

Abstract
C-reactive protein (CRP) is an acute phase protein produced by hepatocytes. A minor elevation in the baseline levels of serum CRP is considered an indicator of chronic inflammation. In hepatoma Hep3B cells, IL-6 induces CRP expression by activating transcription factors STAT3 and C/EBPβ. IL-1 synergistically enhances the effects of IL-6. The first 157 bp of the CRP promoter are sufficient for IL-1 synergy. Previously, NF-κB, a transcription factor activated by IL-1β in Hep3B cells, has been shown to increase endogenous CRP expression. The purpose of this study was to investigate the possible action of NF-κB on the 157 bp of the proximal promoter. In this study we show that NF-κB requires and acts synergistically with C/EBPβ on the CRP-proximal promoter to regulate CRP expression. We located the regulatory element that consisted of overlapping binding sites for NF-κB (p50-p50 and p50-p65) and OCT-1. The κB site was responsible for the synergy between NF-κB and C/EBPβ and was also necessary for the CRP transactivation by C/EBPβ through the C/EBP site. Mutation of the κB site decreased the synergistic effect of IL-1β on IL-6-induced CRP expression. Basal CRP expression increased dramatically when binding of both OCT-1 and NF-κB was abolished. Combined data from luciferase transactivation assays and EMSA lead us to conclude that the binding of OCT-1 to the promoter, facilitated by p50-p50 in a novel way, represses, whereas replacement of OCT-1 by p50-p65 induces CRP transcription in cooperation with C/EBPβ. This model for CRP expression favors the variation seen in baseline serum CRP levels in a normal healthy population.
C-reactive protein (CRP)2 is a multifunctional acute phase protein whose serum concentration increases in chronic and acute inflammation (1– 4). CRP is primarily produced by hepatocytes (5, 6), and its synthesis is regulated at the transcriptional level (7–9). In human hepatoma Hep3B cells, IL-6 induces CRP expression modestly by activating the transcription factors STAT3 and C/EBPβ (10 –14). IL-1, which alone has no effect on CRP expression in Hep3B cells, synergistically enhances the effects of IL-6 (15). The first 157 bp of the CRP promoter are sufficient for synergistic induction of CRP expression by IL-6 and IL-1β (7, 12). Besides Hep3B cells, STAT3 and C/EBPβ have been shown to act on CRP promoter in other hepatic cell lines also (16–20). On the CRP-proximal promoter, within the first 157 bases, C/EBPβ binds to a site centered at −52, and STAT3 binds to a site at −108 (10 –12). A second C/EBP site is located at position −219 (11).
Three other transcription factors, hepatocyte nuclear factor-1 (HNF-1), HNF-3, and OCT-1, are involved in maintaining the constitutive expression of CRP (11, 21). OCT-1 is a broadly expressed, versatile transcription factor of the POU family of homeo domain proteins. OCT-1 performs many divergent roles in cellular transcriptional regulation, partly due to its flexibility in DNA binding and its ability to associate with multiple and varied coregulators. Although generally thought of as an activator of gene transcription, OCT-1 also represses transcription through a variety of mechanisms (22).
The mode of action of IL-1 in CRP expression is not defined. Because IL-1 activates NF-κB in Hep3B cells, it is hypothesized that IL-1 may be acting through activation of NF-κB (23). There are five NF-κB proteins: p50, p52, p65, Rel-B, and c-Rel; they form homodimers or heterodimers with each other, and bind to κB sites on the promoter regions to modulate transcription (24). It has been shown previously that the NF-κB heterodimer p50-p65 induces endogenous CRP expression in Hep3B cells (23); however a binding site for p50-p65 has not been identified in the first 157 bp of the CRP promoter.
In the current study we investigated the possible presence of a κB site within the 157 bp of the proximal promoter. We located a regulatory element, −74/−59, with overlapping binding sites for NF-κB and OCT-1. Our data indicate that the binding of OCT-1 to the promoter, facilitated by p50-p50 in a novel way, represses, whereas replacement of OCT-1 by p50-p65 induces CRP transcription in cooperation with C/EBPβ.
Materials and Methods
Cell culture, cytokine treatment, transfection, and luciferase (Luc) transactivation assay
Hep3B cells were cultured in serum-free medium overnight for cytokine treatments as described previously (14). The confluency of cells was ~60% at the time of treatments. IL-6 and IL-1β (R&D Systems) were used at concentrations of 10 ng/ml and 1 ng/ml, respectively, and the cells were treated for 24 h. For transient transfections, cells were plated into 6-well plates and transfected using FuGene-6 reagent (Roche) as described previously (25). Luc reporter-CRP promoter constructs were used at 1 μg of plasmid/well. Cytokine treatments were started 16 h after transfection. After 40 h of transfection, Luc assays were performed as described previously (25). Luc activity was measured in a luminometer (Molecular Devices), which was programmed for the integration time of 10 s with no postinjection delay time.
