

SUMMARY
The ubiquitin-proteasome and autophagy-lysosome systems are major proteolytic pathways, whereas function of the Ub-independent proteasome pathway is yet to be clarified. Here, we investigated roles of the Ub-independent REGγ-proteasome proteolytic system in regulating metabolism. We demonstrate that mice deficient for the proteasome activator REGγ exhibit dramatic autophagy induction and are protected against high-fat diet (HFD)-induced liver steatosis through autophagy. Molecularly, prevention of steatosis in the absence of REGγ entails elevated SirT1, a deacetylase regulating autophagy and metabolism. REGγ physically binds to SirT1, promotes its Ub-independent degradation and inhibits its activity to deacetylate autophagy-related proteins, thereby inhibiting autophagy under normal conditions. Moreover, REGγ and SirT1 dissociate from each other through a phosphorylation-dependent mechanism under energy-deprived conditions, unleashing SirT1 to stimulate autophagy. These observations provide a function of the REGγ proteasome in autophagy and hepatosteatosis, underscoring mechanistically a cross-talk between the proteasome and autophagy degradation system in the regulation of lipid homeostasis.
INTRODUCTION
Macroautophagy (hereafter referred to as autophagy) is a conserved degradative process that is essential for cellular homeostasis and quality control and mediates degradation of damaged or excess proteins and organelles in lysosomes (Mizushima, 2007). Its dysregulation is involved in many physiological disorders and human diseases (Mizushima, 2007). Recent studies reveal that autophagy is required for breakdown of lipid droplets and inhibition of autophagy leads to steatosis and fatty liver in mice (Czaja, 2010; Singh et al., 2009). Autophagy also regulates adipocyte differentiation and fat storage (Zhang et al., 2009). These findings present autophagy as a novel therapeutic target that could potentially be manipulated to treat diseases accompanied by excess lipid accumulation (Singh and Cuervo, 2012). Nevertheless, regulatory factors linking autophagy and lipid metabolisms urgently await discovery.
SirT1 (yeast Sir2) is a protein deacetylase that acts as a master metabolic sensor of NAD+ and has been reported to modulate life span and cellular metabolism (Guarente, 2000). SirT1 reduces fat accumulation in white adipose (Picard et al., 2004) and promotes browning of white adipose (Qiang et al., 2012). SirT1 overexpression reduces high-fat diet induced steatosis and improves insulin sensitivity (Pfluger et al., 2008), whereas loss of SirT1 leads to liver steatosis and inflammation (Purushotham et al., 2009). In addition, SirT1 provides a cell survival advantage in response to stress by deacetylating a number of substrates, such as p53 (Luo et al., 2001) and FOXOs (Brunet et al., 2004). SirT1 can be regulated by FOXO3a, p53 and HIC1 at the level of transcription (Chen et al., 2005; Nemoto et al., 2004), and is regulated by DBC1 through protein-protein interaction (Zhao et al., 2008). SirT1 expression is augmented following fasting (Nemoto et al., 2004). We previously reported that DNA damaging agents also induce SirT1 expression (Wang et al., 2006). Importantly, overexpression of SirT1 stimulates autophagy, and SirT1 knockout MEF cells cannot fully activate autophagy under starved conditions (Lee et al., 2008). However, molecular factors and mechanisms that control SirT1 autophagic function are largely unexplored.
The proteasome is a large protein complex consisting of a 20S proteolytic core and three proteasomal activators, 19S (or PA700), 11S (or PA28, REG) and PA200. The 19S activator binds to the 20S core and primarily mediates degradation of ubiquitinated proteins. The 11S activator binds to the proteasome and mainly promotes Ub-independent degradation. However, little attention has been paid to the Ub-independent proteolysis in eukaryotes. Our investigations have revealed that REGγ (or PA28γ), one of the 11S proteasomal activators (Dubiel et al., 1992; Ma et al., 1992), promotes Ub-independent degradation of SRC-3 and p21 (Li et al., 2007; Li et al., 2006). In this study, we found that REGγ knockout mice exhibit autophagy and are protected against HFD-induced liver steatosis through enhanced autophagy. REGγ also serves as a master regulator in switch off/on autophagy under normal and energy-deprivation conditions by regulating SirT1. Our findings suggest that REGγ is a potential therapeutic target for disordered lipid metabolism.
RESULTS
REGγ plays a role in regulating lipid metabolism and HFD-induced liver steatosis
REGγ knockout (KO) mice were reported to display reduced body weight and growth retardation (Barton et al., 2004; Murata et al., 1999). This prompted us to investigate the functional role of REGγ in metabolism. To determine if and how REGγ expression impacts lipid metabolism in vivo, we challenged REGγ-KO mice with a high-fat diet. Interestingly, we observed a slightly increased food intake in REGγ-KO mice on HFD (Fig. 1A). Loss of REGγ had a significant beneficial effect in mice, preventing them from developing liver steatosis after 19 weeks of HFD (Fig. 1B–C) in comparison with the wild-type (WT) littermates, despite that they had similar weight gains during HFD feeding (Fig. S1A). Differential lipid metabolism in these mice was further demonstrated by classic Oil Red O staining of livers, showing that the REGγ-KO mice exhibited a marked reduction of HFD-induced hepatic lipid accumulation (Fig. 1D, upper panel); mice with standard chow diet (CD) had little noticeable changes (Fig. 1D, lower panel). Quantification of the liver and serum triglycerides (TG) levels in mice confirmed that REGγ-KO mice were resistant to HFD-induced liver steatosis (Fig. 1E–F). These data support that REGγ-KO mice are protected from HFD-induced hepatic steatosis.
Fig. 1. REGγ regulates fat accumulation and REGγ-deficient mice are protected from high fat diet (HFD)-induced liver steatosis.
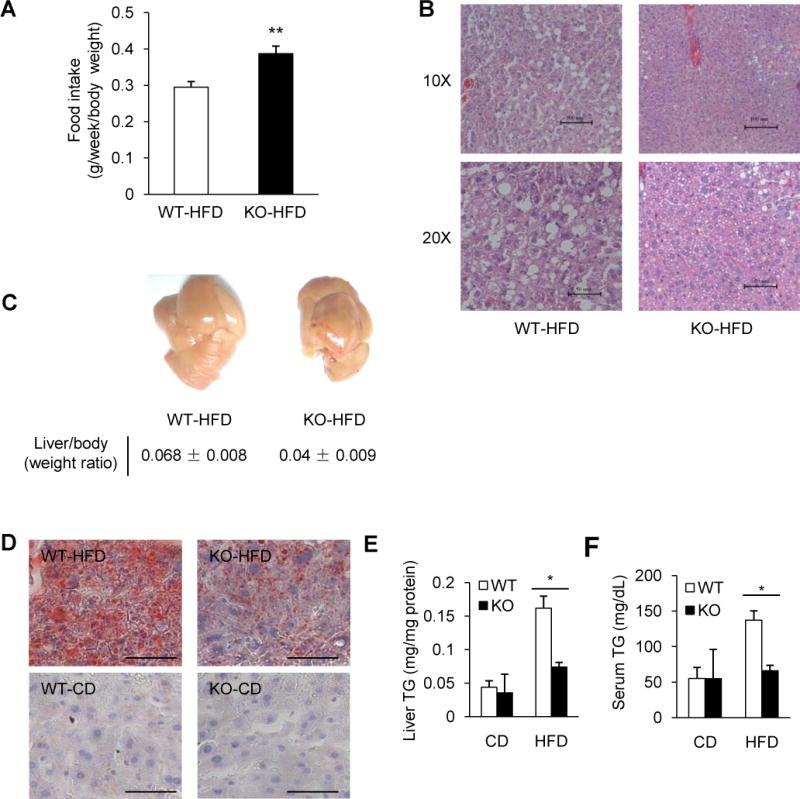
(A) Food intake of wild type (WT) or REGγ-knockout (KO) male mice on HFD (n = 4). REGγ-KO mice exhibited increased food consumption. Data represent mean ± s.d., ** P < 0.01. (B–C) REGγ-KO mice are protected against HFD-induced liver steatosis. Livers were examined in mice fed with HFD for 19 weeks. (B) Liver tissues were stained with hematoxylin and eosin (HE). Pictures were taken using a microscopy with 10x and 20x object lenses. The lipid droplets appeared as uncolored circles in the HE stained tissue slide. (C) Representative liver images (upper panel) and liver/body weight ratios (lower panel) after 19 weeks HFD feeding. (D) Representative images for Oil Red O staining of lipid droplet accumulation in frozen liver sections of mice fed with standard chow diet (CD) or HFD for 19 weeks (scale bar, 50 μm). (E–F) Total triglycerides (TG) levels in liver (E) and serum (F) of mice fed with CD or HFD for 19 weeks. Data represent mean ± s.d. (n = 3), * P < 0.05. See also Fig. S1.
In addition, REGγ overexpression led to an increase of total lipid accumulation as well as an increase in size of lipid droplets in human hepatocellular carcinoma (HepG2) cells, effects that are further exaggerated after oleate treatment (Fig. S1B). Whereas transient overexpression of REGα or REGβ, two homologous proteasome activators (Dubiel et al., 1992; Ma et al., 1992), had no such effect (Fig. S1C). Collectively, the above data revealed a role for REGγ in regulating hepatic lipid metabolism and a protective outcome against HFD-induced liver steatosis in REGγ-KO mice.
