

Abstract
Molecular genetics and genomics are revolutionizing the study and treatment of inherited eye diseases. In recognition of the impact of molecular genetics on vision and ophthalmology, the National Eye Institute established the National Ophthalmic Disease Genotyping and Phenotyping Network (eyeGENE®) as a multidirectional research initiative whereby a clinical component for patients diagnosed with inherited eye disease fosters research into the causes and mechanisms of these ophthalmic diseases. This is accomplished by broadening access to genetic diagnostic testing and maintaining a repository of DNA samples from clinically characterized individuals and their families to allow investigations of the causes, interventions, and management of genetic eye disorders. The eyeGENE® Network currently includes Clinical Laboratory Improvement Amendments (CLIA)-certified diagnostic laboratory partners, over 270 registered clinical organizations with 500 registered users from around the United States and Canada, and is now testing approximately 100 genes representing 35 inherited eye diseases. To date, the Network has received 4400 samples from individuals with rare inherited eye diseases, which are available for access by the vision research community. eyeGENE® is a model partnership between the U.S. federal government, eye health care providers, CLIA-approved molecular diagnostic laboratories, private industry, and scientists who represent a broad research constituency.
Keywords: biorepository, clinical trials, diagnostic genotyping, eyeGENE®, genetic testing, ocular genetics, patient registry
Historically, diseases affecting vision were among the first to gain the attention of Western philosophers. Both Hippocrates and Aristotle recognized the familial transmission of ocular traits (1). In modern times, red–green colorblindness and ocular albinism served to further the understanding of sex-linked transmission and X-chromosome inactivation (2–4). Cloning of the first cancer gene, retinoblastoma, and the first G-protein coupled receptor, opsin, represented watershed events in molecular genetics (5–7). The vision research community has always vigorously worked to uncover ocular disease genes, which has led to great successes in identifying disease-causing genetic variants across the entire spectrum of ocular diseases especially for those inherited in a Mendelian fashion. Yet even for Mendelian traits, the relationship between gene and disease is frequently more complex than a simple one-to-one mapping, which complicates medical diagnostics. For example, X-linked retinitis pigmentosa (XLRP) can result from mutations in either RP2 or RPGR yet manifests as a clinically undistinguishable disease state (8, 9). Consequently, XLRP clinical diagnosis alone cannot distinguish between RP2 or RPGR as the molecular cause. Identifying the cause of disease by genetic analysis is critical, as future molecular therapies may treat these two forms of XLRP differently by targeting the distinct genes or their individual cellular mechanisms. Conversely, different mutations within a single gene can result in remarkably distinct clinical phenotypes, as for TGFBI gene mutations, which yield a spectrum of clinically distinct corneal disorders, including lattice corneal dystrophy, granular, Avellino (combined lattice-granular), Reis–Bückler, and Thiel-Behnke corneal dystrophies (10).
Complex common diseases, specifically age-related macular degeneration (AMD), represent the first success of the human genome HapMap Project with the identification of genetic risk factors for AMD (11–15). This achievement heralded opportunities to explore and reveal the complexities of multifactorial common diseases beyond those affecting vision. The availability of populations with readily quantifiable ocular disease traits supports ascertaining large case–control cohorts for genetic analysis and gene-based clinical trials for complex vision disease.
The successful elucidation of the genetic etiology of ocular diseases paved the way for the development of gene-based therapies. Gene therapy success for Leber congenital amaurosis (LCA) (16–19), a type of childhood blindness, solidified proof-of-concept for gene-based interventions. Vision is found to be severely limited in children who lack the normal RPE65 protein, and provision of a functional RPE65 protein augments visual ability and function (20–24). The success with RPE65-associated LCA has led to the initiation of gene-based clinical trials for other retinal diseases such as MERTK-associated autosomal recessive retinitis pigmentosa, ABCA4-associated Stargardt disease and the exudative form of AMD (25).