Preparation of nuclear extract and EMSA
Nuclear extracts were prepared using the NE-PER nuclear and cytoplasmic kit (Pierce) and were used in EMSA as reported previously (14). The oligonucleotide (oligo) 5′-GATCCGGGGACTTTCCATGGATGGGGACTTTCCATGG-3′ was used as the consensus κB site-containing probe. Oligos were obtained from Integrated DNA Technologies. The gel shift incubation buffer contained 40 mM KCl, 16 mM HEPES (pH 7.9), 1 mM EDTA, 2.5 mM DTT, 0.15% Nonidet P-40, 8% Ficoll, and 1 μg of poly(deoxyinosinic-deoxycytidylic acid). The Ab to C/EBPβ (C19), p50 (H119), p65 (C20), HNF-1 (H205), and OCT-1 (C21) were purchased from Santa Cruz Biotechnology. Unlabeled competitor oligos were used in a 200-fold molar excess. DNA-protein complexes were resolved in native 4.5% polyacrylamide gels containing 2.5% glycerol. Gels were analyzed in a phosphor imager using Image-Quant software (GE Healthcare). Sequences of the top strand of the double-strand oligos, derived from the CRP promoter and used in EMSA, were as follows: oligo 1, 19 bp long, 5′-ATGTTGGAAAATTATTTAC-3′; oligo 2, 25 bp long, 5′-CAATGTTGGAAAATTATTTACATAG-3′; and oligo 3, oligo 2 with mutated κB site, 5′-CAATGTTGGTTAATAATTTACATAG-3′ (the κB sites are underlined, and the mutated bases are in bold).
Engineering of CRP promoter-Luc reporter constructs
The wild-type (WT) construct, Luc-157 WT CRP, has been described previously (13). The Luc-300 WT construct was prepared according to a previously reported method (26). Briefly, genomic DNA (Promega) was used to PCR-amplify a fragment corresponding to nt −300/−1 of the CRP promoter, using the primers 5′-CCTAGATCTAGAGCTACCTCCTCCTGCCTGG-3′ and 5′-CCGACGCGTACCCAGATGGCCACTCGTTTAATATGTTACC-3′. Primers were designed to contain the BglII and MluI restriction sites, respectively. PCR product was cloned into the Luc reporter vector pGL2 basic (Pro-mega), and the DNA sequence was confirmed. These two WT constructs were used as templates for mutagenesis. Constructs containing mutated κB and STAT3 sites were generated using the QuikChange site-directed mutagenesis kit (Stratagene). The κB site was mutated by substituting –72AAAATT–67 with –72TTAATA–67 using mutagenic primers 5′-GCGCCACTATGTAAATTATTAACCAACATTGCTTGTTGGGGC-3′ and 5′-GCCCCAACAAGCAATGTTGGTTAATAATTTACATAGTGGCGC-3′. The STAT3 site was mutated by substituting –111TCCCGA–106 with –111GATATC–106 using mutagenic primers 5′-GCTTCCCCTCTGATATCAGCTCTGACACCTG-3′ and 5′-CAGGTGTCAGAGCTGATATCAGAGGGGAAGC-3′. Mutations were verified by sequencing. Plasmids were purified using Maxiprep plasmid isolation kit (Eppendorf).
Results
NF-κB acts synergistically with C/EBPβ on the CRP-proximal promoter
To determine whether NF-κB could induce CRP transactivation through the proximal promoter under any experimental condition, constructs of the WT promoter regions −300/−1 and −157/+3 linked to Luc reporter (Luc-300 WT and Luc-157 WT) were trans-fected into Hep3B cells along with expression vectors for NF-κB (p50 and p65) and C/EBPβ. NF-κB did induce CRP promoter-driven Luc expression, but only in the presence of C/EBPβ (Fig. 1). The amount of C/EBPβ was critical for inducing the effect of NF-κB on both −300/−1 and −157/+3 promoters. For 200 ng of p50 and p65 plasmids, >20 ng of C/EBPβ plasmid were required to observe the synergistic induction. The data indicated that NF-κB required and acted synergistically with C/EBPβ bound to its proximal, but not the distal, site and that a κB site must be located within the 157 bp on the CRP promoter.
FIGURE 1.
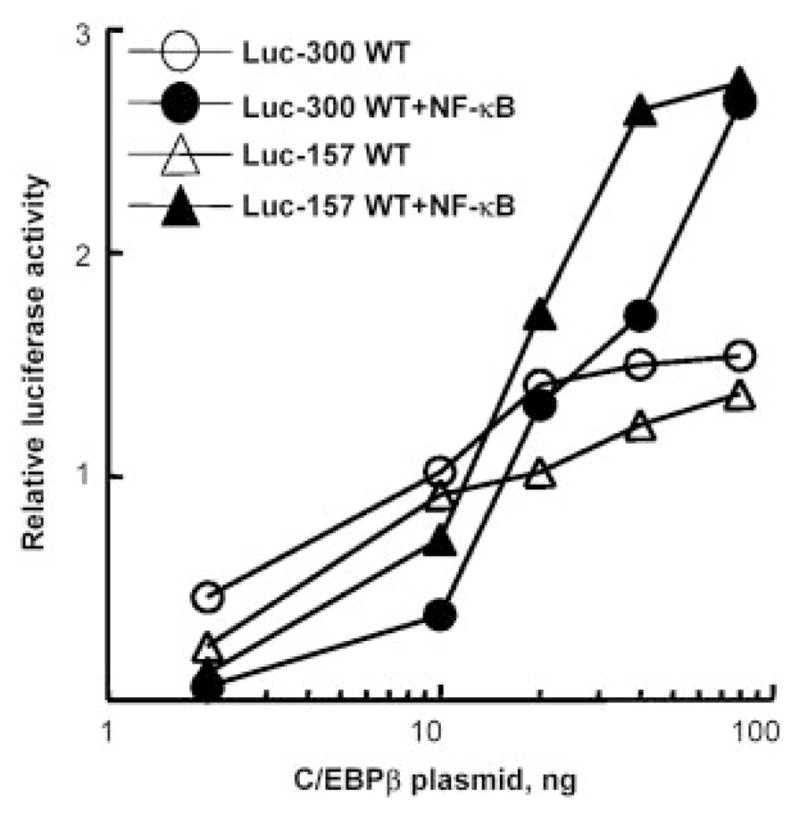
NF-κB acts on first 157 bp of the CRP promoter and synergizes with C/EBPβ to induce CRP promoter (−157/+3 or −300/−1)-driven luciferase expression. A representative experiment is shown; three independent experiments exhibited similar patterns. Cells were transfected with CRP promoter-Luc constructs (Luc-300 WT and Luc-157 WT) and plasmids encoding C/EBPβ (increasing doses) and NF-κB (p50 and p65, 200 ng each). CRP transactivation was represented as the relative luciferase activity.