Deficiency of REGγ triggers autophagy
To clarify the effect of REGγ on lipid metabolism, we examined mRNA expression of a number of lipid transport, lipogenesis and fatty acid oxidation genes in the liver of REGγ-WT and -KO mice (Fig. S1D). However, there was no significant change in the expression of selected genes between REGγ-WT and -KO mouse livers. Next, we examined fatty acid uptake, lipid synthesis and fatty acid β-oxidation in primary hepatic cells from livers of REGγ-WT and -KO mice. The REGγ-deficient hepatic cells showed no significant alterations in fatty acid uptake or lipid synthesis, but exhibited a slight but significant increase in fatty acid β-oxidation after pretreatment with fatty acids (Fig. S1E–G). These data suggest that the protection of HFD-induced hepatic steatosis in REGγ-KO mice is likely associated with increased lipid oxidation.
Recent studies reveal that autophagy promotes lipid droplet breakdown and the rate of fatty acid β-oxidation increased during lipid loading but to a much lesser extent in cells with inhibited autophagy (Singh et al., 2009). Moreover, autophagy is critical for β-oxidation of fatty acids in the liver (Sinha et al., 2012). Therefore, we tested whether REGγ regulates liver steatosis via autophagy. Transmission electron microscopic analysis of liver autophagy in REGγ-WT and -KO mice revealed REGγ KO led to a marked increase of the number of autophagic vacuoles (AVs) in vivo (Fig. 2A). Mice fed a HFD have reduced hepatic autophagy (Yang et al., 2010). Similarly, our data showed the number of AV s in liver sections decreased after HFD-treatment, but the number of AV s in HFD-treated REGγ-KO mice still maintain at a certain level in comparing with REGγ-WT mice on HFD. In addition, we also observed AVs that enwrapped lipid droplets (Fig. 2A–#2, 4) or mixed lipid droplets and AVs (Fig. 2A–#3) in liver sections of REGγ-KO mice on HFD. HFD-treated REGγ-WT mice displayed robust accumulation of large lipid droplets, whereas HFD-fed REGγ-KO mice showed a marked reduction of both the number and the size of lipid droplets (Fig. 2A). These results indicate that REGγ deficiency induces autophagy in mouse liver, and that the protection of HFD-induced hepatic steatosis in REGγ-KO mice may result from increased autophagy.
Fig. 2. REGγ deficiency triggers autophagy.
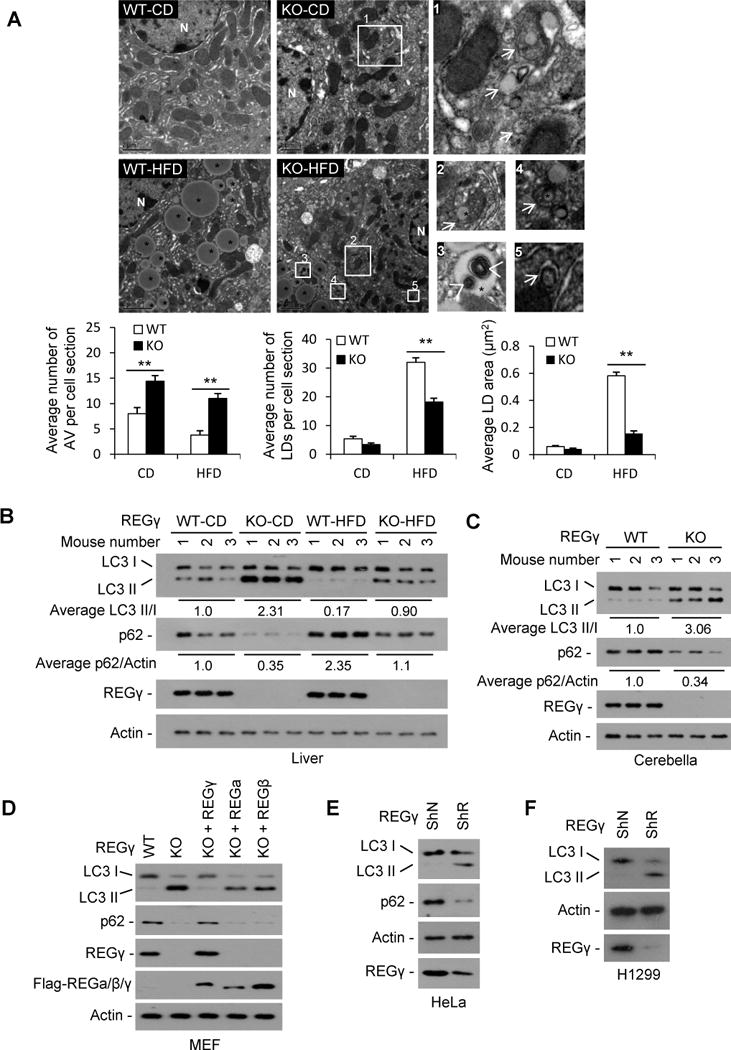
(A) Transmission electron microscopy of liver tissues from REGγ-WT and -KO mice (n=3) with CD or HFD for 19 weeks. * indicates lipid droplet (bright round organelle). Images 1–5 are high magnification of the “white boxes” areas indicated in the images of KO-CD and KO-HFD. The arrows show autophagic vacuoles containing lipid droplets, other cargos or mixed cargos, the arrow heads show autophagic vacuoles in lipid droplets. The average numbers and sizes of autophagic vacuoles (AVs) and lipid droplets (LDs) were quantified (lower panels). The average number of autophagic vacuoles, lipid numbers and lipid size was determined from a randomly selected pool of 5–10 fields under each condition. Data represent mean ± s.d., ** p < 0.01. (B–C) The protein levels of autophagy markers p62 and LC3 in livers (B) and cerebellums (C) of three pairs of REGγ-WT and -KO mice maintained on a standard chow diet (CD) or HFD for 19 weeks were determined by Western blot. The relative average levels of LC3-II/I or p62/actin under each condition was quantified by densitometry of immunoblot signals and presented below the blots. (D) The protein levels of LC3 and p62 in REGγ-WT and -KO MEF cell lines were detected by Western blot. To determine the specific effect for REGγ, REGγ-KO MEF cells were infected with lentiviral vectors expressing Flag-tagged REGγ, α, or β for 48 h respectively. (E–F) Protein levels of LC3 and p62 in HeLa and H1299 cells with stable knockdown of REGγ (ShR) or a vector control (ShN) were determined by Western blot. See also Fig. S2.
Next, we examined the levels of autophagy markers in the livers of REGγ-KO and -WT mice fed with CD or HFD for 19 weeks (Fig. 2B). Results showed a remarkably increased LC3-II and a clearly decreased p62 in REGγ-KO livers with a standard chow diet. HFD led to a marked decrease in LC3-II and an accumulation of p62 in both REGγ-WT and -KO mouse livers, but HFD-treated REGγ-KO mice still maintain higher autophagy levels than REGγ-WT mice with HFD, indicating that autophagy levels are always higher in REGγ-KO mice than in WT at given diet. In addition, REGγ-KO mice exhibited a decreased p62 and an increased LC3-II in cerebellum (Fig. 2C). Reduced p62 and enhanced LC3-II were also observed in REGγ-KO MEF cells as well as HeLa and H1299 cells with REGγ stable knockdown (Fig. 2D–F). Moreover, expression of an RNAi-resistant REGγ, but not REGα/β, restored REGγ deficiency-induced autophagy, indicating the specificity of REGγ in its inhibition of autophagy (Fig. 2D).
To further assess the role of REGγ in autophagy, we evaluated autophagic flux in REGγ-WT and -KO MEFs treated with or without bafilomycin A1 (Ba) which inhibits fusion between autophagosomes and lysosomes (Fig. S2A). Results showed that in the presence of Ba, loss of REGγ also results in an increased accumulation of LC3-II under both normal and starvation conditions, indicating that the differences in LC3-II levels in REGγ-WT and -KO MEF cell lines were due to autophagic induction rather than a blockade at downstream steps. The inhibitory effect of REGγ on autophagy was further confirmed by quantitation of GFP-LC3 puncta formation. REGγ-deficient cells displayed an increase in both the number of punctate LC3 per cell and the GFP-LC3 punctae-positive cells (Fig. S2B–C). Therefore, normal REGγ expression keeps autophagy at a low level under basal conditions.
REGγ inhibits autophagy in a SirT1 dependent manner under normal conditions
Previous studies demonstrate that suppression of the ubiquitin proteasome system induces ER stress, and autophagy can be activated by ER stress (Ding et al., 2007a). Moreover, ER stress contributes to metabolic dysfunction and hepatic steatosis, and a fundamental change is observed in hepatic ER function in obesity from protein to lipid synthesis and metabolism (Fu et al., 2012; Fu et al., 2011). To assess whether ER stress may be involved in REGγ deficiency-induced autophagy and hepatic steatosis resistance, we examined the status of ER homeostasis in REGγ-KO or -knockdown cells in the presence or absence of an ER stress inducer thapsigargin (Fig. S2D–F). Nevertheless, REGγ deficiency exhibited no significant effect to ER stress under normal or thapsigargin treated conditions. These results suggest that REGγ deficiency-induced autophagy is not likely due to ER stress.