These achievements signal exciting times for the fields of ophthalmology and vision. Disease-causing mutations are associated with glaucoma, cataracts, strabismus, corneal dystrophies and retinal degenerations, as well as other related eye conditions. The breadth of genetic information highlights the significant inroads made in understanding the molecular basis of human ophthalmic diseases. As a result, gene-centered therapies are being pursued for diseases that were once considered untreatable. However, testing for ocular genes was for a long time an cottage industry and fragmented in laboratories that provided access to testing primarily on a research basis. Further, the federally mandated requirement for medical laboratory certification through the Clinical Laboratory Improvement Amendments (CLIA) slowed the medical application of genetics information derived from research-based investigations. Also, the general scarcity of genetically characterized individuals with rare inherited eye conditions handicaps the interest of researchers and pharmaceutical companies to develop therapies for these orphan diseases. The National Eye Institute (NEI) recognized the need to deliver on the promise of genetics, while simultaneously stimulating the pace of gene-based discoveries.
The formation of eyeGENE®
The NEI responded by establishing the National Ophthalmic Disease Genotyping and Phenotyping Network (eyeGENE®), a community resource designed to bring together patients, providers, molecular testing laboratories and researchers for the common goal of patient care, education and research (http://www.nei.nih.gov/eyegene). Through participation in eyeGENE®, eligible individuals gain access to the diagnostic testing performed by the eyeGENE® Network and have the option to participate in research studies related to their disease.
The eyeGENE® Network infrastructure consists of a centralized Coordinating Center, a CLIA-certified biospecimen receiving facility and DNA extraction and shipping laboratory, a biorepository, a patient registry and a genotype–phenotype database housed at the NEI (Figure S1, Supporting information) (26). Briefly, eyeGENE® incorporates three essential elements: (i) Community-based eye health care providers may enroll eligible individuals to facilitate research in inherited eye conditions, become involved in potential collaborations, and aid in patient clinical care by obtaining genetic test results; (ii) Individuals with inherited eye disease have the opportunity to receive molecular diagnostic testing and participate in research by contributing their sample and information to a deidentified database. They may also elect to be notified of their eligibility to participate in clinical studies; and (iii) Researchers can be granted access to de-identified DNA samples, clinical information, and/or a pool of participants interested in participating in research studies.
eyeGENE® was designed as a two-stage initiative governed by clinical protocols approved by a National Institutes of Health (NIH) Institutional Review Board (IRB). Stage 1 of the eyeGENE® Initiative (Clinical Trials.gov Identifier NCT00378742), ‘National Ophthalmic Genotyping and Phenotyping Network – Creation of DNA Repository for Inherited Ophthalmic Diseases’, accrues samples from individuals with inherited eye diseases. Stage 2 – ‘Distribution of Data and Biomaterials from the NEI’ allows the allocation of samples, data and the contact of participants recruited through Stage 1. To date, eyeGENE® has approved 10 research proposals from vision researchers through Stage 2. Researchers have requested access to the database, biorepository or both. Others have requested the recontact of enrolled individuals with specific genotypes. Requests to use these resources are varied. Approved studies include the molecular modeling of pathogenic variants, high-throughput genetic variation screening, the identification and recruitment of individuals with specific mutations to develop induced pluripotent stem (iPS) cells, and genotype–phenotype correlation studies. The diversity of these studies showcases the usefulness of the eyeGENE® Network as a resource to the vision community.
Research opportunities through eyeGENE®
Individuals enrolled in eyeGENE® may choose to be contacted for recruitment to clinical or research studies. Patients consent to have their extracted DNA and de-identified clinical data made available to vision researchers who have received approval for an eyeGENE® Stage 2 research study. To apply for access, the principal investigator (PI) submits an eyeGENE® access application including a detailed study proposal and agrees to the eyeGENE® terms of use.