A κB site is located at position −69 on the CRP promoter
We read the DNA sequence of the CRP promoter and found a potential κB site (−74/−63) overlapping the known binding sites for transcription factors HNF-1/OCT-1/HNF-3 (11, 21) (Fig. 2).
FIGURE 2.
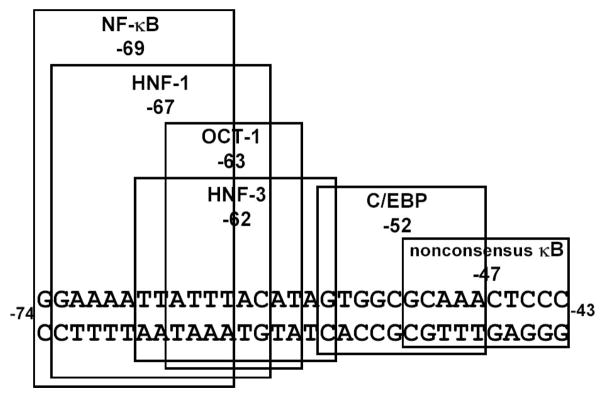
Localization of the κB site on the CRP-proximal promoter. The nucleotide sequence of the CRP promoter between positions −74 and −43, relative to the transcription start site, is shown. The sequences of the putative κB site centered at −69 and known binding sites for other transcription factors are boxed.
Oscillation between the binding of p50-p50/OCT-1 and p50-p65 on the overlapping κB/OCT-1 sites
Binding of NF-κB to the putative κB site was determined by EMSA, using consensus κB site probe and nuclear extracts from cells treated with IL-1β as the source of NF-κB (Fig. 3A). IL-1β treatment induced formation of the NF-κB p50-p65 complex (lanes 1–5). Oligos 1, 2, and 3 derived from CRP promoter and containing the κB site were used as competitors. The 19-bp oligo 1 (−79/−61) did not compete efficiently with the probe for binding to NF-κB (lane 6); however, the 25-bp oligo 2 (−81/−57) competed (lane 7). To confirm that the competition was due to the κB site on oligo 2, the κB site was mutated in oligo 3, and this oligo did not compete for binding NF-κB (lane 8). Thus, the region −74/−63 on the CRP promoter is the κB site, and a certain flanking sequence is necessary for binding NF-κB.
FIGURE 3.
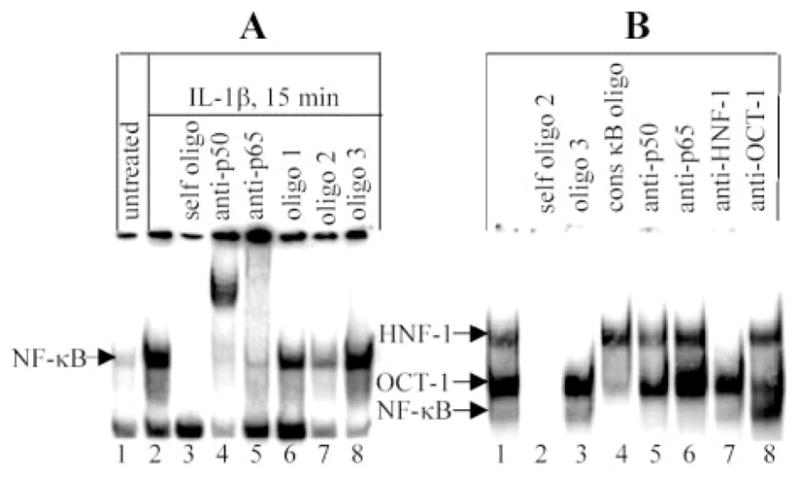
Binding of OCT-1 and NF-κB to the composite OCT-1/κB site. A, CRP promoter’s κB site competes with the consensus κB site for binding to NF-κB. EMSA using radiolabeled consensus κB site probe and nuclear extract from IL-β-treated cells as the source of NF-κB; Competitor oligos 1–3, containing the putative κB site, were derived from CRP promoter. Oligo 1, 19 bp containing the κB site; oligo 2, 25 bp containing the κB site; oligo 3, oligo 2 with mutated κB site. B, Direct binding of NF-κB to CRP promoter’s κB site. EMSA used oligo 2 as a probe and nuclear extract from IL-β-treated cells. The competitors (200-fold excess of unlabeled oligos) and the Ab were added to the reaction mixtures before the addition of probe. Results were analyzed by a phosphor imager. The mobility of the free probe is not shown. A representative of three EMSAs is shown.
