

Abstract
Bilirubin exhibits antioxidant and antimutagenic effects in vitro. Additional tetrapyrroles that are naturally abundant were tested for antigenotoxicity in Salmonella. Un-/conjugated bilirubin (1 and 2), biliverdin (4), bilirubin and biliverdin dimethyl esters (3 and 5), stercobilin (6), urobilin (7), and protoporphyrin (8) were evaluated at physiological concentrations (0.01–2 μmol/plate; 3.5–714 μM) against the metabolically activated food-borne mutagens aflatoxin B1 (9) and 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (10). Compound 8 most effectively inhibited the mutagenic effects of 9 in strain TA102 and 10 in TA98. Compound 7 inhibited 9-induced mutagenesis in strain TA98 most effectively, while 1 and 4 were promutagenic in this strain. This is likely due to their competition with mutagens for phase-II detoxification. Mechanistic investigations into antimutagenesis demonstrate that tetrapyrroles react efficiently with a model epoxide of 9, styrene epoxide (11), to form covalent adducts. This reaction is significantly faster than that of 11 with guanine. Hence, the evaluated tetrapyrroles inhibited genotoxicity induced by poly-/heterocyclic amines found in foods, and novel evidence obtained in the present investigation suggests this may occur via chemical scavenging of genotoxic metabolites of the mutagens investigated. This may have important ramifications for maintaining health, especially with regard to cancer prevention.
Aflatoxin B1 (9) and 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (10) represent food-contaminating mutagens, as found in some nuts/cereals and cooked meat.1,2 Upon absorption and hepatic transport, the molecules undergo oxidation/glucuronidation by cytochrome systems (CYPs) and UDP-glucuronosyltransferases (UGTs),3 in pathways commonly known as phase-I/II detoxification. These processes give rise to short-lived, highly reactive molecules that readily react with DNA,4 thereby producing DNA–metabolite adducts.5,6 Mutagenic intermediate formation can result in DNA strand breaks7 and mutations,8,9 which are relevant precursors to malignant transformation.10,11 Hepatic processing of harmful bioactivated intermediates represents a key event in mutagenesis and involves the action of multiple CYP isoforms to form exo-/endoepoxides12,13 (Figure 1). The efficiency of this processing determines the concentration and half-life of these reactive species.
Figure 1.

Simplified summary of the metabolism of aflatoxin B1 (AfB1, 9), which can lead to DNA–adduct formation. Inset: Structure of styrene epoxide (11) chosen as a mimic for AFB1-exo-8,9-epoxide in reactivity studies.
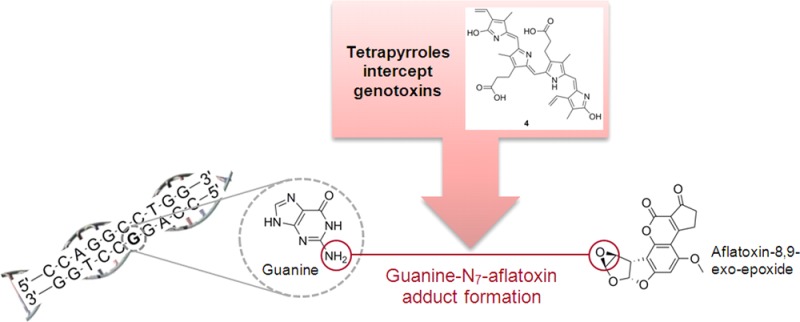
AFB1-exo-8,9-epoxide (Figure 1) is an example of a mutagenic metabolite of 9. This compound reacts with DNA bases, particularly guanine (G), to produce covalent adducts, thereby causing DNA damage. Due to the health risk associated with experimentation with derivatives of 9, and national restrictions regarding its use in Australia, the less reactive styrene epoxide (11; Figure 1) was chosen as a model for investigations into possible inhibitory mechanisms of 9-induced mutagenesis by tetrapyrroles. Compound 11 can react with the G bases (N7 position) of DNA,14,15 in a reaction analogous to that of 9-epoxide with G. The aromatic ring of 11 mimics the aromatic moiety of 9-epoxide, which can participate in π-electron interactions with the π-systems of tetrapyrroles and thus mimic any similar interactions that may occur between tetrapyrroles and derivatives of 9.
The main CYPs involved in activation of 10 are similar to those responsible for the metabolism of 9 and include CYP1A2, which transforms 10 to its 2-hydroxyamino intermediate. Subsequent conjugation/esterification by sulfotransferase/acetyltransferase generates O-sulfonyl/O-acetyl esters.16 During its metabolism, 10, like 9 and tetrapyrroles, undergoes glucuronidation by hepatic UGT-glucuronosyltransferase isoenzymes (UGT1 isoforms),17 to generate glucuronides of 10 from its main 2-hydroxyamino and 4′-hydroxy intermediates.18,19
In addition to xenobiotic detoxification, the liver plays an essential role in the metabolism of potentially protective tetrapyrrolic molecules.20−22 Unconjugated bilirubin (1) and its derivatives are biological heme catabolites formed within the liver and spleen.23 Heme oxygenase converts heme to biliverdin (4), and biliverdin reductase subsequently generates 1, which is glucuronidated in the liver by the UGT1A family of enzymes.24 Deficiency in UGT1A1 activity, as seen in Gilbert’s syndrome, results in a mild unconjugated hyperbilirubinemia,25,26 which may interfere with xenobiotic metabolism.27 After biliary excretion, 1 in its conjugated form is further metabolized in the gut, forming stercobilin (6) and urobilin (7), which are reabsorbed or eliminated via the urogenital and intestinal tracts. Interest in the physiological importance of tetrapyrroles is increasing, due to multiple, large epidemiological studies describing a protective relationship between 1 and lung28 and colorectal cancers.29 An underlying mechanism of protection may be related to the antioxidant capacity of tetrapyrroles; however, no conclusive evidence of their ability to react with electrophilic mutagenic intermediates has thus far been published. The present contribution is the first to document antimutagenic effects of tetrapyrroles against food-derived mutagens, which has explicit implications for understanding why these compounds are so clearly associated with the reduced incidence of colorectal cancer. Due to their enterohepatic abundance, it is hypothesized that specific tetrapyrroles could reduce the risk of carcinogenesis by interacting with food-borne contaminants,30,31 hindering their absorption, or by reacting with mutagenic intermediates in the liver,22 thereby reducing oxidative DNA damage/adduct formation. With the abundance of 1–8 in the gut/enterocytes and bile, revealing novel compound effects could lead to the development of additional therapeutics to reduce cancer in at-risk populations.32
This study aimed to (1) reveal novel effects of endogenous tetrapyrroles on (dietary-derived) mutation induced by 9 and 10 and to (2) establish whether tetrapyrroles react with 11, a model epoxide of 9, to form covalent conjugates.

