

Abstract
The endoplasmic reticulum (ER) is a multifunctional organelle required for lipid biosynthesis, calcium storage, and protein folding and processing. A number of physiological and pathological conditions, as well as a variety of pharmacological agents, are able to disturb proper ER function and thereby cause ER stress, which severely impairs protein folding and therefore poses the risk of proteotoxicity. Specific triggers for ER stress include, for example, particular intracellular alterations (e.g., calcium or redox imbalances), certain microenvironmental conditions (e.g., hypoglycemia, hypoxia, and acidosis), high-fat and high-sugar diet, a variety of natural compounds (e.g., thapsigargin, tunicamycin, and geldanamycin), and several prescription drugs (e.g., bortezomib/Velcade, celecoxib/Celebrex, and nelfinavir/Viracept). The cell reacts to ER stress by initiating a defensive process, called the unfolded protein response (UPR), which is comprised of cellular mechanisms aimed at adaptation and safeguarding cellular survival or, in cases of excessively severe stress, at initiation of apoptosis and elimination of the faulty cell. In recent years, this dichotomic stress response system has been linked to several human diseases, and efforts are underway to develop approaches to exploit ER stress mechanisms for therapy. For example, obesity and type 2 diabetes have been linked to ER stress-induced failure of insulin-producing pancreatic beta cells, and current research efforts are aimed at developing drugs that ameliorate cellular stress and thereby protect beta cell function. Other studies seek to pharmacologically aggravate chronic ER stress in cancer cells in order to enhance apoptosis and achieve tumor cell death. In the following, these principles will be presented and discussed.
1. Introduction
The endoplasmic reticulum (ER) is a vital organelle present in all eukaryotic cells. It consists of interconnected, branching membranous tubules, vesicles, and cisternae that provide a distinct subcellular compartment with a number of functions. The rough ER is studded with ribosomes on its outer surface and plays a key role in protein synthesis and secretion. The smooth ER lacks associated ribosomes and therefore is not primarily involved in protein synthesis, but is central to the synthesis of fatty acids and phospholipids, assembly of lipid bilayers, metabolism of carbohydrates, and regulation of calcium homeostasis. In the liver, enzymes in the smooth ER metabolize and detoxify hydrophobic chemicals, such as drugs and carcinogens, and direct them for secretion from the body. While some cells may have little smooth ER, all eukaryotic cells have conspicuous amounts of rough ER, as the latter is essential for the synthesis of plasma membrane proteins and proteins of the extracellular matrix. Rough ER is particularly abundant in secretory cells, such as antibody-producing plasma cells, insulin-secreting beta cells, or cells of milk-producing glands, where a large fraction of the cytosol is occupied by rough ER. The sarcoplasmic reticulum is a specialized form of the ER in muscle cells and functions to sequester and release large amounts of calcium to effect muscle contractions and relaxation [1].
The ER is a highly dynamic organelle, and its complex functions can be significantly influenced by a multitude of parameters both inside the cell and in its microenvironment. For instance, the availability of oxygen (hypoxia) or glucose (hypoglycemia), hyperthermia, acidosis, calcium levels, the redox milieu, energy levels (modulated by hypoxia and hypoglycemia), and other factors can impact and disturb proper functioning of the ER, resulting in ER stress and impacting protein folding in the lumen of the ER. Protein folding is a complex process that depends on the interaction of chaperone proteins, foldases, and glycosylating enzymes, as well as appropriate calcium levels and an oxidizing environment. ER stress impairs this process and results in the accumulation of unfolded or misfolded proteins, which leads to the activation of a specific cellular process called the unfolded protein response (UPR) [2, 3]. Activation of the UPR represents the defining criterion of ER stress, although oftentimes the terms UPR and ER stress are used interchangeably.
The accumulation of unfolded, misfolded, insoluble, or otherwise damaged proteins can irreparably damage cellular functions and thus pose a proteotoxic threat to the survival of the cell. Several cellular mechanisms coordinately function to ameliorate this risk. Prime among these is the core function of the UPR, which is aimed at correct protein folding and overall proper protein processing. However, terminally misfolded proteins, that is, those that cannot be repaired, will be removed from the cell's inventory by one of two separate processes. One is ER-associated degradation (ERAD), which exports damaged proteins back into the cytoplasm and delivers them to the proteasome for degradation and clearance. The other is aggresome formation, where damaged proteins are compacted together with other cellular debris into juxtanuclear complexes and then recycled via autophagy. Indeed, autophagy, a cellular mechanism for the recycling of surplus or defective cellular components, has been found reciprocally linked to ER stress. Several studies have shown that severe ER stress activates the autophagic process and, conversely, that blockage of autophagy leads to aggravated ER stress and cell death [4–8]. (The molecular details of the autophagic process have been covered in recent reviews [9–12] and will not be presented here.)
A well-documented ultrastructural response to ER stress is displayed by the pronounced dilation of the ER lumen. For example, yeast cells expand their ER volume at least 5-fold under UPR-inducing conditions, and similar effects have also been confirmed in mammalian cells in vitro and in vivo [4, 13–16]. This expansion of ER lumen is thought to be necessary to accommodate increasing amounts of lumenal constituents, in particular those that are being synthesized to manage ER stress, and thus appears to be an adjustment to cope with increasing crowding conditions, which favor protein aggregation and are detrimental for proper protein folding [17].
Due to the cytotoxic risk that the accumulation of misfolded/unfolded proteins poses to the cell, it is not surprising that cellular sensors and pathways have evolved to respond to this threat. This stress response system, called the ER stress response or the UPR, displays a dichotomic yin-yang characteristic (Figure 1), where mild or short-term stress triggers activation of a response module that either leads to the neutralization of the initial stress or adaption to it, but where severe or long-lasting stress favors activation of a proapoptotic module that will lead to cell death. In both cases, the initial signal (e.g., accumulation of misfolded proteins inside the ER) is transmitted across the ER membrane, through the cytoplasm, and into the nucleus, where alterations in gene expression patterns effect the respective resulting phenotypic outcome, that is, adaptation and survival or apoptosis.
Figure 1.
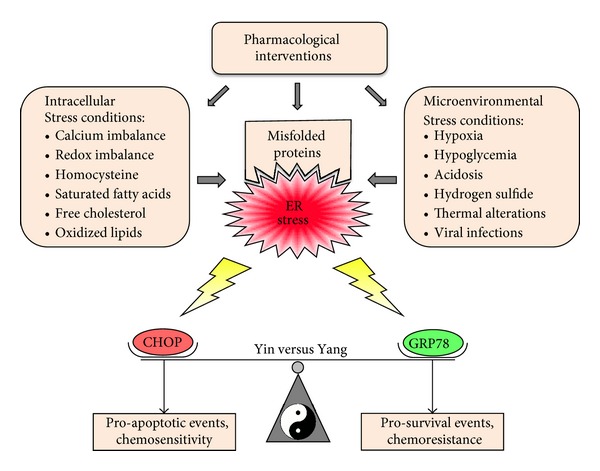
Triggers of ER stress and the yin-yang balance of cell survival versus cell death. A great variety of conditions and pharmacological compounds can disturb ER homeostasis, leading to ER stress and the accumulation of unfolded and misfolded proteins. In response, ER stress signaling pathways stimulate pro-survival efforts to either neutralize the stressful insult or adapt to it. GRP78 plays a key role in the cell's attempt to adapt and survive. In contrast, if ER stress is too severe, the pro-apoptotic module of this cellular system gains dominance and shifts the balance towards cell death. CHOP represents a central executor of this latter process. In essence, these opposing processes of cell death versus survival are reflective of the yin-yang (shadow and light) concept of Chinese philosophy, where seemingly contrary forces are interconnected and interdependent as part of a greater whole. Although many other components participate in balancing the cell's yin-yang response to ER stress, the opposing efforts of prosurvival (yang) GRP78 and proapoptotic (yin) CHOP represent important tenets of this struggle; as well, their expression levels are being used as convenient markers and readouts as to the ER stress status of cells. Details of GRP78 and CHOP functions are presented in Figures 2 and 3, respectively.
2. Key Players in the ER Stress Response
Considering the diversity of functions of the ER, which include quality control and secretion of proteins, lipid and membrane biosynthesis, and control of intracellular calcium homeostasis, it is not unexpected that a large number of regulatory components participate in these processes and, in one way or another, become involved in the ER stress response/UPR. For example, the ER lumen is rich in calcium-dependent molecular chaperones, such as glucose-regulated protein 78 (GRP78, also called BiP: immunoglobulin heavy chain-binding protein [18, 19]), GRP94, calnexin and calreticulin, enzymes involved in posttranslational protein modifications, such as protein disulphide isomerase (PDI), oxidoreductases, and those performing protein glycosylation and lipidation, as well as numerous others involved in lipid and membrane biosynthesis.
A variety of disturbances can interfere with proper processing in the ER and thus trigger the ER stress response/UPR (Figure 1). For instance, under conditions of low glucose supply (hypoglycemia), N-linked glycosylation of proteins is impaired [20, 21]. Imbalanced cellular redox homeostasis, which can be caused by hypoxia and prooxidant or reducing agents, interferes with disulphide bonding of proteins [22]. Aberrant calcium levels impinge on the activity of calcium-dependent chaperones [23, 24]. Impaired removal and degradation of terminally misfolded proteins (by blocked ERAD or compromised autophagy) results in the accumulation of these potentially proteotoxic proteins [5, 25–27]. Viral infections may overload the ER lumen with virus-encoded proteins [28–30]. A diet high in fats and sugars (chronic hyperlipidemia and hyperglycemia) has also been linked to increased ER stress, particularly in the liver and in insulin-secreting β-cells of the pancreas (see the following) [31–34]. Key players of the ER stress system that are involved in responding to these threats to cellular homeostasis are presented in the following subsections.
2.1. Master Regulator GRP78
Among the many ER-resident proteins, the chaperone GRP78 stands out because in addition to its calcium binding and protein processing function, it exerts a key role as a master initiator of early ER stress/UPR signaling. As implied by its name, GRP78 initially has been characterized as a glucose-regulated protein, where restricting the availability of glucose in cell culture medium resulted in pronounced stimulation of GRP78 transcription and translation, and thus provided initial clues as to its activation during cellular stress conditions [35, 36]. A large number of subsequent studies established that a great variety of cellular and microenvironmental disturbances, as well as many pharmacological interventions, can lead to increased GRP78 expression, along with aggravated ER stress. Indeed, the significantly increased amount of GRP78 protein over baseline expression has become an established indicator and marker for the presence of cellular ER stress [37–39].
GRP78 belongs to the heat shock protein 70 (HSP70) family of proteins, where many of its members have been characterized as chaperones within the ER. In recent years, however, it was discovered that GRP78 can also be present outside the ER; for example, the protein was found in the cytosol [40], in mitochondria [41], in the nucleus [42], and at the cell surface of tumor cells [43–47]. It has thus emerged that GRP78, as well as a few other traditional ER-localized chaperones, can function beyond this compartment and are involved in processes not directly connected to posttranslational protein processing [39, 48]. For purposes of this current paper, the focus shall remain on the intra-ER functions of GRP78.
In unstressed cells, a fraction of ER-luminal GRP78 is bound to three different ER transmembrane proteins: (i) inositol-requiring kinase/endoribonuclease 1 (IRE1) [49], (ii) protein kinase activated by double-stranded RNA (PKR)-like ER kinase (PERK) [50], and (iii) activating transcription factor 6 (ATF6) [51]. Binding of GRP78 to the ER-luminal domains of these proteins keeps their activity suppressed and maintains them in an inactive state. Upon ER stress and concomitant accumulation of misfolded and unprocessed proteins, GRP78 is sequestered away from PERK, IRE1, and ATF6 in order to attend to the increased need for protein folding. As a result, dissociation from GRP78 leads to the activation of all three of these transmembrane proteins, thereby unfolding three distinct branches of the ER stress response/UPR (see Figure 2 and general reviews [2, 3, 52]). Among the consequences of these signaling events is increased expression of GRP78, which not only serves to provide the needed additional chaperone capacity, but also eventually will reassociate with PERK, IRE1, and ATF6 in order to return these signaling modules to their inactive modes when homeostasis has been reestablished.
Figure 2.
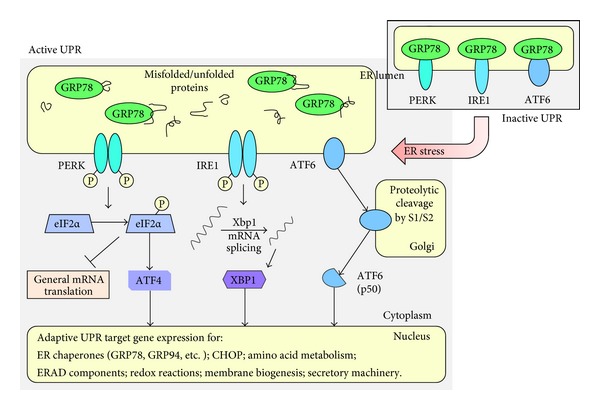
Overview of the three signaling branches of the ER stress response/UPR. In the absence of ER stress, ER luminal GRP78 associates with ER transmembrane proteins PERK, IRE1, and ATF6 to block their activation (shown as inactive UPR in the top right square). Upon ER stress, accumulating unfolded and misfolded proteins inside the ER sequester GRP78, thus dissociating this master regulator from all three transmembrane sensors and relieving their blockage. Activation of PERK entails homodimerization and autophosphorylation, leading to phosphorylation of eIF2α, which terminates global protein translation, but exempts selected ER stress-associated proteins, such as ATF4. Activation of IRE1 also entails homodimerization and autophosphorylation. Endonuclease activity of activated IRE1 removes an intron from Xbp1 mRNA to generate a shorter splice variant that encodes transcription factor XBP1. ATF6 translocates to the Golgi, where it is proteolytically cleaved by S1 and S2 proteases to generate the transcriptionally active p50 fragment. All three transcription factors, ATF4, XBP1, and ATF6-p50 translocate into the nucleus where they regulate the expression of a variety of gene products collectively involved in managing ER stress. (See text for further details and references.)
2.2. The IRE1 Signaling Branch
Activation of IRE1 represents the most conserved signaling branch of the ER stress response/UPR [53, 54]. It is a bifunctional enzyme with serine/threonine protein kinase and endoribonuclease (RNase) activity in its cytosolic domain. Release from suppression by GRP78 triggers its homodimerization and autophosphorylation as part of the activation process [52]. Activated IRE1 cleaves a 26-base fragment from the mRNA encoding X box-binding protein 1 (XBP1), resulting in spliced XBP1s and translation of a potent transcription factor controlling the expression of genes involved in ERAD and protein folding, as well as others directing the synthesis of phospholipids that are required for the expansion of ER membranes during ER stress [49, 55]. IRE1 signaling and XBP1 splicing are particularly important in highly secretory cells where the protein folding machinery is continuously engaged with a high amount of nascent proteins [56]. Therefore, this branch of control serves as a key adaptive mechanism to match ER folding capacity with the demands of protein folding [57, 58].
In addition to splicing a number of mRNAs, a second function of IRE1 is to activate a signaling cascade involved in controlling cell fate with regard to cell death. Here, activated IRE1 recruits tumor-necrosis- factor-receptor- (TNFR-) associated factor 2 (TRAF2), which results in the downstream activation of apoptosis signal-regulated kinase 1 (ASK1) and c-Jun N-terminal kinase (JNK) [59, 60]. On one hand, sustained JNK activity during prolonged ER stress inhibits antiapoptotic members of the Bcl-2 (B cell lymphoma 2) family of proteins. On the other hand, JNK phosphorylates and activates proapoptotic BH3-only proteins, such as Bid (BH3 interacting domain death agonist) and Bim (Bcl-2-interacting mediator of cell death). Combined, these events lead to oligomerization of Bax and Bak, resulting in permeabilization of the outer mitochondrial membrane and execution of the intrinsic apoptotic process [58, 61, 62] (see Figure 3).
Figure 3.
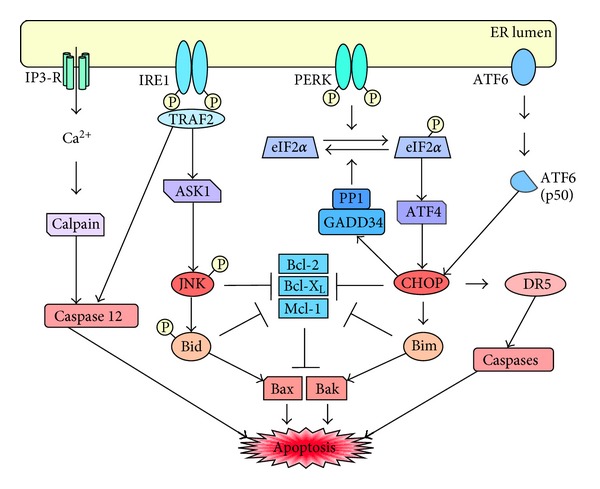
Cell death signaling by the ER stress response/UPR. In case of severe and sustained ER stress, a number of proapoptotic events begin to dominate and lead to apoptosis. Transcription factors ATF4 and ATF6-p50 stimulate CHOP expression. On one hand, CHOP stimulates expression of GADD34, which associates with PP1, resulting in dephosphorylation of eIF2α, thus reactivating global cellular protein synthesis. On the other hand, CHOP inhibits antiapoptotic proteins of the Bcl-2 family and stimulates pro-apoptotic Bim, altogether leading to heterodimerization and activation of pro-apoptotic Bax and Bak. CHOP also stimulates expression of cell surface death receptor DR5, which sensitizes cells to pro-apoptotic stimuli, presumably via calibrating the extrinsic apoptotic pathway involving caspase 12. Similarly, activated JNK complements the pro-apoptotic efforts of CHOP. JNK becomes phosphorylated and activated by protein kinase ASK1 upon association of TRAF2 with activated IRE1. Association of TRAF2 with activated IRE1 also leads to activation of caspase 12. Calcium release from the ER via IP3 receptors can activate calpains, which further stimulate caspase 12 activation via proteolytic cleavage of its inactive procaspase precursor.
