

Abstract
The combination of PPh3/I2 has been shown to be effective for conversion of a range of carboxylic acids to 2°, 3°, and Weinreb amides. Simplification of the procedure was possible with the use of polymer-supported PPh3/I2. Weinreb amides produced via the use of polymer-supported PPh3 could be filtered through a short silica gel plug and used in further transformations. Thus, use of polymer-supported PPh3 offers potential applicability to diversity-oriented reactions. Formal total syntheses of apocynin and pratosine, as well as syntheses of anhydrolychorinone and hippadine, have been achieved via the use of this amide-forming method. An attempt has been made to gain insight into this reaction.
Keywords: Amide, Weinreb amide, triphenylphosphane, polymer-supported triphenylphosphane, iodine
Introduction
Besides being a ubiquitous functionality, the amide linkage is prominent in functional group interconversions during multistep syntheses. This is reflected in a substantial amount of literature that continues to emanate on new methods for formation of the amide bond.[1,2] Halophosphonium salts derived from PPh3/I2 and [(R2N)3]P/I2 have recently found applications in the activation of the amide linkages of hypoxanthine nucleosides for further transformations,[3–7] and dehydration of oximes by PPh3/I2 has been reported.[8] Direct activation of tautomerizable heterocycles for C–C bond-forming reactions by PyBroP has recently been demonstrated.[9] Thus, halophosphonium compounds enjoy wide applications in organic transformations. In the area of amide bond formation, the combination of PPh3 with halogen sources such as NCS,[10] NBS,[11] Br2,[12] BrCCl3,[13] CCl4,[13,14] CBr4[14] and trichloroisocyanuric acid[15] have all been explored. In addition, polymer-supported PPh3 (Pol–Ph3P) has been utilized for amidation in combination with CCl4[16] and Cl3CCN[17].
To our surprise, the simple combination of PPh3 and I2 for generation of the amide linkage seems to have remained unstudied. The simplicity in handling these reagents, and the fact that these are inexpensive, renders them particularly attractive for this purpose. In this paper we have explored the utility of this reagent combination, as well as Pol–Ph3P for the synthesis of various amides and synthetically versatile Weinreb amides. We have applied this method en route to ketones, the formal synthesis of two natural products, and the total synthesis of two others, where amide formation is a key step.
Results and Discussion
By using 31P{1H} NMR spectroscopy we have previously observed that upon mixing PPh3 and I2 in a 1:1 ratio, a new species is produced, presumably (Ph3P+–I)I−. This entity was capable of reaction with the amide group of hypoxanthine nucleosides.[5] Therefore, our first question was whether such a species could react with carboxylic acids as well. Since BroP and PyBroP are well known reagents for amidation,[18] this appeared feasible. For reaction to occur, deprotonation of the carboxylic acid would be necessary and iPr2NEt was selected for this purpose. The overall reaction is depicted in Scheme 1. Here either an acyl phosphonium species or an acyl iodide (produced by substitution of Ph3PO with iodide) could undergo reaction with the amine, producing the amide.
Scheme 1.

A plausible mechanism for the amidation reaction.
With this rationale we set about exploring the general versatility of the method using a range of carboxylic acids and amines. The results from this analysis are presented in Table 1.
Table 1.
Amide and Weinreb amide synthesis using PPh3/I2.[a]
| ||||
|---|---|---|---|---|
|
| ||||
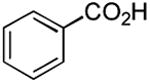
| Entry | Acid | Amine | Time | Yield |
| 1 |
|
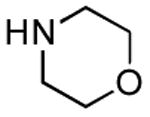
|
4 h | 77%[2g] |
| 2 |
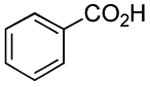
|
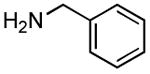
|
1.5 h | 77%[19] |
| 3 |
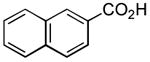
|
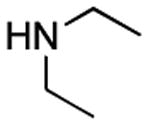
|
12 h | 60%[20] |
| 4 |
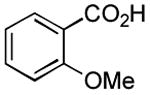
|
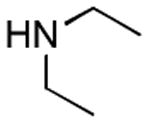
|
1 h | 74%[21] |
| 5 |
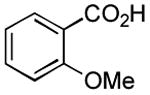
|
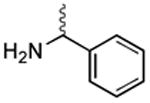
|
2 h | 85%[22] |
| 6 |
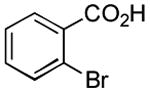
|
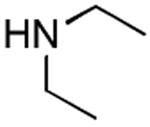
|
2 h | 77%[23] |
| 7 |
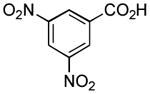
|
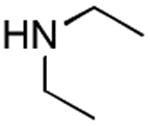
|
2 h | 83%[24] |
| 8 |
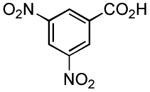
|
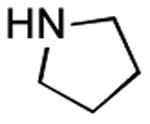
|
1.5 h | 83% |
| 9 |
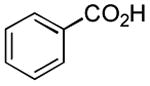
|
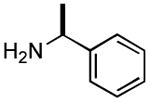
|
0.5 h | 78%[25] |
| 10 |
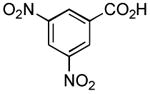
|
|
3 h 3 h |
52%[24] 72%[b] |
| 11 |
|
|
0.5 h | 83%[26] |
| 12 |
|
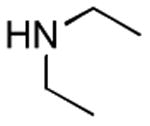
|
1 h 1 h |
63%[27] 72%[c] |
| 13 |
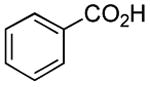
|
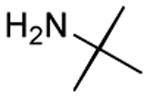
|
12 h | 80%[28] |
| 14 |
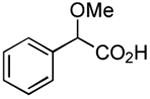
|
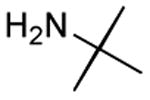
|
1 h 1 h |
56%[29] 73%[c] |
| 15 |
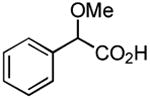
|
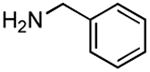
|
2 h 2 h |
45%[29] 67%[c] |
| 16 |
|
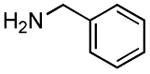
|
13 h 13 h |
44%[30] 53%[c] |
| 17 |
|
|
3 h 1 h |
48%[31] 65%[c] |
| 18 |
|
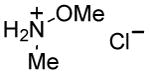
|
1 h | 70%[32,d] |
| 19 |
|
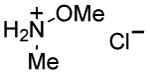
|
1 h | 65%[33,d] |
Reactions were conducted using 1 mmol each of PPh3, I2 carboxylic acid, amine, and 1.5 mmol of iPr2NEt in 4 mL of CH2Cl2, unless noted otherwise.