EMSA, using oligo 2 as the probe, provided direct visualization of binding of NF-κB (Fig. 3B). Three specific complexes were formed (lanes 1 and 2). The complex on the top was HNF-1 (lane 7), the next complex was OCT-1 (lane 8), and the fastest migrating complex contained NF-κB p50-p65 (lanes 5 and 6). Unexpected results are shown in lanes 3, 4, and 8. In lane 3, the mutated oligo 3 (with the OCT-1 site intact) competed with HNF-1 complex only, showing that mutation of the κB site abolished binding to OCT-1 in addition to NF-κB. This result indicated that binding of OCT-1 to the probe required an intact κB site. In lane 4, binding of OCT-1 to the probe, in addition to that of NF-κB, was drastically diminished in the presence of unlabeled consensus κB site oligo, indicating dependence of OCT-1 on NF-κB proteins for binding to its site. In lane 8, binding of p50-p65 to the probe was increased by the addition of anti-OCT-1 Ab, indicating that OCT-1 inhibited binding of p50-p65 to the probe. This result also indicated that the NF-κB proteins required for OCT-1 binding (lane 4) to the probe must be p50-p50 and explained the low intensity of the NF-κB complex seen in lane 1. We confirmed the capability of the κB site to bind p50-p50 by using recombinant p50 (Fig. 4A). We conclude that the region −81/−57 on the CRP promoter binds either OCT-1 or p50-p65, and the binding of OCT-1 to its site requires prior transient binding of p50-p50 to the κB site. Lastly, to confirm the finding that the binding of OCT-1 to its site was dependent on an intact κB site, as seen in lane 3 (Fig. 3B), an EMSA was performed using oligo 3 as the probe (Fig. 4B). Only one specific complex containing HNF-1 was formed.
FIGURE 4.
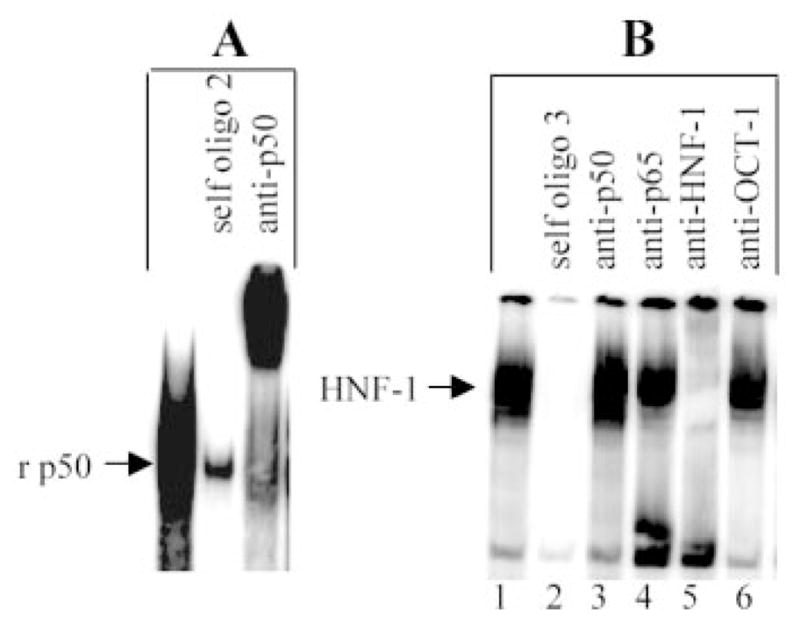
Binding of p50 and HNF-1 to oligo 2 and oligo 3, respectively. A, EMSA using oligo 2 as a probe and recombinant p50 (4 gel shift units/lane). B, EMSA using oligo 3 as a probe and nuclear extract from IL-β-treated cells. The competitors (200-fold excess of unlabeled oligos) and the Ab were added to the reaction mixtures before the addition of probe. Results were analyzed by a phosphor imager. The mobility of the free probe is not shown. A representative of three EMSAs is shown.
The κB site is functional
To determine whether the κB site mediated the synergistic effect of NF-κB on CRP transactivation by C/EBPβ, we conducted trans-activation assays using Luc-300 m-κB and Luc-157 mutated-κB (m-κB) constructs with the mutated κB site (Fig. 5). In Luc-300 WT, NF-κB enhanced the inducing effect of C/EBPβ from 78- to 127-fold. In the Luc-300 m-κB construct, NF-κB did not do so; instead, the effect of C/EBPβ alone was reduced by ~90% (from 78- to 8-fold) compared with the WT construct, indicating that an intact κB site was also necessary for maximum transactivation by C/EBPβ itself. Similar results were obtained with Luc-157 constructs. We conclude that the κB site is responsible for synergy between NF-κB and C/EBPβ and is also necessary for the action of C/EBPβ through the C/EBP site.
FIGURE 5.

The κB site and the C/EBP site act together to regulate CRP expression. The basal Luc activity for each construct is considered 1, and Luc activities in response to C/EBPβ (80 ng) and NF-κB (p50 and p65, 200 ng each) are plotted as fold induction over basal expression. The average ± SEM of three experiments are shown.
Participation of the κB site in the synergy between IL-6 and IL-1β
To determine whether the κB site mediated the synergistic effect of IL-1β on CRP transactivation by IL-6, we conducted transactivation assays using Luc-300 m-κB and Luc-157 m-κB constructs with the mutated κB site (Fig. 6). In Luc-300 WT, IL-1β enhanced the inducing effect of IL-6 5.1-fold. In the Luc-300 m-κB construct, the synergistic effect of IL-1β was reduced by 50% (from 5.1- to 2.6-fold) compared with the WT construct. Similar results were obtained with Luc-157 constructs.