To understand the molecular basis of REGγ-mediated regulation of autophagy, we carried out a large scale proteomic screen using antibody array (Fullmoon Biosystems Inc.) and a reverse phase protein array (RPPA) to identify proteins differentially regulated in REGγ-positive and REGγ-null MEF cells (X.L. & S.H., unpublished work). The Fullmoon arrays contain antibodies against phospho and total proteins (Bernier et al., 2011) and RPPA assays include additional proteins not present in the Fullmoon array, together covering nearly 1400 proteins in more than 30 different pathways. In the RPPA analysis, we discovered that SirT1 enhancement in REGγ-null MEFs was statistically significant, similar to the significant changes in the positive controls of the known REGγ targets, p53 and p21 in REGγ-null MEFs (Fig. S3A). The fact that REGγ-KO triggers autophagy (Fig. 2) and SirT1 overexpression stimulates autophagy motivated us to test whether REGγ affects autophagy by negatively regulating SirT1.
First, we assessed whether REGγ interacts with SirT1. Results showed that SirT1 co-precipitated with REGγ in 293T cells overexpressing GFP-REGγ and Flag-SirT1 (Fig. 3A). Endogenous REGγ binds to SirT1 but not other sirtuins such as SirT2 (Fig. 3B). The direct association between REGγ and SirT1 was verified in vitro (Fig. 3C). Moreover, a region of SirT1 (aa 378 to 458) within the catalytic domain and a region corresponding to amino acids 66–103 in REGγ is essential for REGγ-SirT1 interaction (Fig. S3B–C). These results indicate that REGγ is a SirT1 interacting protein.
Fig. 3. REGγ inhibits autophagy in a SirT1 dependent manner under normal conditions.
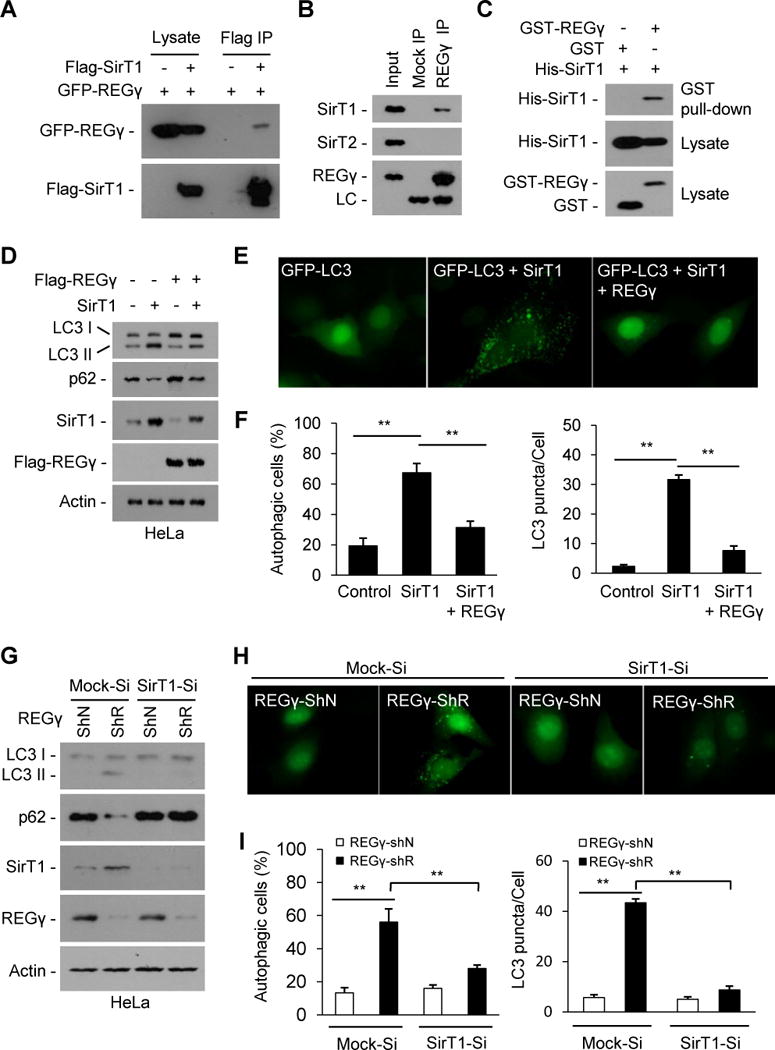
(A–C) REGγ interacts with SirT1. (A) Flag-SirT1 and GFP-REGγ were expressed in the 293T cells and immunoprecipitated with FLAG-M2 agarose beads, and co-precipitated REGγ was detected by Western blot. (B) Endogenous REGγ in HeLa cells was precipitated using anti-REGγ antibody or with IgG (Mock IP), and co-precipitated SirT1 and SirT2 were detected by Western blot. (C) REGγ interacts with SirT1 in vitro. His-tagged SirT1 was incubated with GST-REGγ or GST proteins for 4 h at 4oC. REGγ-SirT1 co-precipitation was determined by GST pulldown and Western blot. (D–F) REGγ inhibits SirT1 autophagic function. (D) HeLa cells were transfected with indicated plasmids for 30 h. The conversion of LC3-I to LC3-II and levels of p62 were determined by Western blot. (E) REGγ inhibits SirT1-stimulated GFP-LC3 punctae formation. HeLa cells seeded on coverslips were transfected with GFP-LC3 and indicated plasmids for 30 h and GFP-LC3 were visualized by fluorescence microscopy. (F) The percentages of GFP-LC3 positive cells with GFP-LC3 punctae and the GFP-LC3 punctae per cell in panel E were quantified. Data represent mean ± s.d., ** P < 0.01. (G–I) SirT1 is required for REGγ-deficient induced autophagy (G) SirT1-knockdown attenuates REGγ knockdown-induced LC3-II accumulation and p62 reduction. HeLa cells with stable knockdown of SirT1 (SirT1-Si) or control vectors (Mock-Si) were infected with pLL3.7 vector (ShN) or REGγ-knockdown (ShR) lentivirus for 72 h. Cell lysates were western blotted for SirT1, REGγ, LC3 and p62. (H) SirT1 knockdown represses REGγ knockdown-induced formation of GFP-LC3 punctae. Cells in panel G were transiently transfected GFP-LC3 for 20 h and visualized by fluorescence microscopy. (I) The percentages of GFP-LC3 positive cells with GFP-LC3 punctae and the GFP-LC3 punctae per cell in panel H were quantified. Data represent mean ± s.d., ** P < 0.01. See also Fig. S3.
Next, we evaluated whether REGγ regulates autophagy via SirT1. Concomitant expression of REGγ inhibited SirT1-induced conversion of LC3-I to LC3-II (Fig. 3D) and formation of GFP-LC3 punctae (Fig. 3E–F). Increased LC3-II and reduced p62 were observed in REGγ-only knockdown cells, but not in cells with REGγ/SirT1 double knockdown (Fig. 3G). In parallel, REGγ knockdown increased the number of LC3 punctae per cell and GFP-LC3 punctae-positive cells, whereas REGγ/SirT1 double knockdown exhibited little change in the number of the LC3 punctae and the percentage of punctae+ cells (Fig. 3H–I). These results indicate that endogenous SirT1 is required for REGγ deficiency-induced autophagy.
REGγ mediates Ub-independent degradation of SirT1
Next, we examined whether REGγ is involved in SirT1 degradation. Results showed a strong negative correlation between REGγ and SirT1 expression in vivo (Fig. 4A–B). Stable knockdown of REGγ results in elevated SirT1 expression (Fig. 4C). We also observed increased SirT1 expression in REGγ-KO MEF cells, and restoration of REGγ expression in these cells repressed SirT1 expression (Fig. 4D). However, REGγ-deficiency had no effect on SirT1 transcript levels (Fig. 4E). Moreover, REGγ overexpression instigated a reduction of endogenous SirT1 protein (Fig. 4F) and increased SirT1 degradation (Fig. 4G). In contrast, the rate of SirT1 decay was markedly delayed in both REGγ-knockdown HeLa cells (Fig. 4H) and REGγ-knockout MEFs (Fig. 4I), indicating that endogenous REGγ expression renders SirT1 unstable. Using a ts85 cell line which harbors a thermolabile ubiquitin-activating enzyme that abolishes the transfer of ubiquitin to target proteins at nonpermissive temperatures (37°C) (Li et al., 2007), we observed a slower degradation rate in SirT1 at 37°C compared to that at 30°C, while REGγ overexpression could still promote SirT1 degradation at 37°C (Fig. S4A). REGγ overexpression had no effect on SirT1 ubiquitination, whereas treatment with proteasome inhibitor MG132 only mildly increased the level of ubiquitinated SirT1 (Fig. S4B). These data indicate that REGγ induces SirT1 degradation in an Ub-independent manner.
Fig. 4. REGγ mediates degradation of SirT1.