Yield obtained by using 2 molar equiv each of PPh3 and I2, and 1.5 molar equiv of o-toluidine.
Yield obtained by using 2 molar equiv each of PPh3 and I2.
2.5 Molar equiv of iPr2NEt was used.
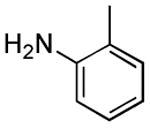
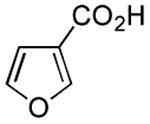
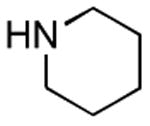
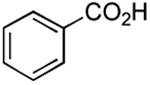
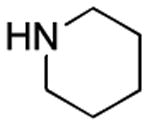
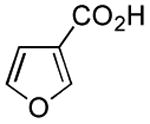
Results from Table 1 indicate that (Ph3P+–I)I− is suitably effective for the conversion of a several of carboxylic acids to the corresponding amides, and at a 1:1:1:1:1.5 stoichiometry of PPh3/I2/carboxylic acid/amine/iPr2NEt, primary and secondary amines react well. Substitution alpha to the amine does not hinder the reaction (entries 5 and 9). Reaction of an o-substituted aryl amine proceeded in acceptable yield (entry 10). 3-Furoic acid reacted efficiently, without complication (entry 11). Reaction of the highly hindered tbutylamine with benzoic acid proceeded well (entry 13). α-Methoxyphenylacetic acid reacted reasonably with both tbutylamine and benzylamine (entries 14 and 15). Acceptable reactions of 3-chloropropanoic acid with benzylamine (entry 16) and trans-3-hexenoic acid with piperidine (entry 17) were observed.
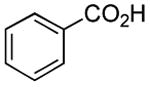
Although many reactions gave moderate to good product yields under the above-mentioned stoichiometry, some reactions could be improved via use of 2 molar equiv each of PPh3 and I2 (entries 10, 12, 14–17). Also, of interest is the fact that N-methoxy-N-methyl (Weinreb[34]) amides could also be prepared by this route (entries 18 and 19). Owing to the synthetic versatility of Weinreb amides, methods for their facile preparation are of continued interest.[35–39]
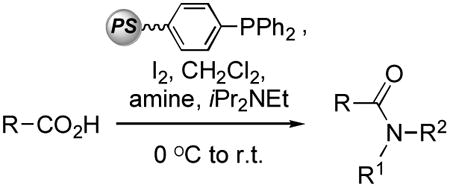
Because Ph3PO is a byproduct in this reaction, we next considered facilitating its easy removal. For this polymer-supported PPh3 (Pol–PPh3) offered a simple solution. As indicated earlier, Pol–PPh3 has been used for amidation,[16,17] and PPh3/CBr4 has been used in Weinreb amide synthesis.[40] However to our knowledge, Pol–PPh3 has not been used in combination with the cheaper I2. Our results with Pol–PPh3/I2 are shown in Table 2.
Table 2.
Use of Pol–PPh3/I2 for synthesis of amides and Weinreb amides.[a]
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | Acid | Amine | Time | Yield |
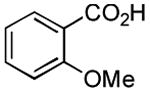
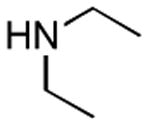
| 1 |
|
|
2 h | 75%[21] |
| 2 |
|
|
2 h | 72%[25] |
| 3 |
|
|
1 h | 65% |
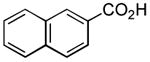
| 4 |
|
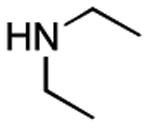
|
11 h 3 h |
57%[20] 71%[b] |
| 5 |
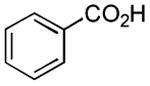
|

|
6 h | 53%[41] |
| 6 |
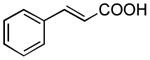
|
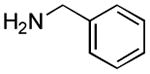
|
50 min 1 h |
53%[42] 78%[b] |
| 7 |
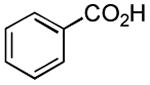
|
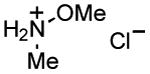
|
1 h | 76%[32,c] |
| 8 |
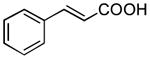
|
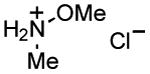
|
1 h | 79%[39h,c] |
Reactions were conducted using 1 mmol each of Pol–PPh3 (2.28 mmol/g loading), I2 carboxylic acid, amine, and 1.5 mmol of iPr2NEt in 4 mL of CH2Cl2, unless specified otherwise.
