

Abstract
Complex diseases (e.g., cardiovascular disease and type 2 diabetes, among many others) pose the biggest threat to human health worldwide and are among the most challenging to investigate. Susceptibility to complex disease may be caused by multiple genetic variants (GVs) and their interaction, by environmental factors, and by interaction between GVs and environment, and large study cohorts with substantial analytical power are typically required to elucidate these individual contributions. Here, we discuss the advantages of both power and feasibility afforded by the use of extended pedigrees of rhesus macaques (Macaca mulatta) for genetic studies of complex human disease based on next-generation sequence data. We present these advantages in the context of previous research conducted in rhesus macaques for several representative complex diseases. We also describe a single, multigeneration pedigree of Indian-origin rhesus macaques and a sample biobank we have developed for genetic analysis of complex disease, including power of this pedigree to detect causal GVs using either genetic linkage or association methods in a variance decomposition approach. Finally, we summarize findings of significant heritability for a number of quantitative traits that demonstrate that genetic contributions to risk factors for complex disease can be detected and measured in this pedigree. We conclude that the development and application of an extended pedigree to analysis of complex disease traits in the rhesus macaque have shown promising early success and that genome-wide genetic and higher order -omics studies in this pedigree are likely to yield useful insights into the architecture of complex human disease.
Keywords: complex disease, pedigree, quantitative trait, rhesus macaque
Introduction
Preventing or curing complex diseases such as cardiovascular disease (CVD), diabetes, addiction, macular degeneration, and arthritis, among many others, poses one of the biggest challenges to human health worldwide. Susceptibility to these diseases is determined by genes, by environmental influences, and by interaction both among genes and between genes and environment. Given this complexity, genetic analysis of complex disease aims to characterize the contribution of additive genetic effects relative to observed variation in the trait studied (i.e., heritability), to identify the comprehensive set of genetic variants (GVs) and their interactions that confer directional effects on the trait, and to describe how GVs and environment interact to influence trait expression. However, achieving any of these goals is no trivial matter because of the substantial analytical power required to detect the many GVs of varying effect size, and the corresponding large number of interactions expected to determine disease susceptibility. The need for sufficient power to support these analyses typically requires very large study cohorts of either unrelated (population-based) or related (family-based) individuals, either of which can be challenging to develop in many human populations. In contrast, large managed colonies of rhesus macaques (Macaca mulatta) with extensive pedigree information and close genetic similarity to humans are readily available at many primate research centers. Despite this obvious opportunity and the widespread use of the macaque as a model for human disease pathology, the application of pedigreed rhesus macaques to genetic discovery in complex human disease is currently nonexistent. In this article, we discuss advantages of feasibility and power gained when using extended pedigrees of rhesus macaques for genetic discovery based on next-generation sequence (NGS) data. We also present highlights of research conducted in rhesus macaques for selected complex human diseases to demonstrate how genome-wide genetic discovery in the rhesus macaque model would add to our knowledge of these diseases. Finally, we describe the development of an extended pedigree and sample biobank of Indian-origin macaques at the Oregon National Primate Research Center (ONPRC) intended for genetic research in complex disease, and the results of initial studies of heritability in this pedigree for several quantitative risk factors for human disease.
Advantages of the Rhesus Macaque Model for Genetic Discovery in Complex Human Disease
Genetic Similarity to Humans and the Feasibility of Developing Extended Rhesus Macaque Pedigrees
Large, managed colonies of rhesus macaques such as those found at many primate research centers have inherent advantages that make developing extended pedigrees in macaques a viable alternative to population-based human study cohorts. These advantages start with a very close genetic and physiologic similarity to humans. The most recent common ancestor between macaque and human occurred approximately 25 million years ago. The rhesus macaque genome exhibits synteny for 89% of human genes and shares 93% mean sequence identity with the human genome (Rhesus Macaque Genome Sequencing and Analysis Consortium et al. 2007). This close degree of genetic similarity to humans, combined with the widespread use of rhesus macaques in physiologic studies of complex disease, makes this species an obvious choice for studying genetic effects on disease that will translate easily to the human condition, particularly for early stages of disease that are difficult or impossible to study in humans.
Rhesus macaques are also polygamous breeders and naturally form multi-male, multi-female groups in which males compete for access to females, a social grouping pattern reproduced in the group-housed macaques at the ONPRC. Male macaques can reproduce as early as 2 years of age, and large paternal half-sibship cohorts are common. Compared with the standard human generation time of 20 years, the breeding strategies and generation times found in rhesus macaques can produce very broad, multi-generational pedigrees over a much shorter time span. Further, because animals may live to their mid-20s in captivity, animals from several successive generations may be available for sampling and phenotyping at any single point in time. Combined with the multiple years of pedigree information available for many primate research center colonies, these advantages of breeding patterns and generation time make it eminently feasible to characterize very large and complex pedigrees with substantial analytical power for genetic studies. Moreover, because animals receive comprehensive veterinary care on an ongoing basis throughout the year, blood samples and whole-body phenotypes of interest may be collected on hundreds of animals per year at nominal cost by working closely with veterinary and animal care personnel during routine exams. In contrast, the human generation time and mobility common to many Westernized countries frequently limit human studies to population-based study designs with unrelated individuals. Such studies may require sample sizes that are orders of magnitude larger in order to reproduce the analytical power found in a smaller number of close relatives.
Power for Analysis of Rare Variants In Extended Rhesus Macaque Pedigrees
In recent years, the focus of most gene mapping studies in humans was on the contribution of common genetic variants to common complex disease, an approach implemented by the International HapMap Project (International HapMap Consortium 2003) and aided by the design of commercial technologies for surveying genetic variation in populations of unrelated individuals. However, the very small proportion of disease or trait heritability ultimately explained by this approach has led to much recent discussion of the basis for this “missing heritability.” Although many hypotheses have been proposed to explain missing heritability, including epigenetic processes (Furrow et al. 2011), methodological deficiencies (Ehret et al. 2012; Zuk et al. 2012), and gene–environment or other interaction (Kaprio 2012), the hypothesis repeated most frequently is the previously neglected contribution of rare or low-frequency variation to heritability of complex disease (Manolio et al. 2009). Concurrent with the realization that rare variants may account for a substantial portion of missing heritability in complex disease, the cost of sequencing a whole genome has plummeted, and the ability to afford sequencing of whole or partial genomes for large numbers of individuals is now within reach. With the rapid approach of the $1000 genome, the field of complex disease genetics is shifting from genome-wide association studies (GWASs) based on common variants assessed using microarray technology to analysis of NGS data (exome and whole-genome), which reveals the entire complement of genetic variation, both common and rare, and allows the testing of contributions from rare variants to complex disease.
Pedigree-based approaches to mapping causal rare variants for complex disease from NGS data have several significant advantages over population-based approaches (Wilson and Ziegler 2011). For variants of approximately 1–5% frequency, population-based cohorts have been shown to have lower power to detect causal variants than same-sized family-based studies. This is due to the dilution of the effect size at the level of the population caused by the low frequency of variants and their effects (even when severe) on relatively small numbers of individuals. Thus, to the extent that rare variants contribute to heritability in complex disease, studies based on thousands or many thousands of unrelated individuals may be required to identify them. In contrast, rare variants are shared by many relatives in pedigrees, thus increasing their frequency in the study. This enrichment for rare variants in pedigrees results in increased locus-specific heritability (a function of both allele frequency and effect size) and improved power to identify causal variants. Moreover, pedigree-based studies are not only able to enrich for rare variants but also may enrich for phenotypes of interest by focusing on lineages with many affected individuals or with substantial trait variation (Wilson and Ziegler 2011).
Family-based studies in rhesus macaques have the potential for even greater power to detect causal rare variants than such studies in humans because of the relative ease of developing very large and complex pedigrees that are not possible in human populations. The increased mobility and generation time of human subjects frequently limits family-based genetic studies in human populations to collections of nuclear families or sibling pairs or groups, a study design with significantly reduced power compared with the use of extended pedigrees (Williams and Blangero 1999). Moreover, rhesus macaque colonies may exhibit a level of coancestry similar to that found in human genetic isolates as a result of limited gene flow, and animals typically experience the same managed environment (e.g., diet, housing), both factors that enhance power to detect genetic signal over noise. Given the advantages outlined above, pedigree-based approaches appear a far more desirable choice than population-based approaches for the study of complex disease genetics in the rhesus macaque model.
Value of Rhesus Macaque Pedigrees to Preclinical and Translational Studies
Large rhesus macaque pedigrees may also provide unique advantages in preclinical and translational studies, such as the ability to estimate the relative contributions to disease risk or response to therapeutics from inherited risk variants, specified environments, and family history. The advantages to conducting such studies in pedigreed rhesus macaques include the usual ones of power, feasibility, and genetic similarity, and add knowledge of inheritance patterns, access to detailed clinical family history, and the ability to rigorously control environmental factors. The potential utility of decomposing response or disease risk into underlying factors is suggested by a recent study by Do and colleagues (2012) that showed that family history may be a better predictor of disease liability than single nucleotide polymorphism (SNP)–based models for high frequency diseases with large heritability (e.g., coronary artery disease, Alzheimer's disease), whereas for diseases of low frequency (e.g., celiac disease, Parkinson disease), SNP-based models perform as well or better than family history. Although differences in DNA sequence and allele frequencies between rhesus macaques and humans may preclude the application of genetic risk models developed in monkeys to all clinical disease in humans, such approaches may have particular utility for diseases influenced by genetic variants that are highly conserved between macaques and humans. Another potential translational application of large rhesus macaque pedigrees is the development of immortalized cell lines from animals of known relationship. Such a resource would allow the estimation of heritability and the application of gene mapping and systems genetics approaches to molecular and cellular response to experimental challenge and drug therapies measured in vitro.
General Challenges to Detecting Causal Rare Variants from NGS Data
Given the interest in the contribution of rare variants to complex disease, the enrichment for rare variants in pedigrees, and the ability to comprehensively survey all genetic variation in the genome at ever-decreasing cost, it is not surprising that there has been renewed interest in family-based designs in genome-wide studies. Both linkage analysis and association methods, as well as approaches that incorporate both types of information, may be applied in pedigrees with genome-wide sequence data to narrow the region of interest and to identify potentially causal variants. However, there are considerably more variants discovered in a NGS study, and because of this, there will be much greater correlation among these variants than seen previously from studies based on more sparse marker data, both within and across linkage disequilibrium blocks (Thomas et al. 2011; Tintle et al. 2011). This increased correlation may be caused by linkage or gametic phase disequilibrium, quality of the sequence alignment, or simple random chance. Failure to address this issue appears to inflate type I error and apparent power for both family- and population-based methods when there is a true but unknown genetic contribution to the phenotype.
To analyze rare sequence variants, it is necessary to aggregate them into new derived variables, and the success of various approaches to solve this problem can rely on assumptions about the direction of effects and may be constrained by variable numbers of sequence variants within a gene or other defined region of analysis (Ye and Engelman 2011). However, if there is prior information available on the variants themselves or on their effects on the trait (e.g., nonsynonymous or synonymous substitution effects, candidate genes, biological pathways, or gene networks) that reflects the true underlying etiology of a trait, the use of this prior information may be used to aggregate variants informatively and improve power to detect causal variants (Chen et al. 2011). The informative aggregation of variants may also alleviate the multiple testing burden in sequence-based studies, a problem that is greatly magnified by the sheer number of variants discovered in these studies relative to the number of markers used in a standard GWAS. A lack of prior information on which to aggregate sequence variants, particularly in intergenic regions, may prove a considerable challenge for studies relying on whole-genome sequence data. These and related problems are currently an active area of research.