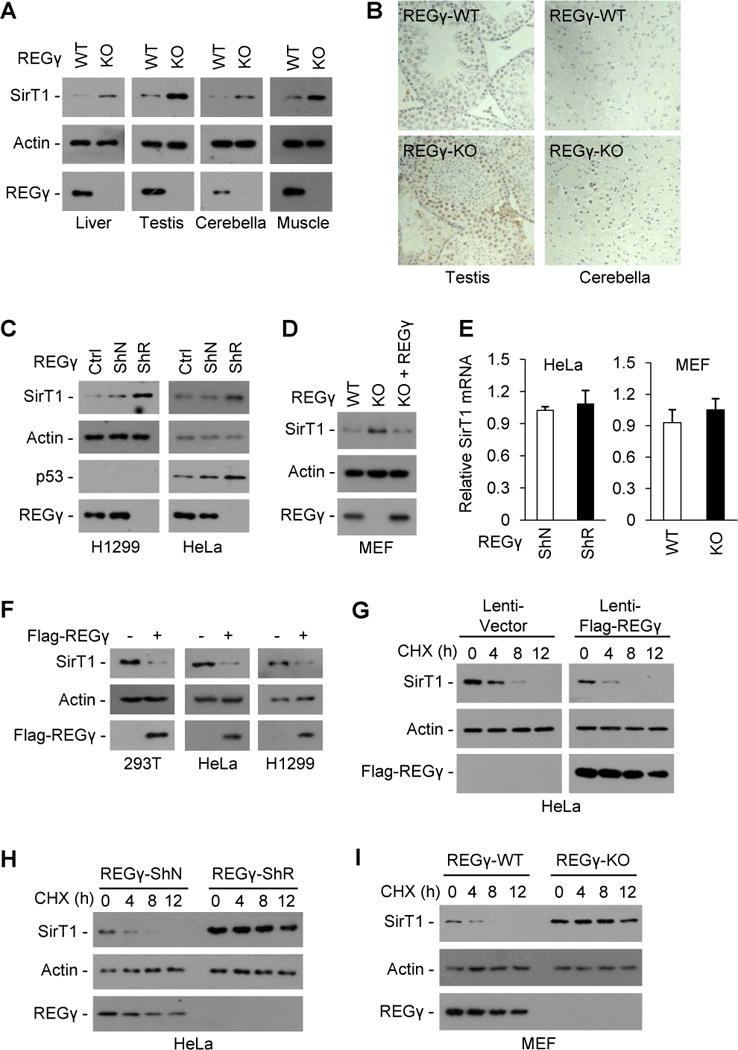
(A–E) REGγ deficiency causes accumulation of endogenous SirT1. (A–B) SirT1 protein levels in liver, testis, cerebellum and muscle tissues of REGγ-WT and -KO mice were examined by Western blot (A) and immunohistochemical staining (B). (C) H1299 and HeLa cells were stably infected with control lentivirus (ShN) or lentivirus expressing REGγ siRNA (ShR) and analyzed for REGγ, SirT1 and p53 expression levels by western blot. (D) MEFs from REGγ-WT and -KO mice were analyzed for REGγ and SirT1 expression levels by Western blot. To restore REGγ expression, REGγ-KO MEFs were infected with a lentiviral vector expressing REGγ for 48 h. (E) Relative mRNA expression of SirT1 in REGγ-deficient cells were examined by quantitative real-time PCR analysis. Data represent mean ± s.d. (n =3). (F) 293T, HeLa and H1299 cells were transfected with a control vector or Flag-REGγ plasmid for 36 h, cell extracts were subjected to Western blotting. (G–I) REGγ promotes SirT1 degradation. HeLa cells infected with control lentivirus or lentivirus overexpressing REGγ for 24 h (G), HeLa cells stably expressing a control lentivirus (ShN) or a lentiviral REGγ shRNA (ShR) (H), or MEFs cells from REGγ-WT and -KO mice (I) were treated with translation inhibitor cycloheximide (CHX, 50 μg/ml) and analyzed for SirT1 stability by Western blot. See also Fig. S4.
REGγ inhibits SirT1-mediated deacetylation of the autophagy machinery
SirT1 stimulates autophagy by deacetylating Atg5, Atg7 and Atg8 (Lee et al., 2008). Since SirT1 is required for REGγ deficiency-induced autophagy (Fig. 3G–I), we hypothesize that REGγ regulates autophagy via SirT1 mediated deacetylation of Atg proteins. As expected, REGγ KO reduced acetylation levels of Atg5/7 in primary hepatocytes, and pharmacological inhibition of SirT1 with nicotinamide (NAM) blocked REGγ deficiency-induced Atg5/7 deacetylation (Fig. 5A). Acetylation of ULK1 (Atg1) (Lin et al., 2012) and Atg3 promote autophagy (Yi et al., 2012). However, Atg5/7 but not Atg1/3 bind to SirT1 (Fig. S5A), and REGγ overexpression could not affect Atg1/3 acetylation (data not shown). Taken together, we believe that SirT1-dependent deacetylation of Atg5/7 contributes to REGγ deficiency-induced autophagy.
Fig. 5. REGγregulates SirT1 autophagic function by inhibiting its binding to and deacetylating autophagy complex.
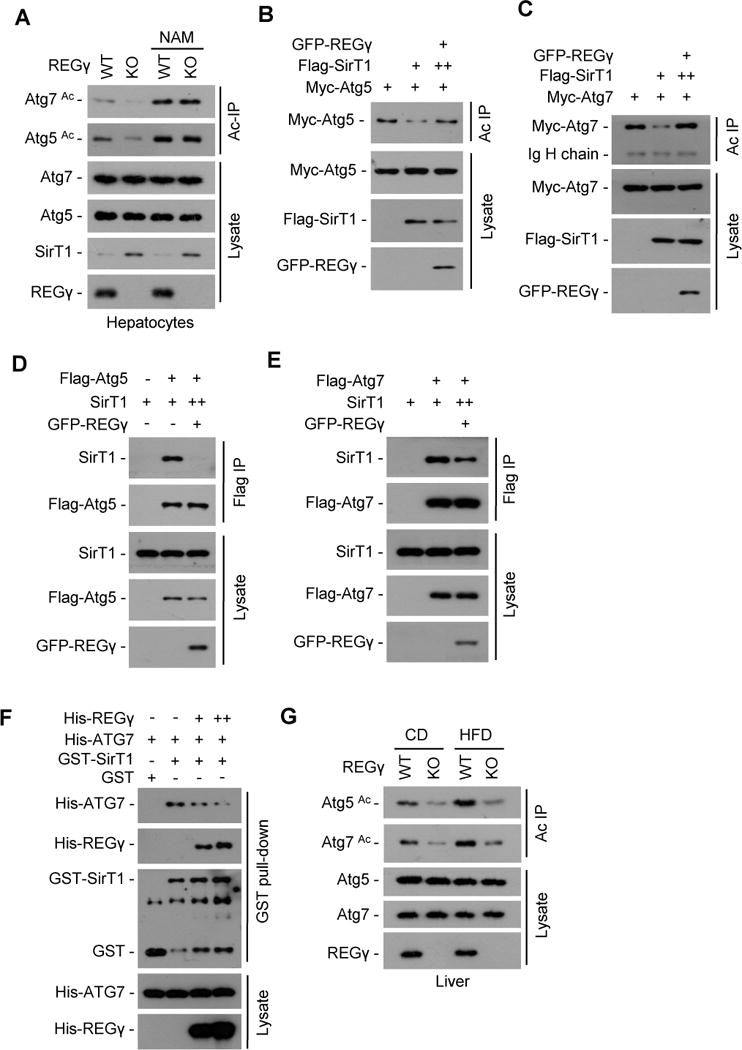
(A) REGγ regulates acetylation states of Atg5 and Atg7 through SirT1. REGγ-WT and -KO primary hepatocytes were treated with or without NAM (5 mM, 12 h), cell extracts were immunoprecipitated by anti-acetyl-lysine antibody (Ac IP). The immunoprecipitates were subjected to Western blotting with anti-Atg5 or -Atg7 antibody. (B–C) 293T cells were cotransfected with indicated plasmids and acetylated proteins were immunoprecipitated by anti-acetyl-lysine antibody (Ac IP). To ensure equal expression of SirT1, more SirT1 plasmid DNA (++) was cotransfected with REGγ. The acetylation levels of Atg5/7 protein were determined by Western blot using anti-Myc tag antibody. (D–E) REGγ inhibits SirT1 binding to Atg5/7. 293T cells transfected with indicated plasmids and Flag-tagged Atg5/7 were immunoprecipitated using FLAG-M2 agarose beads followed by Western blot. To ensure equal expression of SirT1, more SirT1 plasmid DNA (++) was cotransfected with REGγ. (F) REGγ inhibits SirT1-Atg7 binding in vitro. GST-SirT1 was incubated with His-Atg7 in the presence or absence of His-REGγ for 4 h at 4° C, followed by GST pulldown and Western blot. (G) REGγ regulates Atg5 and Atg7 acetylation in vivo. The acetylation status of Atg5 and Atg7 were measured in hepatocytes from REGγ-WT and -KO mice on the CD or HFD for 19 weeks. Liver tissue lysates were immunoprecipitated by anti-acetyl-lysine antibody (Ac IP), and the endogenous acetylation levels of Atg5 and Atg7 proteins were determined by Western blot using anti-Atg5 or -Atg7 antibodies. See also Fig. S5.
Next, we examined whether overexpression of REGγ can alter SirT1 deacetylase activity on Atg5/7. To avoid REGγ-induced degradation of SirT1, increasing amounts of SirT1 expression plasmids (++) were contransfected with REGγ. Results showed that REGγ inhibited SirT1-mediated deacetylation of Atg5/7 (Fig. 5B–C), indicating that REGγ directly inhibits SirT1 activity in deacetylating Atg5/7. Interestingly, REGγ has no effect to SirT1-induced deacetylation of p53 (Fig. S5B), suggesting a specific role for REGγ in regulating of autophagy complex.
To further address why REGγ selectively inhibits SirT1-induced deacetylation of Atg5/7, we checked whether REGγ competes with Atg proteins for SirT1 binding. Overexpression of REGγ inhibited SirT1 binding to Atg5/7 (Fig. 5D–E). Restoring expression of REGγ in REGγ-KO MEF cells reduced endogenous SirT1-Atg7 interaction under low-dose of MG132 treatment (Fig. S5C). REGγ also displaces Atg7 from SirT1 in vitro in a dose dependent manner (Fig. 5F). These results suggest that REGγ-induced SirT1-Atg5/7 dissociation also contributes to its inhibitory effect on autophagy.