FIGURE 6.
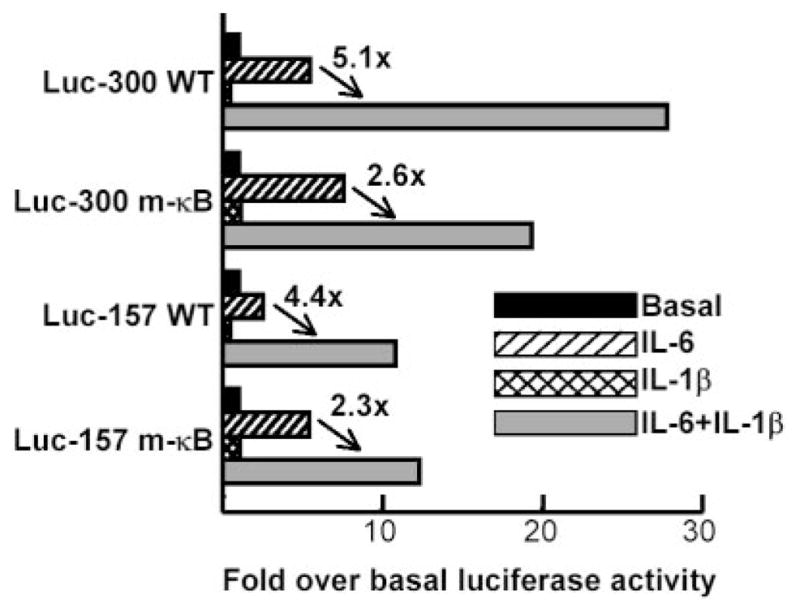
Synergistic effect of IL-1β on IL-6-induced CRP expression is only partially mediated by NF-κB. The basal Luc activity for each construct is considered 1, and Luc activity in response to IL-6 and IL-1β is plotted as the fold induction over basal expression. A representative experiment is shown.
The overlapping κB and OCT-1 sites regulate basal CRP expression
Basal transactivation of the CRP promoter Luc-300 m-κB and Luc-157 m-κB was increased ~15-fold compared with the basal transactivation of the corresponding WT promoters (Fig. 7). As a control, when the STAT3 site was mutated (m-ST), basal activity of the promoter did not increase. Because mutation of the κB site abolished binding of OCT-1 to the promoter (Figs. 3B and 4B), the transactivation results suggest that OCT-1 acts as a repressor of CRP expression.
FIGURE 7.
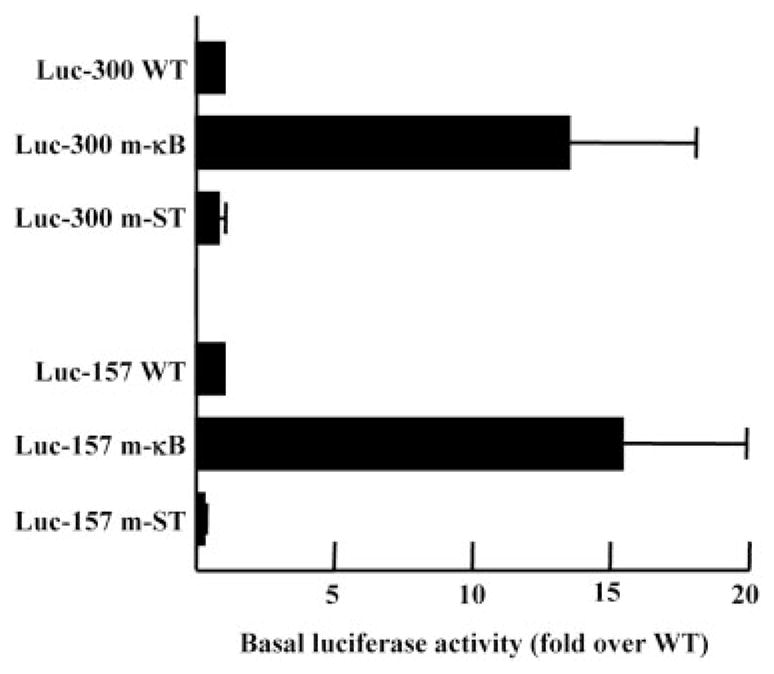
The basal level of CRP expression was elevated from that of the promoter with the mutated κB site. The basal Luc activities of mutated-STAT3 (m-ST) and m-κB constructs are plotted as the fold induction over that of the WT construct whose basal activity was taken as 1. The average ± SEM of five experiments are shown.
Discussion
To explore the mechanism of CRP gene expression, we evaluated the participation of NF-κB in the induction of CRP gene expression through the proximal promoter. Our major findings were as follows: 1) NF-κB p50-p65 acted synergistically with C/EBPβ to induce CRP transactivation through 157 bp of the promoter; 2) a minimum amount of C/EBPβ was critical for NF-κB synergy; 3) a κB site was located at position −69, overlapping the known OCT-1/HNF-1/HNF-3 sites; 4) the κB site was required for the synergism between NF-κB and C/EBPβ; 5) the κB site, in part, contributed to the synergism between IL-6 and IL-1β; 6) basal CRP expression was increased dramatically when the binding of both OCT-1 and NF-κB to their cognate sites was abolished; and 7) a novel interaction between OCT-1 and NF-κB dimers p50-p50 and p50-p65 was observed, indicating that this binding site on the CRP promoter was a key element in regulating CRP gene expression under basal and inflammatory conditions.