2.3. The ATF6 Signaling Branch
ER transmembrane-localized ATF6 harbors a basic leucine zipper (bZIP) motif and transcription factor properties. Upon its release from ER-luminal GRP78, Golgi localization sequences are unmasked, whereupon ATF6 translocates to the Golgi apparatus. Here, it is proteolytically cleaved by Golgi-resident site-1 protease (S1P, a serine-protease) in its ER luminal domain and by site-2 protease (S2P, a metalloprotease) within its region that spans the Golgi phospholipid bilayer, resulting in the release of the cytosolic bZIP transcription factor domain from the Golgi membrane [63]. Upon translocation to the nucleus, ATF6 stimulates expression of a number of genes whose protein products contribute to protein folding, protein secretion, and ERAD, thereby supporting the cell's effort to cope with ER stress and accumulated misfolded/unfolded proteins [37, 64] (Figure 2). Examples of ATF6-regulated genes include GRP78 and GRP94, protein disulphide isomerase (PDI), XBP1, and CHOP [52, 65, 66].
Besides ATF6, a number of other ER-transmembrane bZIP transcription factors have been described in recent years that are also regulated by intramembrane proteolysis. In contrast to ubiquitous ATF6, expression of these factors appears to be tissue specific to variable degrees. Examples are cAMP responsive element-binding protein 3 (CREB3, also called Luman), CREB4/TISP40, CREBH, CREB3L1/OASIS, and CREB3L2/BBF2H2 (see detailed references in reviews: [31, 67]). Although most of these have been found activated upon ER stress and appear to contribute to the general ER stress response/UPR, it is not entirely clear what sets them apart from ATF6 and why cells would require multiple ATF6-like molecules in the ER. It has been surmised that these variant proteins perhaps respond to tissue-specific conditions of ER stress that may require tissue-specific gene expression patterns to resolve that stress [31].
2.4. The PERK Signaling Branch
Activation of PERK involves its homodimerization and autophosphorylation, which is followed by phosphorylation of its main substrate, eukaryotic initiation factor 2 alpha (eIF2α). Phosphorylation of eIF2α attenuates global protein synthesis, thereby decreasing protein influx to the ER in support of resolving the cytotoxic threat from accumulated misfolded proteins [68]. At the same time, phosphorylation of eIF2α changes the efficiency of AUG initiation codon usage and leads to the preferential translation of a small number of mRNAs, including activating transcription factor 4 (ATF4), a transcription factor that stimulates a set of genes involved in supporting recovery and adaptation [50]. Among ATF4-regulated genes is the one encoding CHOP, a key transcription factor that is important to initiate the apoptotic program in case of excessive ER stress [69] (see details in next subsection).
Besides eIF2α, nuclear factor-erythroid 2-related factor 2 (Nrf2) represents a second immediate substrate for phosphorylation by PERK. Upon activation, this basic-leucine zipper transcription factor migrates to the nucleus where it activates genes encoding antioxidant proteins and detoxifying enzymes [70]. Because ER stress may involve the accumulation of reactive oxygen species (ROS), thereby promoting a state of oxidative stress, Nrf2 plays a critical role in fighting such perturbations in redox homeostasis [71]. The importance of this defensive role of Nrf2 has been further emphasized by findings that Nrf2-deficient cells displayed greatly increased cell death following exposure to ER stress [72].
2.5. Proapoptotic CHOP
The expression levels of CHOP (C/EBP homologous protein, also called GADD153: growth arrest and DNA damage inducible gene 153 [73, 74]) are kept very low in nonstressed cells. Upon acute ER stress, however, CHOP expression is strongly stimulated through IRE1- and PERK-mediated signaling and the activities of ATF4 and ATF6 transcription factors. The full proapoptotic effect of CHOP only emerges when ER stress cannot be subdued by the efforts of the prosurvival module of the response system, and the levels of misfolded proteins remain high. In this case, CHOP stimulates a transcriptional profile that facilitates a pro-apoptotic program. It includes expression of proapoptotic Bim and repression of antiapoptotic Bcl-2 [75, 76], which represents a mechanism that is aligned with similar pro-apoptotic efforts of JNK mentioned previously (see Figure 3, and detailed refs. in [62, 69, 77]). As well, CHOP induces death receptor 5 (DR5), which further sensitizes cells to apoptotic stimulation by a variety of conditions that cause ER stress [78].
Another target gene of CHOP is growth arrest and DNA damage inducible protein 34 (GADD34), a regulatory subunit of protein phosphatase type 1 (PP1); CHOP-induced stimulation of GADD34 expression leads to PP1 activation and dephosphorylation of eIF2α, resulting in the resumption of general translation and normal functioning [79, 80] (Figure 3). Thus, while sustained elevation of CHOP expression triggers strong pro-apoptotic signaling, its initial effect on GADD34 may contribute to the restoration of homeostasis—with the caveat that the renewed supply of client proteins to the ER, if taking place too early, that is, under conditions where ER stress is not yet completely resolved, can trigger the generation of reactive oxygen species (ROS) with deleterious consequences for cell survival [77]. In any case, the dissolution of ER stress entails mandatory suppression of CHOP levels as a prerequisite for return to homeostasis [81].
3. The Yin-Yang Principle of ER Stress
The primary goal of the ER stress response/UPR is to reestablish cellular homeostasis either by eliminating the stressful trigger (e.g., via restoring redox or calcium balance) or by adapting to it (e.g., via permanently increasing its folding capacity in case of revved-up protein synthesis). However, if these countermeasures prove unsuccessful and severe imbalances persist, the response system abandons its prosurvival efforts and instead initiates proapoptotic mechanisms that gain dominance and eventually will lead to cell death. Because of these dichotomic efforts between cell survival and cell death, the ER stress response mechanisms can be viewed as a cellular display of yin-yang principles, where the two opposing forces of cell death and survival balance each other for the greater good of ensuring survival of the organism as a whole (Figure 1).
GRP78, the previously introduced master regulator of the ER stress response, represents the perhaps most critical proponent of the prosurvival yang module of this system. As mentioned, GRP78 protein is key in activating the response system in an initial effort to pursue adaptation and cellular survival. Even more so, the robust pro-survival potency of this protein provides significant growth advantage to tumor cells and endows them with the ability to withstand and even thrive under otherwise adverse microenvironmental conditions, such as hypoglycemia and hypoxia that is common within tumor regions with insufficient blood supply. Worse for a patient with cancer, chronically elevated expression of GRP78 in tumor tissue may provide resistance to chemotherapy and may spell worse prognosis [82–86].
At the other end of this yin-yang balance are proteins such as CHOP and IRE1-regulated JNK, which are representatives of the pro-apoptotic yin mechanisms of the stress response. Both proteins exert significant pro-apoptotic efforts in a variety of ways, central among them the suppression of important antiapoptotic proteins and the stimulation of pro-apoptotic components [69, 87] (Figure 3).
The relevance of IRE1-mediated JNK activation for ER stress-induced cell death has been highlighted by experiments where the activity of this module was blocked with small molecule inhibitors or the use of knockout cell lines [59, 69, 87, 88]. For example, besides modulating the balance of Bcl-2 protein family members, IRE1-JNK signaling can also induce autophagy [6]. As well, IRE1 engages pathways involving stress kinase p38, extracellular signal regulated kinase (ERK), and nuclear factor kappa B (NF-κB) (see details in reviews [31, 58, 89, 90]).
Activated by the PERK/ATF4 axis and by ATF6, transcription factor CHOP in particular represents a crucial executor of the yin module via its ability to impinge on mitochondrial events that function to integrate and amplify the cell death pathways [69, 91, 92]. In unstressed cells, CHOP protein levels generally are below detection levels, but are substantially increased upon acute ER stress. In fact, the presence of conspicuous amounts of CHOP protein represents a marker for the acute phase of the activated ER stress response. Prolonged, high-level expression of CHOP indicates that the ER stress response system has exceeded the limits of its protective yang capacity and that it has switched to its pro-apoptotic yin module—despite the continued presence of elevated GRP78 [69, 81]. Conversely, during moderately intense short-term stress, or when the cell is adapting to longer-lasting chronic stress, efforts of the pro-survival yang module include the suppression of CHOP expression as a prerequisite for recovery and survival, which entails reassociation of GRP78 with and inactivation of the ER transmembrane signaling components PERK, IRE1, and ATF6 [81, 93, 94].
Taken together, pro-survival (yang) GRP78 and pro-apoptotic (yin) CHOP and JNK represent key opposing forces of the ER stress response. Although many additional components mediate the respective final outcome of this process, in simplified terms the antagonistic activities of GRP78 and CHOP signify the cell's yin-yang struggle during its efforts to cope with ER stress. For this reason, both proteins are convenient and indeed frequently used markers to distinguish between the chronic and acute phases of ER stress [81, 91, 95].
4. Targeting ER Stress for Therapy
As presented in greater detail later, the activated ER stress response system has been found involved in a number of human diseases and therefore is being recognized as an emerging target for therapy (see Table 1). In accordance with the previously described yin-yang principle of this cellular system, two conceptually opposing approaches are offered in order to therapeutically target ER stress. On one hand, experimental efforts are aimed at supporting the pro-survival yang modules and/or blocking the pro-apoptotic yin components in order to subdue the pro-apoptotic processes and achieve increased cellular survival. This approach is appropriate in cases where severe ER stress threatens proper organ function, as is the case, for example, in type 2 diabetes where chronic ER stress may lead to the destruction of pancreatic β-cells (see below). Table 2 shows a list of compounds with inherent potency to ameliorate ER stress and minimize its apoptotic consequences.
Table 1.
Human diseases linked to ER stress.
| Disease | Linkage to ER stress | References |
|---|---|---|
| Type 2 diabetes | (i) Obesity induces ER stress (ii) Hyperlipidemia and hyperglycemia induce ER stress (iii) Free fatty acids (palmitate) induce beta cell apoptosis (iv) Deletion of CHOP improves beta cell function and survival |
[31, 34, 98, 119, 127, 142, 288] |
|
| ||
| Atherosclerosis | (i) Oxidized lipids induce ER stress (ii) Hyperhomocysteinemia induces ER stress (iii) Cholesterol loading induces ER stress-mediated cell death (iv) Reduced plaque necrosis in mice lacking CHOP |
[289–295] |
|
| ||
| Nonalcoholic fatty liver disease | (i) Forced GRP78 expression reduces hepatic steatosis in mice (ii) ER stress activates cholesterol and triglyceride biosynthesis (iii) Hyperlipidemia and hyperglycemia induce ER stress |
[296–299] |
|
| ||
| Alcoholic liver disease | (i) Alcohol induces ER stress | [32, 300, 301] |
|
| ||
| Heart disease | (i) ER stress contributes to cardiac myocyte apoptosis (ii) Activation of ER stress in infarcted mouse heart (iii) GRP78 and GRP94 protect against ischemic injury |
[302–308] |
|
| ||
| HBV and HCV infection | (i) HBV induces GRP78 and GRP94 (ii) HCV suppresses IRE1/XBP1 pathway |
[309–312] |
|
| ||
| Alzheimer's disease | (i) Mutant presenilin 1 induces ER stress (ii) Mutant presenilin 1 sensitizes to ER stress-induced apoptosis (iii) AD brains show ER stress features |
[313–318] |
|
| ||
| Parkinson's disease | (i) Parkin expression impacts ER stress (ii) ATF4 leads to increase in parkin expression |
[317–321] |
|
| ||
| Huntington disease | (i) Polyglutamine induces ER stress (ii) ER stress markers are present in postmortem HD brain |
[322–327] |
|
| ||
| Amyotrophic lateral sclerosis | (i) Mutant SOD1 activates ER stress (ii) Mutant SOD1 interferes with ERAD (iii) ER stress markers detected in spinal cord of ALS patients |
[318, 328–330] |
|
| ||
| Prion disease | (i) ER stress markers detected in brains affected with prions (ii) ER chaperones are involved in regulation of misfolded prion protein |
[331–334] |
|
| ||
| Cancer | (i) Tumor-specific microenvironment activates ER stress (ii) Cancer cells display chronic display of ER stress markers (iii) Knockdown of GRP78 or of CHOP affects chemosensitivity |
[83, 97, 151, 222, 335–337] |
Table 2.
Compounds with potency to ameliorate ER stress.
| Compound | Target/effect | References |
|---|---|---|
| Chemical chaperones (TUDCA, 4-PBA) | (i) Increased protein folding capacity (ii) Increased ERAD efficiency (iii) Minimized caspase activation |
[129, 338–343] |
|
| ||
| Inducers of chaperone activity (lithium, valproate, BIX) | (i) Increased expression and activity of chaperones (ii) Supportive of cell-protective ER stress mechanism |
[344–350] |
|
| ||
| Benzodiazepines | (i) Inhibition of ASK1 function and IRE1-ASK1 signaling (ii) Obstructive to pro-apoptotic ER stress mechanism |
[88] |
|
| ||
| Inhibitors of eIF2-alpha phosphatase (salubrinal, Guanabenz) | (i) Inhibition of PP1/GADD34 phosphatase activity (ii) Supportive of attenuated global protein synthesis |
[351–353] |
|
| ||
| Antioxidants (BHA, TM2002, and baicalein) | (i) Sequestration of free radicals (ii) Reduction of oxidative stress and apoptosis |
[354–357] |
|
| ||
| Inducers of antioxidant pathways (carnosic acid, triterpenoids) | (i) Stimulation of NRF2 pathway (ii) Protection from oxidative stress and resulting apoptosis |
[358–363] |
|
| ||
| Stress kinase inhibitors (JNK or p38 inhibitors) | (i) Inhibition of pro-apoptotic JNK or p38 pathways (ii) Antagonism to pro-apoptotic CHOP function |
[354, 364–367] |
BHA: butylated hydroxyanisole.
BIX: BiP/GRP78 inducer X (1-(3,4-dihydroxyphenyl)-2-thiocyanate-ethanone).
4-PBA: 4-phenyl butyric acid.
TUDCA: tauroursodeoxycholic acid.
On the other hand, efforts to further aggravate preexisting ER stress and enhance pro-apoptotic processes could be beneficial in the case of malignant neoplasms. For example, pharmacological enhancement of chronic ER stress in cancer cells may exceed the protective yang capacity and may trigger selective tumor cell death [96]. The basis of this principle lies in the observation that many tumor cells, unlike normal cells, harbor chronically activated pro-survival ER stress components, such as elevated levels of GRP78, in order to manage intensified protein synthesis or to adapt to hostile microenvironmental conditions, such as hypoglycemia, hypoxia, acidosis, or chemotherapy [38, 86, 95, 97]. As a consequence of the already engaged ER stress system, fewer contingencies may be left to accommodate additional intensities of ER stress in these cells [25, 96]. In comparison, normal cells without chronic ER stress may harbor greater reserves to withstand intervention with pharmacological agents aimed at aggravating the ER stress response (see below). Table 3 shows a list of compounds that are known to aggravate ER stress and have shown pro-apoptotic potency in tumor cells in vitro and in animal models in vivo.
Table 3.
Representative compounds with potency to trigger and aggravate ER stress.
| Compound | Mechanism linking to induction of ER stress | References |
|---|---|---|
| Thapsigargin, celecoxib, DMC | (i) Inhibition of SERCA activity (ii) Ensuing calcium imbalance |
[209, 246, 250, 368, 369] |
|
| ||
| A-23187, ionomycin | (i) Calcium ionophores: stimulation of Ca2+ flux (ii) Ensuing calcium imbalance |
[208, 370, 371] |
|
| ||
| Tunicamycin | (i) Glycosylation inhibitor (ii) Ensuing obstruction of protein folding |
[372, 373] |
|
| ||
| 2-Deoxyglucose | (i) Hexokinase/glycolysis inhibitor (ii) Ensuing obstruction of protein folding |
[208, 374, 375] |
|
| ||
| 2-Mercaptoethanol, Dithiothreitol | (i) Reducing agents (ii) Disruption of disulphide bonds |
[376, 377] |
|
| ||
| Geldanamycin | (i) HSP90 and GRP94 inhibitor (ii) Ensuing impairment of protein folding |
[378, 379] |
|
| ||
| Brefeldin A | (i) ADP-ribosylation factor inhibitor (ii) Impairment of protein trafficking |
[380, 381] |
|
| ||
| Bortezomib, Nelfinavir | (i) Protease and proteasome inhibitors (ii) Ensuing accumulation of terminally misfolded proteins |
[217, 219, 230, 233, 382–384] |
In the following sections, considerations for targeting the yin-yang principle of ER stress for therapeutic purposes will be presented. On one side, diabetes will be discussed as an example of a disease where the alleviation of ER stress might hold promise for therapy. On the other side, cancer will serve as an example where the opposite approach, namely, the further aggravation of preexisting ER stress, is being explored for therapeutic purposes.
5. ER Stress in Obesity and Diabetes
The pathogenesis of a number of human diseases has been associated with ER protein-folding defects and ER stress/UPR (Table 1). Perhaps the most convincing evidence for the relevance of ER stress to disease development and progression has been collected in the case of type 2 diabetes (T2D), where the combination of in vitro results, animal experiments, and human studies have established ER stress-induced β-cell failure in the pancreas as among the fundamental etiologies of this disease (see detailed refs. in reviews [31, 98–101]).
As of 2010, T2D is estimated to affect about 285 million people worldwide and represents a major cause of morbidity and mortality [102]. The disease is characterized by a complex group of metabolic conditions, including inadequate insulin secretion by pancreatic islet cells and/or peripheral insulin resistance, and dysregulated hepatic glucose production. It has been well recognized that hyperglycemia, saturated free fatty acids, and obesity in general are key risk factors for the development of T2D, and these same conditions are recognized triggers of ER stress, particularly in organs such as liver and pancreas [32, 34, 98, 103]. Progression of T2D places increased demands on pancreatic β-cells for insulin production in order to compensate for spreading insulin resistance. Augmented processing of proinsulin to insulin in the ER, combined with the increased presence of free fatty acids and glucose, is thought to trigger chronic ER stress. If these conditions are maintained for extended periods, as would be the case in obese patients and people with high-fat/high-sugar diets, chronic ER stress conditions eventually may lead to β-cell death, thus initiating a vicious cycle of exacerbated hyperglycemia [104–107]. As a consequence, the progressive decline in pancreatic β-cell function and insulin secretion causes impaired glucose tolerance and established T2D, along with reduced β-cell mass [108–111].
