, and phospho-tyrosine antibodies were from Upstate. The following antibodies were from Sigma: anti-phospho-ERK1/2 (T183/Y185), total ERK, and tubulin. Anti-VEGFR2 antibody was purchased from Millipore. The phospho-4E-BP (T37/46) antibody used for immunofluorescence was purchased from Cell Signaling (rabbit monoclonal antibody [MAb] 326B4). Anti-CD31 was purchased from Dianova.
Kc cell suspensions were placed on coated slides, fixed with cold methanol, blocked, and incubated with PVR antibodies (1:1,000) overnight at 4°C. PVR was visualized using Cy2-conjugated donkey anti-rabbit secondary antibodies (Jackson Laboratories), and the nucleus was stained with propidium iodide (PI) (1 μg/ml; Sigma). Images were taken using a confocal microscope (Zeiss LSM510) at a magnification of ×650.
Wandering larvae were collected, inverted, fixed, permeabilized, and then incubated with PI alone (for differential interference contrast [DIC] imaging) or with anti-P-4E-BP (1:1,000) overnight and subsequently with Cy2-conjugated donkey anti-rabbit antibodies, and the nucleus was stained with PI. Wing discs were isolated, and images were taken using a confocal microscope with or without (for P-4E-BP) a DIC setting at ×200 magnification.
IP and Western blotting were performed according to Vega-Rubin-de-Celis et al. (51).
Stimulation assays.
Conditioned medium (CM) was collected from confluent Kc cells cultures every 3 days, supplemented with 10% FBS, filtered, and used for stimulation. Kc cells were plated in 12-well plates and treated with dsRNA as described above. One day after RNAi, complete medium was changed to serum-free medium, and cells were starved for 2 days. The next day, cells were stimulated with conditioned medium (CM) as indicated (see the figures). In the case of insulin stimulation, cells were treated with RNAi for 3 days and then stimulated with 1 μM insulin from bovine pancreas (Sigma) for the indicated times (see the figures).
Hemocyte experiments.
To treat larvae with sunitinib, drug was mixed with food (standard recipe published in the Bloomington Drosophila Stock Center website) to a final concentration of 70 μM. Because sunitinib was dissolved in DMSO, food containing the corresponding amount of DMSO was used as a control. Twenty-four hours after egg deposit, stage L1 larvae were floated using 20% sucrose in PBS and transferred to food supplemented with either vehicle or sunitinib. When larvae developed to wandering stage, hemolymph was collected for hemocyte counts. Briefly, wandering larvae were washed in PBS three times and dried on Kimwipe papers. Ten to 15 larvae were placed in 100 μl of PBS, and hemolymph was collected by poking holes on larvae without disrupting the gut. Hemocytes were counted using a hemocytometer.
RT-PCR.
Total RNA was isolated from Kc and S2 cells using an RNeasy kit (Qiagen). cDNA was synthesized with random hexamers from 1 μg of total RNA using Moloney murine leukemia virus (MMLV) reverse transcriptase (Invitrogen). Quantitative reverse transcription-PCR (qRT-PCR) of Lobe was performed according to Peña-Llopis et al. (52) and normalized to the expression levels of RpS17 using the primers listed in Table S1 in the supplemental material.
Endothelial cell RNAi and sunitinib treatment.
Approximately 1.5 × 105 cells (either HUVEC or HDMEC) were plated per well of a six-well plate for 2 days; then cells were treated with different concentrations of sunitinib or DMSO overnight before being harvested for Western blotting. To knock down TSC2, cells were plated as described above, and small interfering RNAs (siRNAs; Dharmacon) were transfected using Oligofectamine (Invitrogen) according to the manufacturer's instructions. Cells were incubated for 2 days prior to subsequent manipulation. siRNA sequences are as described in Brugarolas et al. (53). Cells were treated with 10 μM sunitinib overnight before being harvested for Western blotting.
Tumor graft treatment.
Tumor samples were obtained from patients providing written informed consent according to an Institutional Review Board-approved protocol allowing for the use of discarded surgical specimens for research. Animal studies were approved by the UT Southwestern Institutional Animal Care and Use Committee. Tumor graft experiments were performed as previously described (54) except that tumor graft-bearing mice were treated for 3 days. When tumors reached 250 to 300 mm3, mice were treated with either vehicle or sunitinib. Vehicle (consisting of 5% ethanol, 5% polyethylene glycol 400 [PEG400], and 5% Tween 80, and the remaining 85% of 5% dextrose water [D5W]) or sunitinib (dissolved in 0.5% carboxymethyl cellulose [Sigma] in D5W) was administered by oral gavage every 12 h at 10 mg/kg of body weight. Mice were sacrificed ∼3 h after the last treatment, and tumors were collected and fixed in 10% buffered formalin phosphate (Fisher Scientific).
PVR modeling and alignment with mammalian counterparts.
Human and fly RTK sequences closely related to PVR were aligned using the Mafft server (55). A tree was built using distances calculated from the resulting alignment (with the kinase insert loop trimmed) using the MOLPHY package JTT probability matrix with frequencies option. Initial tree topologies were built with njdist, and a maximum-likelihood tree was built using the local rearrangement search (−R option) of the PROTML program in MOLPHY (56). Structure models of the wild-type and sunitinib-resistant A-loop mutant PVR kinase domains were built using the SwissModel alignment mode (57) based on templates of (i) the inactive-state KIT kinase domain bound to sunitinib (Protein Data Bank identifier [PDB ID] 3g0e) and (ii) the activated-state KIT kinase domain bound to an ADP and a peptide product (PDB ID 1pkg).
Statistics.
All statistical analyses were performed using a Student t test. Significant P values are defined in the figure legends.
RESULTS
PVR is essential for TORC1 activity.
We conducted an RNAi screen of kinases and phosphatases for novel regulators of TORC1 in Drosophila Kc167 (Kc) cells. We used Western blotting to assay for phosphorylation of Drosophila S6K at T398 (equivalent to T389 in human S6K1), a site directly phosphorylated by TORC1 (58). We examined 638 dsRNAs targeting all the kinases and phosphatases in the Drosophila genome. Interestingly, a dsRNA targeting pvr markedly inhibited the phosphorylation of S6K (P-S6K). The same results were observed with three nonoverlapping dsRNAs (Fig. 1A).
Fig 1.

PVR is required for TORC1 activation in insect cells. (A, C, and D) Western blot analyses were performed using the indicated cell types (below blots) grown in 10% serum and treated with the stated dsRNAs, and, where shown, transfected with HA-4E-BP (or an empty vector [EV]). UT, untreated; Ra, rapamycin; β-gal, β-galactosidase; Luc, luciferase. (B) Confocal images of Kc cells treated with the indicated dsRNAs and stained using a PVR polyclonal antibody. Magnification, ×650; red, propidium iodide; green, PVR.
We generated a rabbit polyclonal antibody directed against the C-terminal 240 amino acids of PVR. PVR was expressed on the surface of Kc cells, and PVR knockdown substantially depleted PVR (Fig. 1B). Consistent with previous reports (36, 37, 42), ERK phosphorylation (P-ERK) was also abolished by PVR knockdown (Fig. 1A).
To determine how generalized this phenomenon was, we examined a second commonly used cell line, S2 cells. As with Kc cells, depletion of PVR using three nonoverlapping dsRNAs profoundly inhibited P-S6K in this cell type (Fig. 1C). Notably, the inhibition of P-S6K by PVR RNAi was comparable to that observed by depletion of TOR (or S6K) or by treatment with the TORC1 inhibitor rapamycin (Fig. 1A and C). While we observed that PVR depletion reduced total S6K levels, P-S6K inhibition preceded the downregulation of S6K protein (see Fig. S1 in the supplemental material). Thus, PVR is required for baseline S6K activity in several cell types.
To determine whether the effect of PVR on S6K was mediated by TORC1, we examined another TORC1 substrate. Mammalian 4E-BP1 is a canonical TORC1 substrate, and Drosophila 4E-BP phosphorylation (P-4E-BP) is inhibited by rapamycin (see Fig. S2 in the supplemental material). As P-4E-BP levels were undetectable in Kc cells, cells were transfected with HA-4E-BP. Ectopically expressed 4E-BP behaved as expected: rapamycin (or TOR dsRNA) accelerated 4E-BP migration and decreased P-4E-BP (Fig. 1D; see also Fig. S2). PVR depletion with three nonoverlapping dsRNAs gave results similar to TOR ablation (Fig. 1D). In contrast, knockdown of the negative TORC1 regulators Tsc1 and Tsc2 increased P-4E-BP (and P-S6K) (Fig. 1A, C, and D). Thus, PVR is not only important for ERK activation, as previously shown (36, 38, 42), but also necessary to sustain TORC1 activity in Drosophila tissue culture cells.
PVR is required for maximal activation and sustained activity of TORC1 by insulin.
To further characterize the role of PVR, we examined the effects of serum starvation and growth factor stimulation. Kc cells depleted of PVR (or control cells treated with an irrelevant dsRNA) were starved and stimulated with conditioned medium. Conditioned medium, which contains PVR ligands secreted by Kc cells (36), stimulated ERK activation but not S6K (Fig. 2A; see also Fig. S3A in the supplemental material). PVR depletion inhibited baseline S6K and ERK phosphorylation and the activation of ERK by conditioned medium (Fig. 2A; see also Fig. S3A).
Fig 2.

PVR is required for maximal and sustained activation of TORC1 and ERK. Western blot analyses were performed using Kc cells treated with the indicated dsRNA for 2 days, starved for 2 to 3 days, and then stimulated with conditioned medium (A) or with insulin (1 μM) (B and C). R, rapamycin. Short and long indicate the length of exposure time.