The Rhesus Macaque Model for Common Complex Human Disease
The rhesus macaque is a well-established model for pathologic processes in many complex human diseases, and a number of studies have employed a candidate gene approach to explore genetic contributions to these pathologies. However, we currently lack appropriate study populations to enable a move beyond the candidate gene approach toward genome-wide genetic discovery in this species. To underscore the need for pedigreed study populations that will support genome-wide studies in the rhesus macaque model, we briefly summarize key findings from studies of pathology and genetic contributions to pathology in both humans and macaques for a representative set of diseases with significant impact on public health. For these and other human diseases in which significant heritability is undisputed but for which many contributing genetic factors remain unknown, an extended pedigree of phenotyped rhesus macaques can provide a powerful resource for discovering new genetic variants that influence disease risk, and for translating these findings to the human condition.
Atherosclerosis
The rhesus macaque has a lengthy history as a nonhuman primate model for human atherosclerosis, particularly in response to experimental diets rich in fat and cholesterol. Depending on the type and amount of dietary fat and cholesterol and the length of time fed, rhesus macaques develop either mild hypercholesterolemia similar to that commonly seen in the general human population or the more extreme hypercholesterolemia seen in familial human disease (Bhattacharyya and Eggen 1987; Bond et al. 1980; Newman et al. 1974; Pronczuk et al. 1991; Rudel 1997). Coincident with this increase in plasma cholesterol and in direct proportion to the length of time on the experimental diet, rhesus macaques develop increasingly severe atherosclerosis that is strikingly similar to that observed in humans. Observed stages of lesion development include the retention of lipid-rich macrophages (foam cells) and other immune cells in the subendothelium, the appearance of fatty streaks in the aorta and coronary arteries, the progression from fatty streak to uncomplicated fibrous atheroma with proliferation of smooth muscle cells, and eventually the progression to more complicated lesions characterized by intimal necrosis, calcification, and hemorrhage (Armstrong 1976; Bond et al. 1980; Davis and Wissler 1984; Gresham 1976). Of particular note, rhesus macaques fed experimental diets have also been observed to progress to clinically relevant stages of disease (Williams et al. 1991), including ischemia with significant stenosis of the coronary arteries and sudden death resulting from occlusive thrombosis and myocardial infarction (Bond et al. 1980).
Despite these observations and the indisputable contribution of genetics to CVD in humans (Roberts and Stewart 2012; Schunkert et al. 2011), virtually nothing is known about the genetic basis of susceptibility to atherosclerosis or associated risk factors in rhesus macaques. This is unfortunate, given that the rhesus macaque model offers researchers the ability to investigate rigorously aspects of atherogenesis that may be difficult or impossible to study in humans, such as genetic susceptibility to early disease processes and gene-by-environment interaction. The likelihood that such studies would be fruitful may be inferred from the virtually complete identity between macaques and humans for a gene known to influence proatherogenic lipid levels in humans, LDLR, which codes for the low-density lipoprotein (LDL) receptor. The rhesus macaque LDLR exhibits 95–100% identity with the human LDLR and its corresponding protein at multiple levels of organization, including gene length, open reading frame length, protein length, and mRNA sequence, and 85–100% identity across functional domains in the protein (Kassim et al. 2011; Südhof et al. 1985). This degree of genetic similarity in LDLR corresponds well with clinical symptoms shared between macaques and humans with familial hypercholesterolemia (Kusumi et al. 1993), associated with mutations in LDLR in both species (Hummel et al. 1990). Given the increases in circulating lipids, correlated increases in atherosclerosis, and clinical symptoms of CVD in rhesus macaques fed experimental diets, and the similarities observed in macaques and humans for familial disease, it seems likely that genome-wide studies conducted in rhesus macaques will identify both known and novel genetic variants that increase susceptibility to atherosclerosis in both species.
Obesity and Type 2 Diabetes Mellitus
Both spontaneous and induced obesity are well-known in rhesus macaques, and, similar to humans, macaques with increasing adiposity progress from dysregulation of glucose metabolism to insulin resistance and ultimately to clinical type 2 diabetes mellitus (T2DM). In studies of spontaneous (noninduced) obesity in individually housed, male rhesus macaques of middle age or older, only one-third could be described as nonobese, whereas two-thirds were either moderately or very obese, despite maintenance on a low-fat, low-cholesterol chow diet (Kemnitz and Francken 1986; Kemnitz et al. 1989). Obese macaques showed a pattern of abdominal excess fat deposition very similar to that seen in obese humans, with abdominal circumference accounting for the largest differences in body dimensions between obese and nonobese animals. Changes in body size are correlated with age and sex, even in free-ranging macaques; body weight and central fat deposition are greatest in females aged 10 to 14 years and in males aged 15 to 19 years (Schwartz and Kemnitz 1992). These findings suggest that rhesus macaques have a natural propensity toward weight gain and obesity in middle age and beyond, similar to observations in humans.
Obesity in rhesus macaques is associated with metabolic dysfunction. Obese macaques have elevated fasting serum insulin, higher fasting plasma glucose levels, slower glucose disappearance rates, and higher triglycerides than nonobese monkeys (Kemnitz and Francken 1986). In particular, central obesity appears to be the most strongly predictive of metabolic dysfunction; abdominal circumference, which together with body mass index (BMI) explains 96% of variance in body fat, is strongly correlated with fasting plasma insulin levels and with insulin resistance. Central obesity in rhesus macaques also appears to be an excellent predictor of T2DM, which develops following increases in body weight and fat, changes in plasma insulin, and acute response to glucose challenge, and may ultimately include pancreatic islet amyloid deposition and decreases in the number of islet β cells (reviewed in Bauer et al. 2011; Bodkin and Hansen 1988; de Koning et al. 1993; Kahn et al. 2001).
The role of genetic influences on obesity in humans is well-established, as indicated by very large recent GWAS of BMI and waist-to-hip ratio, a measure of central obesity in humans (Heid et al. 2010; Speliotes et al. 2010). One two-stage GWAS conducted in a total 249,796 individuals found new loci and replicated previous results for 32 variants associated with BMI that accounted for 6–11% of genetic variation in this trait. Because the distribution of fat has a different genetic etiology than that influencing BMI, a separate GWAS of waist-to-hip ratio was conducted in 190,803 individuals. This study revealed 14 loci associated with waist-to-hip ratio that explained approximately 1% of total variance in this trait, with several of these loci exhibiting evidence for sexual dimorphism. In rhesus macaques, despite evidence supporting a relationship between central obesity and metabolic dysregulation that predisposes animals to the development of T2DM, no studies have yet investigated a genetic basis for traits related to obesity.
In addition to spontaneous obesity featuring impaired glucose tolerance, insulin resistance, and progression to T2DM, the rhesus macaque also develops additional symptoms of human metabolic syndrome when challenged with experimental diets associated with insulin resistance in humans. Bremer and colleagues (2011) recently tested the hypothesis that rhesus macaques consuming beverages sweetened with fructose (containing approximately 50% sucrose and high-fructose corn syrup) would develop insulin resistance and metabolic syndrome, with some progressing to overt T2DM. After being fed fructose as a supplement to a standard commercial monkey chow diet daily for 1 year, all macaques developed components of the metabolic syndrome, primarily as increased adiposity, insulin resistance, inflammation, and dyslipidemia, and 4 of the 29 macaques developed overt T2DM.
Genetic studies of T2DM in rhesus macaques are only just beginning. Notably, a recent exploratory GWAS conducted in 14 macaques with prediabetes or T2DM and eight healthy control macaques using a human SNP microarray (Hansen et al. 2011) revealed a candidate causal genetic variant associated with T2DM. Although a lack of power prevented results from achieving a standard threshold of significance, Hansen and colleagues (2011) combined results from a lowered significance threshold with analysis of gene expression in liver, heart, and skeletal muscle from 51 macaques to identify CBLN2, a member of a gene family coding for a precursor protein CER, which is expressed in rat pancreas and controls levels of plasma insulin. The association of this gene with T2DM in rhesus macaques received independent support from its association with insulin resistance in 1449 T2DM patients and 1482 matched control subjects from Finland and Sweden. These results underscore the urgent need for more powerful mechanisms of genetic discovery in the rhesus macaque, and provide encouraging evidence that further genetic study of T2DM in this species is likely to be productive.
Macular Degeneration
Given the rapid expansion in the number of adults aged 65 years and older in the United States expected in the next 20 years, common late-onset diseases such as age-related macular degeneration (AMD) will likely be the subject of increasing investigation. Studies of AMD in humans have demonstrated high heritability for both early and late forms of disease and have repeatedly implicated SNPs in both a gene coding for complement factor H (CFH) and in a transcribed region of unknown function (age-related maculopathy susceptibility 2 [ARMS2]) (Holliday et al. 2013). Rhesus macaques are an excellent physiologic model for AMD based on the close resemblance between the macaque and human eye (Francis et al. 2008; Hope et al. 1992). These similarities include a macula with a foveal pit containing structural characteristics important for spatial acuity, a feature shared only among the Old World monkeys, apes, and humans. Macaques spontaneously develop both drusen (yellow-white protein aggregates in the retinal pigment epithelium) that accumulate with age and pigmentary changes in the macula that mimic hallmarks of early to intermediate human macular disease.
In humans, AMD is associated with two SNPs found in ARMS2 and in the adjacent promoter region of HTRA1, which codes for a serine protease and is known to be in complete linkage disequilibrium with LOC3877145/ARMS2 in white, Japanese, and Chinese populations (Dewan et al. 2006; Yang et al. 2006). A recent sequencing study of these two locations in rhesus macaques revealed that, as in humans, these gene regions are adjacent, and additionally described two SNP variants associated with drusen in macaques, one variant in each region. Unlike in humans, these two variants were in high but not complete linkage disequilibrium. In macaques, the HTRA1 variant has functional consequences for gene expression, but the role of the ARMS2 variant is still not fully understood. The replicated association of the ARMS2 and HTRA1 genetic variants with drusen in macaques suggests that this species is likely to be an excellent genetic model for AMD; however, the ability to expand to genome-wide studies of AMD in macaques is currently constrained by the lack of resources to enable such studies, including appropriate study populations.
Alcohol Abuse and Dependence
Rhesus macaques share many features of alcohol abuse disorders with humans, including behavioral and physiologic responses to alcohol and genetic influences on these responses (Barr 2013; Grant and Bennett 2003; Schwandt et al. 2010). For example, chronic alcohol consumption has been shown to alter hypothalamic-pituitary-adrenal (HPA) axis activity, the neuroendocrine stress response pathway (Boschloo et al. 2011). Candidate gene studies testing this hypothesis in rhesus macaques have demonstrated that NPY (Lindell et al. 2010), DRD1 (Newman et al. 2009), and CRH (Barr et al. 2008), genes coding for proteins involved in neuroendocrine regulation of stress, are associated with HPA axis dysfunction and with alcohol consumption in this species. The interaction between HPA axis dysfunction and alcohol consumption in rhesus macaques suggests that, as with humans, there may be common genetic links between neuroendocrine stress response and alcohol use.