A large number of in vitro and in vivo studies, as well as investigations in patients with T2D and obese individuals, some of them postmortem [112, 113], have contributed to and confirmed a key contributing role of ER stress in the development of T2D. For instance, it has been shown that free fatty acids, in particular palmitate, activate the ER stress response in β-cells, as indicated by the phosphorylation of PERK and eIF2α, concomitant inhibition of protein synthesis, activation of IRE1 and ATF6, and overexpression of ATF4 and CHOP [114–119]. As well, high glucose levels have been shown to elevate several ER stress markers in cultured rat islets and β-cells [120, 121]. Phosphorylation of insulin receptor substrate-1 (IRS-1) by JNK leads to the inhibition of insulin signal transduction and contributes to peripheral insulin resistance [122]. Similarly, deficiencies in ER stress pathways, such as impairment of the PERK-eIF2α branch or overly active IRE1 signaling, impair folding capacity and insulin processing within the ER and suffice to trigger β-cell dysfunction and death [104, 123–125]. Other reports provided evidence that CHOP expression critically contributes to ER stress-induced β-cell death under conditions of increased insulin demand [119, 126–128].
Studies with genetically obese (ob/ob) or diet-induced obese mice revealed elevated levels of PERK and eIF2α phosphorylation, IRE1-mediated JNK activation, and higher amounts of GRP78 in the liver and adipose tissue of these animals, as compared to lean controls [34]. With the use of knockout cells and synthetic inhibitors, it was demonstrated that IRE1, JNK, and XBP1 significantly regulated insulin receptor signaling, thus establishing a critical role of these ER stress response components in insulin action and obesity [34]. In a subsequent paper [129], the same group of investigators provided evidence that this link between ER stress and T2D can be exploited for therapeutic purposes with orally active chemical chaperones. Two compounds in particular, 4-phenyl butyric acid (PBA) and taurine-conjugated ursodeoxycholic acid (TUDCA), which are agents with known capacity to reduce ER stress, alleviated ER stress and resulted in normalization of hyperglycemia and restoration of systemic insulin sensitivity in these obese animals, thus altogether acting as potent antidiabetic agents [100, 129].
Similar protective effects were also observed when berberine, an alkaloid that has been part of traditional Chinese medicine (TCM), was administered to diabetic rats [130]. TCM contains a number of herbs and antidiabetic formulas that are usually served as adjuvants to improve diabetic syndromes in combination with routine antidiabetic drugs [131]. Examples are ginseng, garlic, cinnamon, bitter lemon, rehmannia root, dwarf lilyturf tuber, and others. Several of these herbs have shown some potency to improve insulin sensitivity, stimulate insulin secretion, and protect pancreatic islets and have become popular complementary and alternative approaches to the treatment of T2D syndromes [132–134]. While their molecular mechanism of action remains to be established, the example of berberine could indicate that ER stress pathways might be targeted by at least some of these TCMs.
In other studies, PBA and TUDCA [135, 136], as well as fluvoxamine [137], were shown to act as leptin-sensitizing agents. Leptin is a protein that is primarily synthesized by adipose tissue, and its circulating levels are proportional to the total amount of fat in the human body. Binding of this hormone to receptors in the hypothalamus signals inhibition of food intake and thus provides an important rheostat to prevent overeating and obesity [138, 139]. In recent years, leptin resistance has been documented in the majority of the obese population and has helped to explain the difficulties of obese individuals to control food intake and body weight. Intriguingly, a number of recent papers have linked leptin resistance to ER stress, thus providing yet another aspect to the potential treatment of obesity with ER stress-modifying agents [136, 137, 140, 141].
The close dynamic relationship between ER stress and obesity-linked illnesses, such as insulin resistance and T2D, has also been established in human subjects. For example, several reports have demonstrated increased levels of multiple ER stress markers in adipose tissue of obese subjects, and these markers were significantly correlated with percent body fat and body mass index (BMI) [142, 143]. A study of obese individuals before and after weight loss through gastric bypass surgery demonstrated that the presence of ER stress markers in liver and adipose tissue was significantly diminished after weight loss and metabolic improvement [144]. Previously mentioned ER stress minimizers TUDCA and PBA were also tested in obese human subjects. In one study, TUDCA was shown to increase hepatic and muscle insulin sensitivity as compared to placebo control, although markers of ER stress in muscle or adipose tissue did not change (and liver tissue was not analyzed) [145]. Another study provided evidence that PBA may provide health benefits by ameliorating insulin resistance and β-cell dysfunction in overweight or obese subjects [146].
In summary, accumulating evidence indicates a role of chronic ER stress in the development of obesity and T2D and other metabolic diseases (Table 1). Keeping in mind that these types of diseases and their interacting mechanisms are complex, ER stress is probably only one of several contributing factors, although apparently a very important one. This raises the prospect that pharmacological interventions aimed at alleviating ER stress may provide therapeutic benefit, and therefore these possibilities are being intensively investigated. Additional studies need to explore the exact molecular mechanisms of compounds such as TUDCA or PBA, their long-term benefit and safety, and their potential interactions with commonly prescribed diabetes medications, such as the biguanide metformin and peroxisome-proliferator-activated-receptor- (PPAR-) activating thiazolidinediones.
6. ER Stress in Cancer
Cancer cells frequently deal with a set of stressful conditions that are quite different from the metabolic challenges encountered by liver cells or pancreatic β-cells in obese individuals. Cancer cells commonly encounter hostile microenvironmental conditions, such as hypoxia, hypoglycemia, and acidosis, which are fairly typical for many tumor types and which are known triggers of ER stress. However, unlike normal liver cells or pancreatic β-cells, cancer cells oftentimes have a high proliferative index, which effectively supports the selection of cell variants with genetic or phenotypic changes that enable adaptation to and survival under stressful conditions frequently documented in tumor cell lines and primary clinical samples [147–150]. Prominent among these changes is chronic activation of the protective yang module of the ER stress response system, as indicated by the presence of permanently elevated levels of pro-survival GRP78 in most tumor cells [95, 151].
In breast cancer, for example, overexpressed GRP78 is frequently detected in malignant, but not benign, breast cancer tissue and is correlated with poor prognosis for breast cancer patients [83–85]. Unfortunately for the patient, GRP78 not only protects tumor cells from the detrimental impact of a hostile microenvironment, but at the same time also provides chemoresistance. For example, in vitro studies have established that increased GRP78 levels protect cancer cells from the cytotoxic effects of several chemotherapeutic agents commonly used in the clinic, such as paclitaxel (Taxol), doxorubicin (Adriamycin), or temozolomide (Temodar) [85, 86, 151, 152]. In fact, overexpression of GRP78 in breast tumor tissue of patients has been shown to be predictive of their resistance to doxorubicin treatment [82–86]. GRP78 also confers chemoresistance to tumor-associated endothelial cells [152, 153] and supports tumor angiogenesis in mouse models of mammary tumor development [82]. Thus, the critical role of GRP78 in shielding tumor cells from suboptimal microenvironmental conditions and protecting them from chemotherapy has been well recognized [95, 154, 155].
Pro-apoptotic CHOP, located opposite to GRP78 on the yin-yang balance of ER stress (Figure 1), generally is not conspicuously expressed in tumor tissues or tumor cell lines—despite low level, chronic ER stress conditions—because the pro-survival module maintains dominance and GRP78 acts to keep CHOP transcription low [93, 94]. However, if ER stress is acutely aggravated, CHOP transcription will be strongly stimulated, and the duration and extent of this increase has been shown to represent a decisive factor in determining the cells' fate with regard to survival versus death [75, 81].
On one side, the presence of chronic ER stress and permanently elevated levels of GRP78 provides a significant survival advantage to tumor cells exposed to sub-optimal microenvironmental conditions. On the other side, this phenotype distinguishes cancer cells from most normal cells, which generally receive plenty of nutrients and oxygen and therefore do not display symptoms of chronic ER stress. Therefore, this differential may represent an opportunity for therapeutic intervention specifically aimed at the already engaged ER stress response and/or overexpressed GRP78 in cancer cells [25, 37, 156]. In particular, one could envision that tumor-specific blockage of GRP78 function and/or strong stimulation of CHOP expression might serve to provide meaningful therapeutic benefit by tilting the yin-yang balance of ER stress in favor of its pro-apoptotic module. In the following, the current practical state of this principle will be presented.
6.1. Inhibition of GRP78
Several studies have presented different methods to block GRP78 function in vitro and in vivo. For example, knockdown of GRP78 expression by antisense oligonucleotides or siRNA approaches were instrumental in establishing GRP78's pro-survival and chemoprotective roles in a variety of cell types [86, 94, 151, 152, 157, 158]. A very different approach was presented with the use of bacterial subtilase cytotoxin (SubAB), a virulence factor of several major bacterial pathogens, such as Vibrio cholerae, Shigella dysenteriae, Bordetella pertussis, and certain pathogenic strains of Escherichia coli [159]. The catalytic A subunit (SubA) of this toxin was shown to harbor protease function able to cleave GRP78 in a highly specific fashion, where no other cellular target protein could be identified [160]. In an attempt to verify its potential cancer therapeutic value, SubA was fused to epidermal growth factor (EGF) as a targeting vehicle, and this engineered fusion protein was shown to specifically kill tumor cells overexpressing EGF receptor (EGFR) in vitro and in vivo [161]. Additional studies suggested that, although GRP78 cleavage is necessary to trigger ER stress-induced cell death by SubAB, additional signaling pathways, including Akt, mTOR (mammalian target of rapamycin), MAPK (mitogen-activated protein kinase), and NF-κB might participate in these processes [162–166] (see also the following).
There is an increasing number of mostly microbial metabolites that have revealed the unique characteristic of being highly cytotoxic to tumor cells only under hypoglycemic culture conditions, that is, when glucose concentrations were lowered to less than 10% of normal or when glycolysis was blocked via the addition of the hexokinase inhibitor 2-deoxyglucose (2-DG). Examples are arctigenin [167], deoxyverrucosidin [168], efrapeptin J [169], analogs of JBIR [170], piericidin A [171], prunustatin A [172], pyrvinium [173], rottlerin [174], valinomycin [175], and versipelostatin [176]. Collectively, these compounds are considered GRP78 downregulators, because they were shown to block the adaptive induction of GRP78 transcription in response to hypoglycemia. It has been surmised that inhibition of GRP78 stimulation by these compounds prevents hypoglycemic cells from mounting their adaptive survival response, thereby leading to selective apoptosis of sugar-craving tumor cells [176]. Several biguanides (metformin, phenformin, and buformin) also were shown to belong to this group of GRP78 downregulators [177], which is intriguing in the context of metformin's use as an antidiabetic medication [178] and its recently recognized potential for cancer risk reduction [179, 180].
The isoflavone and soy ingredient genistein, as well as the polyphenolic green tea component (–)-epigallocatechin-3-gallate (EGCG), have been shown to inhibit GRP78 expression or activity, although not all studies were consistent in this regard. For example, on one hand genistein was demonstrated to block increased GRP78 transcription during ER stress, implying muted pro-survival responses [181–183], whereas other studies showed increased GRP78 expression in response to genistein, implying activation of the adaptive pro-survival response [184, 185]. However, in these latter reports pro-apoptotic CHOP expression also was greatly increased, and the overall outcome presented significantly diminished tumor cell survival despite increased amounts of GRP78, signifying dominance of the pro-apoptotic yin over the pro-survival yang ER stress module. In comparison to genistein, EGCG has not revealed transcriptional blockage of GRP78, but instead was shown to bind to and inhibit the ATPase activity of GRP78 [186], and this effect is being considered as one of the mechanisms mediating green tea's noted ability to sensitize tumor cells to chemotherapeutic treatment [86, 186, 187].
Altogether however, the previously presented prospects of selectively blocking GRP78 in future clinical applications, with the intent to achieve chemoprevention, chemosensitization, or other cancer therapeutic outcomes, are complicated by two important aspects. The first of these is presented by the recognition of oftentimes multifaceted properties of some of these GRP78-inhibitory compounds. For instance, both EGCG and genistein have revealed a multitude of other biological effects (for a few examples, see refs. [188–191] for EGCG, and refs. [192–194] for genistein), which makes it quite difficult to unequivocally ascribe any anticancer outcome to the inhibition of GRP78. Metformin also is known to affect a number of different cellular targets [179, 195], including direct inhibition of complex I of the mitochondrial respiratory chain [196, 197]. The complexity of cellular responses to some of these purported GRP78 inhibitors was further underscored by transcriptome analysis, where it was demonstrated that metformin, versipelostatin, or pyrvinium affected the expression of well over 100 different glucose-regulated genes, besides GRP78, when glucose was removed from the medium of cultured cells [177]. Therefore, it has been difficult to ascertain the specificity of many compounds reported as GRP78 inhibitors.
The second important aspect complicating the selective targeting of GRP78 arises from the emerging recognition that GRP78 is not solely an ER-luminal protein, but also can be found outside the ER and indeed appears to have functions unrelated to the ER stress response. The presence of GRP78 has been reported for the nucleus [42], mitochondria [41], cytosol [40], and the cell surface of many tumor cells [43, 46, 47, 198]. Cell surface localization in particular, where GRP78 has been found associated with important cell growth-stimulatory partner proteins, has been shown to add to GRP78's pro-survival and proliferation-stimulatory repertoire beyond its ER stress functions [44, 45, 199–202]. Therefore, in view of GRP78's varied subcellular localizations and apparently multiple functions, it has been difficult to determine whether outcomes from the inhibition of this protein can be ascribed to effects on the ER stress response or other processes where GRP78 impacts cell growth and survival. However, the application of antibodies that specifically target cell-surface GRP78 has been tremendously useful in beginning to distinguish between these possibilities and in establishing some of the major functions of cell surface GRP78 [43, 203–205]. As well, the previously mentioned SubA subunit of bacterial subtilase cytotoxin has been found to specifically cleave cell surface-localized GRP78 and therefore also represents a valuable tool in this regard [206].
It has been shown that ER stress can actively promote cell surface localization of GRP78 [46], and it is possible that the presence of chronic ER stress in tumor cells may represent a major factor for preponderant cell surface GRP78 in tumor cells. It is therefore not entirely clear whether the anticancer effects resulting from GRP78 inhibition can be, or even need to be, clearly separated into those mediated via interactions at the cell surface and those mediated via interference with the ER stress response system. For cancer therapeutic purposes, the overriding consideration would be to achieve chemosensitization and eliminate tumor growth via effective blockage of GRP78 function, no matter its localization.
6.2. Stimulation of CHOP
Based on the yin-yang principle of the ER stress response (Figure 1), in order to effect tumor cell death, the complementary tackle to blocking the yang function of GRP78 would be to further aggravate ER stress in order to accomplish a dominance of yin processes, in particular the overexpression of the pro-apoptotic master executor CHOP. In this context it is noteworthy to clarify the recently coined [207] term ER stress aggravator (ERSA), which denotes any compound capable of exacerbating preexisting ER stress (as present in most cancer cells), as opposed to “triggering” ER stress, which refers to the induction of ER stress from nonstressed conditions (which is generally the case for normal cells). Thus, the process of ER stress aggravation forms the basic principle of exploiting ER stress specifically for purposes of cancer therapy.
A large number of pharmacological compounds are known to be able to trigger or aggravate ER stress (Table 3). The classical ER stress triggers, such as thapsigargin, tunicamycin, brefeldin A, ionomycin, or mercaptoethanol have been instrumental over the past two decades in investigating and establishing the cellular processes that govern the ER stress response and UPR [208]. More recently, several clinically used drugs have been found to harbor ERSA activity, even though their original indication was entirely unrelated to ER stress. This latter group includes bortezomib (Velcade), celecoxib (Celebrex), and nelfinavir (Viracept) (see the following).
Among the classical ER stress triggers/aggravators, the sesquiterpene lactone thapsigargin is by far the most widely studied. This compound inhibits sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA), which results in massive leakage of calcium out of its ER storage compartment and severe ER stress [209, 210]. However, potential clinical use of thapsigargin is unfeasible, primarily due to its well-recognized systemic toxicity and its classification as a tumor promoter [211, 212]. Nonetheless, alternative approaches have been developed to circumvent these drawbacks. One such approach entailed the design of a chemically modified molecule, where thapsigargin was coupled to a peptide carrier that is a substrate for prostate-specific antigen (PSA) protease. In mouse models, this prodrug is specifically activated at sites of metastatic prostate cancer and exerts selective anticancer activity [213]. Another thapsigargin-based pro-drug is activated by carboxypeptidase prostate-specific membrane antigen (PSMA) and has demonstrated promising anticancer activity in several mouse xenograft tumor models in vivo, with minimal toxicity to the animals [214]. This latter molecule, called G202, currently is being tested in a phase I dose-escalation clinical trial in patients with advanced cancer in the United States.
The ERSA activities of bortezomib, celecoxib, and nelfinavir were discovered only after these drugs had been approved for clinical use. Bortezomib had been developed as a proteasome inhibitor, and based on this biological activity it was introduced to the market as a treatment of multiple myeloma (MM) and mantle cell lymphoma [215, 216]. Subsequently, several studies established that proteasome inhibition by bortezomib resulted in potent aggravation of ER stress, as indicated by greatly increased expression of ER stress markers, such as GRP78 and CHOP, in vitro and in vivo [97, 217–219]. Secretory cells, such as MM, appear to be particularly sensitive to killing via proteasome inhibition, presumably because revved-up protein synthesis places extraordinary demands on protein processing and highly active ERAD to remove terminally misfolded proteins [219–221]. As well, the key ER stress signaling component XBP-1 has been found overexpressed in MM, and its dysregulation has been implicated in MM pathogenesis [222, 223]. ER stress induced by bortezomib has been linked to blockage of NF-κB function, increased cellular sensitivity to TRAIL (tumor necrosis factor-related apoptosis-inducing ligand), and caspase-mediated apoptosis via extrinsic death receptor-initiated and intrinsic mitochondrially controlled pathways [224–228]. Thus, altogether there is good evidence that aggravated ER stress represents a central mechanism of bortezomib-induced tumor cell death and that drug-treated cells die due to proteotoxicity.