Results and Discussion
AfB1 (9)-Induced Mutagenesis in Salmonella typhimurium Strain TA102
All bilirubinoids including 1–3 significantly reduced the mutagenic effects of 9 (p < 0.01; Figure 2A). Both 4 and 5 also reduced revertant counts significantly versus the positive control (p < 0.001; Figure 2B), with 4 being the more effective compound. Compound 8, however, was the most effective of all tetrapyrroles tested, reducing the revertant counts in strain TA102 (p < 0.001; Figure 2C). Compound 7 attenuated mutation induced by 9 at all concentrations tested, with similar results achieved for 6 (p < 0.05; Figure 2C). The overall order of effectiveness based on IP0.5 values (Table 1) was 8 > 1 = 2 > 4 > 3 > 6 = 7 > 5.
Figure 2.

Antimutagenic effects of (A) bilirubin (1), bilirubin ditaurate (2), bilirubin dimethyl ester (3); (B) biliverdin (4), biliverdin dimethyl ester (5); and (C) stercobilin (6), urobilin (7), and protoporphyrin (8) against metabolically activated aflatoxin B1 (AfB1, 9; 0.24 × 10–6 mol/plate)-induced mutagenesis in Salmonella typhimurium strain TA102.
Table 1. Antimutagenic Behavior of Tetrapyrroles against Metabolically Activated AfB1 (9) and PhIP (10) in Salmonella typhimurium Strains TA102 and TA98.
| strain | mutagen [mol/plate] | S9 | tetrapyrrolea | IP0.5 [pos control inhibition, %]b | significantly different from (p ≤ 0.05) | His+pos ± SDc |
|---|---|---|---|---|---|---|
| TA102 | 9, 0.24 × 10–6 | + | 8 | –68 | 1, 2, 3, 4, 5, 6, 7 | 1142 ± 112 |
| 1 | –54 | 5, 6, 7, 8 | ||||
| 2 | –54 | 5, 6, 7, 8 | ||||
| 4 | –52 | 3, 5, 6, 7, 8 | ||||
| 3 | –45 | 1, 2, 4, 5, 7, 8 | ||||
| 6 | –43 | 1, 2, 4, 5, 8 | ||||
| 7 | –42 | 1, 2, 3, 4, 5, 8 | ||||
| 5 | –27 | 1, 2, 3, 4, 6, 7, 8 | ||||
| TA98 | 9, 0.8 × 10–7 | + | 7 | –79 | 1, 2, 3, 4, 5, 6, 8 | 318 ± 26 |
| 5 | –59 | 1, 2, 3, 4, 6, 7, 8 | ||||
| 3 | –45 | 1, 2, 4, 5, 6, 7, 8 | ||||
| 6 | –37 | 1, 2, 3, 4, 5, 7, 8 | ||||
| 8 | –11 | 1, 2, 3, 4, 5, 6, 7 | ||||
| 2 | +268 | 1, 3, 4, 5, 6, 7, 8 | ||||
| 1 | +352 | 2, 3, 5, 6, 7, 8 | ||||
| 4 | +363 | 2, 3, 5, 6, 7, 8 | ||||
| TA98 | 10, 0.1 × 10–7 | + | 8 | –97 | 1, 2, 3, 4, 5, 6, 7 | 789 ± 77 |
| 1 | –82 | 2, 3, 5, 6, 7, 8 | ||||
| 4 | –78 | 1, 2, 3, 5, 6, 7, 8 | ||||
| 3 | –68 | 1, 2, 4, 5, 6, 7, 8 | ||||
| 2 | –64 | 1, 3, 4, 5, 6, 7, 8 | ||||
| 6 | –57 | 1, 2, 3, 4, 7, 8 | ||||
| 5 | –55 | 1, 2, 3, 4, 7, 8 | ||||
| 7 | –48 | 1, 2, 3, 4, 5, 6, 8 |
1: unconjugated bilirubin, 2: bilirubin ditaurate, 3: bilirubin dimethyl ester, 4: biliverdin, 5: biliverdin dimethyl ester, 6: stercobilin, 7: urobilin, 8: protoporphyrin. AfB1: aflatoxin B1 (9), PhIP: 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (10).
IP0.5: percentage positive control inhibition at 0.5 μmol/plate (highest common sample dose per plate): – indicates mutagen attenuating effect, + amplifying effect.
His+pos: average positive control revertant counts ± SD.
AfB1 (9)-Induced Mutagenesis in Salmonella typhimurium Strain TA98
Compound 7 was the most antimutagenic of all compounds tested in strain TA98 and reduced mutagenesis induced by 9 by up to 75% (p < 0.05; Figure 3C). Significant antimutagenic effects were also demonstrated for 3, 5, and 6 with a lower but still measurable effect observed for 8 (p < 0.05; Figure 3A–C). The overall order of effectiveness based on IP0.5 values (Table 1) was 7 > 5 > 3 > 6 > 8. In contrast to these compounds and to the previous TA102 strain, 1, 2, and 4 did not attenuate mutagenesis provoked by 9 under the test conditions used (Figure 3A), and 1 and 4 had a promutagenic effect (p > 0.05).