The above data strongly endorse a specific inhibitory effect of REGγ on SirT1-mediated Atg5/7 deacetylation. Paradoxically, REGγ is mostly localized in nucleus, while Atg5/7 proteins are largely cytosolic. Therefore, we examined how SirT1 mediates functional links between the REGγ and Atg5/7 in mouse primary hepatocytes. Results from immunostaining showed that both REGγ and SirT1 were localized primarily in the nucleus (Fig. S5D). Most of the previous studies observed REGγ primarily localized in nucleus, whereas a few studies also showed a cytoplasm localized REGγ (Wojcik, 1999). We observed that a small fraction of REGγ localized into the cytoplasm, lack of REGγ leads to an accumulation of SirT1 not only in nucleus but also in cytoplasm, and Atg5/7 are found in both the cytoplasm and nucleus (Fig. S5D). These results suggest that each of these proteins might dynamically shuttle between nucleus and cytoplasm although major localization and competition could be in one or two compartments.
Next, we determined in vivo regulation of Atg5/7 acetylation by REGγ (Fig. 5G). Results showed that REGγ deficiency markedly reduced hepatic acetylation levels of Atg5/7 in both CD- and HFD-treated mice. HFD treatment increased levels of Atg5/7 acetylation, but HFD-induced hepatic accumulation of Acetyl-Atg5/7 is much less in REGγ-KO mice in comparison with wildtype mice. These results indicate that the REGγ-SirT1-Atg-autophagy signaling pathway is involved in regulating HFD-induced hepatic steatosis in vivo. Taken together, our data suggest that REGγ binds to SirT1, promotes SirT1 degradation, disrupts the interaction of SirT1-Atg5/7, and specifically inhibits SirT1 activity for deacetylating Atg proteins, which then keeps cellular autophagy at a basal level under physiological conditions.
REGγ controls hepatic steatosis through a SirT1- and autophagy-dependent mechanism
Next, we tested whether REGγ regulates hepatic cellular steatosis in a SirT1- and autophagy-dependent manner. We observed that absence of REGγ was associated with accumulation of LC3-II/SirT1 and reciprocal reduction of p62 in primary hepatocytes isolated from REGγ-KO mice (Fig. 6A). REGγ-KO led to a significantly reduced accumulation of intracellular lipids in oleate-palmitate treated primary hepatocytes, whereas 3-MA (autophagy inhibitor) and NAM (SirT1 inhibitor) both had a significant inhibitory effect on REGγ deficiency-mediated clearance of cellular lipids (Fig. 6B–C). Similar results were observed in REGγ-knockdown HepG2 cells (Fig. S6A–C). These data suggest that SirT1 and autophagy are both required for the protective effect against hepatic steatosis in REGγ-deficient mice.
Fig. 6. REGγ-deficiency attenuates hepatic steatosis through a SirT1- and autophagy-dependent mechanism.
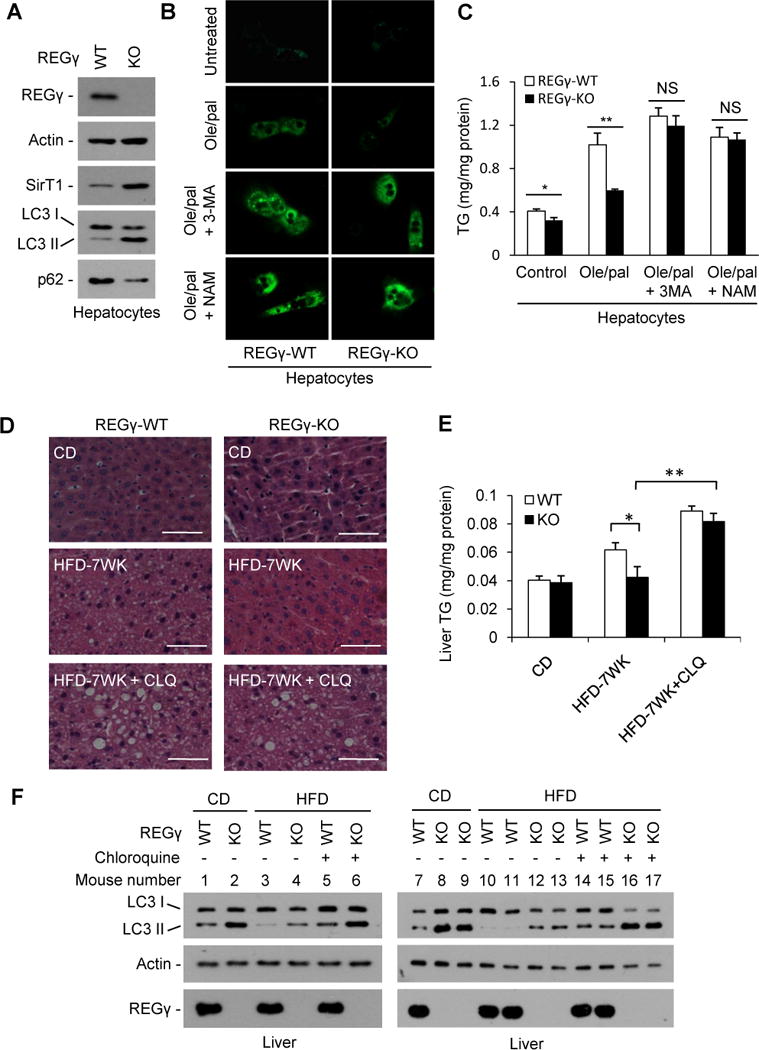
(A) REGγ-KO increased levels of SirT1 and autophagy in hepatocytes. Protein levels of SirT1, p62 and LC3 in primary hepatocytes isolated from REGγ-WT and -KO mice were determined by Western blot. (B) REGγ knockout attenuates fatty-acid induced hepatic cellular steatosis. REGγ-WT and -KO primary hepatocytes were treated with or without 0.3 mM oleate/palmitate (2:1) for 20 h in the presence of autophagy inhibitor 3-methyladenine (3MA, 5 mM) or SirT1 inhibitor nicotinamide (NAM, 5 mM), followed by BODIPY 493/503 staining and visualized by fluorescence microscopy. (C) Cells of panel B were analyzed for total triglycerides levels. Data represent mean ± s.d., * P < 0.05, ** P < 0.01, NS = non-significant. (D–F) REGγ-proteasome regulates hepatic lipid metabolism through inhibition of autophagy. REGγ-WT and REGγ KO mice were fed with standard chow diet (CD) or high fat diet (HFD) for 7 weeks (mice began to feed HFD 3 weeks after birth). Four weeks after HFD, mice (n=3) were treated with intraperitoneal injection of chloroquine (CLQ, 60mg/kg body weight) every other day along with continued HFD for another 3 weeks. (D) Liver tissues were stained with hematoxylin and eosin. Pictures were taken using a microscopy with 20x object lenses. The lipid droplets appeared as uncolored circles in the HE stained tissue slide. (E) Total triglyceride (TG) levels of livers in panle D were measured. Data represent mean ± s.d. (n=3), *P < 0.05, **P < 0.01. (F) Liver tissue lysates in panel D were subjected to Western blot. Chloroquine elicits an accumulation of LC3-II in HFD-treated mice, indicating inhibition of autophagy. See also Fig. S6.
To further address the contributions of autophagy in REGγ deficiency-induced prevention of HFD-induced fatty liver in vivo, REGγ-WT and -KO mice were fed HFD for 7 weeks accompanied with simultaneous intraperitoneal injection of the autophagy inhibitor chloroquine (CLQ) or vehicle during the last 3 weeks. By H&E staining of frozen liver sections and liver TG measurements, we found that 7 weeks of HFD treatment already led to a small but significant increase of lipid droplets and TG levels in livers of REGγ-WT, but not REGγ-KO mice (Fig. 6D–E). Remarkably, CLQ treatment highly increased accumulation of large lipid droplets and TG levels in livers of both REGγ-KO and -WT mice on HFD. More importantly, the differences between REGγ-WT and -KO mice in levels of TG and lipid droplets accumulation on HFD disappeared following co-treatment with CLQ. The effect of CLQ to inhibit autophagy was confirmed by Western blot analysis of LC3-II accumulation (Fig. 6F). These results indicate that inhibiting autophagy eliminates the protective roles of REGγ-deficiency in HFD-induced liver steatosis, substantiating that autophagy is required for REGγ-mediated regulation of hepatic lipid metabolism. Interestingly, increased REGγ-SirT1 association was observed after oleate treatment (Fig. S6D), suggesting that REGγ-SirT1 association may contribute to high-fat induced hepatic steatosis. Collectively, these results demonstrate that the REGγ-proteasome regulates hepatic lipid metabolism through SirT1 and autophagy pathways.
Energy starvation dissociates REGγ-SirT1 and releases SirT1 to stimulate autophagy
Autophagy occurs at relatively low levels under basal conditions but can be induced by a variety of nutrient and intracellular stresses (Kroemer et al., 2010), we therefore examined the impact of starvation on REGγ-SirT1 interactions. As a result, glucose deprivation (GD) significantly reduced REGγ-SirT1 binding (Fig. 7A). In parallel, REGγ-SirT1 binding was reduced in cells treated with 2-deoxy-D-glucose (2DG), a potent inhibitor of glucose metabolism that mimics the effect of GD (Fig. 7B). In a reciprocal fashion, GD increased the binding of SirT1 to Atg7 (Fig. 7C). These results indicate that energy deprivation entails SirT1-REGγ dissociation and thus releases SirT1 to bind to and deacetylates Atg proteins, setting off the autophagic process.