A binding site for NF-κB p50-p65 within the CRP-proximal promoter of the CRP gene was not identified in previous attempts (13, 14, 23). The κB site found previously on the CRP promoter was located at position −2652, although a nonconsensus κB site for binding p50-p50 was present in the proximal promoter at position −48. We found that p50-p65, in the presence of C/EBPβ, acted as an inducer of CRP expression. Moreover, transactivation by C/EBPβ through the C/EBP site located at position −52 also required the κB site, strongly indicating a functional association between the two sites. The physical interaction in vitro and synergism in transcriptional activity between NF-κB and C/EBPβ acting through their adjacent sites is a general phenomenon and has been reported for a number of other gene promoters (27, 28). Previously reported data (13, 14) showed that p65-p65 inhibited the inducing effects of p50-p50 and also of C/EBPβ on CRP expression through the proximal promoter. In those transactivation experiments using the overexpression approach, the amount of C/EBPβ was kept constant, and the amount of p50 or p65 varied. In contrast, in this report, we used constant amounts of p50 and p65 with increasing amounts of C/EBPβ. It was not obvious whether the previously reported inhibitory effect of p65 was due to the sequestration of a limited amount of C/EBPβ by p65 homodimers (13, 14).
The presence of a κB site within the first 157 bp strongly indicated that this site could be the IL-1-response element on the CRP promoter. The results indicated that the activation of p50-p65 and the κB site at −69, contributed only partially to the synergistic effect of IL-1 on IL-6-induced CRP gene expression. Our data support the idea that IL-1, besides activating NF-κB in Hep3B cells, participates in IL-6 synergy via other pathways, as has been shown in the case of other IL-1-regulated genes (29).
We present our data obtained from mutational analysis of the CRP promoter and from the unique interaction of transcription factor on the OCT-1/κB site in the form of a working model (Fig. 8). This model would guide us to the next series of experiments to demonstrate the functions of OCT-1, p50-p50, and p50-p65 in regulating CRP expression. There are four features of this model. 1) The absence of binding of any transcription factor to the OCT-1/κB site enhances basal CRP expression. 2) The binding of OCT-1 to the promoter requires previous transient binding of p50-p50 to the overlapping κB site. 3) OCT-1 represses basal CRP expression, consistent with the known role of OCT-1 as a repressor of gene expression (30). Thus, basal CRP expression may vary depending upon the availability of free p50-p50 and OCT-1 in the hepatocyte nuclei. The importance of the ratio of various transcription factors in regulating gene expression has been documented (31). 4) Under inflammatory conditions, p50-p65 replaces OCT-1 to induce CRP transcription. Such oscillation between nucleoprotein complexes on the gene promoters has been described previously (32). In addition, because the C/EBP site is only 16 bp away from the κB site, C/EBPβ and p50-p65 may form a stable ternary complex.
FIGURE 8.
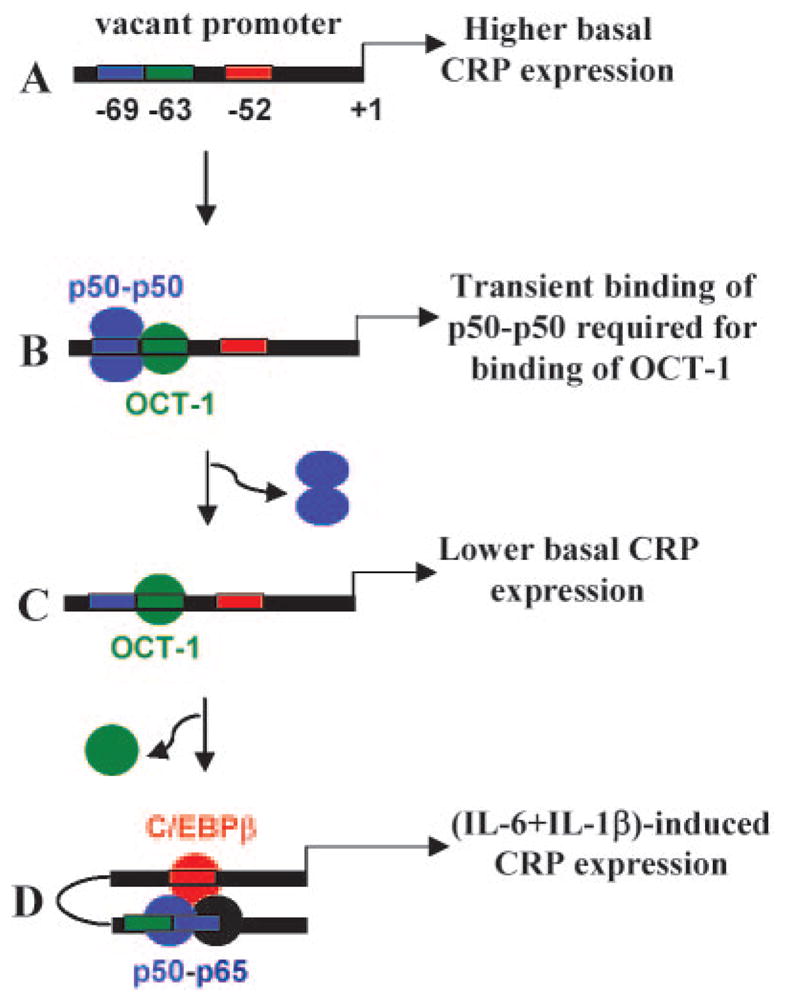
A model showing the role of the overlapping κB/OCT-1 sites functioning with the C/EBP site in regulating basal and induced CRP expression. A, Vacant OCT-1, κB, and C/EBP sites increase basal CRP expression. B, Binding of OCT-1 to its site requires previous transient binding of p50-p50 to the κB site. Once OCT-1 is bound, p50-p50 leaves its site. C, OCT-1-binding represses basal CRP expression. D, Cytokines such as IL-6 and IL-1β activate C/EBPβ and NF-κB p50-p65, respectively. A switch occurs between the repressor OCT-1 and p50-p65. Because the κB site is only 16 bp away from the C/EBP site, a physical interaction between NF-κB and C/EBPβ is possible, resulting in induced CRP expression.