Figure 3.

Antimutagenic effects of (A) bilirubin (1), bilirubin ditaurate (2), bilirubin dimethyl ester (3); (B) biliverdin (4), biliverdin dimethyl ester (5); and (C) stercobilin (6), urobilin (7), and protoporphyrin (8) against metabolically activated aflatoxin B1 (AfB1, 9; 0.8 × 10–7 mol/plate)-induced mutagenesis in Salmonella typhimurium strain TA98.
PhIP (10)-Induced Mutagenesis in Salmonella typhimurium Strain TA98
The most effective tetrapyrrole at inhibiting mutagenesis induced by 10 in TA98 was 8, which attenuated the mutagenic effects by more than 90% and resulted in near complete detoxification of the mutagen (p < 0.001; Figure 4C). All bilirubinoids (1–3) were highly effective against mutagenesis provoked by 10 (p < 0.001; Figure 4A) and attenuated its effects by over 60%. Compounds 4 and 5 also reduced revertant counts over the entire range of concentrations tested (p < 0.001; Figure 4B) as did 6 and 7 (p < 0.05; Figure 4C). The overall order of effectiveness based on IP0.5 values (Table 1) was 8 > 1 > 4 > 3 > 2 > 6 > 5 > 7.
Figure 4.

Antimutagenic effects of (A) bilirubin (1), bilirubin ditaurate (2), bilirubin dimethyl ester (3); (B) biliverdin (4), biliverdin dimethyl ester (5); and (C) stercobilin (6), urobilin (7), and protoporphyrin (8) against metabolically activated 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP, 10; 0.1 × 10–7 mol/plate)-induced mutagenesis in Salmonella typhimurium strain TA98.
Results from the bacterial model of this study demonstrate that tetrapyrroles inhibit the mutagenic effects of 9 in both S. typhimurium strains TA102 and TA98. The tetrapyrroles used, however, vary greatly in the planarity of their structure, aromaticity, and extent of conjugation and in the presence or absence of free carboxylic acid groups. The irregularity in effectiveness of the various tetrapyrroles tested across the two strains, therefore, suggests that the presence of a single, unifying mechanism of action for this inhibition is unlikely. Previous studies have suggested that the planar, aromatic nature of 9 allows for tetrapyrrole complex formation through π-stacking interactions, inhibiting covalent33,34 and noncovalent interaction between DNA and 9.35−38 Similarly, complex formation in the gut between porphyrins and food-borne mutagens, followed by electrophile scavenging and complexing, has been suggested.39,40 If this is the major mechanism of the inhibition of mutagenesis, the more conjugated, planar tetrapyrroles such as 4 and 8 would be expected to be the most potent inhibitors of those tetrapyrroles tested. While 8 showed the highest potency in the 9-TA102 experiment, derivatives of 1 were the next most potent, and these are both not planar nor fully conjugated. Simple π-electron-mediated complexation is therefore unlikely to be the sole explanation for the antimutagenic effects of tetrapyrroles.
Dashwood et al. proposed that the exocyclic amine groups of poly-/heterocyclic mutagens covalently bind to free −COOH groups in chlorophyllin.41 In the 9-TA102 study, 1 and 4 exhibited significantly higher inhibition activity than their respective dimethyl esters (3 and 5), suggesting that the −COOH groups present in 1 and 4 may be key to their activity. It is difficult to draw a solid conclusion, as 6 and 7 both contain −COOH groups and were less potent inhibitors of mutation than 3 in this assay. However, the reduced potency of 6 and 7 may be related to the progressive loss of aromaticity of their terminal pyrrole rings.
Despite the potencies of 1 and 4 in the 9-TA102 experiment, these two compounds were promutagenic in the TA98 strain. This promutagenic effect is likely due to 1 and 4 competing with AfB1 for the same phase-II metabolism enzymes,42 thus resulting in increased concentrations of reactive intermediates of 9. This has a relatively higher impact on strain TA98, because its DNA is particularly sensitive to G-adduct formation.43 From a physiological perspective, this result has potential important ramifications for individuals with Gilbert’s syndrome, who experience impaired UGT1A1 activity44 and mildly elevated concentrations of 1,45 based on a less efficient glucuronidation.
The observation that compound 8 was the most effective agent against 9 using strain TA102 and was moderately effective in strain TA98 confirms the previously reported potent antigenotoxic activity of this substance.22 However, this is the first investigation to demonstrate an inhibitory effect against food-derived genotoxins. A previous study has suggested that 8 is a strong reducing agent based on its extended π-electron system,22 allowing for covalent binding capacity with biomolecules.46,47 This supports the hypothesis that 8 inhibits mutagenesis induced by 9, by direct chemical reaction with the mutagen.