Numerous studies have also found strong similarities between macaques and humans linking alcohol-related behaviors to the function of neurotransmitters associated with mood, emotion, and the experience of reward. Dysfunction in the pathway regulating serotonin has been linked with a low response to alcohol, impaired impulse control, and early-onset alcoholism in humans (Hinckers et al. 2006). In support of a similar effect in macaques, one recent study identified an additive effect on HPA axis dysfunction in macaques from variants in two genes that influence serotonin signaling (TPH2, involved in the biosynthesis of serotonin, and SLC6A4, which codes for the serotonin transport protein), in addition to those found in the CRH gene (Ferguson et al. 2012). Similar to humans, rhesus macaques have a variable number repeat polymorphism in the transcriptional control region of the serotonin transporter gene (SLC6A4), commonly known as 5HTT-LPR, that influences its expression and predicts intoxication in rhesus macaques reared without mothers, suggesting a genotype-by-environment interaction effect on intoxication (Barr et al. 2003). The endogenous opioid system in humans is implicated in the experience of reward and has also been linked to vulnerability to addiction (Oswald and Wand 2004). Similar to humans, macaques carry a nonsynonymous mutation in OPRM1, the mu-opioid receptor gene, that has been associated with increased alcohol-induced stimulation, increased alcohol consumption, and increased frequency of intoxication, particularly in males (Barr et al. 2007), also consistent with findings in humans. These results suggest a substantial genetic component to alcohol-related physiologic response and behavior and highlight the need for genome-wide genetic analysis of traits related to alcohol abuse and dependence.
The Potential of the Rhesus Macaque as a Model for Other Complex Human Disease
Acute and Chronic Colitis in Rhesus Macaques
In addition to serving as models for human disease, captive-bred rhesus macaques also spontaneously develop disease that appears similar to human disease but for which the macaque-specific etiology and overlap with human disease are not well understood. Colitis in rhesus macaques is characterized by dehydration, lethargy, loose stool and/or dysentery, electrolyte imbalance, and metabolic acidosis. Both acute and chronic colitis represent a significant cause of morbidity and mortality in managed colonies of rhesus macaques. In 2010, among the 3181 outdoor-housed rhesus macaques at the ONPRC, 250 animals accounted for more than 700 clinical cases of diarrhea, an incidence rate of 7.8%. Of these, 103 animals were euthanized because of severe, acute colitis or chronic colitis and wasting, resulting in a mortality rate of 3.2% (Prongay et al. 2013). Similar rates have been reported elsewhere (Hird et al. 1984; Schneider et al. 1960).
Macaque colitis may have heterogeneous underlying etiologies, although evidence suggests a significant role for both genetic and environmental factors, the hallmark of complex disease. To date, most research has focused on environmental causes of colitis. Of note, some of the most successful models of environmental colitis are developed around pathogens that can be isolated from both sick and healthy animals. Of these, the rhesus model of acute bacillary dysentery, using oral administration of Shigella flexneri, is perhaps the best characterized (Kinsey et al. 1976; Mulder 1971; Pucak et al. 1977; Takeuchi et al. 1968) and provided early insight into the microscopic features and cellular pathophysiology of this syndrome. Bacterial models of chronic colitis, using Camphylobacter species (Tribe and Fleming 1983) and Escherichia coli (Kang et al. 2001), two bacteria commonly isolated from healthy animals, have also been developed and may prove to be useful models for human inflammatory bowel disease (Sestak et al. 2003). In addition, the association between viral infection and diarrheal disease remains a dynamic area of research. Rhesus macaques experimentally infected with simian immunodeficiency virus may develop chronic colitis, similar to human immunodeficiency virus patients. Disease severity and lesion location varies among individuals, and these differences have recently been correlated with interleukin 6 and SOC-3 gene expression (Mohan et al. 2007). This variation in symptoms is also seen among animals infected with adenovirus. Early studies implicated adenovirus in chronic colitis (Sestak et al. 2003; Stuker et al. 1979), but recent reports suggest the virus is also prevalent in asymptomatic carriers (Roy et al. 2012). Among animals infected with the same bacterial or viral pathogens, substantial variability in symptoms suggests the possibility of genetic variation among individuals in susceptibility or response to infection.
In addition to genetic susceptibility to environmental pathogens, genetic variation in response to other environmental stress may also contribute to colitis. Early weaning and social stress are both strongly correlated with disease. For example, nursery-reared animals have diarrhea rates 7.5 times higher than infants reared with mothers (Elmore et al. 1992), and the rate of diarrhea among nursery-reared animals is 3 to 4 times higher than that for animals reared in any other housing type (Hird et al. 1984). Indoor, group-housed animals and animals in single or paired housing have higher diarrhea rates than outdoor, group-housed animals (Hird et al. 1984; Sestak et al. 2003; Wolfensohn 1998). Historically, these differences have been attributed purely to a lack of maternal or other environmental influence, but associations are increasingly made between genetic variation and environmental factors likely to cause social stress. For example, polymorphisms in the gene coding for the serotonin transporter (SLC6A4) have been implicated in differences in behavioral response to stress between macaque infants with secure attachments to their mother and infants lacking parental attachment (Suomi 2011).
Dietary sensitivities also offer a new and potentially powerful model of genetic susceptibility to colitis. A subset of rhesus macaques with noninfectious chronic colitis appear to have inherited gluten sensitivity, similar to human celiac disease, although genetic analysis is still ongoing (Sestak et al. 2011). This finding suggests that the rhesus macaque may be a good model for genetic susceptibility to human celiac disease, which has been associated with variants in the TAGAP gene (Eyre et al. 2010).
Degenerative and Inflammatory Arthritis in Rhesus Macaques
Rhesus macaques spontaneously develop both degenerative and inflammatory arthritis, and similarities in structural, mechanical loading, and connective tissue properties in the joint make this an attractive model for human disease (Châteauvert et al. 1989; Châteauvert et al. 1990). Naturally occurring osteoarthritis and spondyloarthropathy has been extensively studied in a troop affiliated with the Cayo Santiago colony at the Caribbean Primate Research Center in Cayo Santiago, Puerto Rico. Spondyloarthropathy was identified in 20% of the animals aged 8 years or older and, similar to humans, females had higher disease incidence (Pritzker et al. 1989; Rothschild et al. 1997). A metabolically associated degenerative osteoarthritis similar to human calcium pyrophosphate dihydrate crystal deposition disease was also identified in this population (Pritzker et al. 1989; Rothschild et al. 1999). Additionally, estrogen-depleted bone loss has been confirmed in studies of aging adult female macaques at the Wisconsin National Primate Research Center (Colman et al. 1999). More recently, age-associated disc space narrowing and osteophytosis was confirmed in a cohort of animals followed for 11 years at the same center (Duncan et al. 2011).
The heterogeneous and progressive nature of these various arthropathies suggests significant similarities to human disease, and their pathology has been explored experimentally in rhesus macaques. For example, type II collagen-induced arthritis (CIA), a model for rheumatoid arthritis, is induced in 70% of animals inoculated with type II collagen (Bakker et al. 1990). Genetic susceptibility to CIA has been implicated in rhesus macaques by association of CIA with the major histocompatibility complex (MHC) class I region (Bakker et al. 1992), and further studies demonstrated that the MHC class 1 allele Mamu-B26 was protective for CIA (Vierboom et al. 2005; Vierboom et al. 2007). A similar pattern of immune-mediated resistance to CIA was noted in 2000 at the Wisconsin National Primate Research Center after an outbreak of Shigella bacteria. Similarly, a cohort of macaques was reported to develop reactive arthritis, an inflammatory spondyloarthropathy, in response to infectious enterocolitis (Rothschild 2005), and a protective effect of the MHC A locus allele, Mamu-A*12, was associated with resistance to this arthropathy (Urvater et al. 2000). Further research in rhesus macaques aimed at clarifying the heterogeneity among arthropathies, characterizing genetic susceptibility and environmental influences on disease, and exploring overlap with human disease is urgently needed.
The ONPRC Research Pedigree and Sample Biobank
Development of an Extended Pedigree at the ONPRC for Genetic Analysis of Complex Traits in the Indian-Origin Rhesus Macaque
Based on the advantages described in this paper, we aimed to develop a single, extended pedigree of Indian-origin rhesus macaques for the purpose of genome-wide genetic analysis of quantitative risk factors for complex human disease. This pedigree was characterized by developing custom Python scripts to select a set of animals from the approximately 4500-member colony of rhesus macaques housed at the ONPRC that optimized several criteria. Animals must (1) be of pure Indian ancestry, (2) have parentage assignment based on genotypes at 12 to 28 microsatellite loci, (3) be a member of a minimum three-generation vertical lineage, (4) be available for sampling, (5) have no ancestors in common outside the pedigree, and (6) form a single pedigree configuration. The resulting pedigree contains 1289 rhesus macaques, including 800 females and 489 males, and spans six generations (see Figure 1). This pedigree represents approximately 29% of the total population of rhesus macaques found at the ONPRC. Animals range in age from 1.6 to 28.5 years, corresponding to a developmental age range of 4.8 to 85.5 human years (the distribution of age by sex in this pedigree is summarized in Figure 2). Table 1 summarizes the most frequent relationship classes found within this pedigree; these relationships provide hypotheses of excess alleles shared identical by descent among macaques, and the large number and complexity of these relationships is the source of power in genetic analysis conducted in extended pedigrees.
Figure 1.

Diagram of the 1289-member research pedigree of Oregon National Primate Research Center rhesus macaques
Figure 2.

Distribution of age and sex among the 1289 rhesus macaques in the Oregon National Primate Research Center research pedigree.
Table 1.
Summary of the most frequent relationship classes found within the single, 1289-member pedigree described
| Relationship | No. | Relationship | No. |
|---|---|---|---|
| Unrelated | 890,106 | Half-2nd cousins | 1284 |
| Parent–offspring | 1966 | Half-siblings and half-1st cousins | 371 |
| Siblings | 165 | Half-siblings and half-avuncular | 73 |
| Grandparent–grandchild | 2150 | Double half-avuncular | 193 |
| Avuncular | 222 | Double half-1st cousins | 358 |
| Half-siblings | 6954 | Half-2nd cousins, once removed | 128 |
| Great-grandparent–grandchild | 1027 | Half-1st cousins, twice removed | 209 |
| Grand-avuncular | 56 | Half-great-grand avuncular | 98 |
| Half-avuncular | 13,179 | Half-1st cousins and half-avuncular | 142 |
| 1st cousins | 156 | Half-avuncular and half-grand avuncular | 30 |
| Great-great-grandparent–grandchild | 84 | Half-avuncular and half-1st cousins, once removed | 330 |
| Half-grand avuncular | 3079 | Half-1st cousins and half-2nd cousins | 139 |
| 1st cousins, once removed | 128 | Parent–offspring and half-avuncular | 16 |
| Half-1st cousins | 10,413 | Half-1st cousins, once removed and half-2nd cousins, once removed | 14 |
| Half-1st cousins, once removed | 6740 | Half-sibs and half-2nd cousins | 24 |
| 2nd cousins | 27 |
Sample Biobank and Whole-Body Phenotypes
To enable extensive phenotyping of pedigree members, we collected whole blood samples on these macaques under an approved institutional animal care and use committee protocol. All samples were collected after an overnight fast and at a consistent time of day (approximately 10:00 AM) (see Table 2). Whole blood was processed for serum, plasma, leukocytes, and peripheral blood mononuclear cells and aliquoted separately in RNAlater (Ambion) to support transcriptome analysis and for storage in liquid nitrogen to enable viable cell assays. As of this writing, approximately 95% (n = 1229) of the macaques in this pedigree have at minimum a source of high-quality DNA that will enable analysis of NGS data, and approximately 66% (n = 852) have at least banked serum and plasma to support multiple biomarker assays. We have also collected multiple measures of morphometry and adiposity on approximately 730 of the same animals, including crown–heel, rump–heel, and crown–rump lengths, chest circumference, maximum girth, abdominal circumference, circumference of the upper arm and thigh, and measures of skin-fold thickness at the subscapular, upper arm/triceps, and thigh. Importantly, the vast majority of these samples and phenotypes were collected during routine processing of animals in group housing and did not require the assignment of animals to research. This approach minimizes costs and increases feasibility of large-scale phenotyping efforts and does not interfere with animal assignment to other research projects.