Nelfinavir had been developed as an inhibitor of HIV (human immunodeficiency virus) protease [229]. Due to its protease activity, it is thought to also block cellular proteasome activity [230, 231] and thus to elicit pro-apoptotic activities similar to bortezomib, as indicated by the accumulation of polyubiquitinated proteins, increased expression of ER stress markers GRP78 and CHOP, and caspase activation [232–235]. The discovery of nelfinavir's ERSA activity in vitro and in mouse tumor models in vivo [232, 233] and its potential for chemosensitization [236–238] and radiosensitization [239–241], all of which were characterized a decade after the drug's approval for the treatment of HIV infections, have spurred efforts to repurpose this drug for cancer therapeutic purposes [242, 243]. In the United States, a number of clinical trials are ongoing to establish and verify nelfinavir's usefulness for inclusion in cancer therapeutic regimens.
Celecoxib belongs to the class of nonsteroidal anti-inflammatory drugs (NSAIDs) and had been developed as a selective inhibitor of cyclooxygenase 2 (COX-2). It has been approved for the treatment of inflammatory conditions and pain and as an adjunct for the therapy of familial adenomatous polyposis (FAP) [244]. Subsequently, however, several additional biological activities and targets of this drug emerged [245–249]. Prime among these was its potency to inhibit the ER calcium pump SERCA, thus aggravating ER stress in a manner similar to thapsigargin [250]. Indeed, a number of reports have demonstrated that calcium release from the ER is the most immediate effect of celecoxib treatment and can be detected within seconds of adding the drug to cells in culture [251–254]. As well, celecoxib-treated cells display transiently blocked general protein translation via PERK-mediated phosphorylation of eIF2α [255], along with greatly increased expression of ER stress markers GRP78 and CHOP in vitro and in tumor tissues of drug-treated animals in vivo [97, 252–254].
Quite intriguingly, it could be demonstrated that celecoxib's main feature, that is, the ability to block COX-2 activity, can be separated from this drug's cytotoxic potency, and a number of structurally related analogs of celecoxib were created that displayed only one or the other function [256, 257]. For example, 2,5-dimethyl-celecoxib (DMC) was shown to have lost the ability to block COX-2, yet it fully preserved cytotoxic potency [97, 252, 257–262]. Similar outcomes were reported with several other celecoxib analogs, such as TT101 [263, 264], CEA [265, 266], and OSU-03012 [256, 266–269]. Conversely, unmethylated celecoxib (UMC) represents an analog with further increased COX-2-inhibitory potency, but much decreased cytotoxic efficacy [270, 271]. Altogether, these reports clearly established that celecoxib's COX-2-inhibitory function was neither able to nor required to induce cytotoxic outcomes, but that COX-2-independent features of the molecule were responsible. Indeed, a series of experiments with celecoxib and celecoxib analogs demonstrated that these compounds' apoptosis-inducing and anticancer effects were most closely aligned with their ability to trigger ER stress via inhibition of SERCA activity [97, 252, 253, 265, 270–274]. A causal relationship to ER stress was confirmed with knockdown experiments, where siRNA to GRP78 increased and siRNA to CHOP decreased cell death in response to treatment of cells with celecoxib or its analogs [97, 252–254, 275].
In summary, bortezomib, nelfinavir, and celecoxib are firmly established medications, and their anticancer activity appears to involve, at least in part, aggravation of ER stress. In the case of bortezomib, the optimized pharmacological function is inhibition of the proteasome, which directly relates to ER stress aggravation. Therefore, proteasome-inhibitory activity and ERSA activity emerge in a similar concentration range of the drug, and bortezomib's classification as an ERSA originates from its proteasome-inhibitory activity. However, this is not the case with celecoxib. Here, the optimized pharmacological function is inhibition of COX-2, and ERSA activity merely represents a secondary, COX-2-independent function that requires substantially higher drug concentrations than inhibition of COX-2. It might be for this reason that celecoxib, although efficacious for chemoprevention of colon cancer [276–278], has not revealed impressive anticancer effects when included in therapeutic regimens aimed at advanced cancer types in humans [279–287]. Nonetheless, extensive work with celecoxib analogs, in particular DMC, has revealed that ERSA activity within the celecoxib molecule can be further optimized, as indeed DMC exerts significantly stronger ERSA activity, along with greater cytotoxic efficacy, than its parental compound [97, 252, 270, 271]. As such, DMC and similar compounds represent promising cancer therapeutic drug candidates aimed at the exploitation of ER stress in tumor cells. While phase I clinical trials with OSU-03012 have been initiated, other celecoxib analogs have not yet been considered for clinical investigations.
7. Perspective
The ER stress response system/UPR features yin-yang principles, characterized by the struggle for dominance between pro-survival and pro-apoptotic modules and their most prominent components, GRP78 and CHOP, respectively. There is now compelling evidence that chronic ER stress and concomitant activation of one or more branches of the UPR are important in the pathogenesis of a number of diseases. As an illustrative example, the link between high-fat/high-sugar diet, ER stress, and dysregulation of pancreatic β-cell function has been presented previously. In such cases, therapeutics aimed at ameliorating ER stress by promoting proper protein processing and generally supporting proper ER maintenance may prove useful for prevention and/or therapy. On the other hand, chronic ER stress in cancer may be exploited therapeutically via the opposite approach, namely, via the selective pharmacological aggravation of the ER stress condition in tumor cells.
A detailed understanding of the consequences of pharmacological interference with ER stress responses in a patient is necessary in order to translate the respective approaches into therapeutic opportunities. Here, our knowledge is still far from complete, and more research is urgently needed. Among the principal challenges is the identification of which ER stress signaling module(s) represent(s) the most promising target in each of the diverse diseases that have been linked to ER stress. As well, future studies should investigate and evaluate the requirements for acute versus long-term medical interventions, potential side effects of such approaches, their interference with other cellular processes and signaling pathways, and possible crosstalk with inflammation and metabolism in general.
Conflict of Interests
The author has no financial interest or relation with the commercial identities mentioned in this paper.
Acknowledgments
The author would like to thank his colleagues Thomas C. Chen, Florence M. Hofman, Stan G. Louie, and Nicos A. Petasis for constructive discussions and productive collaborations and former and current members of his laboratory for their dedication and research efforts.
Abbreviations
- ATF:
Activating transcription factor
- CHOP:
CCAAT/enhancer binding protein homologous protein (also called GADD153)
- 2-DG:
2-deoxy-D-glucose
- DMC:
2,5-dimethyl-celecoxib
- DR5:
Death receptor 5
- ER:
Endoplasmic reticulum
- ERAD:
Endoplasmic reticulum-associated degradation
- ERSA:
Endoplasmic reticulum stress aggravator
- GRP78:
Glucose regulated protein of molecular weight 78 (also called BiP)
- IRE1:
Inositol-requiring enzyme
- NF-κB:
Nuclear factor kappa B
- 4-PBA:
4-phenyl butyric acid
- PERK:
Protein kinase activated by double-stranded RNA (PKR)-like ER kinase (also called pancreatic ER kinase)
- SERCA:
Sarcoplasmic/endoplasmic reticulum calcium ATPase
- TRAIL:
Tumor necrosis factor-related apoptosis-inducing ligand
- TUDCA:
Tauroursodeoxycholic acid.
References
- 1.Alberts B, Johnson A, Lewis J, et al. Molecular Biology of the Cell. 5th edition. New York, NY, USA: Garland Science; 2008. [Google Scholar]
- 2.Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Seminars in Cell and Developmental Biology. 2007;18(6):716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature Reviews Molecular Cell Biology. 2007;8(7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 4.Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biology. 2006;4(12, article e423) doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding WX, Ni HM, Gao W, et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. Journal of Biological Chemistry. 2007;282(7):4702–4710. doi: 10.1074/jbc.M609267200. [DOI] [PubMed] [Google Scholar]
- 6.Ogata M, Hino SI, Saito A, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Molecular and Cellular Biology. 2006;26(24):9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. Journal of Biological Chemistry. 2006;281(40):30299–30304. doi: 10.1074/jbc.M607007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yin JJ, Li YB, Wang Y, et al. The role of autophagy in endoplasmic reticulum stressinduced pancreatic beta cell death. Autophagy. 2012;8:158–164. doi: 10.4161/auto.8.2.18807. [DOI] [PubMed] [Google Scholar]
- 9.Schleicher SM, Moretti L, Varki V, Lu B. Progress in the unraveling of the endoplasmic reticulum stress/autophagy pathway and cancer: implications for future therapeutic approaches. Drug Resistance Updates. 2010;13(3):79–86. doi: 10.1016/j.drup.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 10.White EJ, Martin V, Liu JL, et al. Autophagy regulation in cancer development and therapy. American Journal of Cancer Research. 2011;1:362–372. [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou S, Zhao L, Kuang M, et al. Autophagy in tumorigenesis and cancer therapy: Dr. Jekyll or Mr. Hyde? Cancer Letters. 2012;323:115–127. doi: 10.1016/j.canlet.2012.02.017. [DOI] [PubMed] [Google Scholar]
- 12.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nature Reviews Drug Discovery. 2012;11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartley T, Siva M, Lai E, Teodoro T, Zhang L, Volchuk A. Endoplasmic reticulum stress response in an INS-1 pancreatic β-cell line with inducible expression of a folding-deficient proinsulin. BMC Cell Biology. 2010;11, article 59 doi: 10.1186/1471-2121-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raposo G, van Santen HM, Leijendekker R, Geuze HJ, Ploegh HL. Misfolded major histocompatibility complex class I molecules accumulate in an expanded ER-Golgi intermediate compartment. Journal of Cell Biology. 1995;131(6 I):1403–1419. doi: 10.1083/jcb.131.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Welihinda AA, Tirasophon W, Kaufman RJ. The cellular response to protein misfolding in the endoplasmic reticulum. Gene Expression. 1999;7(4–6):293–300. [PMC free article] [PubMed] [Google Scholar]
- 16.Zuber C, Fan JY, Guhl B, Roth J. Misfolded proinsulin accumulates in expanded pre-Golgi intermediates and endoplasmic reticulum subdomains in pancreatic beta cells of Akita mice. The FASEB Journal. 2004;18(7):917–919. doi: 10.1096/fj.03-1210fje. [DOI] [PubMed] [Google Scholar]
- 17.Despa F. Dilation of the endoplasmic reticulum in beta cells due to molecular overcrowding? Kinetic simulations of extension limits and consequences on proinsulin synthesis. Biophysical Chemistry. 2009;140(1–3):115–121. doi: 10.1016/j.bpc.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Haas IG, Wabl M. Immunoglobulin heavy chain binding protein. Nature. 1983;306(5941):387–389. doi: 10.1038/306387a0. [DOI] [PubMed] [Google Scholar]
- 19.Munro S, Pelham HRB. An hsp70-like protein in the ER: identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell. 1986;46(2):291–300. doi: 10.1016/0092-8674(86)90746-4. [DOI] [PubMed] [Google Scholar]
- 20.Csala M, Kereszturi E, Mandl J, Banhegyi G. The endoplasmic reticulum as the extracellular space inside the cell: role in protein folding and glycosylation. Antioxidants and Redox Signaling. 2012;16:1100–1108. doi: 10.1089/ars.2011.4227. [DOI] [PubMed] [Google Scholar]
- 21.Roth J, Zuber C, Park S, et al. Protein N-glycosylation, protein folding, and protein quality control. Molecules and Cells. 2010;30(6):497–506. doi: 10.1007/s10059-010-0159-z. [DOI] [PubMed] [Google Scholar]
- 22.Hagiwara M, Nagata K. Redox-dependent protein quality control in the endoplasmic reticulum: folding to degradation. Antioxidants and Redox Signaling. 2012;16:1119–1128. doi: 10.1089/ars.2011.4495. [DOI] [PubMed] [Google Scholar]
- 23.Ma Y, Hendershot LM. ER chaperone functions during normal and stress conditions. Journal of Chemical Neuroanatomy. 2004;28(1-2):51–65. doi: 10.1016/j.jchemneu.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 24.Krebs J, Groenendyk J, Michalak M. Ca2+-signaling, alternative splicing and endoplasmic reticulum stress responses. Neurochemical Research. 2011;36(7):1198–1211. doi: 10.1007/s11064-011-0431-4. [DOI] [PubMed] [Google Scholar]
- 25.Schönthal AH. Endoplasmic reticulum stress and autophagy as targets for cancer therapy. Cancer Letters. 2009;275(2):163–169. doi: 10.1016/j.canlet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 26.Hideshima T, Bradner JE, Wong J, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(24):8567–8572. doi: 10.1073/pnas.0503221102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115(6):727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 28.von dem Bussche A, Machida R, Li K, et al. Hepatitis C virus NS2 protein triggers endoplasmic reticulum stress and suppresses its own viral replication. Journal of Hepatology. 2010;53:797–804. doi: 10.1016/j.jhep.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death and Differentiation. 2006;13(3):393–403. doi: 10.1038/sj.cdd.4401833. [DOI] [PubMed] [Google Scholar]
- 30.Roy CR, Salcedo SP, Gorvel JPE. Pathogen-endoplasmic-reticulum interactions: in through the out door. Nature Reviews Immunology. 2006;6(2):136–147. doi: 10.1038/nri1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annual Review of Biochemistry. 2012;81:767–793. doi: 10.1146/annurev-biochem-072909-095555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dara L, Ji C, Kaplowitz N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology. 2011;53(5):1752–1763. doi: 10.1002/hep.24279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fu S, Watkins SM, Hotamisligil GS. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metabolism. 2012;15:623–634. doi: 10.1016/j.cmet.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 34.Özcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 35.Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends in Biochemical Sciences. 2001;26(8):504–510. doi: 10.1016/s0968-0004(01)01908-9. [DOI] [PubMed] [Google Scholar]
- 36.Lee AS, Delegeane A, Scharff D. Highly conserved glucose-regulated protein in hamster and chicken cells: preliminary characterization of its cDNA clone. Proceedings of the National Academy of Sciences of the United States of America. 1981;78(8 I):4922–4925. doi: 10.1073/pnas.78.8.4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Healy SJM, Gorman AM, Mousavi-Shafaei P, Gupta S, Samali A. Targeting the endoplasmic reticulum-stress response as an anticancer strategy. European Journal of Pharmacology. 2009;625(1–3):234–246. doi: 10.1016/j.ejphar.2009.06.064. [DOI] [PubMed] [Google Scholar]
- 38.Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. doi: 10.1038/onc.2012.130. Oncogene. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang LH, Zhang X. Roles of GRP78 in physiology and cancer. Journal of Cellular Biochemistry. 2010;110(6):1299–1305. doi: 10.1002/jcb.22679. [DOI] [PubMed] [Google Scholar]
- 40.Ni M, Zhou H, Wey S, Baumeister P, Lee AS. Regulation of PERK signaling and leukemic cell survival by a novel cytosolic isoform of the UPR regulator GRP78/BiP. PLoS ONE. 2009;4(8) doi: 10.1371/journal.pone.0006868.e6868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun FC, Wei S, Li CW, Chang YS, Chao CC, Lai YK. Localization of GRP78 to mitochondria under the unfolded protein response. Biochemical Journal. 2006;396(1):31–39. doi: 10.1042/BJ20051916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matsumoto A, Hanawalt PC. Histone H3 and heat shock protein GRP78 are selectively cross-linked to DNA by photoactivated gilvocarcin V in human fibroblasts. Cancer Research. 2000;60(14):3921–3926. [PubMed] [Google Scholar]
- 43.Arap MA, Lahdenranta J, Mintz PJ, et al. Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell. 2004;6(3):275–284. doi: 10.1016/j.ccr.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 44.Misra UK, Deedwania R, Pizzo SV. Activation and cross-talk between Akt, NF-κB, and unfolded protein response signaling in 1-LN prostate cancer cells consequent to ligation of cell surface-associated GRP78. Journal of Biological Chemistry. 2006;281(19):13694–13707. doi: 10.1074/jbc.M511694200. [DOI] [PubMed] [Google Scholar]