As observed previously in the 9-TA102 experiment of this study, 8 most effectively inhibited the mutagenic effects of 10 in strain TA98. This suggests that 8 is a potent inhibitor of frame-shift mutations at concentrations present within the intestine/enterocytes (0.2–14 μM).48 Considering that frame-shift mutations are an important cause of carcinogenesis in gastric and colon cancer,49,50 these results could be of importance in explaining a possible protective role for 8, due to its enterohepatic distribution. With reference to related tetrapyrroles, this hypothesis is supported by a preceding study that did not include 8,21 but showed that bacterial absorption of 1 and 4 protected against frame-shift mutation in strain TA98. This observation might explain in part the inverse relationship between serum levels of 1 and colorectal cancer in vivo.51
Tetrapyrrole–SO (11) Model Reaction
Mass spectrometry (low-resolution ESIMS) analysis of the initial experiments indicated that G (MW = 151 g/mol) under the test conditions reacted with 11 (MW = 120 g/mol) to produce two detectable products, with the major one (>90% by MS signal intensity) giving a [M + H]+ peak at 272 amu and the minor product (<10%) showing an m/z of 392 ([M + H]+). These correspond to covalent adducts of G with one and two units of 11, respectively. The conversion of G to these adducts over a seven-day period was low, and a large quantity of unreacted G was recovered. A control experiment confirmed that G in the presence of silica gel alone was stable over seven days under these conditions. In the presence of selected tetrapyrroles (reactions 3–5 in Table 2), none of the G–11 adducts were detected; however, several compounds with molecular weights corresponding to tetrapyrrole–11 adducts were detected. In the case of 4 and 8, peaks corresponding to unreacted G and tetrapyrrole were accompanied by peaks assigned to adducts of tetrapyrrole and one to five units of 11. For compound 1, evidence of the conversion of 1 to 4, followed by reaction of both tetrapyrroles with one to four units of 11, was observed. The addition of 11 to the tetrapyrroles was repeated in the absence of G, yielding similar results. The extent of reaction of tetrapyrroles with 11 was high over the seven-day period of study, with only small amounts of unreacted tetrapyrroles remaining, in contrast to the significantly less facile reaction between G and 11. Table 2 summarizes the positive ion ESIMS analysis of the reaction products.
Table 2. Summary of the Reactions Performed to Assess the Reactivity of Tetrapyrroles to a Model Epoxide (11) and the ESIMS Analysis of the Reaction Products.
| reaction | tetrapyrrolea | guanineb (m/z = 151) | SO (m/z = 120) | CHCl3, silica gel | ESIMSc of products (m/z, [M + H]+) | reaction type |
|---|---|---|---|---|---|---|
| 1 | N | Y | N | Y | 152 (G) | controls |
| 2 | N | Y | Y | Y | 152 (G); 272 (G-11); 392 (G-(2×11)) | |
| 3 | 8 | Y | Y | Y | 152 (G); 563 (8); 683 (8–11); 803 (8-(2×11)); 923 (8-(3×11));1043 (8-(4×11)); 1163 (8-(5×11)) | tetrapyrroles with G |
| 4 | 1 | Y | Y | Y | 152 (G); 585 (1); 583 (4); 703, 705 ((4–11), (1–11)); 823, 825 (4-(2×11), 1-(2×11)); 943, 945 (4-(3×11), 1-(3×11)); 1063, 1065 (4-(4×11), 1-(4×11)) | |
| 5 | 4 | Y | Y | Y | 152 (G); 583 (4); 703 (4–11); 823 (4-(2×11)); 943 (4-(3×11)); 1063 (4-(4×11)); 1183 (4-(5×11)) | |
| 6 | 8 | N | Y | Y | 563 (8); 683 (8–11); 803 (8-(2×11)); 923 (8-(3×11)); 1043 (8-(4×11)); 1163 (8-(5×11)) | tetrapyrroles without G |
| 7 | 1 | N | Y | Y | 585 (1); 583 (4); 703, 705 ((4–11), (1–11)); 823, 825 (4-(2×11), 1-(2×11)); 943, 945 (4-(3×11), 1-(3×11)); 1063, 1065 (4-(4×11), 1-(4×11)) | |
| 8 | 4 | N | Y | Y | 583 (4); 703 (4–11); 823 (4-(2×11)); 943 (4-(3×11)); 1063 (4-(4×11)); 1183 (4-(5×11)) | |
| 9 | 8 | N | N | Y | 563 (8) | controls without 11 |
| 10 | 1 | N | N | Y | 585 (1); 583 (4) | |
| 11 | 4 | N | N | Y | 583 (4) |
1: unconjugated bilirubin; 4: biliverdin; 8: protoporphyrin; 11: styrene epoxide (SO).
G: guanine.
ESIMS: low-resolution mass spectrometry.
The negative ion ESIMS analysis showed the corresponding negative ions and thereby fully supported these data. These results confirmed that tetrapyrroles react with activated aromatic epoxides to form covalent adducts more efficiently and more rapidly than G reacts with the same epoxide.
Tetrapyrrole–Epoxide (11) Interactions
To examine the possibility of covalent adduct formation of reactive epoxides of 9 and tetrapyrroles, novel reactions of 1, 4, and 8 with 11 were performed. The data clearly showed tetrapyrroles efficiently react with 11, with several units of 11 being conjugated to each tetrapyrrole. While it was demonstrated that G does react with 11, it was significantly less efficient than the reaction of tetrapyrroles with G. When G was placed in reaction with 11 in the presence of any of the tetrapyrroles, no G adduct was detected. While any compound with a −COOH group would have been expected to react with 11 via a nucleophilic opening of 11 to provide the ester, this would only account for two units of 11 per molecule of tetrapyrrole. The data indicate that more units are being conjugated to the tetrapyrrole, suggesting that there are other sites on the molecules that may be reactive to epoxides. These preliminary data emphasize that the antimutagenic properties of tetrapyrroles are partly a consequence of their ability to react with an activated epoxide (11) more quickly than DNA bases, forming covalent adducts, and thus reducing cellular DNA damage.
In summary, the experiments conducted in this investigation demonstrate that endogenous tetrapyrroles exert antigenotoxic effects, by preventing mutation caused by food-derived mutagens. This is the first study to provide mechanistic insight showing that tetrapyrroles react more readily than G with a model epoxide (11) to form covalent adducts. Therefore, the data suggest endogenous tetrapyrroles may neutralize reactive epoxides before they react with DNA and cause mutation. Although not directly transferable to the human condition, this investigation could have important ramifications for maintaining health. This is particularly pertinent to cancer prevention within the digestive tract, as one of the main loci of tetrapyrrole abundance.