Table 2.
Summary of sample inventory collected on the 1289-member pedigree
| Sample type | Pedigreed macaques with samples/measures |
|---|---|
| Serum | 854 |
| Plasma | 852 |
| Leukocytes (for high-quality genomic DNA) | 852 |
| PBMCs for gene expression | 708 |
| PBMCs for viable cell assays | 713 |
| Measures of body morphometry | 732 |
| Measures of adiposity | 725 |
See text for description of measures of body morphometry and adiposity. PBMC, peripheral blood mononuclear cell.
Power of the Pedigree to Detect Heritability in a Quantitative Trait
Our intended focus is primarily on the study of quantitative endophenotypes related to disease risk because they are closer to genetic regulation than clinical diagnoses and thus likely to be more informative for gene mapping. Given this focus, we assessed the power of this pedigree to detect significant heritability in quantitative traits across a wide range of true heritabilities (0.15–0.80) under the assumption that all individuals are phenotyped, using a maximum likelihood–based variance decomposition approach implemented in SOLAR software (Sequential Oligogenic Linkage Analysis Routines, v.4.2.0, Texas Biomedical Research Institute, San Antonio, TX). These results indicate that the power to detect an additive genetic contribution to quantitative trait variation using this pedigree is excellent, with approximately 85% power to detect trait heritabilities as low as 0.08 and 100% power to detect trait heritabilities as low as 0.16 (see Figure 3).
Figure 3.
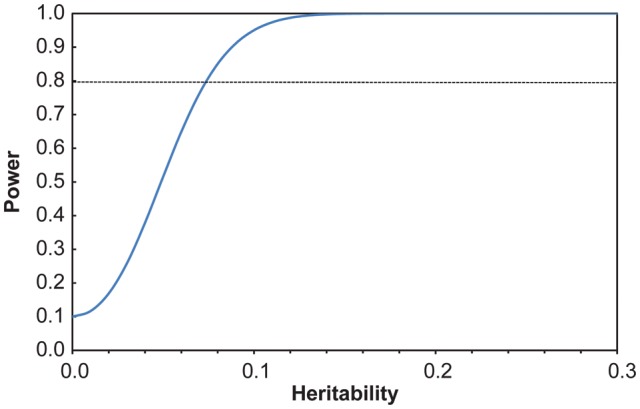
Power of the pedigree to detect heritability for a quantitative trait using a maximum likelihood–based variance decomposition approach. Analysis conducted in SOLAR (v.4.2 Texas Biomedical Research Institute, San Antonio, TX).
Power of the Pedigree to Detect Linkage to a Locus Influencing a Quantitative Trait
Because linkage analysis can reduce the genomic search space, lower the multiple testing burden, and provide prior information that may increase power to fine-map causal genetic variants, we examined the power of the pedigree to detect linkage to a locus influencing a quantitative trait (i.e., a quantitative trait locus [QTL]) using the same variance decomposition approach. Power to detect a locus with a log-of-odds (LOD) score of 3 was examined for QTL heritabilities up to 0.75, assuming that all individuals are phenotyped, in two situations in which total heritability of the trait is 0.25 and 0.75 and residual heritability contributes to the power to detect the QTL (see Figure 4). The power of this pedigree to detect a QTL with an LOD score of 3 (i.e., indicating genome-wide significant evidence) is very good across this range of trait and QTL heritabilities. For example, considering a trait with a total heritability of 0.25, this pedigree provides greater than 80% power to detect a QTL heritability of 0.15; as total trait heritability increases to 0.75, we have greater than 80% power to detect a QTL heritability of 0.13. These QTL heritabilities are smaller or equivalent in magnitude to those reported in several previous QTL and eQTL mapping studies conducted in a large pedigree of baboons for LDL cholesterol levels, lipoprotein-associated phospholipase A2 levels, and transcript levels implicated in immune response (Vinson, Mahaney, Cox et al. 2008; Vinson, Mahaney, Diego et al. 2008; Vinson et al. 2011).
Figure 4.
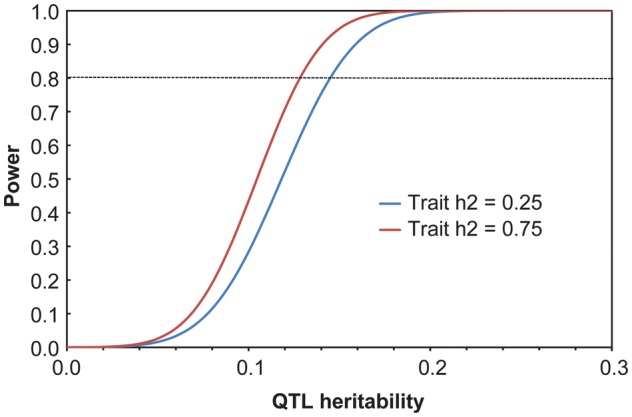
Power of the pedigree to detect linkage to a locus influencing a quantitative trait with a log-of-odds score of 3 across a range of quantitative trait locus heritabilities using a maximum likelihood–based variance decomposition approach. Power for two traits with total heritabilities of 0.25 (blue) and 0.75 (red) is considered. Analyses conducted in SOLAR (v.4.2, Texas Biomedical Research Institute, San Antonio, TX).
Power of the Pedigree to Detect Association between a Quantitative Trait and a Common Causal Variant
We also investigated the power of a measured genotype analysis conducted in our extended pedigree of rhesus macaques to detect association between a quantitative trait and a common, causal genetic variant or a genotyped marker in complete linkage disequilibrium with a causal variant. Simulations were conducted to derive expected χ2 statistics based on a trait with a mean and variance similar to that for high-density lipoprotein (HDL) cholesterol levels in the baboon (Papio hamadrayas, ssp.), a total trait heritability of 0.20, and a SNP with a minor allele frequency of 0.20. The mean effect of the SNP was varied to account for 0.5–5% of the total trait variance at 0.5% intervals, and χ2 was evaluated at each interval. Results from this analysis indicate that the pedigree has 80% power to detect associations accounting for 1.5–2.0% of the total variance in a similar trait (M. Mahaney, Texas Biomedical Research Institute, personal communication, 2013). These results indicate that this pedigree also has substantial power to detect effects of common variants on quantitative traits.
Heritability of Quantitative Risk Factors for Human Disease in the ONPRC Rhesus Macaque Pedigree
To confirm the power of the pedigree to detect genetic influences on quantitative traits, we investigated heritability for multiple quantitative risk factors of human disease, including endophenotypes and whole-organism level traits. Because lipid levels are well-established risk factors for CVD in humans and because they are significantly heritable in both humans and in baboons (Vinson, Mahaney, Cox et al. 2008), we assessed the heritability of fasting plasma lipids in a sample of 193 pedigreed rhesus macaques enriched for paternal half-siblings. We found heritability of similar or greater magnitude than that described in large human populations for all lipids in a standard lipid panel, including levels of total cholesterol, LDL cholesterol, HDL cholesterol, very-low density lipoprotein cholesterol, and triglycerides (Vinson, Mitchell, Toffey, Silver et al. 2013). Moreover, based on the standard use of these measures in studies of human obesity and risk for CVD both in the general population and in T2DM, we assessed heritability for abdominal circumference (normalized by crown–rump length), BMI (based on animal weight at sampling and crown–rump length), and animal weight in kilograms from measures in approximately 475 pedigreed macaques; these traits were also characterized by significant, although more moderate, heritability (Vinson, Mitchell, Toffey, Raboin 2013). Finally, we also assessed heritability for complete blood cell counts and related parameters collected during the course of clinical treatment (i.e., not for research purposes) on 995 pedigreed macaques; white blood cell, red blood cell, and platelet counts, as well as measures of mean cell volume, mean cell hemoglobin, mean cell hemoglobin concentration, hematocrit, and hemoglobin, were all significantly heritable, with heritabilities ranging from modest to substantial (Vinson, Mitchell, Toffey 2012). These results confirm that additive genetic contributions to important risk factors of human disease can be detected and measured in this pedigree. Based on these initial results, analyses of multiple additional endophenotypes implicated in risk for human disease are underway, including insulin, cortisol, vascular cell adhesion molecule 1 (VCAM-1), interferon γ, tumor necrosis factor α, interleukin 2, interleukin 6, interleukin 12p70, ghrelin, osteocalcin, and lipid subparticle size concentrations.
The Future: Next-Generation Sequencing in Pedigreed Rhesus Macaques
Recent advances in next-generation sequencing provide new avenues for genome-wide genotyping in the rhesus macaque, sidestepping the alternative requirement to generate macaque SNP genotyping arrays. As sequencing costs continue to decline rapidly, whole-genome sequencing is becoming an increasingly viable option for large study cohorts, including extended pedigrees, particularly when combined with imputation of genetic variants in unsequenced individuals. The yield of genetic variant data from whole-genome sequencing is expansive; for example, we have recently identified approximately 3.1 million high-quality SNPs per genome in six unrelated Indian rhesus macaques from 30–45X genome coverage (R Ramakrishnan, unpublished data). This amount of data may be collected in a small number of individuals selected from an extended pedigree and potentially combined with low-coverage sequence data to impute genotypes throughout the remainder of the pedigree (Pasaniuc et al. 2012; Uricchio et al. 2012).
As an alternative to whole genome sequencing, selective DNA enrichment techniques can reduce the amount of sequencing required and thus decrease both genotyping costs and data management requirements. Although such approaches will not necessarily identify all causative variants, the application of studies based on selectively reduced sequence data for analysis of complex disease has already been envisioned (Do et al. 2012; Kiezun et al. 2012). A variety of selection techniques is available. A combination of transcriptome sequencing (RNA-seq) and chromatin immunoprecipitation enriched sequencing (ChIP-seq) was recently used to selectively sequence gene-linked genomic regions in rhesus macaques. This approach yielded at least one SNP in each of the 16,797 annotated rhesus macaque genes and a total of 462,802 SNPs in all 14 individuals investigated (Yuan et al. 2012). Alternatively, we and others have found that human-based exon capture designs can recover 80–95% of the macaque-equivalent exons (Jin et al. 2012; Vallender et al. 2011). This “exome-seq” approach can be used for identifying SNPs and insertions/deletions and for defining haplotypes in 95% of coding genes. These genotype datasets can then be leveraged for subsequent genetic studies.