- 45.Shani G, Fischer WH, Justice NJ, Kelber JA, Vale W, Gray PC. GRP78 and cripto form a complex at the cell surface and collaborate to inhibit transforming growth factor β signaling and enhance cell growth. Molecular and Cellular Biology. 2008;28(2):666–677. doi: 10.1128/MCB.01716-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Liu R, Ni M, Gill P, Lee AS. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. Journal of Biological Chemistry. 2010;285(20):15065–15075. doi: 10.1074/jbc.M109.087445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y, Steiniger SCJ, Kim Y, Kaufmann GF, Felding-Habermann B, Janda KD. Mechanistic studies of a peptidic GRP78 ligand for cancer cell-specific drug delivery. Molecular Pharmaceutics. 2007;4(3):435–447. doi: 10.1021/mp060122j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochemical Journal. 2011;434(2):181–188. doi: 10.1042/BJ20101569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ron D, Hubbard SR. How IRE1 reacts to ER stress. Cell. 2008;132(1):24–26. doi: 10.1016/j.cell.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 50.Fels DR, Koumenis C. The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biology and Therapy. 2006;5:723–728. doi: 10.4161/cbt.5.7.2967. [DOI] [PubMed] [Google Scholar]
- 51.Sommer T, Jarosch E. BiP binding keeps ATF6 at bay. Developmental Cell. 2002;3(1):1–2. doi: 10.1016/s1534-5807(02)00210-1. [DOI] [PubMed] [Google Scholar]
- 52.Parmar VM, Schroder M. Sensing endoplasmic reticulum stress. Advances in Experimental Medicine and Biology. 2012;738:153–168. doi: 10.1007/978-1-4614-1680-7_10. [DOI] [PubMed] [Google Scholar]
- 53.Patil C, Walter P. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Current Opinion in Cell Biology. 2001;13(3):349–355. doi: 10.1016/s0955-0674(00)00219-2. [DOI] [PubMed] [Google Scholar]
- 54.Kohno K. Stress-sensing mechanisms in the unfolded protein response: similarities and differences between yeast and mammals. Journal of Biochemistry. 2010;147(1):27–33. doi: 10.1093/jb/mvp196. [DOI] [PubMed] [Google Scholar]
- 55.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21(1):81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 56.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-I. Nature Immunology. 2003;4(4):321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 57.Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annual Review of Biochemistry. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- 58.Jäger R, Bertrand MJ, Gorman AM, et al. The unfolded protein response at the crossroads of cellular life and death during endoplasmic reticulum stress. Biology of the Cell. 2012;104:259–270. doi: 10.1111/boc.201100055. [DOI] [PubMed] [Google Scholar]
- 59.Nishitoh H, Matsuzawa A, Tobiume K, et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes and Development. 2002;16(11):1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 61.Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27(48):6245–6251. doi: 10.1038/onc.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim R, Emi M, Tanabe K, Murakami S. Role of the unfolded protein response in cell death. Apoptosis. 2006;11(1):5–13. doi: 10.1007/s10495-005-3088-0. [DOI] [PubMed] [Google Scholar]
- 63.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Molecular Biology of the Cell. 1999;10(11):3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamamoto K, Sato T, Matsui T, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Developmental Cell. 2007;13(3):365–376. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 65.Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, Mori K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Structure and Function. 2008;33(1):75–89. doi: 10.1247/csf.07044. [DOI] [PubMed] [Google Scholar]
- 66.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 67.Bailey D, O’Hare P. Transmembrane bZIP transcription factors in ER stress signaling and the unfolded protein response. Antioxidants and Redox Signaling. 2007;9(12):2305–2321. doi: 10.1089/ars.2007.1796. [DOI] [PubMed] [Google Scholar]
- 68.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Molecular Cell. 2000;5(5):897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 69.Nishitoh H. CHOP is a multifunctional transcription factor in the ER stress response. Journal of Biochemistry. 2012;151:217–219. doi: 10.1093/jb/mvr143. [DOI] [PubMed] [Google Scholar]
- 70.Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. Journal of Biological Chemistry. 2004;279(19):20108–20117. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]
- 71.Cullinan SB, Diehl JA. Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. International Journal of Biochemistry and Cell Biology. 2006;38(3):317–332. doi: 10.1016/j.biocel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 72.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Molecular and Cellular Biology. 2003;23(20):7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Price BD, Calderwood SK. Gadd45 and gadd153 messenger RNA levels are increased during hypoxia and after exposure of cells to agents which elevate the levels of the glucose- regulated proteins. Cancer Research. 1992;52(13):3814–3817. [PubMed] [Google Scholar]
- 74.Fornace AJ, Jr., Nebert DW, Hollander MC, et al. Mammalian genes coordinately regulated by growth arrest signals and DNA-damaging agents. Molecular and Cellular Biology. 1989;9(10):4196–4203. doi: 10.1128/mcb.9.10.4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bc12 and perturbing the cellular redox state. Molecular and Cellular Biology. 2001;21(4):1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Puthalakath H, O’Reilly LA, Gunn P, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129(7):1337–1349. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 77.Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. Journal of Hepatology. 2011;54(4):795–809. doi: 10.1016/j.jhep.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. Journal of Biological Chemistry. 2004;279(44):45495–45502. doi: 10.1074/jbc.M406933200. [DOI] [PubMed] [Google Scholar]
- 79.Kojima E, Takeuchi A, Haneda M, et al. The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: elucidation by GADD34-deficient mice. The FASEB Journal. 2003;17(11):1573–1575. doi: 10.1096/fj.02-1184fje. [DOI] [PubMed] [Google Scholar]
- 80.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α . Journal of Cell Biology. 2001;153(5):1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rutkowski DT, Arnold SM, Miller CN, et al. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biology. 2006;4(11, article e374) doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dong D, Ni M, Li J, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Research. 2008;68(2):498–505. doi: 10.1158/0008-5472.CAN-07-2950. [DOI] [PubMed] [Google Scholar]
- 83.Fernandez PM, Tabbara SO, Jacobs LK, et al. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Research and Treatment. 2000;59(1):15–26. doi: 10.1023/a:1006332011207. [DOI] [PubMed] [Google Scholar]
- 84.Gazit G, Lu J, Lee AS. De-regulation of GRP stress protein expression in human breast cancer cell lines. Breast Cancer Research and Treatment. 1999;54(2):135–146. doi: 10.1023/a:1006102411439. [DOI] [PubMed] [Google Scholar]
- 85.Lee E, Nichols P, Spicer D, Groshen S, Yu MC, Lee AS. GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Research. 2006;66(16):7849–7853. doi: 10.1158/0008-5472.CAN-06-1660. [DOI] [PubMed] [Google Scholar]
- 86.Wang J, Yin Y, Hua H, et al. Blockade of GRP78 sensitizes breast cancer cells to microtubules-interfering agents that induce the unfolded protein response. Journal of Cellular and Molecular Medicine. 2009;13(9 B):3888–3897. doi: 10.1111/j.1582-4934.2009.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Verma G, Datta M. The critical role of JNK in the ER-mitochondrial crosstalk during apoptotic cell death. Journal of Cellular Physiology. 2012;227:1791–1795. doi: 10.1002/jcp.22903. [DOI] [PubMed] [Google Scholar]
- 88.Kim I, Shu CW, Xu W, et al. Chemical biology investigation of cell death pathways activated by endoplasmic reticulum stress reveals cytoprotective modulators of ASK1. Journal of Biological Chemistry. 2009;284(3):1593–1603. doi: 10.1074/jbc.M807308200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hetz C, Glimcher LH. Fine-tuning of the unfolded protein response: assembling the IRE1α interactome. Molecular Cell. 2009;35(5):551–561. doi: 10.1016/j.molcel.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shore GC, Papa FR, Oakes SA. Signaling cell death from the endoplasmic reticulum stress response. Current Opinion in Cell Biology. 2011;23(2):143–149. doi: 10.1016/j.ceb.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death and Differentiation. 2004;11(4):381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 92.Hitomi J, Katayama T, Eguchi Y, et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. Journal of Cell Biology. 2004;165(3):347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death and Differentiation. 2006;13(3):363–373. doi: 10.1038/sj.cdd.4401817. [DOI] [PubMed] [Google Scholar]
- 94.Suzuki T, Lu J, Zahed M, Kita K, Suzuki N. Reduction of GRP78 expression with siRNA activates unfolded protein response leading to apoptosis in HeLa cells. Archives of Biochemistry and Biophysics. 2007;468(1):1–14. doi: 10.1016/j.abb.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 95.Lee AS. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Research. 2007;67(8):3496–3499. doi: 10.1158/0008-5472.CAN-07-0325. [DOI] [PubMed] [Google Scholar]
- 96.Schönthal AH, Chen TC, Hofman FM, et al. Preclinical development of novel antitumor drugs targeting the endoplasmic reticulum stress response. Current Pharmaceutical Design. 2011;17:2428–2438. doi: 10.2174/138161211797249242. [DOI] [PubMed] [Google Scholar]
- 97.Kardosh A, Golden EB, Pyrko P, et al. Aggravated endoplasmic reticulum stress as a basis for enhanced glioblastoma cell killing by bortezomib in combination with celecoxib or its non-coxib analogue, 2,5-dimethyl-celecoxib. Cancer Research. 2008;68(3):843–851. doi: 10.1158/0008-5472.CAN-07-5555. [DOI] [PubMed] [Google Scholar]
- 98.Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends in Molecular Medicine. 2012;18:59–68. doi: 10.1016/j.molmed.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 99.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with β-cell failure and diabetes. Endocrine Reviews. 2008;29(3):317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ozcan L, Tabas I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annual Review of Medicine. 2012;63:317–328. doi: 10.1146/annurev-med-043010-144749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocrine Reviews. 2008;29(1):42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- 102.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Research and Clinical Practice. 2010;87(1):4–14. doi: 10.1016/j.diabres.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 103.Gentile CL, Frye M, Pagliassotti MJ. Endoplasmic reticulum stress and the unfolded protein response in nonalcoholic fatty liver disease. Antioxidants and Redox Signaling. 2011;15(2):505–521. doi: 10.1089/ars.2010.3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cnop M, Vidal J, Hull RL, et al. Progressive loss of β-cell function leads to worsening glucose tolerance in first-degree relatives of subjects with type 2 diabetes. Diabetes Care. 2007;30(3):677–682. doi: 10.2337/dc06-1834. [DOI] [PubMed] [Google Scholar]
- 105.Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54(supplement 2):S97–S107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- 106.Festa A, Williams K, D’Agostino R, Jr., Wagenknecht LE, Haffner SM. The natural course of β-cell function in nondiabetic and diabetic individuals: the insulin resistance atherosclerosis study. Diabetes. 2006;55(4):1114–1120. doi: 10.2337/diabetes.55.04.06.db05-1100. [DOI] [PubMed] [Google Scholar]
- 107.Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. Journal of Clinical Investigation. 1999;104(6):787–794. doi: 10.1172/JCI7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sakuraba H, Mizukami H, Yagihashi N, Wada R, Hanyu C, Yagihashi S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese type II diabetic patients. Diabetologia. 2002;45(1):85–96. doi: 10.1007/s125-002-8248-z. [DOI] [PubMed] [Google Scholar]
- 109.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 110.Yoon KH, Ko SH, Cho JH, et al. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. The Journal of Clinical Endocrinology & Metabolism. 2003;88:2300–2308. doi: 10.1210/jc.2002-020735. [DOI] [PubMed] [Google Scholar]
- 111.Prentki M, Nolan CJ. Islet β cell failure in type 2 diabetes. Journal of Clinical Investigation. 2006;116(7):1802–1812. doi: 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huang CJ, Lin CY, Haataja L, et al. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress-mediated β-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes. 2007;56(8):2016–2027. doi: 10.2337/db07-0197. [DOI] [PubMed] [Google Scholar]
- 113.Costes S, Huang CJ, Gurlo T, et al. β-cell dysfunctional ERAD/ubiquitin/proteasome system in type 2 diabetes mediated by islet amyloid polypeptide-induced UCH-L1 deficiency. Diabetes. 2011;60(1):227–238. doi: 10.2337/db10-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cnop M, Ladriere L, Hekerman P, et al. Selective inhibition of eukaryotic translation initiation factor 2α dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic β-cell dysfunction and apoptosis. Journal of Biological Chemistry. 2007;282(6):3989–3997. doi: 10.1074/jbc.M607627200. [DOI] [PubMed] [Google Scholar]
- 115.Karaskov E, Scott C, Zhang L, Teodoro T, Ravazzola M, Volchuk A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic β-cell apoptosis. Endocrinology. 2006;147(7):3398–3407. doi: 10.1210/en.2005-1494. [DOI] [PubMed] [Google Scholar]
- 116.Kharroubi I, Ladrière L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL. Free fatty acids and cytokines induce pancreatic β-cell apoptosis by different mechanisms: role of nuclear factor-κB and endoplasmic reticulum stress. Endocrinology. 2004;145(11):5087–5096. doi: 10.1210/en.2004-0478. [DOI] [PubMed] [Google Scholar]
- 117.Komiya K, Uchida T, Ueno T, et al. Free fatty acids stimulate autophagy in pancreatic β-cells via JNK pathway. Biochemical and Biophysical Research Communications. 2010;401(4):561–567. doi: 10.1016/j.bbrc.2010.09.101. [DOI] [PubMed] [Google Scholar]
- 118.Kuo TF, Tatsukawa H, Matsuura T, et al. Free fatty acids induce transglutaminase 2-dependent apoptosis in hepatocytes via ER stress-stimulated PERK pathways. Journal of Cellular Physiology. 2012;227:1130–1137. doi: 10.1002/jcp.22833. [DOI] [PubMed] [Google Scholar]
- 119.Laybutt DR, Preston AM, Åkerfeldt MC, et al. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50(4):752–763. doi: 10.1007/s00125-006-0590-z. [DOI] [PubMed] [Google Scholar]
- 120.Elouil H, Bensellam M, Guiot Y, et al. Acute nutrient regulation of the unfolded protein response and integrated stress response in cultured rat pancreatic islets. Diabetologia. 2007;50(7):1442–1452. doi: 10.1007/s00125-007-0674-4. [DOI] [PubMed] [Google Scholar]
- 121.Hou ZQ, Li HL, Gao L, Pan L, Zhao JJ, Li GW. Involvement of chronic stresses in rat islet and INS-1 cell glucotoxicity induced by intermittent high glucose. Molecular and Cellular Endocrinology. 2008;291(1-2):71–78. doi: 10.1016/j.mce.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 122.Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. Journal of Biological Chemistry. 2002;277(2):1531–1537. doi: 10.1074/jbc.M101521200. [DOI] [PubMed] [Google Scholar]
- 123.Harding HP, Zeng H, Zhang Y, et al. Diabetes mellitus and exocrine pancreatic dysfunction in Perk-/- mice reveals a role for translational control in secretory cell survival. Molecular Cell. 2001;7(6):1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 124.Lipson KL, Fonseca SG, Ishigaki S, et al. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metabolism. 2006;4(3):245–254. doi: 10.1016/j.cmet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 125.Scheuner D, Mierde DV, Song B, et al. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nature Medicine. 2005;11(7):757–764. doi: 10.1038/nm1259. [DOI] [PubMed] [Google Scholar]
- 126.Maris M, Overbergh L, Gysemans C, et al. Deletion of C/EBP homologous protein (Chop) in C57Bl/6 mice dissociates obesity from insulin resistance. Diabetologia. 2012;55:1167–1178. doi: 10.1007/s00125-011-2427-7. [DOI] [PubMed] [Google Scholar]
- 127.Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. Journal of Clinical Investigation. 2008;118(10):3378–3389. doi: 10.1172/JCI34587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.McKimpson WM, Weinberger J, Czerski L, et al. The apoptosis inhibitor ARC alleviates the ER stress response to promote beta-cell survival. doi: 10.2337/db12-0504. Diabetes. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Özcan U, Yilmaz E, Özcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313(5790):1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhou J, Zhou S, Tang J, et al. Protective effect of berberine on beta cells in streptozotocin- and high-carbohydrate/high-fat diet-induced diabetic rats. European Journal of Pharmacology. 2009;606(1–3):262–268. doi: 10.1016/j.ejphar.2008.12.056. [DOI] [PubMed] [Google Scholar]
- 131.Jia W, Gaoz W, Tang L. Antidiabetic herbal drugs officially approved in China. Phytotherapy Research. 2003;17(10):1127–1134. doi: 10.1002/ptr.1398. [DOI] [PubMed] [Google Scholar]
- 132.Xie W, Zhao Y, Zhang Y. Traditional chinese medicines in treatment of patients with type 2 diabetes mellitus. Evidence-Based Complementary and Alternative Medicine. 2011;2011:13 pages. doi: 10.1155/2011/726723.726723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yin J, Zhang H, Ye J. Traditional Chinese medicine in treatment of metabolic syndrome. Endocrine, Metabolic and Immune Disorders. 2008;8(2):99–111. doi: 10.2174/187153008784534330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tong XL, Dong L, Chen L, Zhen Z. Treatment of diabetes using traditional chinese medicine: past, present and future. The American Journal of Chinese Medicine. 2012;40:877–886. doi: 10.1142/S0192415X12500656. [DOI] [PubMed] [Google Scholar]