Experimental Section
General Experimental Procedures
Low-resolution mass spectrometric data were recorded in positive- and negative-electrospray ionization modes, using a Bruker Esquire HCT (high-capacity 3D ion trap) instrument, with a Bruker ESI source. For MS analysis, the solutions were diluted in MeOH to a concentration of 100 nM. Thin-layer chromatography was performed on Merck silica 60 F254 sheets. Flash column chromatography was performed on Lomb silica gel 60 (0.04–0.06 mm; 230–400 mesh), which was also used as the activating agent in the reactivity studies.
Mechanistic Studies: Reactions between Tetrapyrroles and a Model Epoxide (11)
The first experiments were designed to assess the reactivity of selected tetrapyrroles (1, 4, and 8) and G with 11. Compound 11 (5 μmol) was dissolved in CHCl3, and powdered silica gel (3 mg) was added as a proton source in the absence of H2O.52 Guanine (1 μmol) and each tetrapyrrole (1 μmol) were then added separately, with the final reaction volume being 1 mL. Each vial was sealed under an inert atmosphere (Ar gas), protected from light, and was allowed to stand at 37 °C for one week (reactions 3–5 in Table 2). Control reactions containing G and 11 with and without silica gel in CHCl3 were also prepared in the same fashion (reactions 1 and 2 in Table 2). The second experiment directly investigated reactions between tetrapyrroles and 11 without G. Compound 11 (5 μmol), a selected tetrapyrrole (1 μmol), and silica gel (3 mg) were again dissolved in CHCl3 (1 mL), placed under Ar, sealed, wrapped in foil, and allowed to react at 37 °C for one week (reactions 6–8 in Table 2). Control reactions containing a tetrapyrrole and silica gel in the absence of 11 were also performed (reactions 9–11 in Table 2).
After incubation, all reactions were subjected to flash column chromatography, initially using 100% CH2Cl2 as eluent, increasing the percentage of MeOH in the eluting solvent from 0% to 30%. The fractions containing the compounds of interest eluted as mixtures in 5% MeOH to 30% MeOH. These fractions were further purified by a second chromatographic column using toluene/acetone (from 0% acetone to 50% acetone). All fractions from the chromatographic column were examined by low-resolution ESIMS, as described below.
Salmonella typhimurium Reverse Mutation Assay
The Salmonella reverse mutation assay is a well-accepted screening test to evaluate the mutagenic potential of chemicals in vitro.53 Mutagenic substances revert mutated bacteria to their wild-type variant, which allows them to grow. Genotoxicity (or vice versa antigenotoxic effects) of compounds can therefore be assessed by quantifying bacterial growth relative to controls. The experiments performed followed the method of Maron and Ames54 and included 25 min of preincubation. S9 liver homogenate (S9 microsomal fraction from Aroclor 1254 pretreated rats; MP Biomedicals, Illkirch, France) was used as an enzymatic activation system in all of the assays.
Bacterial Strains
Salmonella typhimurium strains were obtained from Dr. Bruce N. Ames (University of California, Berkeley, CA, USA). Frozen permanents were stored at −80 °C until use. Prior to the tests being conducted, the genetic integrity and spontaneous mutation rates of the strains were assessed.53 Two distinct strains were adopted to consider multiple mechanisms of protection from mutation: the DNA repair proficient strain TA102 (hisG428 mutation), for the detection of A–T base pair damage and small deletions provoked by cross-linking agents, which can be reverted by mutagens causing oxidative damage, and TA98 (hisD3052 mutation), which detects primarily G–C base pair and frame-shift mutations. The S. typhimurium strains used in the experiments have different mutations in various genes in the histidine operon; each of these mutations is designed to be responsive to mutagens that act via different mechanisms.53
Salmonella Reverse Mutation Assay: Experimental Design
The assays were performed in a dimly lit laboratory under aseptic working conditions (laminar airflow cabinet Safemate 1.8; Bioair Euroclone, Milan, IT). Samples were protected from light throughout the experiments and were freshly prepared in amber vials before each test. PBS (500 μL) or S9-mix (19.75 mL of distilled H2O, 25 mL of PBS, 500 μL of MgCl2 (0.85 M), 0.5 mL of KCl (1.65 M), 2 mL of NADP (90.8 mM), 250 μL of glucose-6-phosphate (1.08 M), and 2 mL of S9), 100 μL of overnight culture, and 200 μL of a selected tetrapyrrole solution (in DMSO) were mixed in sterile test tubes. For antimutagenic testing, 100 μL of mutagen (in DMSO or DMSO alone for controls; Table 1) was added to each tube. After 25 min of preincubation (37 °C, on a rotary shaker), 2 mL of molten top agar was added to every tube. The mixtures were poured onto minimum glucose agar plates that were incubated at 37 °C for 48 h. His+ revertants were counted manually after having routinely checked the background lawn under a microscope (40× magnification; Olympus CH-2). DMSO final plate concentrations did not exceed 10% v/v.
In the Austrian laboratories, work with mutagens was performed according to directive 2004/37/EC of the European Parliament and the Council of April 29, 2004, on the protection of workers from the risks related to exposure to carcinogens or mutagens at work (Sixth Individual Directive within the meaning of Article 16(1) of Council Directive 89/391/EEC). In Australia, Australia Worksafe by the National Occupational Health and Safety Commission, Approved Criteria for Classifying Hazardous Substances (1994), was followed.