Because insulin stimulation activates insulin receptor (59) leading to S6K activation (60), we examined whether insulin stimulation required PVR. PVR depletion reduced the magnitude of S6K (and ERK) activation by insulin (Fig. 2B; see also Fig. S3B in the supplemental material). Furthermore, PVR loss shortened the time of S6K and, in particular, ERK activation in response to insulin stimulation (Fig. 2C). Thus, PVR is necessary for maximal activation and sustained activity of TORC1 and ERK in response to insulin.
PVR signals through Drk and Ras85D.
We hypothesized that knockdown of the appropriate adaptor protein should phenocopy the loss of P-ERK and P-S6K induced by PVR RNAi. PVR has been shown to physically and genetically interact with Crk (43). However, Crk RNAi, or RNAi of effector proteins downstream of Crk such as Mbc and ELMO, did not affect P-ERK or P-S6K (Fig. 3A), suggesting that Crk is not the mediator of PVR signaling to TORC1 and ERK.
Fig 3.
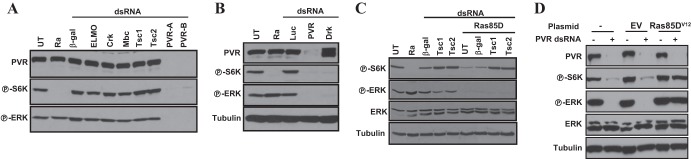
Evaluation of the roles of Drk and Ras85D in PVR-mediated activation of ERK and TORC1. Western blot analyses were performed using Kc cells treated with the indicated dsRNA for 3 days (A, B, and C) or transfected with the indicated plasmids and then treated with dsRNA against either luciferase (−) or PVR (+) (D).
A previous yeast two-hybrid screen identified 15 gene products that interact with the intracellular domain of PVR in a kinase-dependent manner (61). We individually knocked down the expression of all of these genes and examined the effect on S6K and ERK phosphorylation. The summary is shown in Table 1. Among these genes, we found that Drk knockdown markedly downregulated P-S6K and P-ERK (Fig. 3B). These data suggest that Drk, but not Crk, is implicated in transducing signals from PVR to ERK and TORC1. Interestingly, several other adaptor proteins found in the yeast two-hybrid screen were required to sustain S6K activity, including CG32406, Shc, Lnk, and Socs16D (Table 1; see also Fig. 5). Unlike Drk, however, all of these proteins were dispensable for ERK activation.
Table 1.
Summary of PVR adaptor proteins and their effects on ERK and S6K phosphorylationa
| Gene product | P-S6K | P-ERK |
|---|---|---|
| Drk | + | + |
| CG32406 | + | − |
| Shc | + | − |
| Lnk | + | − |
| Socs16D | + | − |
| Dock | − | − |
| Vav | − | − |
| Kurtz | − | − |
| Pellino | − | − |
| Tensin | − | − |
| CG1135 | − | − |
| CG13289 | − | − |
| Socs44A | − | − |
| Dock | − | − |
| CG17168 | − | − |
+, adaptor protein is required for activation; −, depletion has no effect compared to control dsRNA.
Fig 5.

Tsc1/Tsc2-dependent regulation of TORC1 by Drk, CG32406, and other adaptor proteins. Western blot analysis was performed using Kc cells treated with the indicated dsRNA. Ra, rapamycin.
Drk is the ortholog of the adaptor growth factor receptor-bound protein 2 (GRB2), which links multiple RTKs to Ras signaling (62). To determine whether Ras85D was implicated in TORC1 activation, we depleted Ras85D. As shown in Fig. 3C, Ras85D depletion similarly inactivated S6K and ERK phosphorylation. Conversely, expression of a constitutively active form of Ras85D, Ras85DV12, in PVR-depleted Kc cells was sufficient to activate TORC1 and ERK (Fig. 3D). Taken together, these data suggest that PVR signals through Drk (GRB2) and Ras85D to activate TORC1 and ERK.
Reconstitution of TORC1 activation in PVR-depleted cells by the inactivation of Lobe and the Tsc1/Tsc2 complex.
ERK phosphorylates TSC2 leading to the activation of TORC1 (23), and we asked whether inactivation of Tsc1/Tsc2 was sufficient to restore TORC1 activity in PVR-depleted cells. Unexpectedly, Tsc1 (or Tsc2) knockdown in PVR-depleted Kc cells only slightly increased P-S6K (Fig. 4A). This is unlikely to result from an ineffective knockdown as the same dsRNAs substantially increased P-S6K in control cells (Fig. 4A) and fully restored P-S6K in cells depleted of Ras85D (Fig. 3C). Taken together, our results suggest that the loss of Tsc1 or Tsc2 is not sufficient to restore TORC1 activity in PVR-depleted Kc cells. Thus, while Ras85DV12 is sufficient to activate TORC1 downstream of PVR and while Tsc1/Tsc2 loss is sufficient to restore TORC1 activation in cells depleted of Ras85D, our data suggest that PVR regulates TORC1 through a second mechanism that is independent of both Ras85D and Tsc1/Tsc2 complex.
Fig 4.

Restoration of TORC1 activity in Kc cells depleted of PVR by simultaneous inactivation of both the Tsc1/Tsc2 complex and Lobe. Western blotting was performed using Kc cells treated with the indicated dsRNAs (A to C) or after transfection with the empty vector (EV) or myristoylated-HA-Akt (D).
We investigated the role of another inhibitory regulator of TORC1, Lobe (ortholog of PRAS40) (29, 30). Depletion of Lobe in PVR-deficient cells did not increase P-S6K (Fig. 4B). This was not due to insufficient Lobe silencing as, consistent with previous reports (30), depletion of Lobe did restore TORC1 activity in cells simultaneously depleted of Rheb (Fig. 4B).
We hypothesized that the simultaneous inactivation of both the Tsc1/Tsc2 complex and Lobe (PRAS40) might be required to restore TORC1 activity in PVR-depleted Kc cells. We simultaneously depleted both Lobe and Tsc2 and found that, in fact, S6K phosphorylation was restored to levels comparable to those of PVR-expressing wild-type cells (Fig. 4C). Restoration, however, did not reach the same levels as in PVR-expressing cells depleted of Tsc1 (or Tsc2) (Fig. 4C).
Of note, while the inactivation of both Tsc1/Tsc2 and Lobe was required for restoration of TORC1 in Kc cells, in S2 cells, TORC1 activity was restored by simply depleting Tsc1 (or Tsc2) (see Fig. S4A in the supplemental material). These data suggested that Lobe was not implicated in TORC1 regulation in S2 cells. Consistent with these findings, S2 cells expressed lower levels of Lobe than Kc cells (see Fig. S4B). Furthermore, in keeping with the idea that Lobe is not implicated in TORC1 regulation in S2 cells, Lobe knockdown was insufficient to rescue TORC1 activity in Rheb-deficient S2 cells (see Fig. S4C). These data stand in contrast to the role of Lobe in Kc cells, where Lobe depletion was sufficient to activate TORC1 in Rheb-deficient cells (Fig. 4B; see also Fig. S4C). Overall, our data show that PVR activates TORC1 through the Tsc1/Tsc2 complex alone (S2 cells) or synergistically with Lobe (Kc cells), depending upon Lobe expression.
Akt is insufficient to activate TORC1 in PVR-depleted cells.
Because both the TSC2 protein (25–27) and the Lobe ortholog PRAS40 (29, 30) are phosphorylated and inactivated by Akt, we investigated whether Akt would suffice to restore TORC1 activity in PVR-depleted Kc cells. Ectopic expression of constitutively active Akt (myristoylated Akt, myr-Akt) (45) did not restore P-S6K in PVR-depleted cells. Similar results were observed with depletion of PTEN (Fig. 4D). Even when loss of PTEN (or myr-Akt) was combined with the depletion of either Lobe or Tsc2, S6K failed to be activated to baseline levels (Fig. 4D). Only when Tsc2 and Lobe were simultaneously depleted were P-S6K levels restored to baseline (Fig. 4D). Thus, PTEN loss or activated Akt is not sufficient to activate TORC1 in PVR-depleted cells.
Tsc1/Tsc2-dependent activation of TORC1 by Drk, CG32406, and other adaptors.
Overall, our data are consistent with a model whereby PVR signals to TORC1 through both TSC1/TSC2 and Lobe, whereas Ras85D signals to TORC1 through just Tsc1/Tsc2. S6K activity can be restored in Ras85D-depleted Kc cells by knockdown of Tsc1 (or Tsc2) (Fig. 3C), but the simultaneous inactivation of Tsc1/Tsc2 and Lobe is required for the restoration of S6K activity in PVR-depleted cells (Fig. 4C). As for Ras85D, knockdown of Tsc1 (or Tsc2) was sufficient for S6K activation following Drk depletion (Fig. 5). Next, we examined CG32406, Shc, Lnk, and Socs16D. Knockdown of each of these adaptor proteins inactivated S6K but not ERK (as expected), and in every instance, S6K inactivation was reverted by knockdown of Tsc1/Tsc2 (Fig. 5). Thus, our data suggest that the mechanism whereby PVR inactivates Lobe is independent of Drk, CG32406, Shc, Lnk, and Socs16D. In addition, these data again show that all of these proteins, which were previously found to interact with the kinase domain of PVR, are seemingly required for PVR-mediated activation of TORC1.