- 135.Ozcan L, Ergin AS, Lu A, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metabolism. 2009;9(1):35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 136.Won JC, Jang PG, Namkoong C, et al. Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity. 2009;17(10):1861–1865. doi: 10.1038/oby.2009.194. [DOI] [PubMed] [Google Scholar]
- 137.Hosoi T, Miyahara T, Kayano T, et al. Fluvoxamine attenuated endoplasmic reticulum stress-induced leptin resistance. Frontiers in Endocrinology. 2012;3, article 12 doi: 10.3389/fendo.2012.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Coppari R, Bjorbaek C. Leptin revisited: its mechanism of action and potential for treating diabetes. Nature Reviews Drug Discovery. 2012;11:692–708. doi: 10.1038/nrd3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Konner AC, Bruning JC. Selective insulin and leptin resistance in metabolic disorders. Cell Metabolism. 2012;16:144–152. doi: 10.1016/j.cmet.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 140.Hosoi T, Sasaki M, Miyahara T, et al. Endoplasmic reticulum stress induces leptin resistance. Molecular Pharmacology. 2008;74(6):1610–1619. doi: 10.1124/mol.108.050070. [DOI] [PubMed] [Google Scholar]
- 141.Marwarha G, Dasari B, Ghribi O. Endoplasmic reticulum stress-induced CHOP activation mediates the down-regulation of leptin in human neuroblastoma SH-SY5Y cells treated with the oxysterol 27-hydroxycholesterol. Cell Signaling. 2012;24:484–492. doi: 10.1016/j.cellsig.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Boden G, Duan X, Homko C, et al. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes. 2008;57(9):2438–2444. doi: 10.2337/db08-0604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sharma NK, Das SK, Mondal AK, et al. Endoplasmic reticulum stress markers are associated with obesity in nondiabetic subjects. Journal of Clinical Endocrinology and Metabolism. 2008;93(11):4532–4541. doi: 10.1210/jc.2008-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gregor MF, Yang L, Fabbrini E, et al. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes. 2009;58(3):693–700. doi: 10.2337/db08-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kars M, Yang L, Gregor MF, et al. Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes. 2010;59(8):1899–1905. doi: 10.2337/db10-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Xiao C, Giacca A, Lewis GF. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and β-cell dysfunction in humans. Diabetes. 2011;60(3):918–924. doi: 10.2337/db10-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Christofk HR, vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452(7184):230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 148.Hsu PP, Sabatini DM. Cancer cell metabolism: warburg and beyond. Cell. 2008;134(5):703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 149.Prindle MJ, Fox EJ, Loeb LA. The mutator phenotype in cancer: molecular mechanisms and targeting strategies. Current Drug Targets. 2010;11(10):1296–1303. doi: 10.2174/1389450111007011296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shaw RJ. Glucose metabolism and cancer. Current Opinion in Cell Biology. 2006;18:598–608. doi: 10.1016/j.ceb.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 151.Pyrko P, Schöntha AH, Hofman FM, Chen TC, Lee AS. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Research. 2007;67(20):9809–9816. doi: 10.1158/0008-5472.CAN-07-0625. [DOI] [PubMed] [Google Scholar]
- 152.Virrey JJ, Dong D, Stiles C, et al. Stress chaperone GRP78/BiP confers chemoresistance to tumor-associated endothelial cells. Molecular Cancer Research. 2008;6(8):1268–1275. doi: 10.1158/1541-7786.MCR-08-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dong D, Ko B, Baumeister P, et al. Vascular targeting and antiangiogenesis agents induce drug resistance effector GRP78 within the tumor microenvironment. Cancer Research. 2005;65(13):5785–5791. doi: 10.1158/0008-5472.CAN-05-0754. [DOI] [PubMed] [Google Scholar]
- 154.Fu Y, Lee AS. Glucose regulated proteins in cancer progression, drug resistance and immunotherapy. Cancer Biology and Therapy. 2006;5(7):741–744. doi: 10.4161/cbt.5.7.2970. [DOI] [PubMed] [Google Scholar]
- 155.Li J, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Current Molecular Medicine. 2006;6(1):45–54. doi: 10.2174/156652406775574523. [DOI] [PubMed] [Google Scholar]
- 156.de Ridder G, Ray R, Misra UK, Pizzo SV. Modulation of the unfolded protein response by GRP78 in prostate cancer. Methods in Enzymology. 2011;489:245–257. doi: 10.1016/B978-0-12-385116-1.00014-5. [DOI] [PubMed] [Google Scholar]
- 157.Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death and Differentiation. 2008;15(9):1460–1471. doi: 10.1038/cdd.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yeung BHY, Kwan BWY, He QY, Lee AS, Liu J, Wong AST. Glucose-regulated protein 78 as a novel effector of BRCA1 for inhibiting stress-induced apoptosis. Oncogene. 2008;27(53):6782–6789. doi: 10.1038/onc.2008.290. [DOI] [PubMed] [Google Scholar]
- 159.Beddoe T, Paton AW, Le Nours J, Rossjohn J, Paton JC. Structure, biological functions and applications of the AB5 toxins. Trends in Biochemical Sciences. 2010;35(7):411–418. doi: 10.1016/j.tibs.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Paton AW, Beddoe T, Thorpe CM, et al. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature. 2006;443(7111):548–552. doi: 10.1038/nature05124. [DOI] [PubMed] [Google Scholar]
- 161.Backer JM, Krivoshein AV, Hamby CV, et al. Chaperone-targeting cytotoxin and endoplasmic reticulum stress-inducing drug synergize to kill cancer cells. Neoplasia. 2009;11(11):1165–1173. doi: 10.1593/neo.09878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nakajima S, Hiramatsu N, Hayakawa K, et al. Selective abrogation of BiP/GRP78 blunts activation of NF-κB through the ATF6 branch of the UPR: involvement of C/EBPβ and mTOR-dependent dephosphorylation of Akt. Molecular and Cellular Biology. 2011;31(8):1710–1718. doi: 10.1128/MCB.00939-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Tian T, Zhao Y, Nakajima S, et al. Cytoprotective roles of ERK and Akt in endoplasmic reticulum stress triggered by subtilase cytotoxin. Biochemical and Biophysical Research Communications. 2011;410(4):852–858. doi: 10.1016/j.bbrc.2011.06.078. [DOI] [PubMed] [Google Scholar]
- 164.Yamazaki H, Hiramatsu N, Hayakawa K, et al. Activation of the Akt-NF-κB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. Journal of Immunology. 2009;183(2):1480–1487. doi: 10.4049/jimmunol.0900017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zhao Y, Tian T, Huang T, et al. Subtilase cytotoxin activates MAP kinases through PERK and IRE1 branches of the unfolded protein response. Toxicological Sciences. 2011;120(1):79–86. doi: 10.1093/toxsci/kfq368. [DOI] [PubMed] [Google Scholar]
- 166.Yahiro K, Satoh M, Morinaga N, et al. Identification of subtilase cytotoxin (SubAB) receptors whose signaling, in association with SubAB-induced BiP cleavage, is responsible for apoptosis in HeLa cells. Infection and Immunity. 2011;79(2):617–627. doi: 10.1128/IAI.01020-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sun S, Wang X, Wang C, et al. Arctigenin suppresses unfolded protein response and sensitizes glucose deprivation-mediated cytotoxicity of cancer cells. Planta Medica. 2011;77(2):141–145. doi: 10.1055/s-0030-1250179. [DOI] [PubMed] [Google Scholar]
- 168.Choo SJ, Park HR, Ryoo IJ, et al. Deoxyverrucosidin, a novel GRP78/BiP down-regulator, produced by Penicillium sp. Journal of Antibiotics. 2005;58(3):210–213. doi: 10.1038/ja.2005.26. [DOI] [PubMed] [Google Scholar]
- 169.Hayakawa Y, Hattori Y, Kawasaki T, et al. Efrapeptin J, a new down-regulator of the molecular chaperone GRP78 from a marine Tolypocladium sp. Journal of Antibiotics. 2008;61(6):365–371. doi: 10.1038/ja.2008.51. [DOI] [PubMed] [Google Scholar]
- 170.Kozone I, Ueda JY, Takagi M, Shin-Ya K. JBIR-52, a new antimycin-like compound, from Streptomyces sp. ML55. Journal of Antibiotics. 2009;62(10):593–595. doi: 10.1038/ja.2009.79. [DOI] [PubMed] [Google Scholar]
- 171.Hwang JH, Kim JY, Cha MR, et al. Etoposide-resistant HT-29 human colon carcinoma cells during glucose deprivation are sensitive to piericidin A, a GRP78 down-regulator. Journal of Cellular Physiology. 2008;215(1):243–250. doi: 10.1002/jcp.21308. [DOI] [PubMed] [Google Scholar]
- 172.Umeda Y, Chijiwa S, Furihata K, et al. Prunustatin A, a novel GRP78 molecular chaperone down-regulator isolated from Streptomyces violaceoniger . Journal of Antibiotics. 2005;58(3):206–209. doi: 10.1038/ja.2005.25. [DOI] [PubMed] [Google Scholar]
- 173.Yu DH, Mcdonald J, Liu G, et al. Pyrvinium targets the unfolded protein response to hypoglycemia and its anti-tumor activity is enhanced by combination therapy. PLoS ONE. 2008;3(12) doi: 10.1371/journal.pone.0003951.e3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Saito S, Tomida A. Use of chemical genomics in assessment of the UPR. Methods in Enzymology. 2011;491:327–341. doi: 10.1016/B978-0-12-385928-0.00018-3. [DOI] [PubMed] [Google Scholar]
- 175.Ryoo IJ, Park HR, Choo SJ, et al. Selective cytotoxic activity of valinomycin against HT-29 human colon carcinoma cells via down-regulation of GRP78. Biological and Pharmaceutical Bulletin. 2006;29(4):817–820. doi: 10.1248/bpb.29.817. [DOI] [PubMed] [Google Scholar]
- 176.Park HR, Tomida A, Sato S, et al. Effect on tumor cells of blocking survival response to glucose deprivation. Journal of the National Cancer Institute. 2004;96(17):1300–1310. doi: 10.1093/jnci/djh243. [DOI] [PubMed] [Google Scholar]
- 177.Saito S, Furuno A, Sakurai J, et al. Chemical genomics identifies the unfolded protein response as a target for selective cancer cell killing during glucose deprivation. Cancer Research. 2009;69(10):4225–4234. doi: 10.1158/0008-5472.CAN-08-2689. [DOI] [PubMed] [Google Scholar]
- 178.Krentz AJ, Bailey CJ. Oral antidiabetic agents: current role in type 2 diabetes mellitus. Drugs. 2005;65(3):385–411. doi: 10.2165/00003495-200565030-00005. [DOI] [PubMed] [Google Scholar]
- 179.Pierotti MA, Berrino F, Gariboldi M, et al. Targeting metabolism for cancer treatment and prevention: metformin, an old drug with multi-faceted effects. doi: 10.1038/onc.2012.181. Oncogene. In press. [DOI] [PubMed] [Google Scholar]
- 180.Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0033411.e33411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhou Y, Lee AS. Mechanism for the suppression of the mammalian stress response by genistein, an anticancer phytoestrogen from soy. Journal of the National Cancer Institute. 1998;90(5):381–388. doi: 10.1093/jnci/90.5.381. [DOI] [PubMed] [Google Scholar]
- 182.Hong M, Lin MY, Huang JM, et al. Transcriptional regulation of the Grp78 promoter by endoplasmic reticulum stress: role of TFII-I and its tyrosine phosphorylation. Journal of Biological Chemistry. 2005;280(17):16821–16828. doi: 10.1074/jbc.M413753200. [DOI] [PubMed] [Google Scholar]
- 183.Mism UK, Wang F, Pizzo SV. Transcription factor TFII-I causes transcriptional upregulation of GRP78 synthesis in prostate cancer cells. Journal of Cellular Biochemistry. 2009;106(3):381–389. doi: 10.1002/jcb.22016. [DOI] [PubMed] [Google Scholar]
- 184.Lim HA, Kim JH, Sung MK, Kim MK, Park JHY, Kim JS. Genistein induces glucose-regulated protein 78 in mammary tumor cells. Journal of Medicinal Food. 2006;9(1):28–32. doi: 10.1089/jmf.2006.9.28. [DOI] [PubMed] [Google Scholar]
- 185.Yeh TC, Chiang PC, Li TK, et al. Genistein induces apoptosis in human hepatocellular carcinomas via interaction of endoplasmic reticulum stress and mitochondrial insult. Biochemical Pharmacology. 2007;73(6):782–792. doi: 10.1016/j.bcp.2006.11.027. [DOI] [PubMed] [Google Scholar]
- 186.Ermakova SP, Kang BS, Choi BY, et al. (-)-Epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Research. 2006;66(18):9260–9269. doi: 10.1158/0008-5472.CAN-06-1586. [DOI] [PubMed] [Google Scholar]
- 187.Luo T, Wang J, Yin Y, et al. (-)-Epigallocatechin gallate sensitizes breast cancer cells to paclitaxel in a murine model of breast carcinoma. Breast Cancer Research. 2010;12(1, article R8) doi: 10.1186/bcr2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Li Y, Zhang T, Jiang Y, Lee HF, Schwartz SJ, Sun D. (-)-Epigallocatechin-3-gallate inhibits Hsp90 function by impairing Hsp90 association with cochaperones in pancreatic cancer cell line mia paca-2. Molecular Pharmaceutics. 2009;6(4):1152–1159. doi: 10.1021/mp900037p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Nagle DG, Ferreira D, Zhou YD. Epigallocatechin-3-gallate (EGCG): chemical and biomedical perspectives. Phytochemistry. 2006;67(17):1849–1855. doi: 10.1016/j.phytochem.2006.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Nam S, Smith DM, Dou QP. Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. Journal of Biological Chemistry. 2001;276(16):13322–13330. doi: 10.1074/jbc.M004209200. [DOI] [PubMed] [Google Scholar]
- 191.Tachibana H, Koga K, Fujimura Y, Yamada K. A receptor for green tea polyphenol EGCG. Nature Structural and Molecular Biology. 2004;11(4):380–381. doi: 10.1038/nsmb743. [DOI] [PubMed] [Google Scholar]
- 192.Kousidou OC, Tzanakakis GN, Karamanos NK. Effects of the natural isoflavonoid genistein on growth, signaling pathways and gene expression of matrix macromolecules by breast cancer cells. Mini-Reviews in Medicinal Chemistry. 2006;6(3):331–337. doi: 10.2174/138955706776073420. [DOI] [PubMed] [Google Scholar]
- 193.Li H, Xu W, Huang Y, et al. Genistein demethylates the promoter of CHD5 and inhibits neuroblastoma growth in vivo. International Journal of Molecular Medicine. 2012;30:1081–1086. doi: 10.3892/ijmm.2012.1118. [DOI] [PubMed] [Google Scholar]
- 194.Ravindranath MH, Muthugounder S, Presser N, Viswanathan S. Anticancer therapeutic potential of soy isoflavone, genistein. Advances in Experimental Medicine and Biology. 2004;546:121–165. doi: 10.1007/978-1-4757-4820-8_11. [DOI] [PubMed] [Google Scholar]
- 195.Martin-Castillo B, Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Metformin and cancer: doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle. 2010;9(6):1057–1064. doi: 10.4161/cc.9.6.10994. [DOI] [PubMed] [Google Scholar]
- 196.El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. Journal of Biological Chemistry. 2000;275(1):223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 197.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochemical Journal. 2000;348(3):607–614. [PMC free article] [PubMed] [Google Scholar]
- 198.Delpino A, Castelli M. The 78 kda glucose-regulated protein (GRP78/BIP) is expressed on the cell membrane, is released into cell culture medium and is also present in human peripheral circulation. Bioscience Reports. 2002;22(3-4):407–420. doi: 10.1023/a:1020966008615. [DOI] [PubMed] [Google Scholar]
- 199.Davidson DJ, Haskell C, Majest S, et al. Kringle 5 of human plasminogen induces apoptosis of endothelial and tumor cells through surface-expressed glucose-regulated protein 78. Cancer Research. 2005;65(11):4663–4672. doi: 10.1158/0008-5472.CAN-04-3426. [DOI] [PubMed] [Google Scholar]
- 200.Miharada K, Karlsson G, Rehn M, et al. Cripto regulates hematopoietic stem cells as a hypoxic-niche-related factor through cell surface receptor GRP78. Cell Stem Cell. 2011;9:330–344. doi: 10.1016/j.stem.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 201.Misra UK, Deedwania R, Pizzo SV. Binding of activated α2-macroglobulin to its cell surface receptor GRP78 in 1-LN prostate cancer cells regulates PAK-2-dependent activation of LIMK. Journal of Biological Chemistry. 2005;280(28):26278–26286. doi: 10.1074/jbc.M414467200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Misra UK, Pizzo SV. Potentiation of signal transduction mitogenesis and cellular proliferation upon binding of receptor-recognized forms of α2-macroglobulin to 1-LN prostate cancer cells. Cellular Signalling. 2004;16(4):487–496. doi: 10.1016/j.cellsig.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 203.de Ridder GG, Gonzalez-Gronow M, Ray R, Pizzo SV. Autoantibodies against cell surface GRP78 promote tumor growth in a murine model of melanoma. Melanoma Research. 2011;21(1):35–43. doi: 10.1097/CMR.0b013e3283426805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.de Ridder GG, Ray R, Pizzo SV. A murine monoclonal antibody directed against the carboxyl-terminal domain of GRP78 suppresses melanoma growth in mice. Melanoma Research. 2012;22:225–235. doi: 10.1097/CMR.0b013e32835312fd. [DOI] [PubMed] [Google Scholar]
- 205.Misra UK, Pizzo SV. Ligation of cell surface GRP78 with antibody directed against the COOH-terminal domain of GRP78 suppresses Ras/MAPK and PI 3-kinase/AKT signaling while promoting caspase activation in human prostate cancer cells. Cancer Biology and Therapy. 2010;9(2):142–152. doi: 10.4161/cbt.9.2.10422. [DOI] [PubMed] [Google Scholar]
- 206.Ray R, de Ridder GG, Eu JP, et al. The Escherichia coli subtilase cytotoxin a subunit specifically cleaves cell-surface GRP78 and abolishes COOH-terminal-dependent signaling. The Journal of Biological Chemistry. 2012;287:32755–32769. doi: 10.1074/jbc.M112.399808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Thomas S, Sharma N, Golden EB, et al. Preferential killing of triple-negative breast cancer cells in vitro and in vivo when pharmacological aggravators of endoplasmic reticulum stress are combined with autophagy inhibitors. Cancer Letters. 2012;325:63–71. doi: 10.1016/j.canlet.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 208.Pahl HL. Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiological Reviews. 1999;79(3):683–701. doi: 10.1152/physrev.1999.79.3.683. [DOI] [PubMed] [Google Scholar]