Chemicals
Compounds 1 (bilirubin IXα, CAS# 635-65-4), 2 (bilirubin ditaurate, CAS# 89771-93-7), 3 (bilirubin dimethyl ester, CAS# 19792-68-8), 4 (biliverdin IXα, CAS# 55482-27-4), 5 (biliverdin dimethyl ester, CAS# 10035-62-8), 6 (stercobilin, CAS# 34217-90-8), 7 (urobilin, CAS# 28925-89-5), and 8 (protoporphyrin IX, CAS# 553-12-8) were purchased from Frontier Scientific, Carnforth, Lancashire, UK. Compound purity (>98%) and solubility were assessed using HPLC (Hitachi HPLC, equipped with a Shimadzu SPD-M20A detector, a Fortis C18 reversed-phase column (4.6 × 150 mm, 3 μm), and a Phenomenex C18 guard column (4 × 3 mm)) and visible spectrophotometry (Perkin-Elmer Lambda 2 UV/vis spectrophotometer). All other reagents were from Sigma-Aldrich AT and AU (unless otherwise noted), were of the highest analytical grade available, and were stored and used according to the manufacturer’s instructions. Compound 10 was purchased from Toronto Research Chemicals, North York, ON, CA. Tetrapyrrolic compounds were solubilized in DMSO and protected from light using foil, and solutions were used immediately. The composition and preparation of all necessary reagents and solutions have been published elsewhere.55
Sample Preparation
Following procedures that have been previously published,55 six doses of 2, 4, and the previously untested compounds 3 and 5–8 were investigated at 0.01, 0.05, 0.1, 0.5, 1, and 2 μmol/plate (3.5–714 μM). Compound 1 was tested over a range of five doses, including 0.01, 0.05, 0.1, 0.5, and 0.75 μmol/plate (3.5–268 μM). The lowest concentrations tested were selected based on an ability to reduce mutagenicity by 50% and to reflect physiologically relevant concentrations present within the enterohepatic circulation. The respective maximum sample doses were ascertained: (1) by testing the maximum amount of DMSO that did not result in bacterial cytotoxicity (350 μL/plate, ca. 12% v/v) and (2) from the respective maximum solubility of each pigment (supernatant analysis: visible spectrophotometry: 1–3, 455 nm; 4, 5, 380 nm; 6, 7, 400 nm; 8, 410 nm) read on a Perkin-Elmer Lambda 2 UV/vis spectrophotometer after high-speed centrifugation; HPLC analysis was performed as published previously.56,57
Each sample was tested in triplicate, and all of the assays were repeated independently. Every test included three positive control plates (mutagen only) and six negative control plates (no mutagen, no pigment) as well as three no-treatment plates (no mutagen, no pigment, no DMSO). For a substance to be classified as genotoxic in the Salmonella reverse mutation assay, the number of revertant colonies on the test compound plates had to exceed twice the number of colonies grown on the solvent control plates (negative control).53
In addition to investigating the antimutagenic effects of the tetrapyrroles selected for study, the spontaneous mutagenic activities of these compounds were tested; for each sample the respective highest and lowest concentrations were applied without mutagen addition. None of the samples tested caused mutagenesis in either of the bacterial strains (p > 0.05).
Positive and Negative Controls
Aflatoxin B1 (9) induces both base-pair substitution mutation and frame-shift mutation in those strains of S. typhimurium carrying the R-factor plasmid pKM101,58,59 which applies to both of the selected tester strains.53 Metabolism of 9 involves a series of cytochrome enzymatic processes to generate the 8,9-epoxide, which forms covalent DNA and RNA adducts.4 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (10) is a potent mutagen in strain TA98, but is virtually inactive in strain TA102.60 It is metabolized by multiple CYPs, forming a 2-hydroxyamino intermediate, mainly provoking frame-shift and base substitution mutations.61
Table 3. Summary of the Positive Controls Used for Antigenotoxic Testing in the Salmonella Reverse Mutation Assay.
| mutagena | occurrence | class | solvent | strain(s)b | concentration [mol/plate] | S9c |
|---|---|---|---|---|---|---|
| 9 | food borne (agricultural produce, nuts, cereals)62 | planar polycyclic amine; mycotoxin; mutagen | DMSO | TA98 | 0.8 × 10–7 | + |
| TA102 | 0.24 × 10–6 | + | ||||
| 10 | food borne; high-temperature cooking of meat.1,2 Alternative source: tobacco smoke63 | planar heterocyclic amine (HCA), 2-aminoimidazole structure; mutagen | DMSO | TA98 | 0.1 × 10–7 | + |
9: aflatoxin B1 (AfB1), 10: 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP).
TA98/TA102: S. typhimurium strains TA98/TA102.
S9: + indicates metabolic activation was used.
Statistical Analysis
Data were analyzed using IBM SPSS 17.0 for Windows. A p-value ≤ 0.05 was considered significant. All data are presented as means ± SD. Data were tested for normal distribution (Kolmogorov–Smirnov test). Parametric statistical analyses (one-way analysis of variance and the Scheffé post hoc test) were performed on normally distributed data, and corresponding nonparametric tests (Kruskal–Wallis test, Dunnet T3 post hoc test) on not normally distributed data. To assess the order of effectiveness of the test compounds, data were analyzed using IP0.5 values (percentage positive control inhibition at 0.5 μmol/plate test compound), which were determined using the software Derive 6 (Texas Instruments, Dallas, TX, USA).
Acknowledgments
This work was supported by the Austrian Science Fund (Fonds zur Förderung der Wissenschaftlichen Forschung, FWF, grant number P21162-B11).
The authors declare no competing financial interest.