Acknowledgments
We are grateful to Dr. P. Johnson for the gift of expression vector encoding C/EBPβ; Dr. G. Nabel for expression vectors encoding p50 and p65; Drs. N. Goldman, I. Kushner, and D. Samols for Luc-157 WT; and Dr. G. J. Darlington for Hep3B cells. We are also thankful to Mahua Chakraborthy for constructing the CRP promoter Luc-300 WT.
Footnotes
Abbreviations used in this paper: CRP, C-reactive protein; HNF-1, hepatocyte nuclear factor-1; Luc, luciferase; oligo, oligonucleotide; WT, wild type; m, mutated.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Agrawal A. CRP after 2004. Mol Immunol. 2005;42:927–930. doi: 10.1016/j.molimm.2004.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kushner I. The phenomenon of the acute phase response. Ann NY Acad Sci. 1982;389:39–48. doi: 10.1111/j.1749-6632.1982.tb22124.x. [DOI] [PubMed] [Google Scholar]
- 3.Agrawal A, Simpson MJ, Black S, Carey MP, Samols D. A C-reactive protein mutant that does not bind to phosphocholine and pneumococcal C-polysaccharide. J Immunol. 2002;169:3217–3222. doi: 10.4049/jimmunol.169.6.3217. [DOI] [PubMed] [Google Scholar]
- 4.Suresh MV, Singh SK, Agrawal A. Interaction of calcium-bound C-reactive protein with fibronectin is controlled by pH: in vivo implications. J Biol Chem. 2004;279:52552–52557. doi: 10.1074/jbc.M409054200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hurlimann J, Thorbecke GJ, Hochwald GM. The liver as the site of C-reactive protein formation. J Exp Med. 1966;123:365–378. doi: 10.1084/jem.123.2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kushner I, Feldmann G. Control of the acute phase response: demonstration of C-reactive protein synthesis and secretion by hepatocytes during acute inflammation in the rabbit. J Exp Med. 1978;148:466–477. doi: 10.1084/jem.148.2.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang D, Jiang SL, Rzewnicki D, Samols D, Kushner I. The effect of interleukin-1 on C-reactive protein expression in Hep3B cells is exerted at the transcriptional level. Biochem J. 1995;310:143–148. doi: 10.1042/bj3100143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ganter U, Arcone R, Toniatti C, Morrone G, Ciliberto G. Dual control of C-reactive protein gene expression by interleukin-1 and interleukin-6. EMBO J. 1989;8:3773–3779. doi: 10.1002/j.1460-2075.1989.tb08554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li SP, Liu TY, Goldman ND. Cis-acting elements responsible for interleukin-6 inducible C-reactive protein gene expression. J Biol Chem. 1990;265:4136–4142. [PubMed] [Google Scholar]
- 10.Majello B, Arcone R, Toniatti C, Ciliberto G. Constitutive and IL-6-induced nuclear factors that interact with the human C-reactive protein promoter. EMBO J. 1990;9:457–465. doi: 10.1002/j.1460-2075.1990.tb08131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li SP, Goldman ND. Regulation of human C-reactive protein gene expression by two synergistic IL-6 responsive elements. Biochemistry. 1996;35:9060–9068. doi: 10.1021/bi953033d. [DOI] [PubMed] [Google Scholar]
- 12.Zhang D, Sun M, Samols D, Kushner I. STAT3 participates in transcriptional activation of the C-reactive protein gene by interleukin-6. J Biol Chem. 1996;271:9503–9509. doi: 10.1074/jbc.271.16.9503. [DOI] [PubMed] [Google Scholar]
- 13.Cha-Molstad H, Agrawal A, Zhang D, Samols D, Kushner I. The rel family member p50 mediates cytokine-induced C-reactive protein expression by a novel mechanism. J Immunol. 2000;165:4592–4597. doi: 10.4049/jimmunol.165.8.4592. [DOI] [PubMed] [Google Scholar]
- 14.Agrawal A, Cha-Molstad H, Samols D, Kushner I. Transactivation of C-reactive protein by IL-6 requires synergistic interactions of CCAAT/enhancer binding protein b (C/EBPβ) and Rel p50. J Immunol. 2001;166:2378–2384. doi: 10.4049/jimmunol.166.4.2378. [DOI] [PubMed] [Google Scholar]
- 15.Ganapathi MK, Rzewnicki D, Samols D, Jiang SL, Kushner I. Effect of combinations of cytokines and hormones on synthesis of serum amyloid A and C-reactive protein in Hep3B cells. J Immunol. 1991;147:1261–1265. [PubMed] [Google Scholar]
- 16.Wang Y, Ripperger J, Fey GH, Samols D, Kordula T, Wetzler M, Van Etten RA, Baumann H. Modulation of hepatic acute phase gene expression by epidermal growth factor and src protein tyrosine kinases in murine and human hepatic cells. Hepatology. 1999;30:682–697. doi: 10.1002/hep.510300318. [DOI] [PubMed] [Google Scholar]