Conclusion
In this article, we have discussed advantages of both feasibility and analytical power when using extended pedigrees of rhesus macaques for genetic analysis of complex disease traits. These advantages include (1) close genetic and physiological similarity to humans; (2) the availability of very large populations with pedigree information at many primate research centers; (3) the ability to conduct large-scale sampling at nominal cost in managed colonies; (4) mating patterns and short generation times in rhesus macaques that can produce large cohorts of informative relative types, with animals in overlapping generations available for simultaneous sampling; (5) the environmental homogeneity and semi-isolation of managed rhesus macaque colonies that increases power to detect genetic signal over noise; and (6) the enrichment of rare variants in pedigrees, which increases power to detect influential rare variants in pedigrees compared with an equivalent number of unrelated animals. We have also presented short summaries of several representative human diseases investigated using the macaque model that highlight our current limited knowledge of genetic effects on these diseases. Throughout this paper, we have used these discussion points to make the case for the development and use of extended pedigrees for studying genotype–phenotype relationships in the rhesus macaque. Finally, we describe the results of our own efforts to develop a single, extended pedigree and corresponding biobank of Indian-origin rhesus macaques at the ONPRC, including a discussion of the pedigree design and power, and initial results of significant heritability for quantitative risk factors of CVD and metabolic disease. We conclude that the development and application of extended pedigrees to analysis of complex disease traits in the rhesus macaque show great promise for genome-wide genetic and higher order -omics studies in this valuable research model.
Acknowledgments
This project was supported by the Office of the Director/Office of Research Infrastructure Programs (OD/ORIP) of the National Institutes of Health grant number OD 011092, and by support provided to A. Vinson from the Human Genetics Initiative at the Oregon Health & Science University, Portland, Oregon. We gratefully acknowledge the ONPRC veterinary and technical staff for their superb animal care in support of this project; we also thank the reviewers and Michael C. Mahaney, PhD, of the Department of Genetics at the Texas Biomedical Research Institute and the Southwest National Primate Research Center for helpful discussion and for the measured genotype association power analysis.
References
- Armstrong ML. Atherosclerosis in rhesus and cynomolgus monkeys. Primates Med. 1976;9:16–40. [PubMed] [Google Scholar]
- Bakker NP, van Erck MG, Otting N, Lardy NM, Noort RC, t'Hart BA, Jonker M, Bontrop RE. Resistance to collagen-induced arthritis in a nonhuman primate species maps to the major histocompatibility complex class I region. J Exp Med. 1992;175:933–937. doi: 10.1084/jem.175.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker NP, van Erck MG, Zurcher C, Faaber P, Lemmens A, Hazenberg M, Bontrop RE, Jonker M. Experimental immune mediated arthritis in rhesus monkeys. A model for human rheumatoid arthritis? Rheumatol Int. 1990;10:21–29. doi: 10.1007/BF02274777. [DOI] [PubMed] [Google Scholar]
- Barr CS. Non-human primate models of alcohol-related phenotypes: The influence of genetic and environmental factors. Curr Top Behav Neurosci. 2013;13:223–249. doi: 10.1007/7854_2011_142. [DOI] [PubMed] [Google Scholar]
- Barr CS, Dvoskin RL, Yuan Q, Lipsky RH, Gupte M, Hu X, Zhou Z, Schwandt ML, Lindell SG, McKee M, Becker ML, Kling MA, Gold PW, Higley D, Heilig M, Suomi SJ, Goldman D. CRH haplotype as a factor influencing cerebrospinal fluid levels of corticotropin-releasing hormone, hypothalamic-pituitary-adrenal axis activity, temperament, and alcohol consumption in rhesus macaques. Arch Gen Psychiatry. 2008;65:934–944. doi: 10.1001/archpsyc.65.8.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr CS, Newman TK, Becker ML, Champoux M, Lesch KP, Suomi SJ, Goldman D, Higley JD. Serotonin transporter gene variation is associated with alcohol sensitivity in rhesus macaques exposed to early-life stress. Alcohol Clin Exp Res. 2003;27:812–817. doi: 10.1097/01.ALC.0000067976.62827.ED. [DOI] [PubMed] [Google Scholar]
- Barr CS, Schwandt M, Lindell SG, Chen SA, Goldman D, Suomi SJ, Higley JD, Heilig M. Association of a functional polymorphism in the mu-opioid receptor gene with alcohol response and consumption in male rhesus macaques. Arch Gen Psychiatry. 2007;64:369–376. doi: 10.1001/archpsyc.64.3.369. [DOI] [PubMed] [Google Scholar]
- Bauer SA, Arndt TP, Leslie KE, Pearl DL, Turner PV. Obesity in rhesus and cynomolgus macaques: a comparative review of the condition and its implications for research. Comp Med. 2011;61:514–526. [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya AK, Eggen DA. Relationships between dietary cholesterol, cholesterol absorption, cholesterol synthesis, and plasma cholesterol in rhesus monkeys. Atherosclerosis. 1987;67:33–39. doi: 10.1016/0021-9150(87)90262-0. [DOI] [PubMed] [Google Scholar]
- Bodkin NL, Hansen BC. Nonhuman primate studies on diabetes, carbohydrate intolerance and obesity. In: Howard CFJ, editor. Monographs in Primatology. Vol 12. New York: Alan R. Liss; 1988. pp. 7–27. [Google Scholar]
- Bond MG, Bullock BC, Bellinger DA, Hamm TE. Myocardial infarction in a large colony of nonhuman primates with coronary artery atherosclerosis. Am J Pathol. 1980;101:675–692. [PMC free article] [PubMed] [Google Scholar]
- Boschloo L, Vogelzangs N, M CM, Vreeburg SA, Smit JH, van den Brink W, Veltman DJ, de Geus EJC, Beekman ATF, Penninx BWJH. Heavy alcohol use, rather than alcohol dependence, is associated with dysregulation of the hypothalamic-pituitary-adrenal axis and the autonomic nervous system. Drug Alcohol Depend. 2011;116:170–176. doi: 10.1016/j.drugalcdep.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Bremer AA, Stanhope KL, Graham JL, Cummings BP, Wang W, Saville BR, Havel PJ. Fructose-fed rhesus monkeys: A nonhuman primate model of insulin resistance, metabolic syndrome, and type 2 diabetes. Clin Transl Sci. 2011;4:243–252. doi: 10.1111/j.1752-8062.2011.00298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Châteauvert JM, Grynpas MD, Kessler MJ, Pritzker KP. Spontaneous osteoarthritis in rhesus macaques. II. Characterization of disease and morphometric studies. J Rheumatol. 1990;17:73–83. [PubMed] [Google Scholar]
- Châteauvert J, Pritzker KP, Kessler MJ, Grynpas MD. Spontaneous osteoarthritis in rhesus macaques. I. Chemical and biochemical studies. J Rheumatol. 1989;16:1098–1104. [PubMed] [Google Scholar]
- Chen GK, Chen G, Wei P, DeStefano AL. Incorporating biological information into association studies of sequencing data. Genet Epidemiol. 2011;35:S29–S34 doi: 10.1002/gepi.20646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman RJ, Kemnitz JW, Lane MA, Abbott DH, Binkley N. Skeletal effects of aging and menopausal status in female rhesus macaques. J Clin Endocrinol Metab. 1999;84:4144–4148. doi: 10.1210/jcem.84.11.6151. [DOI] [PubMed] [Google Scholar]
- Davis HR, Wissler RW. Apoprotein B quantification in rhesus and cynomolgus monkey atherosclerotic lesions. Atherosclerosis. 1984;50:241–252. doi: 10.1016/0021-9150(84)90072-8. [DOI] [PubMed] [Google Scholar]
- de Koning EJ, Bodkin NL, Hansen BC, Clark A. Diabetes mellitus in Macaca mulatta monkeys is characterised by islet amyloidosis and reduction in beta-cell population. Diabetologia. 1993;36:378–384. doi: 10.1007/BF00402271. [DOI] [PubMed] [Google Scholar]
- Dewan A, Liu M, Hartman S, Zhang SS, Liu DT, Zhao C, Tam PO, Chan WM, Lam DS, Snyder M, Barnstable C, Pang CP, Hoh J. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science. 2006;314:989–992. doi: 10.1126/science.1133807. [DOI] [PubMed] [Google Scholar]
- Do CB, Hinds DA, Francke U, Eriksson N. Comparison of family history and SNPs for predicting risk of complex disease. PLOS Genet. 2012;8 doi: 10.1371/journal.pgen.1002973. e1002973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do R, Kathiresan S, Abecasis GR. Exome sequencing and complex disease: Practical aspects of rare variant association studies. Hum Mol Genet. 2012;21 doi: 10.1093/hmg/dds387. R1–R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan AE, Colman RJ, Kramer PA. Longitudinal study of radiographic spinal osteoarthritis in a macaque model. J Orthop Res. 2011;29:1152–1160. doi: 10.1002/jor.21390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehret GB, Lamparter D, Hoggart CJ, of GI, Whittaker JC, Beckmann JS, Kutalik Z. A multi-SNP locus-association method reveals a substantial fraction of the missing heritability. Am J Hum Genet. 2012;91:863–871. doi: 10.1016/j.ajhg.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore DB, Anderson JH, Hird DW, Sanders KD, Lerche NW. Diarrhea rates and risk factors for developing chronic diarrhea in infant and juvenile rhesus monkeys. Lab Anim Sci. 1992;42:356–359. [PubMed] [Google Scholar]