References
- Choudhuri S.; Klaassen C. D. Int. J. Toxicol. 2006, 25, 231–259. [DOI] [PubMed] [Google Scholar]
- Rohrmann S.; Hermann S.; Linseisen J. Am. J. Clin. Nutr. 2009, 89, 1418–1424. [DOI] [PubMed] [Google Scholar]
- Hanioka N.; Nonaka Y.; Saito K.; Negishi T.; Okamoto K.; Kataoka H.; Narimatsu S. Chemosphere 2012, 89, 526–529. [DOI] [PubMed] [Google Scholar]
- Sotomayor R. E.; Sahu S.; Washington M.; Hinton D. M.; Chou M. Environ. Mol. Mutagen. 1999, 33, 293–302. [PubMed] [Google Scholar]
- Guengerich F. P.; Hosea N. A.; Parikh A.; Bell-Parikh L. C.; Johnson W. W.; Gillam E. M.; Shimada T. Drug Metab. Dispos. 1998, 26, 1175–1178. [PubMed] [Google Scholar]
- Guengerich F. P.; Johnson W. W.; Shimada T.; Ueng Y. F.; Yamazaki H.; Langouet S. Mutat. Res. 1998, 402, 121–128. [DOI] [PubMed] [Google Scholar]
- Park U. S.; Su J. J.; Ban K. C.; Qin L.; Lee E. H.; Lee Y. I. Gene 2000, 251, 73–80. [DOI] [PubMed] [Google Scholar]
- Majer B. J.; Hofer E.; Cavin C.; Lhoste E.; Uhl M.; Glatt H. R.; Meinl W.; Knasmuller S. Food Chem. Toxicol. 2005, 43, 433–441. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K.; Nagao M.; Esumi H.; Sugimura T. Cancer Res. 1992, 52, 2092–2098. [PubMed] [Google Scholar]
- Hagiwara A.; Miyashita K.; Nakanishi T.; Sano M.; Tamano S.; Kadota T.; Koda T.; Nakamura M.; Imaida K.; Ito N.; Shirai T. Cancer Lett. 2001, 171, 17–25. [DOI] [PubMed] [Google Scholar]
- Smela M. E.; Currier S. S.; Bailey E. A.; Essigmann J. M. Carcinogenesis 2001, 22, 535–545. [DOI] [PubMed] [Google Scholar]
- Gallagher E. P.; Kunze K. L.; Stapleton P. L.; Eaton D. L. Toxicol. Appl. Pharmacol. 1996, 141, 595–606. [DOI] [PubMed] [Google Scholar]
- Ueng Y. F.; Shimada T.; Yamazaki H.; Peterguengerich F. J. Toxicol. Sci. 1998, 23Suppl. 2132–135. [DOI] [PubMed] [Google Scholar]
- Vodicka P.; Koskinen M.; Vodicková L.; Štetina R.; Šmerák P.; Bárta I.; Hemminki K. Chem. Biol. Interact. 2001, 137, 213–227. [DOI] [PubMed] [Google Scholar]
- Phillips D. H.; Farmer P. B. Crit. Rev. Toxicol. 1994, 24Suppl. 1S35–S46. [DOI] [PubMed] [Google Scholar]
- Ozawa S.; Chou H. C.; Kadlubar F. F.; Nagata K.; Yamazoe Y.; Kato R. Jpn. J. Cancer Res. 1994, 85, 1220–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strassburg C. P.; Manns M. P.; Tukey R. H. J. Biol. Chem. 1998, 273, 8719–8726. [DOI] [PubMed] [Google Scholar]
- Han J. F.; He X. Y.; Herrington J. S.; White L. A.; Zhang J. F.; Hong J. Y. Drug Metab. Dispos. 2008, 36, 745–752. [DOI] [PubMed] [Google Scholar]
- Nowell S. A.; Massengill J. S.; Williams S.; Radominska-Pandya A.; Tephly T. R.; Cheng Z.; Strassburg C. P.; Tukey R. H.; MacLeod S. L.; Lang N. P.; Kadlubar F. F. Carcinogenesis 1999, 20, 1107–1114. [DOI] [PubMed] [Google Scholar]
- Bulmer A. C.; Ried K.; Blanchfield J. T.; Wagner K. H. Mutat. Res. 2008, 658, 28–41. [DOI] [PubMed] [Google Scholar]
- Mölzer C.; Huber H.; Diem K.; Wallner M.; Bulmer A. C.; Wagner K. H. Toxicol. In Vitro 2012, 27, 433–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mölzer C.; Huber H.; Steyrer A.; Ziesel G.; Wallner M.; Ertl A.; Plavotic A.; Bulmer A.; Wagner K.-H. Free Radical Res. 2012, 46, 1369–1377. [DOI] [PubMed] [Google Scholar]
- Stocker R.; Yamamoto Y.; McDonagh A. F.; Glazer A. N.; Ames B. N. Science 1987, 235, 1043–1046. [DOI] [PubMed] [Google Scholar]
- Klatskin G. Annu. Rev. Med. 1961, 12, 211–250. [DOI] [PubMed] [Google Scholar]
- Vitek L. Front. Pharmacol. 2012, 3551–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner M.; Blassnigg S. M; Marisch K.; Pappenheim M. T.; Müllner E.; Mölzer C.; Nersesyan A.; Marculescu R.; Doberer D.; Knasmüller S.; Bulmer A. C.; Wagner K.-H. Mutagenesis 2012, 27, 731–735. [DOI] [PubMed] [Google Scholar]
- Strassburg C. P. Pharmacogenomics 2008, 9, 703–715. [DOI] [PubMed] [Google Scholar]
- Horsfall L. J.; Rait G.; Walters K.; Swallow D. M.; Pereira S. P.; Nazareth I.; Petersen I. JAMA 2011, 305, 691–697. [DOI] [PubMed] [Google Scholar]
- Jiraskova A.; Novotny J.; Novotny L.; Vodicka P.; Pardini B.; Naccarati A.; Schwertner H. A.; Hubacek J. A.; Puncocharova L.; Smerhovsky Z.; Vitek L. Int. J. Cancer 2012, 131, 1549–1555. [DOI] [PubMed] [Google Scholar]
- Dalezios J. I.; Hsieh D. P.; Wogan G. N. Food Cosmet. Toxicol. 1973, 11, 605–616. [DOI] [PubMed] [Google Scholar]
- Mykkanen H.; Zhu H.; Salminen E.; Juvonen R. O.; Ling W.; Ma J.; Polychronaki N.; Kemilainen H.; Mykkanen O.; Salminen S.; El-Nezami H. Int. J. Cancer 2005, 115, 879–884. [DOI] [PubMed] [Google Scholar]
- Dashwood R.; Guo D. In Proceedings of the International Symposium of the Princess Takamatsu Cancer Research Fund; 26th Princess Takamatsu Symposium, Genomic Instability and Carcinogenesis, Nov 1995; pp 181–189.