- 209.Treiman M, Caspersen C, Christensen SB. A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases. Trends in Pharmacological Sciences. 1998;19(4):131–135. doi: 10.1016/s0165-6147(98)01184-5. [DOI] [PubMed] [Google Scholar]
- 210.Winther AML, Liu H, Sonntag Y, et al. Critical roles of hydrophobicity and orientation of side chains for inactivation of sarcoplasmic reticulum Ca2+-ATPase with thapsigargin and thapsigargin analogs. Journal of Biological Chemistry. 2010;285(37):28883–28892. doi: 10.1074/jbc.M110.136242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hakii H, Fujiki H, Suganuma M. Thapsigargin, a histamine secretagogue, is a non-12-O-tetradecanoylphorbol-13-acetate (TPA) type tumor promoter in two-stage mouse skin carcinogenesis. Journal of Cancer Research and Clinical Oncology. 1986;111(3):177–181. doi: 10.1007/BF00389230. [DOI] [PubMed] [Google Scholar]
- 212.Ohuchi K, Takahashi C, Hirasawa N, Watanabe M, Fujiki H, Tsurufuji S. Stimulation of histamine release and arachidonic acid metabolism in rat peritonel mast cells by thapsigargin a non-TPA-type tumor promoter. Biochimica et Biophysica Acta. 1989;1003(1):9–14. doi: 10.1016/0005-2760(89)90091-x. [DOI] [PubMed] [Google Scholar]
- 213.Denmeade SR, Jakobsen CM, Janssen S, et al. Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. Journal of the National Cancer Institute. 2003;95(13):990–1000. doi: 10.1093/jnci/95.13.990. [DOI] [PubMed] [Google Scholar]
- 214.Denmeade SR, Mhaka AM, Rosen DM, et al. Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Science Translational Medicine. 2012;4(140) doi: 10.1126/scitranslmed.3003886.140ra86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Adams J, Kauffman M. Development of the proteasome inhibitor Velcade (Bortezomib) Cancer Investigation. 2004;22(2):304–311. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- 216.Chauhan D, Hideshima T, Anderson KC. Proteasome inhibition in multiple myeloma: therapeutic implication. Annual Review of Pharmacology and Toxicology. 2005;45:465–476. doi: 10.1146/annurev.pharmtox.45.120403.100037. [DOI] [PubMed] [Google Scholar]
- 217.Fribley A, Zeng Q, Wang CY. Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Molecular and Cellular Biology. 2004;24(22):9695–9704. doi: 10.1128/MCB.24.22.9695-9704.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Nawrocki ST, Carew JS, Pino MS, et al. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Research. 2005;65(24):11658–11666. doi: 10.1158/0008-5472.CAN-05-2370. [DOI] [PubMed] [Google Scholar]
- 219.Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Meister S, Schubert U, Neubert K, et al. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Research. 2007;67(4):1783–1792. doi: 10.1158/0008-5472.CAN-06-2258. [DOI] [PubMed] [Google Scholar]
- 221.Bianchi G, Oliva L, Cascio P, et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood. 2009;113(13):3040–3049. doi: 10.1182/blood-2008-08-172734. [DOI] [PubMed] [Google Scholar]
- 222.Carrasco DR, Sukhdeo K, Protopopova M, et al. "The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell. 2007;11(4):349–360. doi: 10.1016/j.ccr.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Shen Y, Hendershot LM. Identification of ERdj3 and OBF-1/BOB-1/OCA-B as direct targets of XBP-1 during plasma cell differentiation. Journal of Immunology. 2007;179(5):2969–2978. doi: 10.4049/jimmunol.179.5.2969. [DOI] [PubMed] [Google Scholar]
- 224.Chen S, Liu X, Yue P, Schönthal AH, Khuri FR, Sun SY. CCAAT/enhancer binding protein homologous protein-dependent death receptor 5 induction and ubiquitin/proteasome-mediated cellular FLICE-inhibitory protein down-regulation contribute to enhancement of tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by dimethyl-celecoxib in human non-small-cell lung cancer cells. Molecular Pharmacology. 2007;72(5):1269–1279. doi: 10.1124/mol.107.037465. [DOI] [PubMed] [Google Scholar]
- 225.Hideshima T, Ikeda H, Chauhan D, et al. Bortezomib induces canonical nuclear factor-κB activation in multiple myeloma cells. Blood. 2009;114(5):1046–1052. doi: 10.1182/blood-2009-01-199604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Hideshima T, Mitsiades C, Akiyama M, et al. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood. 2003;101(4):1530–1534. doi: 10.1182/blood-2002-08-2543. [DOI] [PubMed] [Google Scholar]
- 227.Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-UP-regulated modulator of apoptosis (PUMA) and NOXA by p53. Journal of Biological Chemistry. 2006;281(11):7260–7270. doi: 10.1074/jbc.M509868200. [DOI] [PubMed] [Google Scholar]
- 228.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Reports. 2006;7(9):880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Perry CM, Frampton JE, McCormack PL, Siddiqui MAA, Cvetković RS. Nelfinavir: a review of its use in the management of HIV infection. Drugs. 2005;65(15):2209–2244. doi: 10.2165/00003495-200565150-00015. [DOI] [PubMed] [Google Scholar]
- 230.Piccinini M, Rinaudo MT, Anselmino A, et al. The HIV protease inhibitors nelfinavir and saquinavir, but not a variety of HIV reverse transcriptase inhibitors, adversely affect human proteasome function. Antiviral Therapy. 2005;10(2):215–223. [PubMed] [Google Scholar]
- 231.Jiang W, Mikochik PJ, Ra JH, et al. HIV protease inhibitor nelfinavir inhibits growth of human melanoma cells by induction of cell cycle arrest. Cancer Research. 2007;67(3):1221–1227. doi: 10.1158/0008-5472.CAN-06-3377. [DOI] [PubMed] [Google Scholar]
- 232.Gills JJ, LoPiccolo J, Tsurutani J, et al. Nelfinavir, a lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and invivo. Clinical Cancer Research. 2007;13(17):5183–5194. doi: 10.1158/1078-0432.CCR-07-0161. [DOI] [PubMed] [Google Scholar]
- 233.Pyrko P, Kardosh A, Wang W, Xiong W, Schönthal AH, Chen TC. HIV-1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Cancer Research. 2007;67(22):10920–10928. doi: 10.1158/0008-5472.CAN-07-0796. [DOI] [PubMed] [Google Scholar]
- 234.Brüning A, Burger P, Vogel M, et al. Nelfinavir induces the unfolded protein response in ovarian cancer cells, resulting in ER vacuolization, cell cycle retardation and apoptosis. Cancer Biology & Therapy. 2009;8(3):226–232. doi: 10.4161/cbt.8.3.7339. [DOI] [PubMed] [Google Scholar]
- 235.Gupta AK, Li B, Cerniglia GJ, Ahmed MS, Hahn SM, Maity A. The HIV protease inhibitor nelfinavir downregulates Akt phosphorylation by inhibiting proteasomal activity and inducing the unfolded protein response. Neoplasia. 2007;9(4):271–278. doi: 10.1593/neo.07124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Brüning A, Friese K, Burges A, Mylonas I. Tamoxifen enhances the cytotoxic effects of nelfinavir in breast cancer cells. Breast Cancer Research. 2010;12(4, article R45) doi: 10.1186/bcr2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Brüning A, Vogel M, Burger P, et al. Nelfinavir induces TRAIL receptor upregulation in ovarian cancer cells. Biochemical and Biophysical Research Communications. 2008;377(4):1309–1314. doi: 10.1016/j.bbrc.2008.10.167. [DOI] [PubMed] [Google Scholar]
- 238.Tian X, Ye J, Alonso-Basanta M, et al. Modulation of CCAAT/enhancer binding protein homologous protein (CHOP)-dependent DR5 expression by nelfinavir sensitizes glioblastoma multiforme cells to tumor necrosis factor-related apoptosisinducing ligand (TRAIL) Journal of Biological Chemistry. 2011;286:29408–29416. doi: 10.1074/jbc.M110.197665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Cuneo KC, Tu T, Geng L, Fu A, Hallahan DE, Willey CD. HIV protease inhibitors enhance the efficacy of irradiation. Cancer Research. 2007;67(10):4886–4893. doi: 10.1158/0008-5472.CAN-06-3684. [DOI] [PubMed] [Google Scholar]
- 240.Gupta AK, Cerniglia GJ, Mick R, McKenna WG, Muschel RJ. HIV protease inhibitors block Akt signaling and radiosensitize tumor cells both in vitro and in vivo. Cancer Research. 2005;65(18):8256–8265. doi: 10.1158/0008-5472.CAN-05-1220. [DOI] [PubMed] [Google Scholar]
- 241.Zeng J, See AP, Aziz K, et al. Nelfinavir induces radiation sensitization in pituitary adenoma cells. Cancer Biology & Therapy. 2011;12:657–663. doi: 10.4161/cbt.12.7.17172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Bernstein WB, Dennis PA. Repositioning HIV protease inhibitors as cancer therapeutics. Current Opinion in HIV and AIDS. 2008;3(6):666–675. doi: 10.1097/COH.0b013e328313915d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Brüning A, Gingelmaier A, Friese K, Mylonas I. New prospects for nelfinavir in non-HIV-related diseases. Current Molecular Pharmacology. 2010;3(2):91–97. doi: 10.2174/1874467211003020091. [DOI] [PubMed] [Google Scholar]
- 244.Koki AT, Masferrer JL. Celecoxib: a specific COX-2 inhibitor with anticancer properties. Cancer Control. 2002;9(2):28–35. doi: 10.1177/107327480200902S04. [DOI] [PubMed] [Google Scholar]
- 245.Grösch S, Maier TJ, Schiffmann S, Geisslinger G. Cyclooxygenase-2 (COX-2)—independent anticarcinogenic effects of selective COX-2 inhibitors. Journal of the National Cancer Institute. 2006;98(11):736–747. doi: 10.1093/jnci/djj206. [DOI] [PubMed] [Google Scholar]
- 246.Schönthal AH. Direct non-cyclooxygenase-2 targets of celecoxib and their potential relevance for cancer therapy. British Journal of Cancer. 2007;97(11):1465–1468. doi: 10.1038/sj.bjc.6604049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Kashfi K, Rigas B. Non-COX-2 targets and cancer: expanding the molecular target repertoire of chemoprevention. Biochemical Pharmacology. 2005;70(7):969–986. doi: 10.1016/j.bcp.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 248.Knudsen JF, Carlsson U, Hammarström P, Sokol GH, Cantilena LR. The cyclooxygenase-2 inhibitor celecoxib is a potent inhibitor of human carbonic anhydrase II. Inflammation. 2004;28(5):285–290. doi: 10.1007/s10753-004-6052-1. [DOI] [PubMed] [Google Scholar]
- 249.Weber A, Casini A, Heine A, et al. Unexpected nanomolar inhibition of carbonic anhydrase by COX-2-selective celecoxib: new pharmacological opportunities due to related binding site recognition. Journal of Medicinal Chemistry. 2004;47(3):550–557. doi: 10.1021/jm030912m. [DOI] [PubMed] [Google Scholar]
- 250.Johnson AJ, Hsu AL, Lin HP, Song X, Chen CS. The cyclo-oxygenase-2 inhibitor celecoxib perturbs intracellular calcium by inhibiting endoplasmic reticulum Ca2+-ATPases: a plausible link with its anti-tumour effect and cardiovascular risks. Biochemical Journal. 2002;366(3):831–837. doi: 10.1042/BJ20020279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Kim SH, Hwang CI, Park WY, Juhnn YS, Lee JH, Song YS. Erratum: GADD153 mediates celecoxib-induced apoptosis in cervical cancer cells. Carcinogenesis. 2007;28(1):223–231. doi: 10.1093/carcin/bgl227. [DOI] [PubMed] [Google Scholar]
- 252.Pyrko P, Kardosh A, Liu YT, et al. Calcium-activated endoplasmic reticulum stress as a major component of tumor cell death induced by 2,5-dimethyl-celecoxib, a non-coxib analogue of celecoxib. Molecular Cancer Therapeutics. 2007;6(4):1262–1275. doi: 10.1158/1535-7163.MCT-06-0629. [DOI] [PubMed] [Google Scholar]
- 253.Tsutsumi S, Gotoh T, Tomisato W, et al. Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death and Differentiation. 2004;11(9):1009–1016. doi: 10.1038/sj.cdd.4401436. [DOI] [PubMed] [Google Scholar]
- 254.Tsutsumi S, Namba T, Tanaka KI, et al. Celecoxib upregulates endoplasmic reticulum chaperones that inhibit celecoxib-induced apoptosis in human gastric cells. Oncogene. 2006;25(7):1018–1029. doi: 10.1038/sj.onc.1209139. [DOI] [PubMed] [Google Scholar]
- 255.Pyrko P, Kardosh A, Schönthal AH. Celecoxib transiently inhibits cellular protein synthesis. Biochemical Pharmacology. 2008;75(2):395–404. doi: 10.1016/j.bcp.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 256.Zhu J, Huang JW, Tseng PH, et al. From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors. Cancer Research. 2004;64(12):4309–4318. doi: 10.1158/0008-5472.CAN-03-4063. [DOI] [PubMed] [Google Scholar]
- 257.Zhu J, Song X, Lin HP, et al. Using cyclooxygenase-2 inhibitors as molecular platforms to develop a new class of apoptosis-inducing agents. Journal of the National Cancer Institute. 2002;94(23):1745–1757. doi: 10.1093/jnci/94.23.1745. [DOI] [PubMed] [Google Scholar]
- 258.Kardosh A, Soriano N, Liu YT, et al. Multitarget inhibition of drug-resistant multiple myeloma cell lines by dimethyl-celecoxib (DMC), a non-COX-2 inhibitory analog of celecoxib. Blood. 2005;106(13):4330–4338. doi: 10.1182/blood-2005-07-2819. [DOI] [PubMed] [Google Scholar]
- 259.Kardosh A, Wang W, Uddin J, et al. Dimethyl-celecoxib (DMC), a derivative of celecoxib that lacks cyclooxygenase-2-inhibitory function, potently mimics the anti-tumor effects of celecoxib on Burkitt’s lymphoma in vitro and in vivo. Cancer Biology and Therapy. 2005;4(5):571–582. doi: 10.4161/cbt.4.5.1699. [DOI] [PubMed] [Google Scholar]
- 260.Kulp SK, Yang YT, Hung CC, et al. 3-phosphoinositide-dependent protein kinase-1/Akt signaling represents a major cyclooxygenase-2-independent target for celecoxib in prostate cancer cells. Cancer Research. 2004;64(4):1444–1451. doi: 10.1158/0008-5472.can-03-2396. [DOI] [PubMed] [Google Scholar]
- 261.Pyrko P, Soriano N, Kardosh A, et al. Downregulation of survivin expression and concomitant induction of apoptosis by celecoxib and its non-cyclooxygenase-2-inhibitory analog, dimethyl-celecoxib (DMC), in tumor cells in vitro and in vivo. Molecular Cancer. 2006;5, article 19 doi: 10.1186/1476-4598-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Song X, Lin HP, Johnson AJ, et al. Cyclooxygenase-2, player or spectator in cyclooxygenase-2 inhibitor-induced apoptosis in prostate cancer cells. Journal of the National Cancer Institute. 2002;94(8):585–591. doi: 10.1093/jnci/94.8.585. [DOI] [PubMed] [Google Scholar]
- 263.Kusunoki N, Ito T, Sakurai N, Handa H, Kawai S. A celecoxib derivative potently inhibits proliferation of colon adenocarcinoma cells by induction of apoptosis. Anticancer Research. 2006;26(5 A):3229–3236. [PubMed] [Google Scholar]
- 264.Kusunoki N, Ito T, Sakurai N, Suguro T, Handa H, Kawai S. A novel celecoxib derivative potently induces apoptosis of human synovial fibroblasts. Journal of Pharmacology and Experimental Therapeutics. 2005;314(2):796–803. doi: 10.1124/jpet.105.086116. [DOI] [PubMed] [Google Scholar]
- 265.Alloza I, Baxter A, Chen Q, Matthiesen R, Vandenbroeck K. Celecoxib inhibits interleukin-12 αβ and β 2 folding and secretion by a novel COX2-independent mechanism involving chaperones of the endoplasmic reticulum. Molecular Pharmacology. 2006;69(5):1579–1587. doi: 10.1124/mol.105.020669. [DOI] [PubMed] [Google Scholar]
- 266.McLaughlin M, Alloza I, Quoc HP, Scott CJ, Hirabayashi Y, Vandenbroeck K. Inhibition of secretion of interleukin (IL)-12/IL-23 family cytokines by 4-trifluoromethyl-celecoxib is coupled to degradation via the endoplasmic reticulum stress protein HERP. Journal of Biological Chemistry. 2010;285(10):6960–6969. doi: 10.1074/jbc.M109.056614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Ding H, Han C, Guo D, et al. Sensitivity to the non-COX inhibiting celecoxib derivative, OSU03012, is p21WAF1/CIP1 dependent. International Journal of Cancer. 2008;123(12):2931–2938. doi: 10.1002/ijc.23895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Johnson AJ, Smith LL, Zhu J, et al. A novel celecoxib derivative, OSU03012, induces cytotoxicity in primary CLL cells and transformed B-cell lymphoma cell line via a caspase- and Bcl-2-independent mechanism. Blood. 2005;105(6):2504–2509. doi: 10.1182/blood-2004-05-1957. [DOI] [PubMed] [Google Scholar]
- 269.Ryan EP, Bushnell TP, Friedman AE, Rahman I, Phipps RP. Cyclooxygenase-2 independent effects of cyclooxygenase-2 inhibitors on oxidative stress and intracellular glutathione content in normal and malignant human B-cells. Cancer Immunology, Immunotherapy. 2008;57(3):347–358. doi: 10.1007/s00262-007-0374-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Chuang HC, Kardosh A, Gaffney KJ, Petasis NA, Schönthal AH. COX-2 inhibition is neither necessary nor sufficient for celecoxib to suppress tumor cell proliferation and focus formation in vitro. Molecular Cancer. 2008;7, article 38 doi: 10.1186/1476-4598-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Chen ST, Thomas S, Gaffney KJ, Louie SG, Petasis NA, Schönthal AH. Cytotoxic effects of celecoxib on Raji lymphoma cells correlate with aggravated endoplasmic reticulum stress but not with inhibition of cyclooxygenase-2. Leukemia Research. 2010;34(2):250–253. doi: 10.1016/j.leukres.2009.09.028. [DOI] [PubMed] [Google Scholar]
- 272.Booth L, Cazanave SC, Hamed HA, et al. OSU-03012 suppresses GRP78/BiP expression that causes PERK-dependent increases in tumor cell killing. Cancer Biology & Therapy. 2012;13:224–236. doi: 10.4161/cbt.13.4.18877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Hamed HA, Yacoub A, Park MA, et al. OSU-03012 enhances Ad.mda-7-induced GBM cell killing via ER stress and autophagy and by decreasing expression of mitochondrial protective proteins. Cancer Biology and Therapy. 2010;9(7):526–536. doi: 10.4161/cbt.9.7.11116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Yacoub A, Park MA, Hanna D, et al. OSU-03012 promotes caspase-independent but PERK-, cathepsin B-, BID-, and AIF-dependent killing of transformed cells. Molecular Pharmacology. 2006;70(2):589–603. doi: 10.1124/mol.106.025007. [DOI] [PubMed] [Google Scholar]