To protect confidentiality, patients are not contacted directly by requesting researchers. Instead, patients and their referring providers are notified by the eyeGENE® Coordinating Center of an available clinical trial or research study for which they might qualify. The decision to participate is left up to the patient. The PI is required to submit an annual progress report and agrees to share information generated from the use of the eyeGENE® Network data and biomaterials with the eyeGENE® Coordinating Center and the vision community.
Collaboration opportunities for the referring provider
The referring provider is able to decide whether she/he wishes to be contacted for potential scientific collaborations through Stage 2 research or for further case study analyses with a Network CLIA laboratory. This provides the referring provider with opportunities for research collaboration.
Measuring the impact of eyeGENE®
Metrics of eyeGENE®’s impact take many forms. The original goals of eyeGENE® have been met through the formation of a: (i) National Ophthalmic Disease Genotyping and Phenotyping Network; (ii) Centralized Ophthalmic Genetic Disease Information and Diagnostic Testing Referral Resource; (iii) Centralized De-identified Biorepository; and (iv) a de-identified Genotype–Phenotype Database.
The significance of eyeGENE® to its participant stakeholders varies with the expectations and needs of the involved parties. The singular benefit to participants with inherited eye disease is that the individual can participate in research. Patients may also learn the genetic cause of their condition. Some participants choose to enroll even though they may already know the genetic cause of their condition. Patient advocacy groups have approached eyeGENE® with well-characterized and already genotyped populations for participation in the Network to broaden access of their group to research. The benefits to referring providers include the confirmation of a clinical diagnosis through molecular testing, which may improve surveillance and disease management for their patients. The Network also provides an avenue for clinicians and their patients to learn about clinical trials for which they might be eligible. Researchers benefit from access to a database and biorepository of well-characterized samples for extensive studies to further research outcomes.
Clinical diagnostic testing and biorepository management science
The original NEI administrative supplements awarded to CLIA diagnostic laboratories and CLIA-associated research laboratories that became part of the Network defined the original disease categories tested (see Supporting information for further discussion). The NEI supplements expanded and enhanced molecular diagnostic testing for ophthalmic diseases by providing resources to build infrastructure necessary for CLIA-approved diagnostic testing; identifying and stratifying cohorts; developing new diagnostic testing methodologies; improving detection of sequence variants; collecting and characterizing phenotype data; demonstrating the clinical validity and utility of screening certain genes prior to their integration into routine testing; developing a standardized set of clinical criteria for each disease category tested through the Network; and working toward standardized reporting of diagnostic test results across participating CLIA laboratories. The latter is a critical issue in molecular diagnostics. eyeGENE® also developed an infrastructure for continent-wide patient/physician/researcher integration, and developed expertize in management of a DNA biorepository, curation of patient genotype and phenotype data, and expanding access to a spectrum of external sources.
eyeGENE® participates in the NIH Rare Disease (RD)-HUB Biospecimen/Biorepository Patient Registry (http://biospecimens.ordr.info.nih.gov/), which lists registered national and international biorepositories and assists interested parties to identify desired biospecimens for research use. There are a number of national and international private and public patient resource collections. Listings can be searched on RDHUB or Orphanet (http://www.orpha.net/consor/cgibin/ResearchTrials.php?lng=EN). Organizations with biorepositories are encouraged to register their collections with RD-HUB as it facilitates collaborations and sharing of material and data among investigators to accelerate research for the discovery of new treatments, therapeutics and eventually cures for rare and common diseases.
The eyeGENE® Network is working to develop and refine common data elements (CDEs) as part of a trans-NIH collaboration with the National Library of Medicine (http://www.nlm.nih.gov/cde/index.html). The NIH supports the use of CDEs in biorepositories, patient registries and clinical research for consistency in reporting, improving data quality and allowing opportunities to compare and combine data across multiple studies.
eyeGENE® growth: by the numbers
eyeGENE® measures its impact using several quantitative factors including the (i) number of patient samples accrued; (ii) number of genes tested; (iii) number of Registered Clinical Organizations (RCOs); (iv) number of diagnostic tests completed and (v) results returned to the referring provider.