Lnk is the Drosophila ortholog of mammalian SH2B proteins and is involved in insulin signaling (63), while Shc is the Drosophila ortholog of mammalian SHC3. Shc is implicated in signaling downstream of Torso and epidermal growth factor receptor (EGFR) (64). Socs16D is similar to mammalian SOCS6 and SOCS7 (65), and SOCS proteins in mammals negatively regulate JAK-STAT signaling (65). However, little is known about CG32406.
PVR interacts with CG32406.
CG32406 depletion abrogated S6K activity (Fig. 5), and CG32406 was found to interact with PVR in a yeast two-hybrid assay in a kinase-dependent manner (61). In CG32406-depleted cells, S6K activity could be restored by simultaneously depleting Tsc1 or Tsc2 (Fig. 5), suggesting that PVR signaling to TORC1 via this adaptor protein involves inhibition of the Tsc1/Tsc2 complex. CG32406 is characterized by an Src homology 2 (SH2) domain and a coiled-coil domain (Fig. 6A). We asked whether CG32406 interacted with PVR. We generated stable Kc cells expressing an HA-tagged form of CG32406 under an inducible promoter and evaluated CG32406 binding to endogenous PVR in PVR immunoprecipitation experiments. As shown in Fig. 6B, anti-PVR antibodies brought down CG32406 in PVR-expressing cells. As expected, anti-PVR antibodies failed to pull down HA-CG32406 in uninduced cells (which do not express HA-CG32406) or when PVR was depleted (Fig. 6B). Thus, CG32406, which is required for TORC1 activation, interacts with PVR by immunoprecipitation and in a yeast two-hybrid assay, and, we renamed it PVR adaptor protein, or PVRAP.
Fig 6.
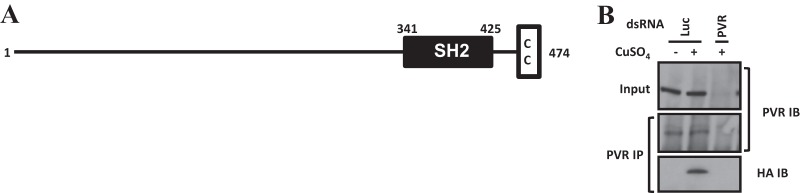
CG32406 protein interacts with PVR. (A) Domain prediction of CG32406 protein. SH2, Src homology 2; CC, coiled coil. (B) Western blot analysis of immunoprecipitated proteins. Kc cells harboring a CuSO4-inducible form of HA-CG32406 were treated with Luc or PVR dsRNA in the absence (−) or presence (+) of CuSO4. IP, immunoprecipitation; IB, immunoblot.
Overall, our data on PVR signaling are consistent with a model whereby PVR signals through multiple adaptor proteins, including Drk and PVRAP, which are individually required for TSC1/TSC2 inactivation and the activation of TORC1. There is a fundamental distinction between these adaptor proteins and PVR. TORC1 activation by PVR requires a second pathway to inhibit Lobe.
PVR loss causes caspase-independent apoptosis.
Consistent with previous observations (38), we noticed a significant decrease in cell numbers following PVR dsRNA treatment. Compared to Kc cells treated with β-galactosidase (β-Gal) dsRNA, cell numbers were markedly reduced by PVR dsRNA (Fig. 7A). Kc cells are a hemocyte cell line (38), and we sought to examine the role of PVR in circulating hemocyte numbers in vivo. We expressed a dominant negative form of PVR (PVRDN) using a hemocyte-specific driver (He-Gal4) and evaluated its effects on blood cells in Drosophila larvae. By comparison to driver alone, PVRDN markedly downregulated hemocyte counts (Fig. 7B). As PVRDN could conceivably affect other processes besides PVR, additional experiments were performed using RNAi. Using the same driver, we expressed two nonoverlapping PVR dsRNAs in hemocytes. As shown in Fig. 7B, PVR RNAi similarly reduced hemocyte numbers in Drosophila larvae. These data show that PVR is required for hemocyte expansion not just in vitro but also in vivo.
Fig 7.

PVR is required for cell number expansion in vitro and in vivo, and TORC1 is implicated in this process. (A) Quantitation of the number of Kc cells treated with the indicated dsRNA. β, β-Galactosidase. (B) Blood cell numbers in larvae expressing PVR DN (DN), PVR RNAi, TOR RNAi, or Scylla/Charybdis using the hemocyte-specific driver He-Gal4. Wt, wild type (driver alone); DN, UAS-PVR DN; PVR RNAi, UAS-PVR RNAi; Scylla/Charybdis, UAS-Scylla/UAS-Charybdis. Data are means ± standard errors of the means (for each genotype, n ≥ 4). **, P < 0.01; ***, P < 0.001. (C, D, and E) TUNEL assay of cells treated as indicated. (C) Kc cells were treated with Luc, PVR, or Diap1 dsRNA followed by vehicle (DMSO) or the pan-caspase inhibitor Z-VAD. Maroon, Luc dsRNA plus vehicle; orange, PVR dsRNA plus vehicle; lime green, PVR dsRNA plus Z-VAD; dark green, Diap1 dsRNA plus vehicle; blue, Diap1 dsRNA plus Z-VAD. Results normalized to Luc, in relative units (RU), are shown in the bar graph. (D) Bar graph representation of apoptosis induction (in RU) in Kc cells treated with the indicated dsRNAs. (E) TUNEL assay of Kc or S2 cells treated with vehicle (Ve) or 25 nM rapamycin (Ra) for 48 h. Max, maximum; FITC-A, fluorescein-12-dUTP.
To get insight into the mechanism whereby loss of PVR reduced hemocyte numbers, we asked whether PVR loss caused apoptosis and performed TUNEL assays. As a positive control, we used a dsRNA targeting the Drosophila inhibitor of apoptosis protein 1 (Diap1). As shown in Fig. 7C, Diap1 knockdown markedly increased apoptosis. Apoptosis was also increased by PVR depletion (Fig. 7C). Previous studies suggest that PVR loss induces caspase-dependent apoptosis (38). To determine whether PVR depletion in Kc cells induced caspase-dependent apoptosis, we treated cells with the pan-caspase inhibitor Z-VAD. However, treatment with Z-VAD did not reduce apoptosis in PVR-depleted cells (Fig. 7C). In contrast, Z-VAD completely rescued the effect of Diap1 loss on apoptosis (Fig. 7C). These data suggested that apoptosis resulting from PVR loss is caspase independent. To further evaluate this notion, we knocked down Drice simultaneously with either PVR or Diap1. While combination knockdown of Diap1 and Drice suppressed Diap1-mediated apoptosis, a similar effect was not observed when the expression PVR and Drice was simultaneously ablated (Fig. 7D; see also Fig. S5 in the supplemental material). Thus, the apoptosis induced by PVR loss is resistant to caspase inhibition.
Constitutively active PVR results in larval hemocyte expansion and tumor-like structures in wing discs that show TORC1 activation.
We asked whether a constitutively active PVR would increase hemocyte numbers. We used a form of PVR in which the extracellular domain is replaced by the dimerization domain of the cI repressor protein from bacteriophage λ, resulting in a stabilized receptor dimer (λ-PVR). Expression of λ-PVR in Drosophila hemocytes using the He-Gal4 driver increased blood cell numbers 3-fold compared to ectopic expression of wild-type PVR, which does not affect hemocyte numbers (Fig. 8A).
Fig 8.

Constitutively active PVR induces hemocyte expansion and tumor-like structures with active TORC1. (A) Blood cell numbers in larvae expressing UAS-PVR (PVR) or UAS-λ-PVR (λ-PVR) using the hemocyte-specific driver He-Gal4. Data are means plus standard errors of the means (for each genotype, n ≥ 4). **, P < 0.01. (B) Differential interference contrast (DIC) confocal images of imaginal wing discs from larvae expressing UAS-PVR (PVR) or UAS-λ-PVR (λ-PVR) using the imaginal wing disc-specific driver MS1096-Gal4. Control, driver alone. (C) Confocal images of imaginal wing discs with the larvae described in panel B stained with P-4E-BP or propidium iodide (PI).
Constitutive activation of PVR in larval imaginal wing discs has been shown to induce hypertrophic tumor-like structures (66) (Fig. 8B), and we asked whether these structures were associated with TORC1 activation. Compared to wing discs expressing wild-type PVR, imaginal discs expressing λ-PVR showed a substantial increase in 4E-BP phosphorylation (Fig. 8C). Consistent with the in vitro results, PVR activates TORC1 in larvae, and this may contribute to the tumor-like phenotype observed. Thus, as for mammalian VEGF and PDGF receptors, constitutively active PVR drives cell proliferation and the formation of tumor-like structures that show TORC1 activation.
TORC1 inhibition phenocopies PVR loss and induces apoptosis.
Rapamycin treatment suppressed cell survival, similar to PVR knockdown (Fig. 7A). We examined whether inhibition of TORC1 phenocopied the loss of PVR. First, we evaluated hemocyte numbers from larvae expressing UAS-TOR RNAi driven by He-Gal4. Similar to PVRDN overexpression (or PVR RNAi), blood cell numbers were significantly decreased in larvae expressing TOR RNAi (Fig. 7B). These findings were extended by the evaluation of larvae overexpressing Scylla and Charybdis, the Drosophila homologs of mammalian REDD1, a negative regulator of TORC1 (47, 53). Similar to TOR RNAi, overexpression of Scylla and Charybdis suppressed blood cell numbers in Drosophila larvae (Fig. 7B). Next, we determined whether inhibition of TORC1 by rapamycin led to apoptosis. As observed for PVR knockdown, exposing Kc and S2 cells to rapamycin increased apoptosis (Fig. 7E). TORC1 inhibition phenocopied the effects of PVR loss on cell survival and apoptosis, suggesting that TORC1 is an important effector downstream of PVR.