Yield obtained by using 2 molar equiv each of Pol–PPh3 and I2.
2.5 Molar equiv of iPr2NEt was used.
Results from Table 2 indicate that use of (Pol–Ph3P+–I)I− is just about as effective as solution chemistry and that this combination can be used for synthesis of Weinreb amides as well. The operational simplicity is exemplified by the fact that the Weinreb amides obtained could be simply filtered through a short silica gel plug and then subjected to reactions with organometallics (Scheme 2, yields were not optimized but examples are to demonstrate utility for high-throughput synthesis). Thus, N-methoxy-N-methylbenzamide could be converted to benzophenone and nbutylphenyl ketone, whereas N-methoxyl-N-methyl-3-phenylacrylamide was converted to chalcone and (E)-1-phenyl-1-hepten-3-one.
Scheme 2.

Synthesis of ketones from Weinreb amides obtained via use of Pol–PPh3/I2, followed by filtration through a silica gel plug.
Next, we considered evaluating use of the methodology described for the synthesis of natural products where a key step is amide formation. The first compound selected was apocynin (acetovanillin), which possesses interesting physiological activities. For example, it has been shown to have vasodialatory properties possibly via inhibition of Rho kinase activity,[43] anti-metastatic activity against human lung cancer cells,[44] and inhibition of cartilage damage caused by inflammation.[45] This compound has previously been prepared by Grignard addition to vanillin acetate followed by oxidation with DDQ,[46a] and Yb(OTf)3 mediated Friedel-Crafts reaction-demethylation of aryl methyl ethers,[46b] As shown in Scheme 3, our formal synthesis of apocynin involved the conversion of veratric acid (1) to the corresponding Weinreb amide 2 using 2 molar equiv each of PPh3 and I2, 1 molar equiv each of carboxylic acid and Me(MeO)NH•HCl, and 2.5 molar equiv of iPr2NEt (69% yield). Addition of MeMgBr to 2 then yielded 3′,4′-dimethoxyacetophenone (3, 97% yield). The conversion of 3 to apocynin by selective demethylation using NaSEt is known in the literature.[47]
Scheme 3.

Formal total syntheses of apocynin and pratosine.
The second formal synthesis was that of pratosine, which belongs to the family of Amaryllidaceae alkaloids, and several members of this family possess high biological activity (e.g. reversible inhibition of fertility in male rats[48a] and antitumor activity[48b]). N-Acylindoline 4 has previously been prepared by a Pd-catalyzed amidation.[49] In our approach, starting from veratric acid (1), amide 4 was synthesized by using of 1 equiv. each of PPh3/I2/carboxylic acid/ indoline and 1.5 equiv. of iPr2NEt (83% yield). Conversion of 4 to pratosine has been reported.[49]
As a third example to showcase the amidation step, we completed the synthesis of anhydrolychorinone and hippadine (Scheme 4). Here again lynchpin N-acyl amide 6 has previously been synthesized by a Pd-catalyzed amidation.[49] We conducted amide formation using 1 molar equiv each of PPh3/I2/piperonylic acid/indoline and 1.5 molar equiv iPr2NEt, or with the use of Pol–PPh3 in place of PPh3 under otherwise identical conditions. Consistent with results in Tables 1 and 2, yields from both methods were comparable, 70% from solution chemistry and 74% with Pol–PPh3. Further conversions were conducted as reported.[49] Oxidation of 6 to anhydrolychorinone proceeded in 23% yield using PhI(OCOCF3)2 and BF3•Et2O (83% reported in the literature[49]), and DDQ olefination proceeded in 78% yield (80% reported in the literature[49]) to yield hippadine.
Scheme 4.

Syntheses of anhydrolychorinone and hippadine.
During the course of our investigations we encountered some unusual 1H NMR characteristics of amides 4 and 6. Although the 1H NMR spectra of our products in CDCl3 matched those in the literature, only six aromatic protons have been reported for each,[49] and this corresponded to our observations. We wanted to ensure that no undesired electrophilic reactions had occurred on the aromatic ring in our cases. Therefore, we sought additional data. When the 1H NMR spectra of 4 and 6 were obtained in C6D6, an additional broad downfield proton resonance was observed in each case (ca δ = 8.1 ppm, see spectra in the Supporting Information). Further, in the case of 4, four distinct methoxy resonances could be observed at δ = 3.31, 3.30, 3.28 and 3.27 ppm. Heating the C6D6 solutions to 70 °C resulted in a significant sharpening of the broad, downfield aromatic resonances in 4 and 6. In the case of 4, coalescence of the methoxy resonances to two major ones was also seen. The COSY spectra for 4 and 6 in C6D6 at 70 °C are fully consistent with their respective structures, and in each case long-range coupling of the benzylic CH2 to the ortho aromatic proton was observed (see data in the Supporting Information).
Some of these properties are likely due to restricted rotation induced by the amide linkage. Although, this in itself is not surprising, it does raise a question about the efficiency of the cyclization reaction by hypervalent iodine reagents (e.g. 6 → anhydrolychorinone), which are conducted at subambient temperatures. That is, in such cases an unfavourably disposed rotamer could potentially influence the yield of the cyclization step.