The eyeGENE® Network received its first sample in September 2006. It received its 100th sample in 2007 and its 1000th sample in 2009. The rate of sample accrual is shown in Fig. 1. Currently, between 700 and 1000 samples are accrued each year. The disease category that contains the largest number of patient samples submitted is the retinitis pigmentosa (RP) and other retinal degenerations category with over 1300 samples (Fig. 2), which corresponds to around 2% of the US population estimated to be diagnosed with RP (Table S1), consistent with the large number of causative genes identified so far [Online Mendelian Inheritance in Man (OMIM®) http://www.ncbi.nlm.nih.gov/omim; Retinal Information Network (RetNET), https://sph.uth.edu/Retnet/]. eyeGENE® contains more than 900 individuals with a diagnosis of Stargardt disease, most of whom have been genotyped. There are six additional disease categories with over 100 individuals in each (See Supporting information for further discussion of efforts to increase representation in other disease categories). To date, there are 4400 samples in the Network. It is even more important to note that over 55% of patients in the Network have received diagnostic results. Given that the Network caters to many rare eye diseases, the accrual rate has surpassed original expectations. As the accrual numbers continue to increase, this resource may have broad future potential.
Fig. 1.

Graph showing the rate of sample accrual from September 2006 through December 2012.
Fig. 2.

Chart depicting the number of samples accrued in each disease category.
The Network initially tested about 20 genes in 9 disease categories; today it tests over 100 genes in 35 disease categories (Table S2). eyeGENE® accrues samples through RCO partnerships. In 2007, there were approximately 30 member RCOs. By 2012, this number increased more than eightfold. RCOs represent both private practices and academic institutions from around the country and Canada. In the United States, there are partner RCOs in all but seven states (Alaska, Delaware, Iowa, Mississippi, Rhode Island, Vermont, and Wyoming). In 2007, there were 11 RCOs who obtained independent IRB approval of the eyeGENE® protocol. By 2012, this number increased more than twofold.
Basic research and clinical application
Perhaps the best measurement of impact is increased knowledge leading to scientific advancement. Although most of the Network laboratories test only for known genes, several Network partners utilize technology that allows for the identification of new genes. Network participation facilitated the discovery of 15 new genes, and the identification of over 250 novel mutations in known genes. Only a few examples will be cited as eyeGENE® is in the process of incorporating the data into the Leiden Open Variation Database (LOVD), a publicly available tool for gene and DNA variant collections. Variants identified through eyeGENE® will be deposited in LOVD and updated periodically.
New genes and mutations identified include those involved in ocular motor disorders. A heterozygous missense mutation in CHN1, encoding α2-chimaerin, a Rac guanosine triphosphatase-activating protein (Rac-GAP) signaling protein implicated in axonal pathfinding, was identified in families with a form of Duane’s retraction syndrome, a complex congenital eye movement disorder (27).
Mutations in certain tubulin isotypes result in ocular motility defects and nervous system disorders. Over 20 unique human heterozygous missense mutations have been identified in genes that encode β-tubulin isotypes TUBB2B and TUBB3 , and analyses of genotype–phenotype correlations support association between the mutated isotype and the ensuing clinical phenotypes. Mutations in TUBB3, a neuron-specific β-tubulin isotype III expressed in post-mitotic neurons, cause the ocular motility disorder congenital fibrosis of the extraocular muscles (CFEOM3), as well as the cortical dysplasia, polymicrogyria (PMG), or a spectrum of cortical dysplasias called malformations of cortical development (MCD) (28). A novel heterozygous missense mutation p.Glu421Lys was reported in TUBB2B that results in an amino acid substitution in a family who segregates CFEOM with PMG. TUBB2B mutations do not typically cause CFEOM, making this p. Glu421Lys substitution unique, providing insight into the divergence between resulting oculomotor phenotypes (29).