Fig. 7. Energy starvation dissociates REGγ-SirT1, unleashing SirT1 to associate with Atg proteins for autophagy activation.
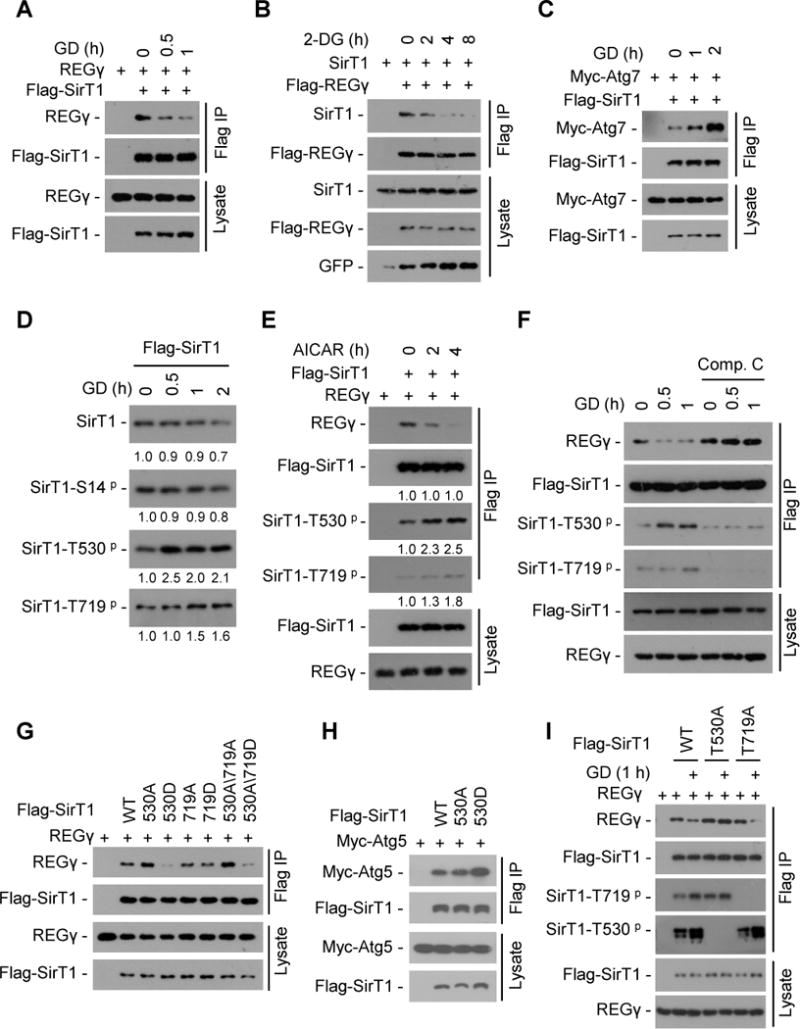
(A) 293T cells were transiently transfected with Flag-SirT1 and REGγ followed by glucose free medium (glucose deprivation, GD) treatment. SirT1 was immunoprecipitated using FLAG-M2 beads and co-precipitation of REGγ was detected by Western blot. (B) 293T cells were transfected with Flag-REGγ, SirT1 and GFP plasmids followed by 2-DG (12.5 mM) treatment. REGγ was immunoprecipitated using FLAG-M2 beads and co-precipitation of SirT1 was detected by Western blot. (C) 293T cells were transiently transfected with Flag-SirT1 and Myc-Atg7 followed by GD treatment. SirT1 was immunoprecipitated using FLAG-M2 beads and co-precipitated Atg7 was detected by Western blot with anti-Myc antibody. (D) Time course of SirT1 phosphorylation in response to glucose starvation. Flag-SirT1 transfected 293T cells were starved for GD for indicated times. SirT1 was immunoprecipitated with FLAG-M2 beads and phosphorylation of SirT1 were determined by Western blot with antibodies against SirT1 phosphorylation at S14, T530 or T719. Relative levels of phosphorylated and total SirT1 were quantified by densitometry of bands and are presented below the blots. (E) Activation of AMPK induces SirT1-phosphorylation coupled with REGγ-SirT1 dissociation. Flag-SirT1 and REGγ were transfected into 293T cells for 24 h, and then cells were treated with or without AMPK activator AICAR (200 μM). SirT1 was immunoprecipitated with FLAG-M2 beads and the immunoprecipitated complex were examined by Western blot. Relative protein levels were quantified by densitometry of bands and are presented below the blots. (F) AMPK inhibitor Compound C prevents glucose starvation-induced dissociation of REGγ-SirT1. Flag-SirT1 transfected 293T cells were treated with Compound C (10 μM) for 1 h followed by a 0.5–1 h GD treatment. SirT1 was immunoprecipitated by FLAG-M2 beads and co-precipitated REGγ were determined by Western blot. (G–H) Phosphorylation-mimetic mutation (T530D) in SirT1 regulates its association with REGγ and Atg5 proteins. Interaction of Flag-SirT1 mutants with REGγ (G) and Atg5 (H) were determined by cotransfection of indicated plasmids into 293T cells followed by immunoprecipitation using FLAG-M2 beads and Western blot. (I) SirT1 Thr530 phosphorylation is responsible for GD-induced REGγ-SirT1 dissociation. Flag-SirT1 mutants and REGγ cotransfected 293T cells were treated with or without GD for 1 h. SirT1 was immunoprecipitated using FLAG-M2 beads and the immunoprecipitated complex were examined by Western blotting. See also Fig. S7.
Phosphorylation was found to modulate the function of SirT1 in cell proliferation (Nasrin et al., 2009). To test whether phosphorylation is involved in starvation-induced SirT1-REGγ dissociation, we generated SirT1 phospho-Ser14, -Thr530, and -Thr719 specific antibodies (Fig. S7A) and examined the effect of GD on SirT1 phosphorylation. Interestingly, GD stimulated SirT1 phosphorylation at T530 and T719, but not S14 (Fig. 7D). The AMP-activated protein kinase (AMPK) is activated upon glucose starvation. We found AICAR (AMPK activator) treatment led to a decreased REGγ-SirT1 binding and a concomitant increase in SirT1 phosphorylation at T530 and T719 (Fig. 7E). In contrast, treatment of AMPK inhibitor Compound C inhibited GD-induced SirT1 phosphorylation and REGγ-SirT1 dissociation (Fig. 7F). These data suggest that AMPK is responsible for glucose starvation induced SirT1 phosphorylation and REGγ-SirT1 dissociation.
Sequence analysis reveals that SirT1 T530 but not T719 is highly conserved in human and other species (Fig. S7B). We found that SirT1 mutation in T530D but not in T719D led to a decreased SirT1-REGγ interaction (Fig. 7G), while the SirT1-T530D mutant had an increased association with Atg5 (Fig. 7H). Dephosphorylation of SirT1 enhanced its association with REGγ in vitro (Fig. S7C). Moreover, the phosphorylation defective T530A but not T719A SirT1 mutant significantly blocked GD-induced REGγ-SirT1 dissociation (Fig. 7I). These results demonstrate that glucose starvation stimulates SirT1 phosphorylation mainly at T530 via AMPK, and this modification causes dissociation of SirT1-REGγ coupled with increased association of SirT1-Atg5/7, contributing to liberation of SirT1 to activate autophagy.
DISCUSSION
Results described in this study identify a previously unknown function of the REGγ proteasome in regulating autophagy and lipid metabolism. REGγ-deficiency leads to autophagy in vivo and protects mice from HFD-induced liver steatosis. Under normal conditions, REGγ directly binds to SirT1, promotes SirT1 degradation, displaces Atg proteins from SirT1, and increases acetylation of Atg proteins, thereby maintaining autophagy at a lower basal level. Upon energy deprivation, REGγ dissociates from SirT1 in a phosphorylation-dependent manner, unleashing SirT1 to interact with Atg proteins and stimulate autophagy. These observations lead us to propose a role for REGγ as a molecular switch regulating autophagy between normal and stress conditions (Fig. S7D). Furthermore, we revealed that REGγ regulates fatty acid-induced hepatic steatosis through a SirT1- and autophagy-dependent mechanism, and autophagy is essential for REGγ-deficiency mediated prevention of HFD-induced fatty liver.
Cells are equipped with two major proteolytic systems (proteasome and autophagy). Inhibiting autophagy compromises degradation of Ub-proteasome substrates (Korolchuk et al., 2009), whereas proteasome inhibitors activate autophagy, suggesting that the two pathways are functionally coupled (Ding et al., 2007b). The discovery that REGγ deficiency triggers autophagy not only identifies a factor linking proteasomal activity and autophagy, but also defines a function for a proteasome activator in shifting the balance between autophagy and the proteasome system. Since REGγ directs a pathway to degrade certain proteins via an Ub-independent mechanism, our results suggest that cells may use autophagy as an alternative proteolytic pathway when the Ub-independent degradation mechanism is compromised. Recently, we reported that REGγ can be acetylated by CBP, whereas SirT1 can bind with REGγ and deacetylates REGγ, which inhibits heptamerization of REGγ, leading to reduced REGγ capacity (Liu et al., 2013). These observations suggest a mutual regulation between REGγ and SirT1. Previous studies revealed that starvation could induce a slight but significant increase of proteasome activity with no upregulation of ubiquitin-proteasome system (Salem et al., 2007). Given starvation markedly reduced REGγ-SirT1 interaction, we suggest that the dissociation of SirT1 from REGγ may contribute to starvation-induced increase of proteasome activity through a post-translational modifications regulatory effect. Identification of REGγ-SirT1 complex has established strong bases for continued study of the interplays between proteasome and autophagy under various conditions.