Attempts at understanding the reaction intermediates
As shown in Scheme 1, an acyl phosphonium species and/or an acyl iodide could be potential intermediates in this amidation. Acyl phosphonium intermediates have been invoked previously in the synthesis of esters, amides, and acyl azides.[10,14,40,50] On the other hand, acyl chlorides and acyl bromides have been prepared by reaction of acids with PPh3/Cl3CCN[51] and PPh3/Br3CCO2Et,[52] respectively. Thus, we questioned whether acyl phosphonium intermediates could be directly observed by 31P{1H} NMR spectroscopy.
A solution of PPh3 in CD2Cl2 at room temperature produced a sharp singlet at δ = −5.0 ppm. Addition of 1 molar equiv of I2 led to rapid disappearance of the phosphane signal and appearance of a new resonance at δ = −18.4 ppm, presumably due to the formation of (Ph3P+–I)I−.[5] Addition of PhCO2H to this mixture led to no observable change in the 31P resonance. However, upon addition of 1.5 molar equiv of iPr2NEt, a new resonance was produced at δ = 30.1 ppm. Finally, addition of 1 molar equiv pyrrolidine to this mixture led to a very small upfield shift of the resonance (δ = 28.9 ppm). A similar result was obtained using morpholine. Thus, it was not possible to establish exactly what type of intermediate was involved.
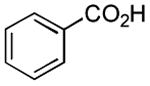
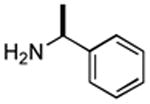
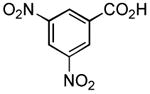
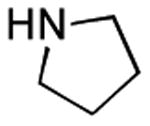
Therefore, in a second line of experimentation we exposed 3,5-dinitrobenzoic acid to the Pol–PPh3/I2/iPr2NEt combination in the absence of pyrrolidine. After 1 h, the polymer was filtered, pyrrolidine was added to the filtrate, and the reaction was continued for an additional 1 h. The amide from this reaction was isolated in 11% yield, which is very low compared to entry 3 in Table 2 (65% yield in a reaction time of 1 h). One possible explanation for the diminished yield could be the loss of the carboxylic acid as a polymer-bound acyl phosphonium species. Had acyl iodide been produced rapidly in the reaction, this should have remained in solution, leading to a better outcome. On the basis of this experiment, it is conceivable that the amidation reaction described herein proceeds largely via intermediacy of an acyl phosphonium species, at least when Pol–PPh3 is utilized.
Conclusions
In summary, we have demonstrated that the combination of PPh3/I2 or Pol–PPh3/I2 can be effectively utilized for the synthesis of a wide range of amides. The advantage of the PPh3/I2 combination is that the reagents are cheap and easily handled, and the reactions are straightforward to conduct. In addition, the synthetically valuable N-methoxyl-N-methyl (Weinreb) amides can also be synthesized using this mild conversion, and simple purification allows for their rapid use in further transformations. The usefulness of this procedure has been demonstrated via the formal total synthesis of two natural products, apocynin and pratosine as well as to the total synthesis of anhydrolychorinone and hippadine, where amide formation is an important step. No undesired problems were evident in any of the cases reported.
Experimental Section
General Experimental Considerations
Thin layer chromatography was performed on 200 μm silica gel plates and column chromatographic purifications were performed on 200–300 mesh silica gel. CH2Cl2 and iPr2NEt were distilled over CaH2, THF was distilled over LiAlH4 and freshly distilled over Na prior to use. All other reagents were obtained from commercial sources and were used without further purification. Pol–PPh3 (PS-triphenylphosphane, 2.28 mmol/g) was obtained from Biotage. 1H NMR spectra were recorded at 500 MHz and are referenced to the residual protonated solvent. 31P{1H} NMR spectra were recorded at 202 MHz and are referenced to 85% H3PO4 as external standard. Chemical shifts (δ) are reported in parts per million and coupling constants (J) are in hertz. Some representative synthetic procedures are given below.
Synthesis of Amides Using PPh3/I2
In a clean, dry 10 mL round bottom flask equipped with a stirring bar were placed PPh3 (1.0 mmol) and I2 (1.0 mmol) in dry CH2Cl2 (4 mL). The reaction mixture was flushed with nitrogen gas and allowed to stir at 0 °C for 5 minutes. At this temperature the carboxylic acid (1.0 mmol) was added, followed by dropwise addition of iPr2NEt (1.5 mmol) and the appropriate amine (1.0 mmol). The reaction mixture was slowly brought to room temperature and allowed to stir until no starting material could be seen on tlc (see Table 1 for reaction times). The reaction mixture was diluted with water, and extracted with CH2Cl2. The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure to afford the crude product, which was purified by chromatography on a silica gel column using hexanes/EtOAc. Any deviations from this procedure are noted under Table 1.
Synthesis of Amides Using Pol–PPh3/I2
To a stirring solution of I2 (1.0 mmol) in dry CH2Cl2 (5 mL) at 0 °C was added Pol–PPh3 (1.0 mmol). The reaction mixture was flushed with nitrogen gas and allowed to stir at 0 °C for 5 minutes. At this temperature the carboxylic acid (1.0 mmol) was added followed by dropwise addition of iPr2NEt (1.5 mmol) and the appropriate amine (1.0 mmol). The reaction mixture was slowly brought to room temperature and allowed to stir until no starting material could be seen on tlc (see Table 2 for reaction times). The reaction mixture was filtered and evaporated to the dryness. The crude product was purified through a short silica gel plug using hexanes/EtOAc. Any deviations from this procedure are noted under Table 2.