- 17.Ochrietor JD, Harrison KA, Zahedi K, Mortensen RF. Role of STAT3 and C/EBP in cytokine-dependent expression of the mouse serum amyloid P-component (SAP) and C-reactive protein (CRP) genes. Cytokine. 2000;12:888–899. doi: 10.1006/cyto.2000.0668. [DOI] [PubMed] [Google Scholar]
- 18.May P, Schniertshauer U, Gerhartz C, Horn F, Heinrich PC. Signal transducer and activator of transcription STAT3 plays a major role in gp130-mediated acute phase protein gene activation. Acta Biochim Polo. 2003;50:595–601. [PubMed] [Google Scholar]
- 19.Castell JV, Gomez-Lechon MJ, David M, Fabra R, Trullenque R, Heinrich PC. Acute-phase response of human hepatocytes: regulation of acute-phase protein synthesis by interleukin-6. Hepatology. 1990;12:1179–1186. doi: 10.1002/hep.1840120517. [DOI] [PubMed] [Google Scholar]
- 20.Taylor AW, Ku NO, Mortensen RF. Regulation of cytokine-induced human C-reactive protein production by transforming growth factor-β. J Immunol. 1990;145:2507–2513. [PubMed] [Google Scholar]
- 21.Toniatti C, Demartis A, Monaci P, Nicosia A, Ciliberto G. Synergistic trans-activation of the human C-reactive promoter by transcription factor HNF-1 binding at two distinct sites. EMBO J. 1990;9:4467–4475. doi: 10.1002/j.1460-2075.1990.tb07897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wysocka J, Herr W. The herpes simplex virus VP16-induced complex: the makings of a regulatory switch. Trends Biochem Sci. 2003;28:294–304. doi: 10.1016/S0968-0004(03)00088-4. [DOI] [PubMed] [Google Scholar]
- 23.Agrawal A, Cha-Molstad H, Samols D, Kushner I. Overexpressed NF-κB can participate in endogenous C-reactive protein induction, and enhances the effects of C/EBPκ and signal transducer and activator of transcription-3. Immunology. 2003;108:539–547. doi: 10.1046/j.1365-2567.2003.01608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayden MS, Ghosh S. Signaling to NF-κB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 25.Agrawal A, Samols D, Kushner I. Transcription factor c-Rel enhances C-reactive protein expression by facilitating the binding of C/EBPβ to the promoter. Mol Immunol. 2003;40:373–380. doi: 10.1016/s0161-5890(03)00148-2. [DOI] [PubMed] [Google Scholar]
- 26.Kleemann R, Gervois PP, Verschuren L, Staels B, Princen HMG, Kooistra T. Fibrates down-regulate IL-1-stimulated C-reactive protein gene expression in hepatocytes by reducing nuclear p50-NF-κB-C/EBPβ complex formation. Blood. 2003;101:545–551. doi: 10.1182/blood-2002-06-1762. [DOI] [PubMed] [Google Scholar]
- 27.LeClair KP, Blanar MA, Sharp PA. The p50 subunit of NF-κB associates with the NF-IL6 transcription factor. Proc Natl Acad Sci USA. 1992;89:8145–8149. doi: 10.1073/pnas.89.17.8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stein B, Cogswell PC, Baldwin AS., Jr Functional and physical associations between NF-κB and C/EBP family members: a Rel domain-bZIP interaction. Mol Cell Biol. 1993;13:3964–3974. doi: 10.1128/mcb.13.7.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang XP, Albrecht U, Zakowski V, Sobota RM, Haussinger D, Heinrich PC, Ludwig S, Bode JG, Schaper F. Dual function of interleukin-1β for the regulation of interleukin-6-induced suppressor of cytokine signaling 3 expression. J Biol Chem. 2004;279:45279–45289. doi: 10.1074/jbc.M313072200. [DOI] [PubMed] [Google Scholar]
- 30.Osborne AR, Zhang H, Fejer G, Palubin KM, Niesen MI, Blanck G. Oct-1 maintains an intermediate, stable state of HLA-DRA promoter repression in Rb-defective cells: an Oct-1 containing repressosome that prevents NF-Y binding to the HLA-DRA promoter. J Biol Chem. 2004;279:28911–28919. doi: 10.1074/jbc.M403118200. [DOI] [PubMed] [Google Scholar]
- 31.Boudreau F, Zhu Y, Traber PG. Sucraseisomaltase gene transcription requires the hepatocyte nuclear factor-1 (HNF-1) regulatory element and is regulated by the ratio of HNF-1α to HNF-1β. J Biol Chem. 2001;276:32122–32128. doi: 10.1074/jbc.M102002200. [DOI] [PubMed] [Google Scholar]
- 32.Gonzalez-Gil G, Kahmann R, Muskhelishvili G. Regulation of crp transcription by oscillation between distinct nucleoprotein complexes. EMBO J. 1998;17:2877–2885. doi: 10.1093/emboj/17.10.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]