Sorsby fundus dystrophy (SFD) is an autosomal dominant maculopathy with early onset of central vision loss due to mutations in the tissue inhibitor of metal-loproteinase 3 gene (TIMP3 ). A novel TIMP3 mutation in exon 1, p.Ser38Cys, was identified in several patients diagnosed with SFD. TIMP3 missense mutations had been previously described in exon 5 in the C-terminal domain. The novel mutation was found in the N-terminal domain, where it generates an unpaired cysteine residue. It is thought to promote dimerization of the TIMP3 protein causing its accumulation in Bruch’s membrane and leading to choroidal neovascularization (30).
Choroideremia (CHM) is an X-linked disorder caused by the CHM gene and is associated with progressive vision loss. Analysis of a large CHM pedigree identified a novel nonsense mutation (p.Tyr398*), which resulted in a pre-mature stop and the loss of a portion of the C-terminal end of the protein. The mutation was found to cause a range of phenotypes in this family, with older female carriers having a tendency to severe phenotypes, (31) thereby nuancing the long-held belief that CHM carrier females only show mild signs of the condition in general (http://www.ncbi.nlm.nih.gov/books/NBK1337/).
Direct patient benefit
Confirming clinical diagnoses through eyeGENE® molecular testing has aided in establishing correlations between cone abnormalities and vision loss in both Stargardt disease and X-linked retinoschisis (32, 33). The eyeGENE® Network has also provided meaningful results for patients. In one example, a Network laboratory demonstrated the importance of testing X-linked genes as the cause of retinal disease in families with a provisional diagnosis of autosomal dominant retinitis pigmentosa (adRP) (34), and identified factors contributing to clinical variation in affected males and carrier females with XLRP (35).
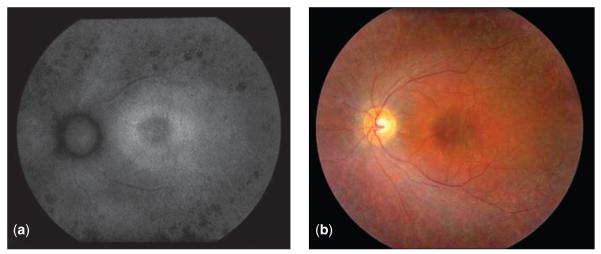
In another case (Fig. 3), an individual was diagnosed with Stargardt disease in adulthood. Molecular testing of the ABCA4 gene yielded inconclusive results: one heterozygous variant of unknown significance (VUS) was reported by an independent CLIA laboratory in 2004. The family history showed three out of six siblings being affected, and in 2011, testing of another affected sibling through eyeGENE® for the full Stargardt disease panel (ABCA4 , PRPH2 and ELOVL4) did not detect the VUS initially reported. However, a known splice site mutation with variable expressivity was found in the PRPH2 gene. This mutation is associated with adRP and autosomal dominant macular dystrophy. In this case, eyeGENE® allowed for the recategorization of the family’s diagnosis to a PRPH2-related maculopathy, and the recognition of an autosomal dominant inheritance pattern in the family.
Fig. 3.
Autofluorescence (a) and color fundus (b) photographs of older adult with PRPH2-associated maculopathy. Ocular history reveals a slowly progressive condition, with relative sparing of the fovea, and reduced full-field electroretinography responses.