Synthesis of Weinreb Amides Using Pol–PPh3/I2
To a stirring solution of I2 (1.0 mmol) in dry CH2Cl2 (5 mL) at 0 °C was added Pol–PPh3 (1.0 mmol). The reaction mixture was flushed with nitrogen gas and allowed to stir at 0 °C for 5 minutes. At this temperature the carboxylic acid (1.0 mmol) was added followed by dropwise addition of iPr2NEt (2.5 mmol) and N,O-dimethylhydroxylamine hydrochloride (1.0 mmol). The reaction mixture was slowly brought to room temperature and allowed to stir until no starting material could be seen on tlc (see Table 2 for reaction times). The reaction mixture was filtered and evaporated to the dryness. The crude product was purified through a short silica gel plug using hexanes/EtOAc.
N-(3,5-Dinitro)benzoylpyrrolidine
Purification of the crude product obtained by the procedure described above on a silica gel column using 60% EtOAc in hexanes afforded 84.3 mg (83% yield) of the title compound as a yellow foam. Rf (40% EtOAc in hexanes) = 0.35. 1H NMR (500 MHz, CDCl3, ambient temperature): δ = 9.13 (s, 1 H, Ar–H), 8.76 (s, 2 H, Ar–H), 3.74 (t, 3JH,H = 6.6, 2 H, pyrrolidinyl–H), 3.52 (t, 3JH,H = 6.6, 2 H, pyrrolidinyl–H), 2.09–2.01 (m, 4 H, pyrrolidinyl–H). 13C NMR (125 MHz, CDCl3, ambient temperature): δ = 164.3, 148.3, 140.3, 127.5, 119.6, 49.5, 46.8, 26.4, 24.2. HRMS calculated for C11H12N3O5 [M + H] +: 266.0771, found 266.0774.
3′,4′-Dimethoxyacetophenone (3)
Step1: preparation of Weinreb amide 2
In a clean, dry 10 mL round bottom flask equipped with a stirring bar, were placed PPh3 (526.0 mg, 2.0 mmol) and I2 (507.6 mg, 2.0 mmol) in dry CH2Cl2 (6 mL). The reaction mixture was flushed with nitrogen gas and allowed to stir at 0 °C for 5 minutes. At this temperature 3,4-dimethoxybenzoic acid (182.1 mg, 1.0 mmol) was added, followed by dropwise addition of iPr2NEt (434 μL, 2.5 mmol) and N,O-dimethylhydroxylamine hydrochloride (97.5 mg, 1.0 mmol). The reaction mixture was slowly brought to room temperature and allowed to stir for 1 h. The reaction mixture was diluted with water, and extracted with CH2Cl2. The organic layer was dried over anhydrous Na2SO4 and evaporated under reduced pressure to afford the crude product. Chromatographic purification on a silica gel column using 40% EtOAc in hexanes afforded 155.2 mg (69% yield) of 2 as a viscous liquid. Rf (40% EtOAc in hexanes) = 0.55. 1H NMR (500 MHz, CDCl3, ambient temperature): δ = 7.33 (d, 3JH,H = 8.4, 1 H, Ar–H), 7.22 (s, 1 H, Ar–H), 6.68 (d, 3JH,H = 8.4, 1 H, Ar–H), 3.86 (s, 3 H, OMe), 3.85 (s, 3 H, OMe), 3.52 (s, 3 H, N–OMe), 3.30 (s, 3 H, N–Me).
Step 2: addition of MeMgBr to 2
In a clean, dry 10 mL round-bottomed flask equipped with a stirring bar, were placed 2 (125.0 mg, 0.554 mmol) in dry THF (5.0 mL) under nitrogen gas, and the mixture was cooled to −10 °C. At this temperature MeMgBr (554 μL, 1.66 mmol) was added dropwise, with stirring. The reaction mixture was slowly brought to 0 °C and allowed to stir for 1 h. The reaction was quenched with saturated aqueous NH4Cl (5 mL) and extracted with EtOAc (20 mL). The organic layer was separated, dried over Na2SO4 and evaporated to dryness. Chromatographic purification on a silica gel column using 35% EtOAc in hexanes afforded 96.2 mg (97% yield) of 3 as a colorless, viscous liquid. Rf (40% EtOAc in hexanes) = 0.62. 1H NMR (500 MHz, CDCl3): δ = 7.49 (d, 3JH,H = 8.3, 1 H, Ar–H), 7.44 (s, 1 H, Ar–H), 6.81 (d, 3JH,H = 8.3, 1 H, Ar–H), 3.86 (s, 3 H, OMe), 3.85 (s, 3 H, OMe), 2.48 (s, 3 H, COCH3). This compound is commercially available.
N-(3,4-dimethoxybenzoyl)indoline (4)[49]
1H NMR (500 MHz, CDCl3, ambient temperature): δ = 7.21 (d, 3JH,H = 7.4, 1 H, Ar–H), 7.16 (dd, 3JH,H = 1.8, 8.2, 1 H, Ar–H), 7.14 (s, 1 H, Ar–H), 7.11 (br s, 1 H, Ar–H), 7.00 (t, 3JH,H= 7.1, 1 H, Ar–H), 6.89 (d, 3JH,H = 8.2, 1 H, Ar–H), 3.66 (t, 3JH,H = 8.3, 2 H, NCH2), 3.41 (s, 3 H, OCH3), 3.38 (s, 3 H, OCH3), 2.51 (t, 3JH,H = 8.3, 2 H, CH2). 1H NMR (500 MHz, C6D6, 70 °C): δ = 7.94 (br s, 1 H, Ar–H), 7.09 (s, 1 H, Ar–H), 7.04 (d, 3JH,H = 7.8, 1 H, Ar–H), 7.00 (t, 3JH,H = 7.8, 1 H, Ar–H), 6.91 (d, 3JH,H = 7.3, 1 H, Ar–H), 6.83 (t, 3JH,H = 7.3, 1 H, Ar–H), 3.66 (t, 3JH,H = 8.3, 2 H, NCH2), 3.41 (s, 3 H, OCH3), 3.38 (s, 3 H, OCH3), 2.51 (t, 3JH,H = 8.3, 2 H, CH2).