Sunitinib inhibits PVR signaling in Drosophila cells.
Sunitinib inhibits VEGF and PDGF receptors, and we wondered whether sunitinib might inhibit PVR. We aligned the kinase domain of PVR with that from its mammalian counterparts and other RTKs that are also inhibited by sunitinib including KIT. Substantial conservation between PVR and mammalian RTKs was observed (Fig. 9). This conservation extends to the activation loop (Fig. 9, A-loop), which plays a critical role in sunitinib binding. The DFG motif of the activation loop adopts two alternate conformations that determine the activation state of the kinase and affect sunitinib binding. In the inactive conformation (DFG-out), the phenylalanine ring points toward the ATP binding site, where it forms a binding pocket for sunitinib (67). Upon activation, the phenylalanine flips to an alternate conformation (DFG-in), which alters the sunitinib binding site and decreases affinity for the drug (Fig. 10A). The phenylalanine in the DFG-in position appears to be in competition with a hydrophobic residue from the juxtamembrane region (JMR), and residues of the JMR are frequently mutated in tumors (68–70), resulting in a shift toward the active kinase. Structure studies of KIT (Fig. 10B) and VEGFR2 (Fig. 10C) illustrate this JMR autoinhibition that may apply to the entire PDGF and VEGF receptor family (67, 71). Thus, kinase activation requires the displacement of the JMR. The importance of the JMR in maintaining the autoinhibitory state is supported by the finding that the majority of oncogenic mutations found in GIST destroy the function of the JMR in KIT or PDGFRα (72–74). We modeled the PVR kinase domain (Fig. 10D) after KIT bound to sunitinib. Our structure model is limited to the kinase domain and lacks a less confidently aligned N-terminal JMR. This model showed that as for VEGFR2 and KIT, PVR may adopt a DFG-out conformation that sunitinib could bind to. While sunitinib was developed to target VEGF and PDGF receptors, remarkable conservation between mammalian and Drosophila receptors exists in the kinase domain and the structural model.
Fig 9.

Alignment of the intracellular kinase domain of PVR and its successor RTKs or other RTKs known to be inhibited by sunitinib. *, mutations in KIT protein leading to sunitinib resistance. The conservation spectrum is represented from dark blue to white for identical to different. Red box, DFG motif.
Fig 10.

Evaluation of PVR inhibition by sunitinib. (A) PVR model of transition from inactive to active state. (B and C) Structure of KIT (B) or VEGFR2 (C) bound to sunitinib. A conserved residue (depicted in stick form) within the JMR (shown in magenta) prevents activation of VEGFR2 and KIT. (D) Model structure of PVR bound to sunitinib. The A-loop (blue) adopts a DFG-out inactive conformation, forming the binding site for sunitinib (black stick). (E) A structure-based sequence alignment of the JMR (highlighted in magenta) and the A-loop (highlighted in blue). The DFG motif F residue and the hydrophobic JMR residue positions are indicated with an asterisk above the sequences. The conserved hydrophobic residue in the JMR competes with the F residues in the DGF motif to keep the receptor in the inactive conformation.
Next, we asked whether sunitinib inhibited PVR. For these experiments, we treated Kc cells with sunitinib and assayed for PVR tyrosine phosphorylation. As a control, the same experiments were performed in cells depleted of PVR. In immunoprecipitation experiments of PVR, we recovered a protein of the expected molecular weight that was absent in PVR-depleted cells (Fig. 11A). This protein was recognized by phosphotyrosine antibodies, and phosphotyrosine levels were reduced following sunitinib treatment (Fig. 11A). These data show that sunitinib inhibits PVR tyrosine phosphorylation.
Fig 11.

Sunitinib inhibits PVR and phenocopies PVR loss. (A and B) Western blot analyses of inputs or PVR immunoprecipitates (IP) were performed using Kc cells treated with the indicated dsRNAs for 3 days and then exposed to sunitinib for 6 h (A). Arrow, PVR. Western blot analyses were performed using Kc or S2 cells treated with the indicated concentrations of sunitinib overnight (B). (C) Cell number analyses of the indicated cells treated with vehicle (Ve), sunitinib (Su; 10 μM) or rapamycin (Ra; 25 nM). (D) TUNEL assay of Kc and S2 cells treated with vehicle or sunitinib for 48 h. (E) Hemocyte numbers from wandering larvae fed with vehicle or sunitinib (70 μM)-supplemented food starting at L1. Data are means ± standard errors of the means (n ≥ 4). **, P < 0.01.
As PVR depletion inhibits S6K and ERK phosphorylation, we asked whether sunitinib had the same effect. As shown in Fig. 11B, sunitinib inhibited P-S6K and P-ERK in both Kc and S2 cells. This inhibition occurred within 5 min of sunitinib addition (see Fig. S6 in the supplemental material). Thus, sunitinib inhibits PVR phosphorylation, leading to the suppression of ERK and TORC1 activity in insect cells.
Sunitinib inhibits cell expansion and reduces hemocyte numbers in Drosophila larvae.
Because blocking PVR function inhibited cell expansion in vitro and decreased blood cell numbers in vivo, we asked whether sunitinib had a similar effect. When Drosophila Kc and S2 cells were treated with sunitinib, cell expansion was inhibited (Fig. 11C). We also determined whether sunitinib induced apoptosis. Exposure to sunitinib increased apoptosis in both Kc and S2 cells (Fig. 11D). To determine whether sunitinib affected hemocyte numbers in Drosophila larvae, we added sunitinib to the fly food. Wandering larvae that had been exposed to sunitinib since stage L1 had lower hemocyte numbers than larvae grown in food supplemented with vehicle (Fig. 11E). While these data show that sunitinib induces apoptosis, sunitinib also appears to inhibit cell proliferation in Kc cells (see Fig. S7 in the supplemental material). Thus, sunitinib largely phenocopies the loss of PVR in vitro and in vivo. Taken together, these data show that sunitinib inhibits not only VEGF/PDGF receptors but also their ortholog, PVR.
Investigating sunitinib resistance mutations using PVR.
Treatment with sunitinib in humans results in the emergence of secondary mutations that render the oncogenic RTK resistant to the effects of sunitinib. Secondary mutations in GIST that render KIT resistant to sunitinib typically map to the A-loop (68–70) and are thought to shift the kinase toward the active state (67, 75, 76). Residues that are mutated in KIT include D816, D820, N822, Y823, and A829 (68–70). D816, D820, and A829 are not conserved in PVR (Fig. 9), but we made mutations in residues corresponding to N822 and Y823. A829 in PVR is a glycine, and it was mutated as well. The residues were mutated to the corresponding activating residues in KIT, and mutant PVR was introduced into Kc cells. Because knocking down PVR inhibits cell expansion, mutant PVR cDNAs (lacking the 3′ untranslated region [UTR]) were transfected into Kc cells before endogenous PVR was knocked down using dsRNA targeting the 3′ UTR. We compared the sensitivities of wild-type and mutant PVR proteins to sunitinib. Unexpectedly, none of these mutations conferred resistance (Fig. 12). In fact, while the sunitinib sensitivity of PVR with an N1159K mutation (PVRN1159K) was similar to that of the wild-type PVR, PVRY1160D and PVRG1166P appeared to be hypersensitive (Fig. 12).
Fig 12.

Mutant PVR fails to confer resistance to sunitinib. Western blot analyses were performed of immunoprecipitated PVR from Kc cells transfected with the indicated plasmids and treated with PVR dsRNA for 2 days to deplete endogenous PVR followed by exposure to sunitinib for the indicated number of hours.
These results either suggest that a preexisting activating mutation (for example, in the JMR) is required for the secondary mutation to confer resistance or point to differences between PVR and mammalian RTKs that cause these mutations to have different effects. Consistent with the latter notion, G1166P in PVR, which possesses different surrounding residues than A829P in KIT, exhibits interactions that may have stabilized the inactive state, causing thereby hypersensitivity to sunitinib (see Fig. S8 in the supplemental material). Because the JMR of PVR is not significantly conserved (although it is conserved among insects) (see Fig. S9 in the supplemental material), the role of the JMR in this process is unknown. However, we suspect that a lack of a preexisting activating mutation as well as differences in context contributed to the differential effects of these resistance mutants in insect cells.
Constitutively dimerized PVR confers resistance to sunitinib in vitro and in vivo.
We tested the constitutively dimerized λ-PVR for sunitinib resistance. In contrast to the PVR mutants examined, λ-PVR was phosphorylated to a significantly greater extent than endogenous PVR, and sunitinib failed to reduce the levels of tyrosine phosphorylation (Fig. 13A). Thus, in contrast to the A-loop mutants, λ-PVR was resistant to sunitinib (Fig. 13A).
Fig 13.