- Eyre S, Hinks A, Bowes J, Flynn E, Martin P, Wilson AG, Morgan AW, Emery P, Steer S, Hocking LJ, Reid DM, Harrison P, Wordsworth P; Yorkshire Early Arthritis Consortium; Biologics in RA Control Consortium, Thomson W, Worthington J, Barton A. Overlapping genetic susceptibility variants between three autoimmune disorders: rheumatoid arthritis, type 1 diabetes and coeliac disease. Arthritis Res Ther. 2010;12 doi: 10.1186/ar3139. R175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Wang S, Chen C-C, Lan L. GWAPower: a statistical power calculation software for genome-wide association studies with quantitative traits. BMC Genetics. 2011;12:12–15. doi: 10.1186/1471-2156-12-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson B, Hunter JE, Luty J, Street SL, Woodall A, Grant KA. Genetic load is associated with hypothalamic-pituitary-adrenal axis dysregulation in macaques. Genes Brain Behav. 2012;11:949–957. doi: 10.1111/j.1601-183X.2012.00856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis PJ, Appukuttan B, Simmons E, Landauer N, Stoddard J, Hamon S, Ott J, Ferguson B, Klein M, Stout JT, Neuringer M. Rhesus monkeys and humans share common susceptibility genes for age-related macular disease. Hum Mol Genet. 2008;17:2673–2680. doi: 10.1093/hmg/ddn167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furrow RE, Christiansen FB, Feldman MW. Environment-sensitive epigenetics and the heritability of complex diseases. Genetics. 2011;189:1377–1387. doi: 10.1534/genetics.111.131912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant KA, Bennett AJ. Advances in nonhuman primate alcohol abuse and alcoholism research. Pharmacol Ther. 2003;100:235–255. doi: 10.1016/j.pharmthera.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Gresham GA. Primate atherosclerosis. In: Kritchevsky D, Pollak OJ, Simms HS, Karger S, editors. Monographs on Atherosclerosis. Vol 7. Basel, Switzerland: S. Karger AG.; 1976. pp. 24–85. [Google Scholar]
- Hansen BC, Shamekh R, Shamekh R, Hansson O, Almgren P, Budagov T, Linden E, Pessin J, Atzmon G. The rhesus monkey: A nonhuman primate model for T2DM- associated gene screening. J Diabetes. 2011;Metab 2:150 [Google Scholar]
- Heid IM, Jackson AU, Randall JC, Winkler TW, Qi L, Steinthorsdottir V, Thorleifsson G, Zillikens MC, Speliotes EK, Mägi R, Workalemahu T, White CC, Bouatia-Naji N, Harris TB, Berndt SI, Ingelsson E, Willer CJ, Weedon MN, Luan J, Vedantam S, Esko T, Kilpeläinen TO, Kutalik Z, Li S, Monda KL, Dixon AL, Holmes CC, Kaplan LM, Liang L, Min JL, Moffatt MF, Molony C, Nicholson G, Schadt EE, Zondervan KT, Feitosa MF, Ferreira T, Lango Allen H, Weyant RJ, Wheeler E, Wood AR; MAGIC, Estrada K, Goddard ME, Lettre G, Mangino M, Nyholt DR, Purcell S, Smith AV, Visscher PM, Yang J, McCarroll SA, Nemesh J, Voight BF, Absher D, Amin N, Aspelund T, Coin L, Glazer NL, Hayward C, Heard-Costa NL, Hottenga JJ, Johansson A, Johnson T, Kaakinen M, Kapur K, Ketkar S, Knowles JW, Kraft P, Kraja AT, Lamina C, Leitzmann MF, McKnight B, Morris AP, Ong KK, Perry JR, Peters MJ, Polasek O, Prokopenko I, Rayner NW, Ripatti S, Rivadeneira F, Robertson NR, Sanna S, Sovio U, Surakka I, Teumer A, van Wingerden S, Vitart V, Zhao JH, Cavalcanti-Proença C, Chines PS, Fisher E, Kulzer JR, Lecoeur C, Narisu N, Sandholt C, Scott LJ, Silander K, Stark K, Tammesoo ML, Teslovich TM, Timpson NJ, Watanabe RM, Welch R, Chasman DI, Cooper MN, Jansson JO, Kettunen J, Lawrence RW, Pellikka N, Perola M, Vandenput L, Alavere H, Almgren P, Atwood LD, Bennett AJ, Biffar R, Bonnycastle LL, Bornstein SR, Buchanan TA, Campbell H, Day IN, Dei M, Dörr M, Elliott P, Erdos MR, Eriksson JG, Freimer NB, Fu M, Gaget S, Geus EJ, Gjesing AP, Grallert H, Grässler J, Groves CJ, Guiducci C, Hartikainen AL, Hassanali N, Havulinna AS, Herzig KH, Hicks AA, Hui J, Igl W, Jousilahti P, Jula A, Kajantie E, Kinnunen L, Kolcic I, Koskinen S, Kovacs P, Kroemer HK, Krzelj V, Kuusisto J, Kvaloy K, Laitinen J, Lantieri O, Lathrop GM, Lokki ML, Luben RN, Ludwig B, McArdle WL, McCarthy A, Morken MA, Nelis M, Neville MJ, Paré G, Parker AN, Peden JF, Pichler I, Pietiläinen KH, Platou CG, Pouta A, Ridderstråle M, Samani NJ, Saramies J, Sinisalo J, Smit JH, Strawbridge RJ, Stringham HM, Swift AJ, Teder-Laving M, Thomson B, Usala G, van Meurs JB, van Ommen GJ, Vatin V, Volpato CB, Wallaschofski H, Walters GB, Widen E, Wild SH, Willemsen G, Witte DR, Zgaga L, Zitting P, Beilby JP, James AL, Kähönen M, Lehtimäki T, Nieminen MS, Ohlsson C, Palmer LJ, Raitakari O, Ridker PM, Stumvoll M, Tönjes A, Viikari J, Balkau B, Ben-Shlomo Y, Bergman RN, Boeing H, Smith GD, Ebrahim S, Froguel P, Hansen T, Hengstenberg C, Hveem K, Isomaa B, Jørgensen T, Karpe F, Khaw KT, Laakso M, Lawlor DA, Marre M, Meitinger T, Metspalu A, Midthjell K, Pedersen O, Salomaa V, Schwarz PE, Tuomi T, Tuomilehto J, Valle TT, Wareham NJ, Arnold AM, Beckmann JS, Bergmann S, Boerwinkle E, Boomsma DI, Caulfield MJ, Collins FS, Eiriksdottir G, Gudnason V, Gyllensten U, Hamsten A, Hattersley AT, Hofman A, Hu FB, Illig T, Iribarren C, Jarvelin MR, Kao WH, Kaprio J, Launer LJ, Munroe PB, Oostra B, Penninx BW, Pramstaller PP, Psaty BM, Quertermous T, Rissanen A, Rudan I, Shuldiner AR, Soranzo N, Spector TD, Syvanen AC, Uda M, Uitterlinden A, Völzke H, Vollenweider P, Wilson JF, Witteman JC, Wright AF, Abecasis GR, Boehnke M, Borecki IB, Deloukas P, Frayling TM, Groop LC, Haritunians T, Hunter DJ, Kaplan RC, North KE, O'Connell JR, Peltonen L, Schlessinger D, Strachan DP, Hirschhorn JN, Assimes TL, Wichmann HE, Thorsteinsdottir U, van Duijn CM, Stefansson K, Cupples LA, Loos RJ, Barroso I, McCarthy MI, Fox CS, Mohlke KL, Lindgren CM. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet. 2010;42:949–960. doi: 10.1038/ng.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinckers AS, Laucht M, Schmidt MH, Mann KF, Schumann G, Schuckit MA, Heinz A. Low level of response to alcohol as associated with serotonin transporter genotype and high alcohol intake in adolescents. Biol Psychiatry. 2006;60:282–287. doi: 10.1016/j.biopsych.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Hird DW, Anderson JH, Bielitzki JT. Diarrhea in nonhuman primates: a survey of primate colonies for incidence rates and clinical opinion. Lab Anim Sci. 1984;34:465–470. [PubMed] [Google Scholar]
- Holliday EG, Smith AV, Cornes BK, Buitendijk GH, Jensen RA, Sim X, Aspelund T, Aung T, Baird PN, Boerwinkle E, Cheng CY, van Duijn CM, Eiriksdottir G, Gudnason V, Harris T, Hewitt AW, Inouye M, Jonasson F, Klein BE, Launer L, Li X, Liew G, Lumley T, McElduff P, McKnight B, Mitchell P, Psaty BM, Rochtchina E, Rotter JI, Scott RJ, Tay W, Taylor K, Teo YY, Uitterlinden AG, Viswanathan A, Xie S. Vingerling JR, Klaver CC, Tai ES, Siscovick D, Klein R, Cotch MF, Wong TY, Attia J, Wang JJ, editors. Wellcome Trust Case Control Consortium 2. Insights into the genetic architecture of early stage age-related macular degeneration: a genome-wide association study meta-analysis. PLoS One. 2013;8(1) doi: 10.1371/journal.pone.0053830. e53830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope GM, Dawson WW, Engel HM, Ulshafer RJ, Kessler MJ, Sherwood MB. A primate model for age related macular drusen. Br J Ophthalmol. 1992;76:11–16. doi: 10.1136/bjo.76.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel M, Li ZG, Pfaffinger D, Neven L, Scanu AM. Familial hypercholesterolemia in a rhesus monkey pedigree: molecular basis of low density lipoprotein receptor deficiency. Proc Natl Acad Sci U S A. 1990;87:3122–3126. doi: 10.1073/pnas.87.8.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- Jin X, He M, Ferguson B, Meng Y, Ouyang L, Ren J, Mailund T, Sun F, Sun L, Shen J, Zhuo M, Song L, Wang J, Ling F, Zhu Y, Hvilsom C, Siegismund H, Liu X, Gong Z, Ji F, Wang X, Liu B, Zhang Y, Hou J, Wang J, Zhao H, Wang Y, Fang X, Zhang G, Wang J, Zhang X, Schierup MH, Du H, Wang J, Wang X. An effort to use human-based exome capture methods to analyze chimpanzee and macaque exomes. PLoS One. 2012;7 doi: 10.1371/journal.pone.0040637. e40637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn SE, Prigeon RL, Schwartz RS, Fujimoto WY, Knopp RH, Brunzell JD, Porte D Jr. Obesity, body fat distribution, insulin sensitivity and Islet beta-cell function as explanations for metabolic diversity. J Nutr. 2001;131:354S–360S. doi: 10.1093/jn/131.2.354S. [DOI] [PubMed] [Google Scholar]
- Kang G, Pulimood AB, Koshi R, Hull A, Acheson D, Rajan P, Keusch GT, Mathan VI, Mathan MM. A monkey model for enterohemorrhagic Escherichia coli. J Infect Dis. 2001;184:206–210. doi: 10.1086/322011. infection. [DOI] [PubMed] [Google Scholar]
- Kaprio J. Twins and the mystery of missing heritability: The contribution of gene–environment interactions. J Intern Med. 2012;272:440–448. doi: 10.1111/j.1365-2796.2012.02587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassim SH, Vandenberghe LH, Hovhannisyan R, Wilson JM, Rader DJ. Identification and functional characterization in vivo of a novel splice variant of LDLR in rhesus macaques. Physiol Genomics. 2011;43:911–916. doi: 10.1152/physiolgenomics.00006.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemnitz JW, Francken GA. Characteristics of spontaneous obesity in male rhesus monkeys. Physiol Behav. 1986;38:477–483. doi: 10.1016/0031-9384(86)90414-2. [DOI] [PubMed] [Google Scholar]
- Kemnitz JW, Goy RW, Flitsch TJ, Lohmiller JJ, Robinson JA. Obesity in male and female rhesus monkeys: fat distribution, glucoregulation, and serum androgen levels. J Clin Endocrinol Metab. 1989;69:287–293. doi: 10.1210/jcem-69-2-287. [DOI] [PubMed] [Google Scholar]
- Kiezun A, Garimella K, Do R, Stitziel NO, Neale BM, McLaren PJ, Gupta N, Sklar P, Sullivan PF, Moran JL, Hultman CM, Lichtenstein P, Magnusson P, Lehner T, Shugart YY, Price AL, de Bakker PI, Purcell SM, Sunyaev SR. Exome sequencing and the genetic basis of complex traits. Nat Genet. 2012;44:623–630. doi: 10.1038/ng.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsey MD, Formal SB, Dammin GJ, Giannella RA. Fluid and electrolyte transport in rhesus monkeys challenged intracecally with Shigella flexneri 2a. Infect Immun. 1976;14:368–371. doi: 10.1128/iai.14.2.368-371.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumi Y, Scanu AM, McGill HC, Wissler RW. Atherosclerosis in a rhesus monkey with genetic hypercholesterolemia and elevated plasma Lp(a) Atherosclerosis. 1993;99:165–174. doi: 10.1016/0021-9150(93)90019-q. [DOI] [PubMed] [Google Scholar]