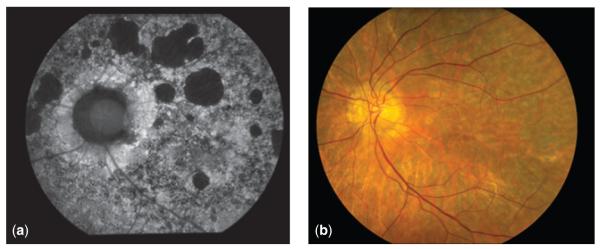
Another instance involved a family enrolled in eyeGENE® in 2008, with a provisional clinical diagnosis of adRP (Fig. 4). No molecular cause had been identified despite previous repeated independent testing. Because the family history could not exclude an X-linked mode of inheritance, RPGR and RP2 were tested next. A VUS was detected in RPGR in two affected individuals. Network laboratories often update their testing schemes and rescreen negative or inconclusive samples for newly discovered genes. In this case, the laboratory proceeded to test the newly added KLHL7 gene. A previously reported mutation was ascertained in this gene (36). Here, the systematic retesting of samples through eyeGENE® led to the identification of the underlying cause of disease in this family.
Fig. 4.
Autofluorescence (a) and color fundus (b) photographs of young adult with KLHL7-associated retinitis pigmentosa. Exam is positive for pigmentary changes, bilateral epiretinal membranes with macular edema, and significantly reduced full-field electroretinography responses.
Diagnostic test development
One objective of the eyeGENE® Network is to develop better methods for gene discovery and mutation detection. High-throughput genotyping assays are critical to the future of diagnostics and research, and members of the eyeGENE® Network are at the forefront in developing microarray-based mutation detection technologies, next-generation sequencing and whole exome/whole genome sequencing for detection of disease-causing mutations (37–39).
eyeGENE® Network: looking ahead
Global interest in eyeGENE® has led the NEI to consider the formation of eyeGENE® International, an umbrella organization of international efforts using similar protocols and operating under similar standards and policies. eyeGENE® has received inquiries for participation from over 20 countries, and has initiated a pilot international partnership. eyeGENE® is a model for collaboration and is visible proof that public-private partnerships can be successful.
Summary
eyeGENE® and its partners are developing important paradigms that may increase understanding of how to maximize access to genetic testing, ensure appropriate utilization of testing by clinicians, optimize high quality genetics-based services, and maintain a well-characterized biorepository that allows researchers access to resources for studying rare inherited eye conditions. It has proven the feasibility of broad genetic testing services, set testing standards for the testing community, and has demonstrated proof of principal of the value of diagnostic genetic testing for clinical ophthalmic care and its contribution to increased scientific knowledge. eyeGENE® integrates genetics into the knowledge base of eye health professionals, and is facilitating the evolution of ophthalmic standard of care to incorporate molecular genetics. eyeGENE® provides a rich intellectual resource that creates important connections between clinical expertise, molecular expertise, institutions, and sample collections, which are necessary to form the collaborative teams required for current investigations in human genetics and genomics and the development of gene-based personalized medicine. It is easy to imagine that the role and value of eyeGENE® will continue to grow as advances in targeted gene-based therapies develop.
Supplementary Material
Acknowledgements
The eyeGENE® Network is supported by the Department of Health and Human Services/National Institutes of Health/National Eye Institute intramural program under eyeGENE® – Protocol 06-EI-0236 which has been funded in part under Contract No. HHSN-260-2007-00001-C. The authors would also like to thank Dr. Wadih Zein, National Eye Institute, for providing the data and the photographs presented in the case studies and the eyeGENE® Network participants for their valuable contributions to this research.
All authors, except R. A., work at the National Eye Institute at the National Institutes of Health and are involved in daily operations of the eyeGENE® Network. R. A.’s clinical laboratory has been supported by the eyeGENE® Network under Contract No. HHS-N-260-2007-00001-C and she has received samples for clinical diagnostic testing.
Footnotes
Supporting Information The following Supporting information is available for this article:
Fig. S1. Schematic illustration of the centralized infrastructure components of eyeGENE®.
Table S1. Accrual of eyeGENE® samples by disease category and comparison to estimated US prevalence.
Table S2. Diagnoses eligible for inclusion and some of the genes tested through eyeGENE®.
Appendix S1. Additional eyeGENE® enrollment details and a look at the Network’s limitations.
Additional Supporting information may be found in the online version of this article.