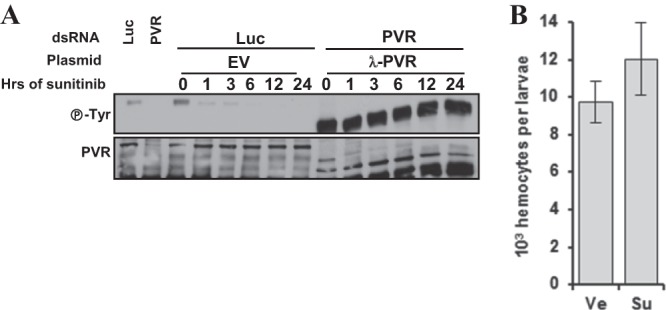
Constitutive PVR dimerization is sufficient to confer resistance to sunitinib in cells in culture as well as in hemocytes in vivo. Western blot analyses were performed of immunoprecipitated PVR from Kc cells transfected with the indicated plasmids, treated with either Luc or PVR dsRNA, and exposed to sunitinib (10 μM) for the indicated amounts of time. (B) Hemocyte numbers from wondering larvae expressing UAS-λ-PVR in blood cells using the He-Gal4 driver grown on food supplemented with either vehicle (Ve) or sunitinib (Su) starting from L1. Ve, vehicle; Su, sunitinib. Data are means ± standard errors of the means (n ≥ 4).
Next, we assessed the ability of λ-PVR to confer sunitinib resistance in vivo. Interestingly, expression of λ-PVR in hemocytes suppressed the ability of sunitinib to reduce blood cell numbers in Drosophila larvae (Fig. 13B). Thus, λ-PVR is resistant to sunitinib in vitro and in vivo. Overall, these data suggest that one mechanism whereby RTK may become resistant to sunitinib is by forming constitutive and stable dimers.
Sunitinib-induced TORC1 inhibition is Tsc1/Tsc2 dependent in Drosophila cells.
We sought to use the insect cells to further study how sunitinib acts. Our work suggested that PVR activates TORC1 through the inactivation of Tsc1/Tsc2 and Lobe, and we asked what the role was of these proteins in sunitinib-mediated TORC1 inhibition. We had determined that restoration of TORC1 activity in PVR-depleted Kc cells required the simultaneous depletion of both Tsc1/Tsc2 and Lobe, whereas in S2 cells, Tsc1/Tsc2 inactivation was enough (Fig. 4C; see also Fig. S4A in the supplemental material). Unexpectedly, depletion of Tsc2 alone in both Kc and S2 cells was sufficient to restore TORC1 in sunitinib-treated cells (Fig. 14). Thus, while the inactivation of both Tsc1/Tsc2 and Lobe was required to restore TORC1 in PVR-depleted cells, Tsc1/Tsc2 inactivation alone was sufficient for restoration in cells treated with sunitinib. The restoration of TORC1 activity by simply inactivating Tsc1/Tsc2 was similar to that observed following depletion of Drk and the other adaptor proteins.
Fig 14.
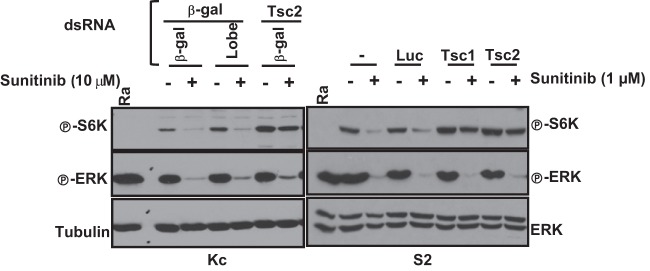
Tsc1/Tsc2-dependent inhibition of TORC1 by sunitinib in Drosophila cells. Western blot analysis was performed using the stated cell types pretreated with the indicated dsRNAs for 2 days and then treated with sunitinib for 24 h. UT, untreated; Ra, rapamycin; β-gal, β-galactosidase.
As expected, Tsc1/Tsc2 inactivation was not sufficient to activate ERK in sunitinib-treated cells. This result is similar also to that with Drk-depleted cells (Fig. 5).
One possibility is that in Kc cells, PVR regulates Lobe in a manner that is at least in part independent of its kinase function. This would be consistent also with the phenotype observed with the adaptor proteins, which would be expected to depend on PVR phosphorylation. More importantly, these results show that sunitinib-mediated inhibition of TORC1 is Tsc1/Tsc2 dependent.
Sunitinib inhibits TORC1 in endothelial cells in vitro and in vivo.
We tested whether sunitinib inhibits TORC1 in mammalian cells. TORC1 was inhibited in primary human endothelial cells (HUVEC and HDMEC) as determined by the loss of P-S6K, P-S6, and P-4E-BP (Fig. 15A). In addition, sunitinib similarly inhibited TORC1 in endothelial cells (CD31 positive) in vivo (Fig. 15B) and in a renal cell cancer tumor graft model (54).
Fig 15.
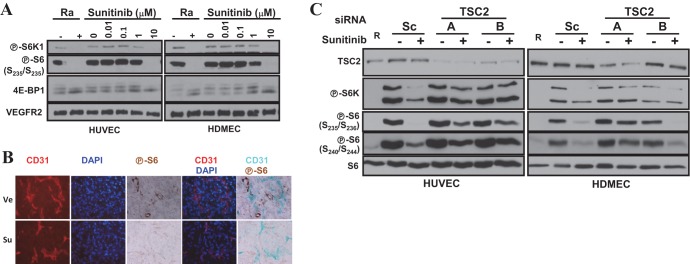
Sunitinib-mediated inhibition of TORC1 in primary human endothelial cells requires the TSC1/TSC2 complex. (A and C) Western blot analyses were performed using HUVEC or HDMEC treated with the indicated concentrations of sunitinib (A) or pretreated with the indicated siRNAs for 2 days and then exposed to sunitinib (10 μM) overnight (C). Ra or R, rapamycin. (B) Immunofluorescence and immunohistochemistry images of orthotopic renal cancer tumor grafts treated with vehicle (Ve) or sunitinib (Su) for 3 days.
The TSC1/TSC2 complex is required for sunitinib-mediated TORC1 inhibition in human endothelial cells.
Our results show that the Tsc1/Tsc2 complex is a critical mediator of sunitinib effects on TORC1 in insect cells, and we asked what the role was of TSC1/TSC2 in primary human endothelial cells. We depleted TSC2 from endothelial cells using siRNA and evaluated the effects of sunitinib. Interestingly, TSC2 depletion largely blocked the effects of sunitinib on TORC1 in both HUVEC and HDMEC (Fig. 15C). Thus, sunitinib-mediated TORC1 inhibition requires an intact TSC1/TSC2 complex. These data illustrate how experiments in lower organisms may provide insights into the mechanism of drug action in higher, more complex, systems.
DISCUSSION
Herein, we show that PVR is essential for ERK and TORC1 activity in insect cells. Furthermore, PVR is also important for maximal effector activation in response to insulin. We show that the activation of TORC1 and ERK requires Ras85D and Drk. In contrast, the other adaptor proteins, Lnk, Shc, Socs16D, and CG32406 (which we named PVRAP), are implicated only in TORC1 activation. Restoring TORC1 activity in PVR-depleted cells requires the inactivation of Tsc1/Tsc2 and Lobe in a cell-type-dependent manner. In contrast, TORC1 activity can be restored in cells depleted of Ras85D, Drk, and the other adaptor proteins simply by inactivating the Tsc1/Tsc2 complex. Constitutively active PVR mutants induce the expansion of hemocytes in Drosophila larvae and promote tumor-like structures that exhibit TORC1 activation. Conversely, PVR loss (or TORC1 inhibition) reduces hemocyte numbers and causes apoptosis. A similar phenotype is observed by treatment with the VEGF/PDGF receptor inhibitor sunitinib. Sunitinib inhibits PVR phosphorylation in insect cells, leading to the inhibition of ERK and TORC1. Sunitinib induces apoptosis in insect cells in vitro and blocks hemocyte expansion in vivo. The effects of sunitinib can be overcome by a constitutively dimerized PVR mutant, which causes hemocyte expansion. As in insect cells, sunitinib inhibits TORC1 in endothelial cells, both in vitro and in vivo, and TORC1 inhibition is TSC1/TSC2 dependent.
PVR is required for baseline TORC1 and ERK activity. It appears that among receptor tyrosine kinases, PVR plays a particularly important role as the activity of both TORC1 and ERK is linked to the state of PVR in both Kc and S2 cells. Furthermore, PVR was also required for maximal and sustained activation of both TORC1 and ERK in response to insulin. In mammals, there is cross talk between VEGF and insulin pathways. Insulin and VEGF receptors share common downstream effectors, and insulin receptor substrate 1 (IRS-1) is recruited to VEGFR2 in response to VEGF stimulation (77). In addition, insulin was shown to induce the expression of VEGF in different cell types (78–80). Given that PVR was required to sustain insulin-mediated activation of TORC1 and ERK, we speculate whether, akin to mammalian cells, insulin induces the expression of PVR ligands, leading to PVR activation and sustained activation of effector pathways. Furthermore, inasmuch as S6 kinase protein expression requires PVR, PVR may be necessary for sustained S6K activation by other RTKs besides the insulin receptor.
Our data show that Drk (Grb2) and Ras85D are important effectors implicated in the activation of both TORC1 and ERK. PVR loss was phenocopied by depletion of Drk or Ras85D. In contrast, depletion of Crk or Mbc had no effect on TORC1 or ERK. In cells expressing high levels of Lobe, TORC1 reactivation following PVR loss required the simultaneous inactivation of both Lobe and the Tsc1/Tsc2 complex. In contrast, TORC1 reactivation in cells depleted of Drk or Ras85D was accomplished by simply inactivating Tsc1/Tsc2. These data are consistent with the notion that PVR signals to Lobe in a Drk- and Ras85D-independent manner.