- Lindell SG, Schwandt ML, Sun H, Sparenborg JD, Björk K, Kasckow JW, Sommer WH, Goldman D, Higley JD, Suomi SJ, Heilig M, Barr CS. Functional NPY variation as a factor in stress resilience and alcohol consumption in rhesus macaques. Arch Gen Psychiatry. 2010;67:423–431. doi: 10.1001/archgenpsychiatry.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan M, Aye PP, Borda JT, Alvarez X, Lackner AA. Gastrointestinal disease in simian immunodeficiency virus-infected rhesus macaques is characterized by proinflammatory dysregulation of the interleukin-6-Janus kinase/signal transducer and activator of transcription3 pathway. Am J Pathol. 2007;171:1952–1965. doi: 10.2353/ajpath.2007.070017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder JB. Shigellosis in nonhuman primates: A review. Lab Anim. 1971;Sci 21:734–738. [PubMed] [Google Scholar]
- Newman WPI, Eggen DA, Strong JP. Comparison of arterial lesions and serum lipids in spider and rhesus monkeys on an egg and butter diet. Atherosclerosis. 1974;19:75–86. doi: 10.1016/0021-9150(74)90045-8. [DOI] [PubMed] [Google Scholar]
- Newman TK, Parker CC, Suomi SJ, Goldman D, Barr CS, Higley JD. DRD1 5'UTR variation, sex and early infant stress influence ethanol consumption in rhesus macaques. Genes Brain Behav. 2009;8:626–630. doi: 10.1111/j.1601-183X.2009.00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald LM, Wand GS. Opioids and alcoholism. Physiol Behav. 2004;81:339–358. doi: 10.1016/j.physbeh.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Pasaniuc B, Rohland N, McLaren PJ, Garimella K, Zaitlen N, Li H, Gupta N, Neale BM, Daly MJ, Sklar P, Sullivan PF, Bergen S, Moran JL, Hultman CM, Lichtenstein P, Magnusson P, Purcell SM, Haas DW, Liang L, Sunyaev S, Patterson N, de Bakker PI, Reich D, Price AL. Extremely low-coverage sequencing and imputation increases power for genome-wide association studies. Nat Genet. 2012;44:631–635. doi: 10.1038/ng.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritzker KP, Châteauvert J, Grynpas MD, Renlund RC, Turnquist J, Kessler MJ. Rhesus macaques as an experimental model for degenerative arthritis. P R Health Sci J. 1989;8:99–102. [PubMed] [Google Scholar]
- Pronczuk A, Patton GM, Stephan ZF, Hayes KC. Species variation in the atherogenic profile of monkeys: Relationship between dietary fats, lipoproteins, and platelet aggregation. Lipids. 1991;26:213–222. doi: 10.1007/BF02543974. [DOI] [PubMed] [Google Scholar]
- Prongay K, Park B, Murphy SJ. Risk factor analysis may provide clues to diarrhea prevention in outdoor-housed rhesus macaques (Macaca mulatta) Am. J. Primatol. 75. 2013:872–882. doi: 10.1002/ajp.22150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucak GJ, Orcutt RP, Judge RJ, Rendon F. Elimination of the Shigella carrier state in rhesus monkeys (Macaca mulatta) by trimethoprim-sulfamethoxazole. J Med Primatol. 1977;6:127–132. doi: 10.1159/000459732. [DOI] [PubMed] [Google Scholar]
- Gibbs RA, Rogers J, Katze MG, Bumgarner R, Weinstock GM, Mardis ER, Remington KA, Strausberg RL, Venter JC, Wilson RK, Batzer MA, Bustamante CD, Eichler EE, Hahn MW, Hardison RC, Makova KD, Miller W, Milosavljevic A, Palermo RE, Siepel A, Sikela JM, Attaway T, Bell S, Bernard KE, Buhay CJ, Chandrabose MN, Dao M, Davis C, Delehaunty KD, Ding Y, Dinh HH, Dugan-Rocha S, Fulton LA, Gabisi RA, Garner TT, Godfrey J, Hawes AC, Hernandez J, Hines S, Holder M, Hume J, Jhangiani SN, Joshi V, Khan ZM, Kirkness EF, Cree A, Fowler RG, Lee S, Lewis LR, Li Z, Liu YS, Moore SM, Muzny D, Nazareth LV, Ngo DN, Okwuonu GO, Pai G, Parker D, Paul HA, Pfannkoch C, Pohl CS, Rogers YH, Ruiz SJ, Sabo A, Santibanez J, Schneider BW, Smith SM, Sodergren E, Svatek AF, Utterback TR, Vattathil S, Warren W, White CS, Chinwalla AT, Feng Y, Halpern AL, Hillier LW, Huang X, Minx P, Nelson JO, Pepin KH, Qin X, Sutton GG, Venter E, Walenz BP, Wallis JW, Worley KC, Yang SP, Jones SM, Marra MA, Rocchi M, Schein JE, Baertsch R, Clarke L, Csürös M, Glasscock J, Harris RA, Havlak P, Jackson AR, Jiang H, Liu Y, Messina DN, Shen Y, Song HX, Wylie T, Zhang L, Birney E, Han K, Konkel MK, Lee J, Smit AF, Ullmer B, Wang H, Xing J, Burhans R, Cheng Z, Karro JE, Ma J, Raney B, She X, Cox MJ, Demuth JP, Dumas LJ, Han SG, Hopkins J, Karimpour-Fard A, Kim YH, Pollack JR, Vinar T, Addo-Quaye C, Degenhardt J, Denby A, Hubisz MJ, Indap A, Kosiol C, Lahn BT, Lawson HA, Marklein A, Nielsen R, Vallender EJ, Clark AG, Ferguson B, Hernandez RD, Hirani K, Kehrer-Sawatzki H, Kolb J, Patil S, Pu LL, Ren Y, Smith DG, Wheeler DA, Schenck I, Ball EV, Chen R, Cooper DN, Giardine B, Hsu F, Kent WJ, Lesk A, Nelson DL, O'brien WE, Prüfer K, Stenson PD, Wallace JC, Ke H, Liu XM, Wang P, Xiang AP, Yang F, Barber GP, Haussler D, Karolchik D, Kern AD, Kuhn RM, Smith KE, Zwieg AS Rhesus Macaque Genome Sequencing and Analysis Consortium. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222–234. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- Roberts R, Stewart AFR. Genes and coronary artery disease: where are we? J Am Coll Cardiol. 2012;60:1715–1721. doi: 10.1016/j.jacc.2011.12.062. [DOI] [PubMed] [Google Scholar]
- Rothschild BM. Primate spondyloarthropathy. Curr Rheumatol Rep. 2005;7:173–181. doi: 10.1007/s11926-996-0036-0. [DOI] [PubMed] [Google Scholar]
- Rothschild BM, Hong N, Turnquist JE. Naturally occurring inflammatory arthritis of the spondyloarthropathy variety in Cayo Santiago rhesus macaques (Macaca mulatta) Clin Exp Rheumatol. 1997;15:45–51. [PubMed] [Google Scholar]
- Rothschild BM, Hong N, Turnquist JE. Skeletal survey of Cayo Santiago rhesus macaques: Osteoarthritis and articular plate excrescences. Semin Arthritis Rheum. 1999;29:100–111. doi: 10.1016/s0049-0172(99)80041-9. [DOI] [PubMed] [Google Scholar]
- Roy S, Sandhu A, Medina A, Clawson DS, Wilson JM. Adenoviruses in fecal samples from asymptomatic rhesus macaques, United States. Emerg Infect Dis. 2012;18:1081–1088. doi: 10.3201/eid1807.111665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudel LL. Genetic factors influence the atherogenic response of lipoproteins to dietary fat and cholesterol in nonhuman primates. J Am Coll Nutr. 1997;16:306–312. doi: 10.1080/07315724.1997.10718691. [DOI] [PubMed] [Google Scholar]
- Schneider NJ, Prather EC, Lewis AL, Scatterdy AV. Enteric bacteriological studies in a large colony of primates. Ann N Y Acad Sci. 1960;85:935–941. doi: 10.1111/j.1749-6632.1960.tb50013.x. [DOI] [PubMed] [Google Scholar]
- Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, Preuss M, R AF, Barbalic M, Gieger C, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwandt ML, Lindell SG, Chen S, Higley JD, Suomi SJ, Heilig M, Barr CS. Alcohol response and consumption in adolescent rhesus macaques: Life history and genetic influences. Alcohol. 2010;44:67–80. doi: 10.1016/j.alcohol.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz SM, Kemnitz JW. Age- and gender-related changes in body size, adiposity, and endocrine and metabolic parameters in free-ranging rhesus macaques. Am J Phys Anthropol. 1992;89:109–121. doi: 10.1002/ajpa.1330890110. [DOI] [PubMed] [Google Scholar]
- Sestak K, Conroy L, Aye PP, Mehra S, Doxiadis GG, Kaushal D. Improved xenobiotic metabolism and reduced susceptibility to cancer in gluten-sensitive macaques upon introduction of a gluten-free diet. PLoS One. 2011;6:e18648 doi: 10.1371/journal.pone.0018648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sestak K, Merritt CK, Borda J, Saylor E, Schwamberger SR, Cogswell F, Didier ES, Didier PJ, Plauche G, Bohm RP, Aye PP, Alexa P, Ward RL, Lackner AA. Infectious agent and immune response characteristics of chronic enterocolitis in captive rhesus macaques. Infect Immun. 2003;71:4079–4086. doi: 10.1128/IAI.71.7.4079-4086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Lango Allen H, Lindgren CM, Luan J, Mägi R, Randall JC, Vedantam S, Winkler TW, Qi L, Workalemahu T, Heid IM, Steinthorsdottir V, Stringham HM, Weedon MN, Wheeler E, Wood AR, Ferreira T, Weyant RJ, Segrè AV, Estrada K, Liang L, Nemesh J, Park JH, Gustafsson S, Kilpeläinen TO, Yang J, Bouatia-Naji N, Esko T, Feitosa MF, Kutalik Z, Mangino M, Raychaudhuri S, Scherag A, Smith AV, Welch R, Zhao JH, Aben KK, Absher DM, Amin N, Dixon AL, Fisher E, Glazer NL, Goddard ME, Heard-Costa NL, Hoesel V, Hottenga JJ, Johansson A, Johnson T, Ketkar S, Lamina C, Li S, Moffatt MF, Myers RH, Narisu N, Perry JR, Peters MJ, Preuss M, Ripatti S, Rivadeneira F, Sandholt C, Scott LJ, Timpson NJ, Tyrer JP, van Wingerden S, Watanabe RM, White CC, Wiklund F, Barlassina C, Chasman DI, Cooper MN, Jansson JO, Lawrence RW, Pellikka N, Prokopenko I, Shi J, Thiering E, Alavere H, Alibrandi MT, Almgren P, Arnold AM, Aspelund T, Atwood LD, Balkau B, Balmforth AJ, Bennett AJ, Ben-Shlomo Y, Bergman RN, Bergmann S, Biebermann H, Blakemore AI, Boes T, Bonnycastle LL, Bornstein SR, Brown MJ, Buchanan TA, Busonero F, Campbell H, Cappuccio FP, Cavalcanti-Proença C, Chen YD, Chen CM, Chines PS, Clarke R, Coin L, Connell J, Day IN, den Heijer M, Duan J, Ebrahim S, Elliott P, Elosua R, Eiriksdottir G, Erdos MR, Eriksson JG, Facheris MF, Felix SB, Fischer-Posovszky P, Folsom AR, Friedrich N, Freimer NB, Fu M, Gaget S, Gejman PV, Geus