Conflict of interest None of the authors have any other conflicts to report.
References
- 1.Waardenburg PJ, Franceschetti A, Klein D. Genetics and ophthalmology. Royal VanGorcum, Publisher Assen/Blackwell Scientific Publications, Ltd./Charles C. Thomas; Netherlands/Oxford/Springfield, IL: 1961. [Google Scholar]
- 2.Falls HF. Sex-linked ocular albinism displaying typical fundus changes in female heterozygote. Am J Ophthalmol. 1951;34:41–50. doi: 10.1016/0002-9394(51)90007-4. [DOI] [PubMed] [Google Scholar]
- 3.Falls HF. The role of heredity in ophthalmology. N Y State J Med. 1951;22:2624. [PubMed] [Google Scholar]
- 4.Horner J. Amtlicher Bericht überdie Verwaltung des Medizinalwesens des Kantons Zurich vom Jahr 1876. Druck der Genossenschaftsbuchdruckerei; Zurich: 1876. Die Erblichkeit des Daltonismus; pp. 208–211. [Google Scholar]
- 5.Friend SH, Bernards R, Rogelj S, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323:643–646. doi: 10.1038/323643a0. [DOI] [PubMed] [Google Scholar]
- 6.Nathans J, Piantanida TP, Eddy RL, Shows TB, Hogness DS. Molecular genetics of inherited variation in human color vision. Science. 1986;232:203–210. doi: 10.1126/science.3485310. [DOI] [PubMed] [Google Scholar]
- 7.Nathans J, Thomas D, Hogness DS. Molecular genetics of human color vision: the genes encoding blue, green, and red pigments. Science. 1986;232:193–202. doi: 10.1126/science.2937147. [DOI] [PubMed] [Google Scholar]
- 8.Breuer DK, Yashar BM, Filippova E, et al. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet. 2002;70:1545–1554. doi: 10.1086/340848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharon D, Sandberg MA, Rabe VW, Stillberger M, Dryja TP, Berson EL. RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am J Hum Genet. 2003;73:1131–1146. doi: 10.1086/379379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warren JF, Abbott RL, Yoon MK, Crawford JB, Spencer WH, Margolis TP. A new mutation (Leu569Arg) within exon 13 of the TGFBI (BIGH3) gene causes lattice corneal dystrophy type I. Am J Ophthalmol. 2003;136:872–878. doi: 10.1016/s0002-9394(03)00541-5. [DOI] [PubMed] [Google Scholar]
- 11.Edwards AO, Ritter R, 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 12.Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 14.Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zareparsi S, Branham KE, Li M, et al. Strong association of the Y402H variant in complement factor H at 1q32 with susceptibility to age-related macular degeneration. Am J Hum Genet. 2005;77:149–153. doi: 10.1086/431426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358:2231–2239. doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- 17.Cideciyan AV, Aleman TS, Boye SL, et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci USA. 2008;105:15112–15117. doi: 10.1073/pnas.0807027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hauswirth WW, Aleman TS, Kaushal S, et al. Treatment of Leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008;19:979–990. doi: 10.1089/hum.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358:2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cideciyan AV, Hauswirth WW, Aleman TS, et al. Human RPE65 gene therapy for Leber congenital amaurosis: persistence of early visual improvements and safety at 1 year. Hum Gene Ther. 2009;20:999–1004. doi: 10.1089/hum.2009.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cideciyan AV, Hauswirth WW, Aleman TS, et al. Vision 1 year after gene therapy for Leber’s congenital amaurosis. N Engl J Med. 2009;361:725–727. doi: 10.1056/NEJMc0903652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maguire AM, High KA, Auricchio A, et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet. 2009;374:1597–1605. doi: 10.1016/S0140-6736(09)61836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simonelli F, Maguire AM, Testa F, et al. Gene therapy for Leber’s congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol Ther. 2010;18(3):643–650. doi: 10.1038/mt.2009.