N-(piperonyl)indoline (6)[49]
To a stirring solution of I2 (253.8 mg, 1.0 mmol) in dry CH2Cl2 (5 mL) at 0 °C was added Pol–PPh3 (438.0 mg, 1.0 mmol). The reaction mixture was flushed with nitrogen gas and allowed to stir at 0 °C for 5 minutes. At this temperature piperonylic acid (166.1 mg, 1.0 mmol) was added, followed by dropwise addition of iPr2NEt (260.0 μL, 1.5 mmol) and indoline (112 μL, 1.0 mmol). The reaction mixture was slowly brought to room temperature and allowed to stir for 2 h. The mixture was filtered and evaporated to the dryness. Chromatographic purification on a silica gel column using 30% EtOAc in hexanes afforded 199.2 mg (74% yield) of 6 as a colorless solid. Rf (40% EtOAc in hexanes) = 0.60. 1H NMR (500 MHz, CDCl3, ambient temperature): δ = 7.21 (d, 3JH,H = 7.4, 1 H, Ar–H), 7.18–7.09 (br s, 1 H, Ar–H), 7.09 (dd, 3JH,H = 1.5, 8.0, 1 H, Ar–H), 7.04 (s, 1 H, Ar–H), 7.01 (t, 3JH,H = 7.4, 1 H, Ar–H), 6.85 (d, 3JH,H = 8.0, 1 H, Ar–H), 6.03 (s, 2 H, OCH2), 4.10 (t, 3JH,H = 7.9, 2 H, indolinyl–H), 3.11 (t, 3JH,H = 7.9, 2 H, indolinyl–H). 1H NMR (500 MHz, C6D6, 70 °C): δ = 7.93 (br s, 1 H, Ar–H), 6.99 (t, 3JH,H = 8.0, 1 H, Ar–H), 6.96 (s, 1 H, Ar–H), 6.89 (d, 3JH,H = 7.8, 1 H, Ar–H), 6.82 (t, 3JH,H = 7.3, 1 H, Ar–H), 6.51 (d, 3JH,H = 7.8, 1 H, Ar–H), 5.29 (s, 2 H, OCH2), 3.51 (t, 3JH,H = 8.0, 2 H, NCH2), 2.45 (t, 3JH,H = 8.0, 2 H, CH2).
Supplementary Material
Acknowledgments
Partial support of this work by NSF Grant CHE–0640417 and a PSC CUNY award to M.K.L. is acknowledged. Mr. Tom Melninkaitis and Mr. Scott Klepfer (Biotage) are thanked for a sample of Pol–PPh3 (PS–triphenylphosphane) used in this work and Dr. Padmanava Pradhan is thanked for his assistance with some NMR experiments. Infrastructural support was provided by Research Centers at Minority Institutions Grant NIH/NCRR/RCMI Grant G12 RR03060.
Footnotes
Supporting information for this article is available on the WWW under http://www.eurjoc.org/or from the author.
Supporting Information (see footnote on the first page of this article): Copies of 1H NMR spectra of all amides and Weinreb amides shown in Tables 1 and 2, 13C NMR spectrum of N-(3,5-dinitro)benzoylpyrrolidine, 1H NMR spectra of 3, 4, 6, anhydrolychorinonine and hippadine, 1H–1H COSY spectra of 4 and 6.