EJ, Gieger C, Gjesing AP, Goel A, Goyette P, Grallert H, Grässler J, Greenawalt DM, Groves CJ, Gudnason V, Guiducci C, Hartikainen AL, Hassanali N, Hall AS, Havulinna AS, Hayward C, Heath AC, Hengstenberg C, Hicks AA, Hinney A, Hofman A, Homuth G, Hui J, Igl W, Iribarren C, Isomaa B, Jacobs KB, Jarick I, Jewell E, John U, Jørgensen T, Jousilahti P, Jula A, Kaakinen M, Kajantie E, Kaplan LM, Kathiresan S, Kettunen J, Kinnunen L, Knowles JW, Kolcic I, König IR, Koskinen S, Kovacs P, Kuusisto J, Kraft P, Kvaløy K, Laitinen J, Lantieri O, Lanzani C, Launer LJ, Lecoeur C, Lehtimäki T, Lettre G, Liu J, Lokki ML, Lorentzon M, Luben RN, Ludwig B; MAGIC, Manunta P, Marek D, Marre M, Martin NG, McArdle WL, McCarthy A, McKnight B, Meitinger T, Melander O, Meyre D, Midthjell K, Montgomery GW, Morken MA, Morris AP, Mulic R, Ngwa JS, Nelis M, Neville MJ, Nyholt DR, O'Donnell CJ, O'Rahilly S, Ong KK, Oostra B, Paré G, Parker AN, Perola M, Pichler I, Pietiläinen KH, Platou CG, Polasek O, Pouta A, Rafelt S, Raitakari O, Rayner NW, Ridderstråle M, Rief W, Ruokonen A, Robertson NR, Rzehak P, Salomaa V, Sanders AR, Sandhu MS, Sanna S, Saramies J, Savolainen MJ, Scherag S, Schipf S, Schreiber S, Schunkert H, Silander K, Sinisalo J, Siscovick DS, Smit JH, Soranzo N, Sovio U, Stephens J, Surakka I, Swift AJ, Tammesoo ML, Tardif JC, Teder-Laving M, Teslovich TM, Thompson JR, Thomson B, Tönjes A, Tuomi T, van Meurs JB, van Ommen GJ, Vatin V, Viikari J, Visvikis-Siest S, Vitart V, Vogel CI, Voight BF, Waite LL, Wallaschofski H, Walters GB, Widen E, Wiegand S, Wild SH, Willemsen G, Witte DR, Witteman JC, Xu J, Zhang Q, Zgaga L, Ziegler A, Zitting P, Beilby JP, Farooqi IS, Hebebrand J, Huikuri HV, James AL, Kähönen M, Levinson DF, Macciardi F, Nieminen MS, Ohlsson C, Palmer LJ, Ridker PM, Stumvoll M, Beckmann JS, Boeing H, Boerwinkle E, Boomsma DI, Caulfield MJ, Chanock SJ, Collins FS, Cupples LA, Smith GD, Erdmann J, Froguel P, Grönberg H, Gyllensten U, Hall P, Hansen T, Harris TB, Hattersley AT, Hayes RB, Heinrich J, Hu FB, Hveem K, Illig T, Jarvelin MR, Kaprio J, Karpe F, Khaw KT, Kiemeney LA, Krude H, Laakso M, Lawlor DA, Metspalu A, Munroe PB, Ouwehand WH, Pedersen O, Penninx BW, Peters A, Pramstaller PP, Quertermous T, Reinehr T, Rissanen A, Rudan I, Samani NJ, Schwarz PE, Shuldiner AR, Spector TD, Tuomilehto J, Uda M, Uitterlinden A, Valle TT, Wabitsch M, Waeber G, Wareham NJ, Watkins H; Procardis Consortium, Wilson JF, Wright AF, Zillikens MC, Chatterjee N, McCarroll SA, Purcell S, Schadt EE, Visscher PM, Assimes TL, Borecki IB, Deloukas P, Fox CS, Groop LC, Haritunians T, Hunter DJ, Kaplan RC, Mohlke KL, O'Connell JR, Peltonen L, Schlessinger D, Strachan DP, van Duijn CM, Wichmann HE, Frayling TM, Thorsteinsdottir U, Abecasis GR, Barroso I, Boehnke M, Stefansson K, North KE, McCarthy MI, Hirschhorn JN, Ingelsson E, Loos RJ. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuker G, Oshiro LS, Schmidt NJ, Holmberg CA, Anderson JH, Glaser CA, Henrickson RV. Virus detection in monkeys with diarrhea: The association of adenoviruses with diarrhea and the possible role of rotaviruses. Lab Anim Sci. 1979;29:610–616. [PubMed] [Google Scholar]
- Südhof TC, Goldstein JL, Brown MS, Russell DW. The LDL receptor gene: A mosaic of exons shared with different proteins. Science. 1985;228:815–822. doi: 10.1126/science.2988123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomi SJ. Risk, resilience, and gene-environment interplay in primates. J Can Acad Child Adolesc Psychiatry. 2011;20:289–297. [PMC free article] [PubMed] [Google Scholar]
- Takeuchi A, Formal SB, Sprinz H. Exerimental acute colitis in the rhesus monkey following peroral infection with Shigella flexneri. An electron microscope study. Am J Pathol. 1968;52:503–529. [PMC free article] [PubMed] [Google Scholar]
- Thomas A, Abel HJ, Di Y, Faye LL, Jin J, Liu J, Wu Z, Paterson AD. Effect of linkage disequilibrium on the identification of functional variants. Genet Epidemiol. 2011;35:S115–S119 doi: 10.1002/gepi.20660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tintle N, Aschard H, Hu I, Nock N, Wang H, Pugh E. Inflated type I error rates when using aggregation methods to analyze rare variants in the 1000 Genomes Project exon sequencing data in unrelated individuals: Summary results from Group 7 at Genetic Analysis Workshop 17. Genet Epidemiol. 2011;35:S56–S60 doi: 10.1002/gepi.20650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tribe GW, Fleming MP. Biphasic enteritis in imported cynomolgus (Macaca fascicularis) monkeys infected with Shigella, Salmonella and Campylobacter species. Lab Anim. 1983;17:65–69. doi: 10.1258/002367783781070957. [DOI] [PubMed] [Google Scholar]
- Uricchio LH, Chong JX, Ross KD, Ober C, Nicolae DL. Accurate imputation of rare and common variants in a founder population from a small number of sequenced individuals. Genet Epidemiol. 2012;36:312–319. doi: 10.1002/gepi.21623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urvater JA, McAdam SN, Loehrke JH, Allen TM, Moran JL, Rowell TJ, Rojo S, López de Castro JA, Taurog JD, Watkins DI. A high incidence of Shigella-induced arthritis in a primate species: Major histocompatibility complex class I molecules associated with resistance and susceptibility, and their relationship to HLA-B27. Immunogenetics. 2000;51:314–325. doi: 10.1007/s002510050625. [DOI] [PubMed] [Google Scholar]
- Vallender EJ. Expanding whole exome resequencing into non-human primates. Genome Biol. 2011;12 doi: 10.1186/gb-2011-12-9-r87. R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierboom MPM, Jonker M, Bontrop RE, 't Hart B. Modeling human arthritic diseases in nonhuman primates. Arthritis Res Ther. 2005;7:145–154. doi: 10.1186/ar1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierboom MPM, Jonker M, Tak PP, 't Hart BA. Preclinical models of arthritic disease in non-human primates. Drug Discov Today. 2007;12:327–335. doi: 10.1016/j.drudis.2007.02.012. [DOI] [PubMed] [Google Scholar]
- Vinson A, Curran JE, Johnson MP, Dyer TD, Moses EK, Blangero J, Cox LA, Rogers J, Havill LM, VandeBerg JL, Mahaney MC. Genetical genomics of Th1 and Th2 immune response in a baboon model of atherosclerosis risk factors. Atherosclerosis. 2011;217:387–394. doi: 10.1016/j.atherosclerosis.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson A, Mahaney MC, Cox LA, Rogers J, VandeBerg JL, Rainwater DL. A pleiotropic QTL on 2p influences serum Lp-PLA2 activity and LDL cholesterol concentration in a baboon model for the genetics of atherosclerosis risk factors. Atherosclerosis. 2008;196:667–673. doi: 10.1016/j.atherosclerosis.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson A, Mahaney MC, Diego VP, Cox LA, Rogers J, VandeBerg JL, Rainwater DL. Genotype-by-diet effects on co-variation in Lp-PLA2 activity and LDL cholesterol concentration in baboons fed an atherogenic diet. J Lipid Res. 2008;49:1295–1302. doi: 10.1194/jlr.M800020-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson A, Mitchell AD, Toffey D. Peripheral blood cell counts are heritable in a large, unselected rhesus macaque pedigree. 2012 Abstract. 62nd Annual Meeting of the American Society of Human Genetics, November 6–10, San Francisco, CA. [Google Scholar]
- Vinson A, Mitchell AD, Toffey D, Raboin MJ. Sex-specific heritability of traits related to human obesity in rhesus macaques. 2013 doi: 10.1371/journal.pone.0072241. Abstract. 63rd Annual Meeting of the American Society of Human Genetics, October 22–26, Boston, MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson A, Mitchell AD, Toffey D, Silver J, Raboin MJ. Sex-specific heritability of spontaneous lipid levels in an extended pedigree of Indian-origin rhesus macaques (Macaca mulatta) PLoS ONE. 8(8):e72241. doi: 10.1371/journal.pone.0072241. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JK, Anthony MS, Clarkson TB. Coronary heart disease in rhesus monkeys with diet-induced coronary artery atherosclerosis. Arch Pathol Lab Med. 1991;115:784–790. [PubMed] [Google Scholar]
- Williams JT, Blangero J. Power of variance component linkage analysis to detect quantitative trait loci. Ann Hum Genet. 1999;63:545–563. doi: 10.1017/S0003480099007848. [DOI] [PubMed] [Google Scholar]
- Wilson AF, Ziegler A. Lessons learned from Genetic Analysis Workshop 17: Transitioning from genome-wide association studies to whole-genome statistical genetic analysis. Genet Epidemiol. 2011;35:S107–S114 doi: 10.1002/gepi.20659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfensohn S. Shigella infection in macaque colonies: Case report of an eradication and control program. Lab Anim Sci. 1998;48:330–333. [PubMed] [Google Scholar]
- Yang Z, Camp NJ, Sun H, Tong Z, Gibbs D, Cameron DJ, Chen H, Zhao Y, Pearson E, Li X, Chien J, Dewan A, Harmon J, Bernstein PS, Shridhar V, Zabriskie NA, Hoh J, Howes K, Zhang K. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science. 2006;314:992–993. doi: 10.1126/science.1133811. [DOI] [PubMed] [Google Scholar]
- Ye KQ, Engelman CD. Detecting multiple causal rare variants in exome sequence data. Genet Epidemiol. 2011;35:S18–S21 doi: 10.1002/gepi.20644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q, Zhou Z, Lindell SG, Higley JD, Ferguson B, Thompson RC, Lopez JF, Suomi SJ, Baghal B, Baker M, Mash DC, Barr CS, Goldman D. The rhesus macaque is three times as diverse but more closely equivalent in damaging coding variation as compared to the human. BMC Genet. 2012;13:52. doi: 10.1186/1471-2156-13-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci U S A. 2012;109:1193–1198. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]