While Drk was required for the activation of both Ras–mitogen-activated protein kinase (MAPK) and TORC1 pathways, we identified other adaptor proteins that were selectively required for TORC1 activation. We found that Lnk, Shc, Socs16D, and PVRAP were all individually required for TORC1 activation. Why TORC1 activation would require each adaptor protein is unclear. However, 10 other adaptor proteins shown to interact with PVR by a yeast two-hybrid screen did not play a role in either Ras-MAPK or TORC1 activation. Interestingly, Lnk, Shc, Socs16D, and PVRAP were dispensable for Ras-MAPK activation. Lnk is the Drosophila homolog of mammalian SH2B proteins and was previously shown to be involved in insulin signaling (63). While in Kc and S2 cells TORC1 activity is coupled to PVR, we cannot exclude the possibility that Lnk ablation inhibits TORC1 by downregulating both PVR and insulin signaling. Shc is the Drosophila homolog of mammalian SHC3, and Shc was implicated in signaling downstream of Torso and EGFR (64). Interestingly, Shc was later found to associate with PVR in an independent study identifying the interacting networks in Drosophila cells (81). Socs16D is similar to mammalian SOCS6 and SOCS7 (65). In mammals, SOCS proteins are important negative regulators of the JAK-STAT pathway (65). In Drosophila, three Socs proteins have been reported, Socs16D, Socs36E, and Socs44A. While both Socs44A and Socs16D interacted with PVR in the yeast two-hybrid screen (61), only Socs16D was required for TORC1 activity. This suggests that Socs16D possesses unique features.
Among four adaptor proteins involved in TORC1 activation, PVRAP (CG32406) was the least studied. We showed that, as for the other adaptor proteins, PVRAP depletion abrogated TORC1 signaling. We generated inducible cells that expressed PVRAP and showed that PVRAP interacts with endogenous PVR. Together with experiments in yeast that show that PVRAP interacts with PVR in a kinase-dependent manner, our experiments show that PVRAP is an essential PVR adaptor protein implicated in TORC1 activation.
While the restoration of TORC1 activity in Lobe-expressing cells depleted of PVR required the simultaneous inactivation of Tsc1/Tsc2 and Lobe, in cells depleted for any of the adaptor proteins (or Ras85D), TORC1 activity was restored by the inactivation of Tsc1/Tsc2. Thus, the data suggest that PVR is linked to Lobe in a manner that is independent of all of these proteins, that these adaptor proteins work together, and, indeed, that each is essential for the activation of TORC1.
PVR is required for cell expansion in vitro and in vivo. PVR depletion reduced cell numbers in vitro and induced apoptosis. Similarly, hemocyte numbers were reduced by PVR inhibition in larvae. In contrast, Mondal et al. (82) showed that blockage of PVR function does not affect lymph gland-derived hemocyte numbers. This discrepancy may result from the examination of different hemocyte populations. Our study examined the effect of PVR on circulating hemocytes in Drosophila larvae. Drosophila hemocytes arise from two sources: the head mesoderm during early embryogenesis (embryonic hemocytes) and the mesoderm lymph glands at later stages of development (lymph gland-derived hemocytes) (83). While circulating hemocytes in adult flies represent a mixed population from both sources, circulating hemocytes at the larval stage arise exclusively from the embryonic hemocytes (84). Our data are consistent with the notion that PVR is required for embryonic hemocyte expansion. Furthermore, we show that PVR depletion caused apoptosis through a process that appears to be caspase independent. Conversely, constitutive PVR activation increased hemocyte numbers and led to the formation of tumor-like structures. These hypertrophic structures in imaginal discs were associated with TORC1 activation, and TORC1 inhibition reduced hemocyte numbers and induced apoptosis in Kc and S2 cells.
We performed experiments with the VEGF/PDGF receptor inhibitor sunitinib. Sunitinib inhibits PVR in Drosophila cells. Sunitinib blocked receptor tyrosine phosphorylation and the activity of ERK and TORC1. Furthermore, sunitinib suppressed the expansion of Kc and S2 cells in vitro and of hemocytes in vivo. Interestingly, however, whereas TORC1 reactivation in Lobe-expressing cells depleted of PVR requires the simultaneous inactivation of both Lobe and Tsc1/Tsc2, sunitinib-mediated inhibition of PVR could be overcome by inactivation of Tsc1/Tsc2 alone. The reconstitution of TORC1 by inactivation of Tsc1/Tsc2 was similar to that observed in cells depleted of Drk (or all other adaptor proteins). We show that sunitinib inhibits PVR tyrosine phosphorylation, which would be required for the binding of adaptor proteins. Thus, the rescue of TORC1 by Tsc1/Tsc2 inactivation in both instances (sunitinib treatment and adaptor depletion) is internally consistent. However, given the requirement for Lobe inactivation in cells depleted of PVR, these data raise the intriguing possibility that PVR loss activates Lobe in a kinase independent manner. As for PVR, sunitinib treatment induced apoptosis and reduced the number of hemocytes in vivo, and similar effects were observed upon inactivation of TORC1, suggesting that TORC1 is an important effector. Sunitinib failed to inhibit constitutively dimerized PVR, and our results suggest that similar mutations causing stable receptor dimerization in human tumors may render them refractory to sunitinib.
As in insect cells, sunitinib treatment inhibited TORC1 in endothelial cells in vitro and in vivo, and sunitinib-mediated TORC1 inhibition required a functional TSC1/TSC2 complex. TORC1 is an important regulator of cell growth and proliferation, and TORC1 inhibition may contribute to explain the antitumor effects of sunitinib. There are two modes of sunitinib action against tumors, and our results have implications for both. First, sunitinib is prescribed to target tumor cells, such as those with mutations in RTK that can be bound by sunitinib. Second, sunitinib is used in renal cancer, not because it targets renal cancer cells, which lack mutations in RTK and are in fact unaffected by sunitinib in vitro (35), but, rather, because sunitinib inhibits endothelial cells and renal cancers are particularly dependent on angiogenesis. In the first case, tumors with somatically acquired mutations in TSC1 (or TSC2) may be less responsive to sunitinib inhibition. Our findings may, in fact, explain why sunitinib was inactive against xenografts derived from Tsc2-deficient fibroblasts (85). The same notion may explain the lack of effect of a second clinically utilized VEGF and PDGF receptor inhibitor, sorafenib, against Tsc2-deficient xenografts (86). Thus, despite the fact that sunitinib inhibits ERK in a TSC1/TSC2-independent manner, these xenograft experiments suggest that, at least in some contexts, TORC1 inhibition may be required for its antiproliferative effects. In the second case, and particularly given that a modest knockdown of TSC2 is sufficient to block sunitinib-induced TORC1 inhibition, sunitinib may be less effective in patients with tuberous sclerosis complex who have germ line mutations in the TSC1 or TSC2 genes. These germ line mutations may reduce the effect of sunitinib on endothelial cells and thereby lower the effectiveness of sunitinib against renal cancer, a cancer type that these patients are predisposed to.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Brugarolas lab for helpful discussions, Amanda Stram for assistance with the dsRNA screen, and N. Perrimon for the dsRNA library. We also thank the following for providing reagents: Pernile Rörth, Denise Montell, Ernest Hafen, Jing Jiang, Nahum Sonenberg, Lily Huang, T. Megraw, and John Abrams. We thank Blanka Kucejova, Sharanya Sivanand, Andrea Pavia-Jimenez, and Patrick Spence for providing tumor sections of vehicle and sunitinib-treated tumor grafts.
The RNAi screen was supported in part by 5R01CA068490 to William G. Kaelin. Other funding includes T32CA124334 and F32CA1360872 to T.A.T and the following grants to J.B.: K08NS051843 and RO1CA129387, as well as a Basil O'Connor Starter Scholar Research Award (5-FY06-582) from the March of Dimes Foundation. J.B. is a Virginia Murchison Linthicum Scholar in Medical Research at UT Southwestern.
The content is solely the responsibility of the authors and does not represent official views from any of the granting agencies.
Footnotes
Published ahead of print 22 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01570-12.