References
- 1.Larock RC. Comprehensive Organic Transformation. 2nd. Wiley-VCH Publishers; New York: 1999. [Google Scholar]
- 2.a) Rao Y, Li X, Danishefsky SJ. J Am Chem Soc. 2009;131:12924–12926. doi: 10.1021/ja906005j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gooβen LJ, Ohlmann DM, Lange PP. Synthesis. 2009:160–164. [Google Scholar]; c) Burés J, Martin M, Urpi F, Vilarrasa J. J Org Chem. 2009;74:2203–2206. doi: 10.1021/jo802825e. [DOI] [PubMed] [Google Scholar]; d) Nordstrøm LU, Vogt H, Madsen R. J Am Chem Soc. 2008;130:17672–17673. doi: 10.1021/ja808129p. [DOI] [PubMed] [Google Scholar]; e) Veitch GE, Bridgwood KL, Ley SV. Org Lett. 2008;10:3623–3625. doi: 10.1021/ol801398z. [DOI] [PubMed] [Google Scholar]; f) Seo SY, Marks TJ. Org Lett. 2008;10:317–319. doi: 10.1021/ol702788j. [DOI] [PubMed] [Google Scholar]; g) Reddy KR, Maheswari CU, Venkateshwar M, Kantam ML. Eur J Org Chem. 2008:3619–3622. [Google Scholar]; h) Gunanathan C, Ben–David Y, Milstein D. Science. 2007;317:790–792. doi: 10.1126/science.1145295. [DOI] [PubMed] [Google Scholar]; i) Maki T, Ishihara K, Yamamoto H. Tetrahedron. 2007;63:8645–8657. [Google Scholar]; j) Matsugi M, Hasegawa M, Sadachika D, Okamoto S, Tomioka M, Ikeya Y, Masuyama A, Mori Y. Tetrahedron Lett. 2007;48:4147–4150. [Google Scholar]; k) Valeur E, Bradley M. Tetrahedron. 2007;63:8855–8871. [Google Scholar]; l) Novak A, Humphreys LD, Walker MD, Woodward S. Tetrahedron Lett. 2006;47:5767–5769. [Google Scholar]; m) Zhang M, Vedantham P, Flynn DL, Hanson PR. J Org Chem. 2004;69:8340–8344. doi: 10.1021/jo048870c. [DOI] [PubMed] [Google Scholar]; n) Nery MS, Ribeiro RP, Lopes CC, Lopes RSC. Synthesis. 2003:272–276. [Google Scholar]; o) Li P, Xu JC. Tetrahedron. 2000;56:4437–4445. [Google Scholar]; p) Katritzky AR, He HY, Suzuki K. J Org Chem. 2000;65:8210–8213. doi: 10.1021/jo000792f. [DOI] [PubMed] [Google Scholar]
- 3.Lin X, Robins MJ. Org Lett. 2000;2:3497–3499. doi: 10.1021/ol000255h. [DOI] [PubMed] [Google Scholar]
- 4.Zlatko J, Lin X, Robins MJ. Nucleosides, Nucleotides Nucleic Acids. 2004;23:137–147. doi: 10.1081/ncn-120027823. [DOI] [PubMed] [Google Scholar]
- 5.Bae S, Lakshman MK. J Org Chem. 2008;73:1311–1319. doi: 10.1021/jo7021795. [DOI] [PubMed] [Google Scholar]
- 6.Bae S, Lakshman MK. J Org Chem. 2008;73:3707–3713. doi: 10.1021/jo702558n. [DOI] [PubMed] [Google Scholar]
- 7.Lakshman MK, Choudhury A, Bae S, Rochttis E, Pradhan P, Kumar A. Eur J Org Chem. 2009:152–159. doi: 10.1002/ejoc.200800752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Narasaiah AV, Sreenu D, Nagaiah K. Synth Commun. 2006;36:137–140. [Google Scholar]
- 9.a) Kang FA, Sui Z, Murray WV. Eur J Org Chem. 2009:461–479. [Google Scholar]; b) Kang FA, Sui Z, Murray WV. J Am Chem Soc. 2008;130:11300–11302. doi: 10.1021/ja804804p. [DOI] [PubMed] [Google Scholar]
- 10.Frøyen P. Synth Commun. 1995;25:959–968. [Google Scholar]
- 11.Bandgar BP, Bettigeri SV. Synth Commun. 2004;34:2917–2924. [Google Scholar]
- 12.Garcia J, Urpí F, Vilarrasa J. Tetrahedron Lett. 1984;25:4841–4844. [Google Scholar]
- 13.Barstow LE, Hruby VJ. J Org Chem. 1971;36:1305–1306. [Google Scholar]
- 14.Yamada Si, Takeuchi T. Tetrahedron Lett. 1971;12:3595–3598. [Google Scholar]
- 15.Rodriguez Rdc, Barros IMA, Lima ELS. Tetrahedron Lett. 2005;46:5945–5947. [Google Scholar]
- 16.Harrison CR, Hodge P, Hunt BJ, Koshdel E, Richardson G. J Org Chem. 1983;48:3721–3728. [Google Scholar]
- 17.Buchstaller HP, Ebert HM, Anlauf U. Synth Commun. 2001;31:1001–1005. [Google Scholar]
- 18.Han SY, Kim YA. Tetrahedron. 2004;60:2447–2467. [Google Scholar]
- 19.Yang X, Birman VB. Org Lett. 2009;11:1499–1502. doi: 10.1021/ol900098q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salvio R, Moisan L, Ajami D, Rebek J., Jr Eur J Org Chem. 2007:2722–2728. [Google Scholar]
- 21.Lysen M, Kelleher S, Begtrup M, Kristensen JL. J Org Chem. 2005;70:5342–5343. doi: 10.1021/jo050459h. [DOI] [PubMed] [Google Scholar]
- 22.Tetsuaki A, Xie F, Tsutomu A, Shuji U, Toshimasa I, Yasuko I, Chuzo I, Naoyoshi M. J Chin Chem Soc (Taipei) 2000;47:865–868. [Google Scholar]
- 23.MacNeil SL, Gray M, Gusev DG, Briggs LE, Snieckus V. J Org Chem. 2008;73:9710–9719. doi: 10.1021/jo801856n. [DOI] [PubMed] [Google Scholar]