This study also provides insights into the function of the REGγ proteasome in the regulation of lipid homeostasis. We have presented compelling evidence demonstrating the role of REGγ in regulating autophagy via SirT1 under different metabolic conditions, and pinpointed the contribution of REGγ in hepatic steatosis and the pathogenesis of fatty liver during HFD feeding. Since inhibiting autophagy by chloroquine markedly eliminates the protective roles of REGγ-deficiency in HFD-induced liver steatosis in vivo, and SirT1 is required for autophagy stimulation in REGγ-deficient cells, we believe that lipid metabolism can be regulated through the REGγ-SirT1-autophagy pathway. However, our study does not exclude other additional mechanisms mediating REGγ function in lipid homeostasis. Interestingly, we found that fatty acid stimulates REGγ-SirT1 association. Further study on the correlations between SirT1 and REGγ under different metabolic conditions may provide guidance for the treatment of certain types of metabolic disease.
The REG (or PA28) proteasome activators comprise three members α, β and γ. REGα/β primarily localize in cytoplasm and regulate immuno-peptide generation, whereas REGγ primarily localizes in nucleus and mainly regulates Ub-independent degradation of subsets of proteins. A recent study described that removal of all three PA28/REG subunits (Triple-REG-KO) decreased hepatic insulin signaling and resulted in minor hepatic steatosis under normal diet condition (Otoda et al., 2013). In contrast, our study using REGγ-KO mice revealed a hepatic steatosis resistance phenotype under HFD conditions. Triple-REG-KO activates an unfolded protein response and increases ER stress in the liver, whereas REGγ-KO does not affect ER homeostatsis. Overexpression of REGγ but not REGα/β led to lipid accumulation. Furthermore, REGγ-KO led to a markedly increased LC3-II accumulation and reduced p62, whereas the effect of REGγ deficiency is not compensated by REGα/β overexpression. Since REGα/β and REGγ have been reported to play distinct biological roles with many functions still unknown, it is likely that simultaneous knockout of three genes may be too complicated and too early for us to understand the full mechanisms.
Our study demonstrates that REGγ promotes SirT1 degradation in an Ub-independent manner. A previous report suggested that JNK1 phosphorylates SirT1 and promotes its Ub-dependent degradation (Gao et al., 2011). Therefore, SirT1 can be degraded via both Ub-dependent and -independent pathways. In addition, we also revealed that glucose deprivation induces SirT1 phosphorylation on T530. We estimated the status of SirT1–T530 phosphorylation under physical conditions in mouse hepatocytes and revealed that ~25% of total SirT1 can be phosphorylated on T530 (T522 in mouse) (data not shown). These observations provide a possible explanation why overexpression of a phospho-deficient SirT1–T530A mutant exhibited no significant effect to its association with REGγ, whereas phosphorylation-mimetic SirT1-T530D mutant markedly reduced its association with REGγ (Fig. 7G, I). Furthermore, we found that HFD-treatment led to a reduction in hepatic levels of SirT1–T530 phosphorylation coupled with a reduction in total SirT1 expression (Fig. S7E). Further study on posttranslational modification of SirT1 under different conditions may provide new insights into the SirT1 regulatory network.
Acetylation is emerging as an important autophagy regulatory mechanism. Tip60 (yeast Esa1) acetylase-mediated Atg1 and Atg3 acetylation can induce autophagy (Lin et al., 2012; Yi et al., 2012), P300 acetylase-mediated acetylation of Atg5/7 led to autophagy inhibition (Lee and Finkel, 2009), SirT1 deacetylase induces autophagy via deacetylation of Atg5/7/8 proteins (Lee et al., 2008). Upon nitrogen starvation, autophagy can be induced by Esa1 via Atg3 acetylation (Yi et al., 2012). Serum starvation triggers signaling from GSK3 to TIP60 to stimulate Atg1 acetylation and autophagy (Lin et al., 2012). In our study, glucose starvation induces AMPK-dependent SirT1 phosphorylation on T530, which entails SirT1-REGγ dissociation and thus releases SirT1 to stimulate autophagy. Interestingly, we detected no significant reduction of REGγ-SirT1 binding after serum starvation (data not shown). These observations proved that cells use distinct acetylation/deacetylation signaling pathways to regulate autophagy in response to different metabolism changes. Collectively, our observations indicate that REGγ forms a complex with SirT1 and serves as an important molecular switch for the off/on regulation of autophagy under normal and energy-deprivation conditions.
In conclusion, the present study establishes a molecular cross-talk between the proteasome activator REGγ and SirT1 signaling in the regulation of autophagy and hepatocyte lipid metabolism. Furthermore, unpublished data in our laboratory also revealed that REGγ-deficient mice exhibit enhanced insulin sensitivity. Thus, it is tempting to speculate that downregulation of REGγ also may protect against diabetes. Our findings that REGγ regulates autophagy and lipid metabolism make it an attractive drug target since pharmacological manipulation of REGγ could have potential benefits on metabolic disorders such as liver steatosis, diabetes and age related diseases.
EXPERIMENTAL PROCEDURES
Animal Experimentation
REGγ KO mice were kindly provided by Dr. John J. Monaco at University of Cincinnati (Barton et al., 2004). All mice used were C57BL/6 male mice. REGγ WT and KO mice were fed either with a standard chow diet, or with a high-fat diet (D12492 with 60 kcal% fat; Research Diets, Inc., NJ, USA) starting when the mice were 3 weeks of age and for a total of 19 additional weeks. Animals are treated according to high ethical and scientific standards.
Measurement of liver/body weight and food intake
Mice had ad libitum access to either normal chow or high fat diet and water. Food intake of wild type (WT) or REGγ-KO male mice (n = 4) on HFD were monitored weekly at 17–18 weeks (one pair of REGγ-WT and -KO littermate mice monitored one week, the other 3 pairs mice monitored for 2 weeks) after HFD. Body weight was determined every 2 weeks. Mice Liver/body weight was measured after being sacrificed.
Cell lines, plasmids and reagents
HeLa, H1299, HepG2, HEK293T, REGγ-WT and -KO MEF cells were maintained in DMEM medium with 10% fetal bovine serum. REGγ-knockdown, SirT1-knockdown or vector control stable cell lines were established by transient transfection and selected with puromycin. Primary hepatocytes were isolated as described previously (Liu et al., 2012). GFP-LC3 and Myc-Atg5/7/8 plasmids were kind gifts from Toren Finkel (NIH, Bethesda, MD). SirT1 knockdown plasmids were previously generated (Wang et al., 2006). Cycloheximide, 2-Deoxy-D-glucose, AMPK activator (AICAR) and inhibitor (Compound C) were obtained from Sigma. Antibodies were purchased from Sigma (LC3, p62, REGγ, Flag-tag and β-actin), Santa Cruz Biotechnology (GFP, SirT1 and Myc-tag), Cell Signaling Technology (Ac-K-103-lysine) and Goat anti-rabbit IgG (Vector Laboratories, USA).
Analysis of autophagy by microscopy
Autophagy was visualized by GFP-LC3 transfection followed by confocal microscopy. For electron microscopy investigations, cells or liver tissues were fixed with 2.5% glutaraldehyde for 3 h, rinsed in PBS, post-fixed in 1% OsO4, dehydrated and embedded in Epon. Ultrathin sections were contrasted with uranyl acetate and lead citrate and photographed in a JEM-2100 transmission electron microscope.
Immunostaining
Immunohistochemical staining in tissues were performed using anti-SirT1 antibody and visualize as previously described (Liu et al., 2010).
Immunoprecipitation and GST pull-down assay
Cell lysate preparation, protein immunoprecipitation and GST pull-down assay were performed as previously described (Hu et al., 2012).
Lipid accumulation and measurement
Cells were incubated in the absence or presence of sodium oleate or oleate/palmitate (2:1) in culture medium supplemented with 1% fatty acid-free BSA. To measure triglyceride (TG), the lipids were extracted from cell lysates using a chloroform/methanol (2:1) mixture. TG was determined using Serum Triglyceride Determination Kit (TR0100, Sigma). For lipid droplet staining, cells were fixed with 10% formalin and stained with Oil Red O working solution and visualized under the bright field microscope. Alternatively, BODIPY 493/503 (0.01 mg/ml) in PBS was applied to lipid droplets stain for 15 minutes and visualized by fluorescence microscopy. Hepatic triglycerides were extracted as previously described (Liu et al., 2012). Blood samples were obtained for serum TG measurements.
Statistical Analyses
Results shown as mean ± SD. Differences were considered significant with a p value < 0.05 using Student’s two-tailed t test.
Supplementary Material
Highlights.