REFERENCES
- 1.Hoch RV, Soriano P. 2003. Roles of PDGF in animal development. Development 130:4769–4784 [DOI] [PubMed] [Google Scholar]
- 2.Heldin CH, Westermark B. 1999. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 79:1283–1316 [DOI] [PubMed] [Google Scholar]
- 3.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. 2006. VEGF receptor signalling—in control of vascular function. Nat. Rev. Mol. Cell Biol. 7:359–371 [DOI] [PubMed] [Google Scholar]
- 4.Ferrara N, Gerber HP, LeCouter J. 2003. The biology of VEGF and its receptors. Nat. Med. 9:669–676 [DOI] [PubMed] [Google Scholar]
- 5.Cross MJ, Dixelius J, Matsumoto T, Claesson-Welsh L. 2003. VEGF-receptor signal transduction. Trends Biochem. Sci. 28:488–494 [DOI] [PubMed] [Google Scholar]
- 6.Gerber HP, Malik AK, Solar GP, Sherman D, Liang XH, Meng G, Hong K, Marsters JC, Ferrara N. 2002. VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature 417:954–958 [DOI] [PubMed] [Google Scholar]
- 7.Kissa K, Herbomel P. 2010. Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature 464:112–115 [DOI] [PubMed] [Google Scholar]
- 8.Bertrand JY, Chi NC, Santoso B, Teng S, Stainier DY, Traver D. 2010. Haematopoietic stem cells derive directly from aortic endothelium during development. Nature 464:108–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boisset JC, van Cappellen W, Andrieu-Soler C, Galjart N, Dzierzak E, Robin C. 2010. In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature 464:116–120 [DOI] [PubMed] [Google Scholar]
- 10.Andrae J, Gallini R, Betsholtz C. 2008. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 22:1276–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones AV, Cross NC. 2004. Oncogenic derivatives of platelet-derived growth factor receptors. Cell. Mol. Life Sci. 61:2912–2923 [DOI] [PubMed] [Google Scholar]
- 12.Otrock ZK, Makarem JA, Shamseddine AI. 2007. Vascular endothelial growth factor family of ligands and receptors: review. Blood Cells Mol. Dis. 38:258–268 [DOI] [PubMed] [Google Scholar]
- 13.Tallquist M, Kazlauskas A. 2004. PDGF signaling in cells and mice. Cytokine Growth Factor Rev. 15:205–213 [DOI] [PubMed] [Google Scholar]
- 14.Makinen T, Veikkola T, Mustjoki S, Karpanen T, Catimel B, Nice EC, Wise L, Mercer A, Kowalski H, Kerjaschki D, Stacker SA, Achen MG, Alitalo K. 2001. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. EMBO J. 20:4762–4773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Padro T, Bieker R, Ruiz S, Steins M, Retzlaff S, Burger H, Buchner T, Kessler T, Herrera F, Kienast J, Muller-Tidow C, Serve H, Berdel WE, Mesters RM. 2002. Overexpression of vascular endothelial growth factor (VEGF) and its cellular receptor KDR (VEGFR-2) in the bone marrow of patients with acute myeloid leukemia. Leukemia 16:1302–1310 [DOI] [PubMed] [Google Scholar]
- 16.Antonescu CR, Yoshida A, Guo T, Chang NE, Zhang L, Agaram NP, Qin LX, Brennan MF, Singer S, Maki RG. 2009. KDR activating mutations in human angiosarcomas are sensitive to specific kinase inhibitors. Cancer Res. 69:7175–7179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin DE, Hall MN. 2005. The expanding TOR signaling network. Curr. Opin. Cell Biol. 17:158–166 [DOI] [PubMed] [Google Scholar]
- 18.Avruch J, Long X, Ortiz-Vega S, Rapley J, Papageorgiou A, Dai N. 2009. Amino acid regulation of TOR complex 1. Am. J. Physiol. Endocrinol. Metab. 296:E592–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu Y, Sato JD. 1999. MAP kinases, phosphatidylinositol 3-kinase, and p70 S6 kinase mediate the mitogenic response of human endothelial cells to vascular endothelial growth factor. J. Cell. Physiol. 178:235–246 [DOI] [PubMed] [Google Scholar]
- 20.Seko Y, Takahashi N, Tobe K, Ueki K, Kadowaki T, Yazaki Y. 1998. Vascular endothelial growth factor (VEGF) activates Raf-1, mitogen-activated protein (MAP) kinases, and S6 kinase (p90rsk) in cultured rat cardiac myocytes. J. Cell. Physiol. 175:239–246 [DOI] [PubMed] [Google Scholar]
- 21.Ming XF, Burgering BM, Wennstrom S, Claesson-Welsh L, Heldin CH, Bos JL, Kozma SC, Thomas G. 1994. Activation of p70/p85 S6 kinase by a pathway independent of p21ras. Nature 371:426–429 [DOI] [PubMed] [Google Scholar]
- 22.Chung J, Grammer TC, Lemon KP, Kazlauskas A, Blenis J. 1994. PDGF- and insulin-dependent pp70S6k activation mediated by phosphatidylinositol-3-OH kinase. Nature 370:71–75 [DOI] [PubMed] [Google Scholar]
- 23.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. 2005. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121:179–193 [DOI] [PubMed] [Google Scholar]
- 24.Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. 2004. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. U. S. A. 101:13489–13494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Potter CJ, Pedraza LG, Xu T. 2002. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 4:658–665 [DOI] [PubMed] [Google Scholar]
- 26.Inoki K, Li Y, Zhu T, Wu J, Guan KL. 2002. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4:648–657 [DOI] [PubMed] [Google Scholar]
- 27.Dan HC, Sun M, Yang L, Feldman RI, Sui XM, Ou CC, Nellist M, Yeung RS, Halley DJ, Nicosia SV, Pledger WJ, Cheng JQ. 2002. Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J. Biol. Chem. 277:35364–35370 [DOI] [PubMed] [Google Scholar]
- 28.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. 2002. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/Akt pathway. Mol. Cell 10:151–162 [DOI] [PubMed] [Google Scholar]
- 29.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. 2007. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 9:316–323 [DOI] [PubMed] [Google Scholar]
- 30.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. 2007. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25:903–915 [DOI] [PubMed] [Google Scholar]
- 31.Wang L, Harris TE, Lawrence JC., Jr 2008. Regulation of proline-rich Akt substrate of 40 kDa (PRAS40) function by mammalian target of rapamycin complex 1 (mTORC1)-mediated phosphorylation. J. Biol. Chem. 283:15619–15627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oshiro N, Takahashi R, Yoshino K, Tanimura K, Nakashima A, Eguchi S, Miyamoto T, Hara K, Takehana K, Avruch J, Kikkawa U, Yonezawa K. 2007. The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J. Biol. Chem. 282:20329–20339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dancey J. 2010. mTOR signaling and drug development in cancer. Nat. Rev. Clin. Oncol. 7:209–219 [DOI] [PubMed] [Google Scholar]
- 34.Sun L, Liang C, Shirazian S, Zhou Y, Miller T, Cui J, Fukuda JY, Chu JY, Nematalla A, Wang XY, Chen H, Sistla A, Luu TC, Tang F, Wei J, Tang C. 2003. Discovery of 5-5-fluoro-2-oxo-1,2-dihydroindol-(3Z)-ylidenemethyl-2,4-dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J. Med. Chem. 46:1116–1119 [DOI] [PubMed] [Google Scholar]
- 35.Huang D, Ding Y, Li Y, Luo WM, Zhang ZF, Snider J, Vandenbeldt K, Qian CN, Teh BT. 2010. Sunitinib acts primarily on tumor endothelium rather than tumor cells to inhibit the growth of renal cell carcinoma. Cancer Res. 70:1053–1062 [DOI] [PubMed] [Google Scholar]
- 36.Duchek P, Somogyi K, Jekely G, Beccari S, Rorth P. 2001. Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell 107:17–26 [DOI] [PubMed] [Google Scholar]
- 37.Cho NK, Keyes L, Johnson E, Heller J, Ryner L, Karim F, Krasnow MA. 2002. Developmental control of blood cell migration by the Drosophila VEGF pathway. Cell 108:865–876 [DOI] [PubMed] [Google Scholar]
- 38.Bruckner K, Kockel L, Duchek P, Luque CM, Rorth P, Perrimon N. 2004. The PDGF/VEGF receptor controls blood cell survival in Drosophila. Dev. Cell 7:73–84 [DOI] [PubMed] [Google Scholar]
- 39.Munier AI, Doucet D, Perrodou E, Zachary D, Meister M, Hoffmann JA, Janeway CA, Jr, Lagueux M. 2002. PVF2, a PDGF/VEGF-like growth factor, induces hemocyte proliferation in Drosophila larvae. EMBO Rep. 3:1195–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zettervall CJ, Anderl I, Williams MJ, Palmer R, Kurucz E, Ando I, Hultmark D. 2004. A directed screen for genes involved in Drosophila blood cell activation. Proc. Natl. Acad. Sci. U. S. A. 101:14192–14197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Learte AR, Forero MG, Hidalgo A. 2008. Gliatrophic and gliatropic roles of PVF/PVR signaling during axon guidance. Glia 56:164–176 [DOI] [PubMed] [Google Scholar]
- 42.Jekely G, Sung HH, Luque CM, Rorth P. 2005. Regulators of endocytosis maintain localized receptor tyrosine kinase signaling in guided migration. Dev. Cell 9:197–207 [DOI] [PubMed] [Google Scholar]
- 43.Ishimaru S, Ueda R, Hinohara Y, Ohtani M, Hanafusa H. 2004. PVR plays a critical role via JNK activation in thorax closure during Drosophila metamorphosis. EMBO J. 23:3984–3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bond D, Foley E. 2009. A quantitative RNAi screen for JNK modifiers identifies Pvr as a novel regulator of Drosophila immune signaling. PLoS Pathog. 5:e1000655. 10.1371/journal.ppat.1000655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miron M, Lasko P, Sonenberg N. 2003. Signaling from Akt to FRAP/TOR targets both 4E-BP and S6K in Drosophila melanogaster. Mol. Cell. Biol. 23:9117–9126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stocker H, Andjelkovic M, Oldham S, Laffargue M, Wymann MP, Hemmings BA, Hafen E. 2002. Living with lethal PIP3 levels: viability of flies lacking PTEN restored by a PH domain mutation in Akt/PKB. Science 295:2088–2091 [DOI] [PubMed] [Google Scholar]
- 47.Reiling JH, Hafen E. 2004. The hypoxia-induced paralogs Scylla and Charybdis inhibit growth by down-regulating S6K activity upstream of TSC in Drosophila. Genes Dev. 18:2879–2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Worby CA, Simonson-Leff N, Dixon JE. 2001. RNA interference of gene expression (RNAi) in cultured Drosophila cells. Sci. STKE 2001:pl1. 10.1126/scisignal.952001pl1 [DOI] [PubMed] [Google Scholar]