- Hayatsu H. In Proceedings of the International Symposium of the Princess Takamatsu Cancer Research Fund; 26th Princess Takamatsu Symposium, Genomic Instability and Carcinogenesis, Nov 1995; pp 172–180.
- Hayatsu H.; Sugiyama C.; Arimoto-Kobayashi S.; Negishi T. Cancer Lett. 1999, 143, 185–187. [DOI] [PubMed] [Google Scholar]
- Arimoto S.; Hayatsu H. Mutat. Res. 1989, 213, 217–226. [DOI] [PubMed] [Google Scholar]
- Arimoto S.; Negishi T.; Hayatsu H. Cancer Lett. 1980, 11, 29–33. [DOI] [PubMed] [Google Scholar]
- Newmark H. L. Nutr. Cancer 1984, 6, 58–70. [DOI] [PubMed] [Google Scholar]
- Mölzer C.; Huber H.; Steyrer A.; Ziesel G. V.; Wallner M.; Goncharova I.; Orlov S.; Urbanová M.; Ahlfors C. E.; Vítek L.; Bulmer A. C.; Wagner K.-H.. J. Porphyrins Phthalocyanines 2013, in press, doi: 10.1142/S1088424613500995. [DOI] [Google Scholar]
- Breinholt V.; Schimerlik M.; Dashwood R.; Bailey G. Chem. Res. Toxicol. 1995, 8, 506–514. [DOI] [PubMed] [Google Scholar]
- Hayatsu H.; Negishi T.; Arimoto S.; Hayatsu T. Mutat. Res. 1993, 290, 79–85. [DOI] [PubMed] [Google Scholar]
- Dashwood R. H.; Breinholt V.; Bailey G. S. Carcinogenesis 1991, 12, 939–942. [DOI] [PubMed] [Google Scholar]
- Bulmer A. C.; Coombes J. S.; Blanchfield J. T.; Toth I.; Fassett R. G.; Taylor S. M. Br. J. Pharmacol. 2011, 164, 1857–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santella R. M. IATREIA 2007, 20Suppl. 238–39. [Google Scholar]
- Kundur A. R.; Bulmer A. C.; Singh I.. Platelets 2013, in press, doi: 10.3109/09537104.2013.764405. [DOI] [Google Scholar]
- Boon A. C.; Hawkins C. L.; Bisht K.; Coombes J. S.; Bakrania B.; Wagner K. H.; Bulmer A. C. Free Radical Biol. Med. 2012, 52, 2120–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anslyn E. V., Dougherty D. A.. Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, 2005. [Google Scholar]
- Meyer E. A.; Castellano R. K.; Diederich F. Angew. Chem., Int. Ed. 2003, 42, 1210–1250. [DOI] [PubMed] [Google Scholar]
- Beukeveld G. J.; Wolthers B. G.; van Saene J. J.; de Haan T. H.; de Ruyter-Buitenhuis L. W.; van Saene R. H. Clin. Chem. 1987, 33, 2164–2170. [PubMed] [Google Scholar]
- Garbe Y.; Maletzki C.; Linnebacher M. PLoS One 2011, 6, e26517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. R.; Chung N. G.; Kang M. R.; Yoo N. J.; Lee S. H. Tumori 2010, 96, 1004–1009. [PubMed] [Google Scholar]
- Zucker S. D.; Horn P. S.; Sherman K. E. Hepatology 2004, 40, 827–835. [DOI] [PubMed] [Google Scholar]
- Kotsuki H.; Hayashida K.; Shimanouchi T.; Nishizawa H. J. Org. Chem. 1996, 61, 984–990. [Google Scholar]
- Mortelmans K.; Zeiger E. Mutat. Res. 2000, 455, 29–60. [DOI] [PubMed] [Google Scholar]
- Maron D. M.; Ames B. N. Mutat. Res. 1983, 113, 173–215. [DOI] [PubMed] [Google Scholar]
- Bulmer A. C.; Ried K.; Coombes J. S.; Blanchfield J. T.; Toth I.; Wagner K. H. Mutat. Res. 2007, 629, 122–132. [DOI] [PubMed] [Google Scholar]
- Brower J. O.; Lightner D. A.; McDonagh A. F. Tetrahedron 2001, 57, 7813–7827. [Google Scholar]
- Bulmer A. C.; Blanchfield J. T.; Coombes J. S.; Toth I. Bioorg. Med. Chem. 2008, 16, 3616–3625. [DOI] [PubMed] [Google Scholar]
- McCann J.; Spingarn N. E.; Kobori J.; Ames B. N. Proc. Natl. Acad. Sci. U.S.A. 1975, 72, 979–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skopek T. R.; Liber H. L.; Kaden D. A.; Thilly W. G. Proc. Natl. Acad. Sci. U.S.A. 1978, 75, 4465–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felton J. S, Knize M. G. In Handbook of Experimental Pharmacology; Cooper C. S., Grover P. L., Eds.; Springer-Verlag: Berlin, 1990; Vol. 94, pp 471–502. [Google Scholar]
- Felton J. S.; Knize M. G.; Dolbeare F. A.; Wu R. Environ. Health Perspect. 1994, 102Suppl. 6201–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapsford K. E.; Taitt C. R.; Fertig S.; Moore M. H.; Lassman M. E.; Maragos C. M.; Shriver-Lake L. C. Biosens. Bioelectron. 2006, 21, 2298–2305. [DOI] [PubMed] [Google Scholar]
- Hecht S. S. Carcinogenesis 2002, 23, 907–922. [DOI] [PubMed] [Google Scholar]