REGγ knockout mice are resistant to high fat diet-induced liver steatosis
REGγ proteasome negatively regulates autophagy
REGγ proteasome regulates lipid metabolism via autophagy
REGγ finely regulates autophagy off/on under normal and starvation conditions
Acknowledgments
This work was supported in part by National Institutes of Health (1R01CA131914 to X. L., R01HL51586 to L.C., and NICHD-HD8188 to B.W.O), Norman Hackerman Advanced Research Program (1082318401) and the Pilot/Feasibility Program of the Diabetes Research Center at Baylor College of Medicine (P30-DK079638) to X. L., Cancer Prevention & Research Institute of Texas Core Facility Support Awards (RP120092) and NCI Cancer Center Support Grant to the Proteomics Shared Resource (P30CA125123) to D.P.E., and grants from National Natural Science Foundation of China (31201044, 31071248, 31171361, 81261120555 and 81071657), National Basic Research Program of China (2009CB918401, 2009CB918402, 2011CB504200) and the Science and Technology Commission of Shanghai Municipality (11DZ2260300). The authors would like to thank Mrs. Myra Costello at the Proteomics Shared Resource for her technical support. None of the authors have competing financial interests related to this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barton LF, Runnels HA, Schell TD, Cho Y, Gibbons R, Tevethia SS, Deepe GS, Jr, Monaco JJ. Immune defects in 28-kDa proteasome activator gamma-deficient mice. J Immunol. 2004;172:3948–3954. doi: 10.4049/jimmunol.172.6.3948. [DOI] [PubMed] [Google Scholar]
- Bernier M, Paul RK, Martin-Montalvo A, Scheibye-Knudsen M, Song S, He HJ, Armour SM, Hubbard BP, Bohr VA, Wang L, et al. Negative regulation of STAT3 protein-mediated cellular respiration by SIRT1 protein. Journal of Biological Chemistry. 2011;286:19270–19279. doi: 10.1074/jbc.M110.200311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science (New York, NY. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell. 2005;123:437–448. doi: 10.1016/j.cell.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Czaja MJ. Autophagy in health and disease. 2. Regulation of lipid metabolism and storage by autophagy: pathophysiological implications. Am J Physiol Cell Physiol. 2010;298:C973–978. doi: 10.1152/ajpcell.00527.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. Journal of Biological Chemistry. 2007a;282:4702–4710. doi: 10.1074/jbc.M609267200. [DOI] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. American Journal of Pathology. 2007b;171:513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubiel W, Pratt G, Ferrell K, Rechsteiner M. Purification of an 11 S regulator of the multicatalytic protease. Journal of Biological Chemistry. 1992;267:22369–22377. [PubMed] [Google Scholar]
- Fu S, Fan J, Blanco J, Gimenez-Cassina A, Danial NN, Watkins SM, Hotamisligil GS. Polysome profiling in liver identifies dynamic regulation of endoplasmic reticulum translatome by obesity and fasting. PLoS Genet. 2012;8:e1002902. doi: 10.1371/journal.pgen.1002902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, Hotamisligil GS. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473:528–531. doi: 10.1038/nature09968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Zhang J, Kheterpal I, Kennedy N, Davis RJ, Ye J. Sirtuin 1 (SIRT1) protein degradation in response to persistent c-Jun N-terminal kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity. Journal of Biological Chemistry. 2011;286:22227–22234. doi: 10.1074/jbc.M111.228874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev. 2000;14:1021–1026. [PubMed] [Google Scholar]
- Hu C, Zhang S, Gao X, Xu X, Lv Y, Zhang Y, Zhu Z, Zhang C, Li Q, Wong J, et al. Roles of Kruppel-associated Box (KRAB)-associated Co-repressor KAP1 Ser-473 Phosphorylation in DNA Damage Response. J Biol Chem. 2012;287:18937–18952. doi: 10.1074/jbc.M111.313262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Molecular Cell. 2009;33:517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Marino G, Levine B. Autophagy and the Integrated Stress Response. Molecular Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IH, Finkel T. Regulation of autophagy by the p300 acetyltransferase. Journal of Biological Chemistry. 2009;284:6322–6328. doi: 10.1074/jbc.M807135200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Amazit L, Long W, Lonard DM, Monaco JJ, O’Malley BW. Ubiquitin- and ATP-independent proteolytic turnover of p21 by the REGgamma-proteasome pathway. Molecular Cell. 2007;26:831–842. doi: 10.1016/j.molcel.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Li X, Lonard DM, Jung SY, Malovannaya A, Feng Q, Qin J, Tsai SY, Tsai MJ, O’Malley BW. The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REGgamma proteasome. Cell. 2006;124:381–392. doi: 10.1016/j.cell.2005.11.037. [DOI] [PubMed] [Google Scholar]
- Lin SY, Li TY, Liu Q, Zhang C, Li X, Chen Y, Zhang SM, Lian G, Ruan K, Wang Z, et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science (New York, NY. 2012;336:477–481. doi: 10.1126/science.1217032. [DOI] [PubMed] [Google Scholar]
- Liu J, Wang Y, Li L, Zhou L, Wei H, Zhou Q, Wang W, Ji L, Shan P, Yang Y, et al. Site-specific Acetylation of the Proteasome Activator REGgamma Directs Its Heptameric Structure and Functions. Journal of Biological Chemistry. 2013;288:16567–16578. doi: 10.1074/jbc.M112.437129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhou D, Zhang F, Tu Y, Xia Y, Wang H, Zhou B, Zhang Y, Wu J, Gao X, et al. Liver Patt1 deficiency protects male mice from age-associated but not high-fat diet-induced hepatic steatosis. J Lipid Res. 2012;53:358–367. doi: 10.1194/jlr.M019257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- Ma CP, Slaughter CA, Demartino GN. Identification, Purification, and Characterization of a Protein Activator (Pa28) of the 20-S Proteasome (Macropain) Journal of Biological Chemistry. 1992;267:10515–10523. [PubMed] [Google Scholar]
- Mizushima N. Autophagy: process and function. Gene Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Murata S, Kawahara H, Tohma S, Yamamoto K, Kasahara M, Nabeshima Y, Tanaka K, Chiba T. Growth retardation in mice lacking the proteasome activator PA28gamma. Journal of Biological Chemistry. 1999;274:38211–38215. doi: 10.1074/jbc.274.53.38211. [DOI] [PubMed] [Google Scholar]
- Nasrin N, Kaushik VK, Fortier E, Wall D, Pearson KJ, de Cabo R, Bordone L. JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. Plos One. 2009;4:e8414. doi: 10.1371/journal.pone.0008414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science (New York, NY. 2004;306:2105–2108. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- Otoda T, Takamura T, Misu H, Ota T, Murata S, Hayashi H, Takayama H, Kikuchi A, Kanamori T, Shima KR, et al. Proteasome dysfunction mediates obesity-induced endoplasmic reticulum stress and insulin resistance in the liver. Diabetes. 2013;62:811–824. doi: 10.2337/db11-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschop MH. Sirt1 protects against high-fat diet-induced metabolic damage. P Natl Acad Sci USA. 2008;105:9793–9798. doi: 10.1073/pnas.0802917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, de Oliveira RM, Leid M, McBurney MW, Guarente L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma (vol 429, pg 771, 2004) Nature. 2004;430:921–921. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo XM, Li XL. Hepatocyte-Specific Deletion of SIRT1 Alters Fatty Acid Metabolism and Results in Hepatic Steatosis and Inflammation. Cell Metab. 2009;9:327–338. doi: 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang L, Wang LH, Kon N, Zhao WH, Lee S, Zhang YY, Rosenbaum M, Zhao YM, Gu W, Farmer SR, et al. Brown Remodeling of White Adipose Tissue by SirT1-Dependent Deacetylation of Ppar gamma. Cell. 2012;150:620–632. doi: 10.1016/j.cell.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem M, Silverstein J, Rexroad CE, 3rd, Yao J. Effect of starvation on global gene expression and proteolysis in rainbow trout (Oncorhynchus mykiss) BMC Genomics. 2007;8:328. doi: 10.1186/1471-2164-8-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Cuervo AM. Lipophagy: connecting autophagy and lipid metabolism. Int J Cell Biol. 2012;2012:282041. doi: 10.1155/2012/282041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha RA, You SH, Zhou J, Siddique MM, Bay BH, Zhu X, Privalsky ML, Cheng SY, Stevens RD, Summers SA, et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. Journal of Clinical Investigation. 2012;122:2428–2438. doi: 10.1172/JCI60580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Chen L, Hou X, Li Z, Kabra N, Ma Y, Nemoto S, Finkel T, Gu W, Cress WD, et al. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nature cell biology. 2006;8:1025–1031. doi: 10.1038/ncb1468. [DOI] [PubMed] [Google Scholar]
- Wojcik C. Proteasome activator subunit PA28 alpha and related Ki antigen (PA28 gamma) are absent from the nuclear fraction purified by sucrose gradient centrifugation. Int J Biochem Cell Biol. 1999;31:273–276. doi: 10.1016/s1357-2725(98)00117-4. [DOI] [PubMed] [Google Scholar]
- Yang L, Li P, Fu SN, Calay ES, Hotamisligil GS. Defective Hepatic Autophagy in Obesity Promotes ER Stress and Causes Insulin Resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C, Ma M, Ran L, Zheng J, Tong J, Zhu J, Ma C, Sun Y, Zhang S, Feng W, et al. Function and molecular mechanism of acetylation in autophagy regulation. Science (New York, NY. 2012;336:474–477. doi: 10.1126/science.1216990. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin SK. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. P Natl Acad Sci USA. 2009;106:19860–19865. doi: 10.1073/pnas.0906048106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451:587–590. doi: 10.1038/nature06515. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.