- 49.Wolff NC, Vega-Rubin-de-Celis S, Xie XJ, Castrillon DH, Kabbani W, Brugarolas J. 2011. Cell-type-dependent regulation of mTORC1 by REDD1 and the tumor suppressors TSC1/TSC2 and LKB1 in response to hypoxia. Mol. Cell. Biol. 31:1870–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crevel I, Crevel G, Gostan T, de Renty C, Coulon V, Cotterill S. 2011. Decreased MCM2-6 in Drosophila S2 cells does not generate significant DNA damage or cause a marked increase in sensitivity to replication interference. PLoS One 6:e27101. 10.1371/journal.pone.0027101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vega-Rubin-de-Celis S, Abdallah Z, Kinch L, Grishin NV, Brugarolas J, Zhang X. 2010. Structural analysis and functional implications of the negative mTORC1 Regulator REDD1. Biochemistry 49:2491–2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pena-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, Wolff NC, Tran TA, Zou L, Xie XJ, Corey DR, Brugarolas J. 2011. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 30:3242–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG., Jr 2004. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 18:2893–2904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sivanand S, Pena-Llopis S, Zhao H, Kucejova B, Spence P, Pavia-Jimenez A, Yamasaki T, McBride DJ, Gillen J, Wolff NC, Morlock L, Lotan Y, Raj GV, Sagalowsky A, Margulis V, Cadeddu JA, Ross MT, Bentley DR, Kabbani W, Xie XJ, Kapur P, Williams NS, Brugarolas J. 2012. A validated tumorgraft model reveals activity of dovitinib against renal cell carcinoma. Sci. Transl. Med. 4:137ra175. 10.1126/scitranslmed.3003649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Katoh K, Toh H. 2008. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 9:286–298 [DOI] [PubMed] [Google Scholar]
- 56.Adachi J, Hasegawa M. 1992. MOLPHY, programs for molecular phylogenetics, I: PROTML, maximum likelihood inference of protein phylogeny. Institute of Statistical Mathematics, Tokyo, Japan [Google Scholar]
- 57.Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. 2009. The SWISS-MODEL repository and associated resources. Nucleic Acids Res. 37:D387–D392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pearson RB, Dennis PB, Han JW, Williamson NA, Kozma SC, Wettenhall RE, Thomas G. 1995. The principal target of rapamycin-induced p70s6k inactivation is a novel phosphorylation site within a conserved hydrophobic domain. EMBO J. 14:5279–5287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fernandez-Almonacid R, Rosen OM. 1987. Structure and ligand specificity of the Drosophila melanogaster insulin receptor. Mol. Cell. Biol. 7:2718–2727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lizcano JM, Alrubaie S, Kieloch A, Deak M, Leevers SJ, Alessi DR. 2003. Insulin-induced Drosophila S6 kinase activation requires phosphoinositide 3-kinase and protein kinase B. Biochem. J. 374:297–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sears HCR. 2004. Identification and characterization of proteins that bind the intracellular domain of PVR. PhD. thesis. Massachusetts Institute of Technology, Cambridge, MA [Google Scholar]
- 62.Tari AM, Lopez-Berestein G. 2001. GRB2: a pivotal protein in signal transduction. Semin. Oncol. 28:142–147 [DOI] [PubMed] [Google Scholar]
- 63.Werz C, Kohler K, Hafen E, Stocker H. 2009. The Drosophila SH2B family adaptor Lnk acts in parallel to chico in the insulin signaling pathway. PLoS Genet. 5:e1000596. 10.1371/journal.pgen.1000596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luschnig S, Krauss J, Bohmann K, Desjeux I, Nusslein-Volhard C. 2000. The Drosophila SHC adaptor protein is required for signaling by a subset of receptor tyrosine kinases. Mol. Cell 5:231–241 [DOI] [PubMed] [Google Scholar]
- 65.Rawlings JS, Rennebeck G, Harrison SM, Xi R, Harrison DA. 2004. Two Drosophila suppressors of cytokine signaling (SOCS) differentially regulate JAK and EGFR pathway activities. BMC Cell Biol. 5:38. 10.1186/1471-2121-5-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rosin D, Schejter E, Volk T, Shilo BZ. 2004. Apical accumulation of the Drosophila PDGF/VEGF receptor ligands provides a mechanism for triggering localized actin polymerization. Development 131:1939–1948 [DOI] [PubMed] [Google Scholar]
- 67.Gajiwala KS, Wu JC, Christensen J, Deshmukh GD, Diehl W, DiNitto JP, English JM, Greig MJ, He YA, Jacques SL, Lunney EA, McTigue M, Molina D, Quenzer T, Wells PA, Yu X, Zhang Y, Zou A, Emmett MR, Marshall AG, Zhang HM, Demetri GD. 2009. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc. Natl. Acad. Sci. U. S. A. 106:1542–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liegl B, Kepten I, Le C, Zhu M, Demetri GD, Heinrich MC, Fletcher CD, Corless CL, Fletcher JA. 2008. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J. Pathol. 216:64–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, Town A, McKinley A, Ou WB, Fletcher JA, Fletcher CD, Huang X, Cohen DP, Baum CM, Demetri GD. 2008. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J. Clin. Oncol. 26:5352–5359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, Leversha MA, Jeffrey PD, Desantis D, Singer S, Brennan MF, Maki RG, DeMatteo RP. 2005. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin. Cancer Res. 11:4182–4190 [DOI] [PubMed] [Google Scholar]
- 71.McTigue M, Murray BW, Chen JH, Deng YL, Solowiej J, Kania RS. 2012. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. U. S. A. 109:18281–18289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y. 1998. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 279:577–580 [DOI] [PubMed] [Google Scholar]
- 73.Chompret A, Kannengiesser C, Barrois M, Terrier P, Dahan P, Tursz T, Lenoir GM, Bressac-De Paillerets B. 2004. PDGFRA germline mutation in a family with multiple cases of gastrointestinal stromal tumor. Gastroenterology 126:318–321 [DOI] [PubMed] [Google Scholar]
- 74.Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den Abbeele AD, Druker BJ, Kiese B, Eisenberg B, Roberts PJ, Singer S, Fletcher CD, Silberman S, Dimitrijevic S, Fletcher JA. 2003. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J. Clin. Oncol. 21:4342–4349 [DOI] [PubMed] [Google Scholar]
- 75.Laine E, Chauvot de Beauchene I, Perahia D, Auclair C, Tchertanov L. 2011. Mutation D816V alters the internal structure and dynamics of c-KIT receptor cytoplasmic region: implications for dimerization and activation mechanisms. PLoS Comput. Biol. 7:e1002068. 10.1371/journal.pcbi.1002068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang HM, Yu X, Greig MJ, Gajiwala KS, Wu JC, Diehl W, Lunney EA, Emmett MR, Marshall AG. 2010. Drug binding and resistance mechanism of KIT tyrosine kinase revealed by hydrogen/deuterium exchange FTICR mass spectrometry. Protein Sci. 19:703–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Senthil D, Ghosh Choudhury G, Bhandari BK, Kasinath BS. 2002. The type 2 vascular endothelial growth factor receptor recruits insulin receptor substrate-1 in its signalling pathway. Biochem. J. 368:49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Miele C, Rochford JJ, Filippa N, Giorgetti-Peraldi S, Van Obberghen E. 2000. Insulin and insulin-like growth factor-I induce vascular endothelial growth factor mRNA expression via different signaling pathways. J. Biol. Chem. 275:21695–21702 [DOI] [PubMed] [Google Scholar]
- 79.Poulaki V, Qin W, Joussen AM, Hurlbut P, Wiegand SJ, Rudge J, Yancopoulos GD, Adamis AP. 2002. Acute intensive insulin therapy exacerbates diabetic blood-retinal barrier breakdown via hypoxia-inducible factor-1α and VEGF. J. Clin. Invest. 109:805–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bermont L, Lamielle F, Lorchel F, Fauconnet S, Esumi H, Weisz A, Adessi GL. 2001. Insulin up-regulates vascular endothelial growth factor and stabilizes its messengers in endometrial adenocarcinoma cells. J. Clin. Endocrinol. Metab. 86:363–368 [DOI] [PubMed] [Google Scholar]
- 81.Guruharsha KG, Rual JF, Zhai B, Mintseris J, Vaidya P, Vaidya N, Beekman C, Wong C, Rhee DY, Cenaj O, McKillip E, Shah S, Stapleton M, Wan KH, Yu C, Parsa B, Carlson JW, Chen X, Kapadia B, VijayRaghavan K, Gygi SP, Celniker SE, Obar RA, Artavanis-Tsakonas S. 2011. A protein complex network of Drosophila melanogaster. Cell 147:690–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mondal BC, Mukherjee T, Mandal L, Evans CJ, Sinenko SA, Martinez-Agosto JA, Banerjee U. 2011. Interaction between differentiating cell- and niche-derived signals in hematopoietic progenitor maintenance. Cell 147:1589–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Traver D, Zon LI. 2002. Walking the walk: migration and other common themes in blood and vascular development. Cell 108:731–734 [DOI] [PubMed] [Google Scholar]
- 84.Holz A, Bossinger B, Strasser T, Janning W, Klapper R. 2003. The two origins of hemocytes in Drosophila. Development 130:4955–4962 [DOI] [PubMed] [Google Scholar]
- 85.Woodrum C, Nobil A, Dabora SL. 2010. Comparison of three rapamycin dosing schedules in A/J. Tsc2+/− mice and improved survival with angiogenesis inhibitor or asparaginase treatment in mice with subcutaneous tuberous sclerosis related tumors. J. Transl. Med. 8:14. 10.1186/1479-5876-8-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee N, Woodrum CL, Nobil AM, Rauktys AE, Messina MP, Dabora SL. 2009. Rapamycin weekly maintenance dosing and the potential efficacy of combination sorafenib plus rapamycin but not atorvastatin or doxycycline in tuberous sclerosis preclinical models. BMC Pharmacol. 9:8. 10.1186/1471-2210-9-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.