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cideciyan AV, Jacobson SG, Beltran WA, et al. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc Natl Acad Sci USA. 2013;110(6):E517–E525. doi: 10.1073/pnas.1218933110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boye SE, Boye SL, Lewin AS, Hauswirth WW. A comprehensive review of retinal gene therapy. Mol Ther. 2013;21(3):509–519. doi: 10.1038/mt.2012.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goetz KE, Reeves MJ, Tumminia SJ, Brooks BP. eyeGENE®: a novel approach to combine clinical testing and researching genetic ocular disease. Curr Opin Ophthalmol. 2012;23(5):355–363. doi: 10.1097/ICU.0b013e32835715c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noriko Miyake N, Chilton J, Psatha M, et al. Human CHN1 mutations hyperactivate α2-chimaerin and cause Duane’s retraction syndrome. Science. 2008;321(5890):839–843. doi: 10.1126/science.1156121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tischfield MA, Baris HN, Wu C, et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell. 2010;140(1):74–87. doi: 10.1016/j.cell.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cederquist GY, Luchniak A, Tischfield MA, et al. An inherited TUBB2B mutation alters a kinesin-binding site and causes polymicrogyria, CFEOM and axon dysinnervation. Hum Mol Genet. 2012;21(26):5484–5499. doi: 10.1093/hmg/dds393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schoenberger SD, Agarwal A. A novel mutation at the N-terminal domain of the TIMP3 gene in Sorsby fundus dystrophy. Retina. 2013;33(2):429–435. doi: 10.1097/IAE.0b013e318263d3b4. [DOI] [PubMed] [Google Scholar]
- 31.Huang AS, Kim LA, Fawzi AA. Clinical characteristics of a large choroideremia pedigree carrying a novel CHM mutation. Arch Ophthalmol. 2012;130(9):1184–1189. doi: 10.1001/archophthalmol.2012.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Y, Ratnam K, Sundquist SM, et al. Cone photoreceptor abnormalities correlate with vision loss in patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2011;52(6):3281–3292. doi: 10.1167/iovs.10-6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duncan JL, Ratnam K, Birch DG, et al. Abnormal cone structure in foveal schisis cavities in X-linked retinoschisis from mutations in exon 6 of the RS1 gene. Invest Ophthalmol Vis Sci. 2011;52(13):9614–9623. doi: 10.1167/iovs.11-8600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Churchill JD, Bowne SJ, Sullivan LS, et al. Mutations in the X-linked retinitis pigmentosa genes RPGR and RP2 found in 8.5% of families with a provisional diagnosis of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013;54:1411–1416. doi: 10.1167/iovs.12-11541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fahim AT, Bowne SJ, Sullivan LS, et al. Allelic heterogeneity and genetic modifier loci contribute to clinical variation in males with X-linked retinitis pigmentosa due to mutations in RPGR. PLoS One. 2011;6(8):e23021. doi: 10.1371/journal.pone.0023021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedman JS, Ray JW, Waseem N, et al. Mutations in a novel BTBKelch protein, KLHL7, cause autosomal dominant retinitis pigmentosa. Am J Hum Genet. 2009;84:792–800. doi: 10.1016/j.ajhg.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song J, Smaoui N, Ayyagari R, et al. High-throughput retina-array for screening 93 genes involved in inherited retinal dystrophy. Invest Ophthalmol Vis Sci. 2011;52:9053–9060. doi: 10.1167/iovs.11-7978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bowne SJ, Sullivan LS, Koboldt DC, et al. Identification of disease-causing mutations in autosomal dominant retinitis pigmentosa (adRP) using next-generation DNA sequencing. Invest Ophthalmol Vis Sci. 2011;52:494–503. doi: 10.1167/iovs.10-6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bowne SJ, Humphries MM, Sullivan LS, et al. A dominant-acting mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur J Hum Genet. 2011;10:1–8. doi: 10.1038/ejhg.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.