- 24.Mohammadpoor–Baltork I, Sadeghi MM, Esmayilpour K. Synth Commun. 2003;33:953–959. [Google Scholar]
- 25.Karnik AV, Kamath SS. Tetrahedron: Asymmetry. 2008;19:45–48. [Google Scholar]
- 26.Zanatta N, Faoro D, Silva SC, Bonacorso HG, Martins MAP. Tetrahedron Lett. 2004;45:5689–5691. [Google Scholar]
- 27.Grieco PA, Clark DS, Withers GP. J Org Chem. 1979;44:2945–2947. [Google Scholar]
- 28.Luca LD, Giacomelli G, Porcheddu A. J Org Chem. 2002;67:6272–6274. doi: 10.1021/jo025960d. [DOI] [PubMed] [Google Scholar]
- 29.Hoffman RV, Nayyar NK. J Org Chem. 1995;60:7043–7046. [Google Scholar]
- 30.Okawara T, Matsuda T, Furukawa M. Chem Pharm Bull. 1982;30:1225–1233. [Google Scholar]
- 31.Kafka S, Kytner J, Silhankova A, Ferles M. Collect Czech Chem Comm. 1987;52:2035–2046. [Google Scholar]
- 32.Glynn D, Bernier D, Woodward S. Tetrahedron Lett. 2008;49:5687–5688. [Google Scholar]
- 33.Kinoshita T, Ichinari D, Sinya J. J Heterocycl Chem. 1996;33:1313–1317. [Google Scholar]
- 34.Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3815–3818. [Google Scholar]
- 35.a) Balasubramaniam S, Aidhen IS. Synthesis. 2008:3707–3738. [Google Scholar]; b) Singh J, Satyamurthi N, Aidhen IS. J Prakt Chem. 2000;342:340–347. [Google Scholar]
- 36.Mentzel M, Hoffmann HMR. J Prakt Chem. 1997;339:517–524. [Google Scholar]
- 37.Khlestkin VK, Mazhukin DG. Curr Org Chem. 2003;7:967–993. [Google Scholar]
- 38.Sibi MP. Org Prep Proc Int. 1993;25:15–40. [Google Scholar]
- 39.One pot methods: Niu T, Zhang W, Huang D, Xu C, Wang H, Hu Y. Org Lett. 2009;11:4474–4477. doi: 10.1021/ol901886u.Deagostino A, Larini P, Occhiato EG, Pizzuto L, Prandi C, Venturello P. J Org Chem. 2008;73:1941–1945. doi: 10.1021/jo7024898.Lee IJ. Bull Korean Chem Soc. 2007;28:695–697.Han KJ, Kim M. Lett Org Chem. 2007;4:20–22.Martnelli JR, Freckmann DMM, Buchwald SL. Org Lett. 2006;8:4843–4846. doi: 10.1021/ol061902t.Woo JCS, Fenster E, Dake GR. J Org Chem. 2004;69:8984–8986. doi: 10.1021/jo048385h.De Luca L, Giacomelli G, Taddei M. J Org Chem. 2001;66:2534–2537. doi: 10.1021/jo015524b.Banwell M, Smith J. Synth Commun. 2001;31:2011–2019.Tunoori AR, White JM, Georg GI. Org Lett. 2000;2:4091–4093. doi: 10.1021/ol000318w.
- 40.Einhorn J, Einhorn C, Luche JL. Synth Commun. 1990;20:1105–1112. [Google Scholar]
- 41.Zhang Z, Yu Y, Liebeskind LS. Org Lett. 2008;10:3005–3008. doi: 10.1021/ol8009682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saito Y, Ouchi H, Takahata H. Tetrahedron. 2008;64:11129–11135. [Google Scholar]
- 43.Schlüter T, Steinbach AC, Steffen A, Rettig R, Grisk O. Cardiovasc Res. 2008;80:271–279. doi: 10.1093/cvr/cvn185. [DOI] [PubMed] [Google Scholar]
- 44.Lirdprapamongkol K, Kramb JP, Suthiphongchai T, Surarit R, Srisomsap C, Dannhardt G, Svasti J. J Agri Food Chem. 2009;57:3055–3063. doi: 10.1021/jf803366f. [DOI] [PubMed] [Google Scholar]
- 45.Lafeber FPJG, Beukelman CJ, van den Worm E, van Roy JLAM, Vianen ME, van Roon JAG, van Dijk H, Bijlsma JWJ. Rheumatology. 1999;38:1088–1093. doi: 10.1093/rheumatology/38.11.1088. [DOI] [PubMed] [Google Scholar]
- 46.a) Suri OP, Bindra RS, Katti NK, Khajuria RK. Indian J Chem Sec B. 1987;26B:587–588. [Google Scholar]; b) Weike S, Jin C. Synth Commun. 2004;34:4199–4205. [Google Scholar]
- 47.Dodge JA, Stocksdale MG, Fahey KJ, Jones CD. J Org Chem. 1995;60:739–741. [Google Scholar]
- 48.a) Chattopadhyaya S, Chattopadhyaya U, Mathur PP, Saini KS, Ghosal S. Planta Med. 1983;49:252–254. doi: 10.1055/s-2007-969864. [DOI] [PubMed] [Google Scholar]; b) Ghosal S, Lochan R, Kumar AY, Srivastava RS. Phytochemistry. 1985;24:1825–1828. [Google Scholar]
- 49.Ganton MD, Kerr MA. Org Lett. 2005;7:4777–4779. doi: 10.1021/ol052086c. [DOI] [PubMed] [Google Scholar]
- 50.a) Frøyen P. Phosphorus Sulfur Silicon Relat Elem. 1994;91:145–151. [Google Scholar]; b) Frøyen P. Phosphorus Sulfur Silicon Relat Elem. 1994;89:57–61. [Google Scholar]
- 51.Jang DO, Park DJ, Kim J. Tetrahedron Lett. 1999;40:5323–5326. [Google Scholar]
- 52.Kang DO, Joo TY, Lee EH, Chaysripongkul S, Chavasiri W, Jang DO. Tetrahedron Lett. 2006;47:5693–5696. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.