- 275.Cho HY, Thomas S, Golden EB, et al. Enhanced killing of chemo-resistant breast cancer cells via controlled aggravation of ER stress. Cancer Letters. 2009;282(1):87–97. doi: 10.1016/j.canlet.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 276.Arber N, Eagle CJ, Spicak J, et al. Celecoxib for the prevention of colorectal adenomatous polyps. The New England Journal of Medicine. 2006;355(9):885–895. doi: 10.1056/NEJMoa061652. [DOI] [PubMed] [Google Scholar]
- 277.Bertagnolli MM, Eagle CJ, Zauber AG, et al. Celecoxib for the prevention of sporadic colorectal adenomas. The New England Journal of Medicine. 2006;355(9):873–884. doi: 10.1056/NEJMoa061355. [DOI] [PubMed] [Google Scholar]
- 278.Steinbach G, Lynch PM, Phillips RKS, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. The New England Journal of Medicine. 2000;342(26):1946–1952. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 279.Cerchietti LCA, Bonomi MR, Navigante AH, Castro MA, Cabalar ME, Roth BMC. Phase I/II study of selective cyclooxygenase-2 inhibitor celecoxib as a radiation sensitizer in patients with unresectable brain metastases. Journal of Neuro-Oncology. 2005;71(1):73–81. doi: 10.1007/s11060-004-9179-x. [DOI] [PubMed] [Google Scholar]
- 280.Crane CH, Mason K, Janjan NA, Milas L. Initial experience combining cyclooxygenase-2 inhibition with chemoradiation for locally advanced pancreatic cancer. American Journal of Clinical Oncology. 2003;26(4):S81–S84. doi: 10.1097/00000421-200308002-00009. [DOI] [PubMed] [Google Scholar]
- 281.Gadgeel SM, Ruckdeschel JC, Heath EI, Heilbrun LK, Venkatramanamoorthy R, Wozniak A. Phase II study of gefitinib, an epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI), and celecoxib, a cyclooxygenase-2 (COX-2) inhibitor, in patients with platinum refractory non-small cell lung cancer (NSCLC) Journal of Thoracic Oncology. 2007;2(4):299–305. doi: 10.1097/01.JTO.0000263712.61697.69. [DOI] [PubMed] [Google Scholar]
- 282.Gaffney DK, Winter K, Dicker AP, et al. A Phase II study of acute toxicity for Celebrex™ (celecoxib) and chemoradiation in patients with locally advanced cervical cancer: primary endpoint analysis of RTOG 0128. International Journal of Radiation Oncology Biology Physics. 2007;67(1):104–109. doi: 10.1016/j.ijrobp.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 283.Gridelli C, Gallo C, Ceribelli A, et al. Factorial phase III randomised trial of rofecoxib and prolonged constant infusion of gemcitabine in advanced non-small-cell lung cancer: the GEmcitabine-COxib in NSCLC (GECO) study. The Lancet Oncology. 2007;8(6):500–512. doi: 10.1016/S1470-2045(07)70146-8. [DOI] [PubMed] [Google Scholar]
- 284.Liao Z, Komaki R, Milas L, et al. A phase I clinical trial of thoracic radiotherapy and concurrent celecoxib for patients with unfavorable performance status inoperable/unresectable non-small cell lung cancer. Clinical Cancer Research. 2005;11(9):3342–3348. doi: 10.1158/1078-0432.CCR-04-1741. [DOI] [PubMed] [Google Scholar]
- 285.O’Byrne KJ, Danson S, Dunlop D, et al. Combination therapy with gefitinib and rofecoxib in patients with platinum-pretreated relapsed non-small-cell lung cancer. Journal of Clinical Oncology. 2007;25(22):3266–3273. doi: 10.1200/JCO.2006.09.2791. [DOI] [PubMed] [Google Scholar]
- 286.Reardon DA, Quinn JA, Vredenburgh J, et al. Phase II trial of irinotecan plus celecoxib in adults with recurrent malignant glioma. Cancer. 2005;103(2):329–338. doi: 10.1002/cncr.20776. [DOI] [PubMed] [Google Scholar]
- 287.Smith MR, Manola J, Kaufman DS, Oh WK, Bubley GJ, Kantoff PW. Celecoxib versus placebo for men with prostate cancer and a rising serum prostate-specific antigen after radical prostatectomy and/or radiation therapy. Journal of Clinical Oncology. 2006;24(18):2723–2728. doi: 10.1200/JCO.2005.03.7804. [DOI] [PubMed] [Google Scholar]
- 288.Oyadomari S, Koizumi A, Takeda K, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. Journal of Clinical Investigation. 2002;109(4):525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Li Y, Schwabe RF, DeVries-Seimon T, et al. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-α and interleukin-6: model of NF-κB- and map kinase-dependent inflammation in advanced atherosclerosis. Journal of Biological Chemistry. 2005;280(23):21763–21772. doi: 10.1074/jbc.M501759200. [DOI] [PubMed] [Google Scholar]
- 290.Kedi X, Ming Y, Yongping W, Yi Y, Xiaoxiang Z. Free cholesterol overloading induced smooth muscle cells death and activated both ER- and mitochondrial-dependent death pathway. Atherosclerosis. 2009;207(1):123–130. doi: 10.1016/j.atherosclerosis.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 291.Tsukano H, Gotoh T, Endo M, et al. The endoplasmic reticulum stress-C/EBP homologous protein pathway-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(10):1925–1932. doi: 10.1161/ATVBAHA.110.206094. [DOI] [PubMed] [Google Scholar]
- 292.Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe-/- and Ldlr-/- mice lacking CHOP. Cell Metabolism. 2009;9(5):474–481. doi: 10.1016/j.cmet.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Sanson M, Augé N, Vindis C, et al. Oxidized low-density lipoproteins trigger endoplasmic reticulum stress in vascular cells: prevention by oxygen-regulated protein 150 expression. Circulation Research. 2009;104(3):328–336. doi: 10.1161/CIRCRESAHA.108.183749. [DOI] [PubMed] [Google Scholar]
- 294.Werstuck GH, Khan MI, Femia G, et al. Glucosamine-induced endoplasmic reticulum dysfunction is associated with accelerated atherosclerosis in a hyperglycemic mouse model. Diabetes. 2006;55(1):93–101. [PubMed] [Google Scholar]
- 295.Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circulation Research. 2010;107(7):839–850. doi: 10.1161/CIRCRESAHA.110.224766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Kammoun HL, Chabanon H, Hainault I, et al. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. Journal of Clinical Investigation. 2009;119(5):1201–1215. doi: 10.1172/JCI37007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Werstuck GH, Lentz SR, Dayal S, et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. Journal of Clinical Investigation. 2001;107(10):1263–1273. doi: 10.1172/JCI11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Pagliassotti MJ. Endoplasmic reticulum stress in nonalcoholic fatty liver disease. Annual Review of Nutrition. 2012;32:17–33. doi: 10.1146/annurev-nutr-071811-150644. [DOI] [PubMed] [Google Scholar]
- 299.Imrie D, Sadler KC. Stress management: how the unfolded protein response impacts fatty liver disease. Journal of Hepatology. 2012;57:1147–1151. doi: 10.1016/j.jhep.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Ji C, Mehrian-Shai R, Chan C, Hsu YH, Kaplowitz N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcoholism. 2005;29(8):1496–1503. doi: 10.1097/01.alc.0000174691.03751.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Kaplowitz N, Tin AT, Shinohara M, Ji C. Endoplasmic reticulum stress and liver injury. Seminars in Liver Disease. 2007;27(4):367–377. doi: 10.1055/s-2007-991513. [DOI] [PubMed] [Google Scholar]
- 302.Glembotski CC. The role of the unfolded protein response in the heart. Journal of Molecular and Cellular Cardiology. 2008;44(3):453–459. doi: 10.1016/j.yjmcc.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Okada KI, Minamino T, Tsukamoto Y, et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110(6):705–712. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- 304.Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circulation Research. 2006;99(3):275–282. doi: 10.1161/01.RES.0000233317.70421.03. [DOI] [PubMed] [Google Scholar]
- 305.Shintani-Ishida K, Nakajima M, Uemura K, Yoshida KI. Ischemic preconditioning protects cardiomyocytes against ischemic injury by inducing GRP78. Biochemical and Biophysical Research Communications. 2006;345(4):1600–1605. doi: 10.1016/j.bbrc.2006.05.077. [DOI] [PubMed] [Google Scholar]
- 306.Pan YX, Lin L, Ren AJ, et al. HSP70 and GRP78 induced by endothelin-1 pretreatment enhance tolerance to hypoxia in cultured neonatal rat cardiomyocytes. Journal of Cardiovascular Pharmacology. 2004;44(supplement 1):S117–S120. doi: 10.1097/01.fjc.0000166234.11336.a9. [DOI] [PubMed] [Google Scholar]
- 307.Vitadello M, Penzo D, Petronilli V, et al. Overexpression of the stress protein Grp94 reduces cardiomyocyte necrosis due to calcium overload and simulated ischemia. The FASEB Journal. 2003;17(8):923–925. doi: 10.1096/fj.02-0644fje. [DOI] [PubMed] [Google Scholar]
- 308.Groenendyk J, Sreenivasaiah PK, Kim DH, Agellon LB, Michalak M. Biology of endoplasmic reticulum stress in the heart. Circulation Research. 2010;107(10):1185–1197. doi: 10.1161/CIRCRESAHA.110.227033. [DOI] [PubMed] [Google Scholar]
- 309.Tardif KD, Mori K, Kaufman RJ, Siddiqui A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. Journal of Biological Chemistry. 2004;279(17):17158–17164. doi: 10.1074/jbc.M312144200. [DOI] [PubMed] [Google Scholar]
- 310.Xu Z, Jensen G, Yen TS. Activation of hepatitis B virus S promoter by the viral large surface protein via induction of stress in the endoplasmic reticulum. Journal of Virology. 1997;71(10):7387–7392. doi: 10.1128/jvi.71.10.7387-7392.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Waris G, Tardif KD, Siddiqui A. Endoplasmic reticulum (ER) stress: hepatitis C virus induces an ER-nucleus signal transduction pathway and activates NF-κB and STAT-3. Biochemical Pharmacology. 2002;64(10):1425–1430. doi: 10.1016/s0006-2952(02)01300-x. [DOI] [PubMed] [Google Scholar]
- 312.Wang HC, Huang W, Lai MD, Su IJ. Hepatitis B virus pre-S mutants, endoplasmic reticulum stress and hepatocarcinogenesis. Cancer Science. 2006;97(8):683–688. doi: 10.1111/j.1349-7006.2006.00235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Unterberger U, Höftberger R, Gelpi E, Flicker H, Budka H, Voigtländer T. Endoplasmic reticulum stress features are prominent in Alzheimer disease but not in prion diseases in vivo. Journal of Neuropathology and Experimental Neurology. 2006;65(4):348–357. doi: 10.1097/01.jnen.0000218445.30535.6f. [DOI] [PubMed] [Google Scholar]
- 314.Katayama T, Imaizumi K, Sato N, et al. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nature Cell Biology. 1999;1(8):479–485. doi: 10.1038/70265. [DOI] [PubMed] [Google Scholar]
- 315.Milhavet O, Martindale JL, Camandola S, et al. Involvement of Gadd153 in the pathogenic action of presenilin-1 mutations. Journal of Neurochemistry. 2002;83(3):673–681. doi: 10.1046/j.1471-4159.2002.01165.x. [DOI] [PubMed] [Google Scholar]
- 316.Niwa M, Sidrauski C, Kaufman RJ, Walter P. A role for presenilin-1 in nuclear accumulation of Ire1 fragments and induction of the mammalian unfolded protein response. Cell. 1999;99(7):691–702. doi: 10.1016/s0092-8674(00)81667-0. [DOI] [PubMed] [Google Scholar]
- 317.Bernales S, Soto MM, McCullagh E. Unfolded protein stress in the endoplasmic reticulum and mitochondria: a role in neurodegeneration. Frontiers in Aging Neuroscience. 2012;4, article 5 doi: 10.3389/fnagi.2012.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Doyle KM, Kennedy D, Gorman AM, et al. Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. Journal of Cellular and Molecular Medicine. 2011;15:2025–2039. doi: 10.1111/j.1582-4934.2011.01374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Imai Y, Soda M, Takahashi R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. Journal of Biological Chemistry. 2000;275(46):35661–35664. doi: 10.1074/jbc.C000447200. [DOI] [PubMed] [Google Scholar]
- 320.Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003;302(5646):819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- 321.Bouman L, Schlierf A, Lutz AK, et al. Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death and Differentiation. 2011;18(5):769–782. doi: 10.1038/cdd.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Kouroku Y, Fujita E, Jimbo A, et al. Polyglutamine aggregates stimulate ER stress signals and caspase-12 activation. Human Molecular Genetics. 2002;11(13):1505–1515. doi: 10.1093/hmg/11.13.1505. [DOI] [PubMed] [Google Scholar]
- 323.Zuleta A, Vidal RL, Armentano D, et al. AAV-mediated delivery of the transcription factor XBP1s into the striatum reduces mutant Huntingtin aggregation in a mouse model of Huntington's disease. Biochemical and Biophysical Research Communications. 2012;420:558–563. doi: 10.1016/j.bbrc.2012.03.033. [DOI] [PubMed] [Google Scholar]
- 324.Noh JY, Lee H, Song S, et al. SCAMP5 links endoplasmic reticulum stress to the accumulation of expanded polyglutamine protein aggregates via endocytosis inhibition. Journal of Biological Chemistry. 2009;284(17):11318–11325. doi: 10.1074/jbc.M807620200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Carnemolla A, Fossale E, Agostoni E, et al. Rrs1 is involved in endoplasmic reticulum stress response in huntington disease. Journal of Biological Chemistry. 2009;284(27):18167–18173. doi: 10.1074/jbc.M109.018325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Lee H, Noh JY, Oh Y, et al. IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Human Molecular Genetics. 2012;21:101–114. doi: 10.1093/hmg/ddr445. [DOI] [PubMed] [Google Scholar]
- 327.Fernandez-Fernandez MR, Ferrer I, Lucas JJ. Impaired ATF6α processing, decreased Rheb and neuronal cell cycle re-entry in Huntington’s disease. Neurobiology of Disease. 2011;41(1):23–32. doi: 10.1016/j.nbd.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 328.Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nature Neuroscience. 2009;12(5):627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- 329.Nishitoh H, Kadowaki H, Nagai A, et al. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes and Development. 2008;22(11):1451–1464. doi: 10.1101/gad.1640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Atkin JD, Farg MA, Walker AK, McLean C, Tomas D, Horne MK. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiology of Disease. 2008;30(3):400–407. doi: 10.1016/j.nbd.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 331.Hetz C, Russelakis-Carneiro M, Maundrell K, Castilla J, Soto C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. The EMBO Journal. 2003;22(20):5435–5445. doi: 10.1093/emboj/cdg537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Hetz C, Russelakis-Carneiro M, Wälchli S, et al. The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. Journal of Neuroscience. 2005;25(11):2793–2802. doi: 10.1523/JNEUROSCI.4090-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Wang SB, Shi Q, Xu Y, et al. Protein disulfide isomerase regulates endoplasmic reticulum stress and the apoptotic process during prion infection and PrP mutantinduced cytotoxicity. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0038221.e38221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Xu K, Zhu XP. Endoplasmic reticulum stress and prion diseases. Reviews in the Neurosciences. 2012;23:79–84. doi: 10.1515/rns.2011.062. [DOI] [PubMed] [Google Scholar]
- 335.Jamora C, Dennert G, Lee AS. Inhibition of tumor progression by suppression of stress protein GRP78/BiP induction in fibrosarcoma B/C10ME. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(15):7690–7694. doi: 10.1073/pnas.93.15.7690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Shuda M, Kondoh N, Imazeki N, et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. Journal of Hepatology. 2003;38(5):605–614. doi: 10.1016/s0168-8278(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 337.Schönthal AH. Targeting endoplasmic reticulum stress for cancer therapy. Frontiers in Bioscience. 2012;4:412–431. doi: 10.2741/s276. [DOI] [PubMed] [Google Scholar]
- 338.Choi SE, Lee YJ, Jang HJ, et al. A chemical chaperone 4-PBA ameliorates palmitate-induced inhibition of glucose-stimulated insulin secretion (GSIS) Archives of Biochemistry and Biophysics. 2008;475(2):109–114. doi: 10.1016/j.abb.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 339.Lee YY, Hong SH, Lee YJ, et al. Tauroursodeoxycholate (TUDCA), chemical chaperone, enhances function of islets by reducing ER stress. Biochemical and Biophysical Research Communications. 2010;397(4):735–739. doi: 10.1016/j.bbrc.2010.06.022. [DOI] [PubMed] [Google Scholar]
- 340.Lenin R, Maria MS, Agrawal M, et al. Amelioration of glucolipotoxicity-induced endoplasmic reticulum stress by a, “chemical chaperone” in human THP-1 monocytes. Experimental Diabetes Research. 2012;2012:10 pages. doi: 10.1155/2012/356487.356487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Xie Q, Khaoustov VI, Chung CC, et al. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatology. 2002;36(3):592–601. doi: 10.1053/jhep.2002.35441. [DOI] [PubMed] [Google Scholar]
- 342.Zhang JY, Diao YF, Kim HR, Jin DI. Inhibition of endoplasmic reticulum stress improves mouse embryo development. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0040433.e40433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Zhu Q, Zhong JJ, Jin JF, et al. Tauroursodeoxycholate, a chemical chaperone, prevents palmitate-induced apoptosis in pancreatic beta-cells by reducing ER stress. doi: 10.1055/s-0032-1321787. Experimental and Clinical Endocrinology & Diabetes. In press. [DOI] [PubMed] [Google Scholar]
- 344.Zhang Z, Tong N, Gong Y, et al. Valproate protects the retina from endoplasmic reticulum stress-induced apoptosis after ischemia-reperfusion injury. Neuroscience Letters. 2011;504:88–92. doi: 10.1016/j.neulet.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 345.Penas C, Verdú E, Asensio-Pinilla E, et al. Valproate reduces CHOP levels and preserves oligodendrocytes and axons after spinal cord injury. Neuroscience. 2011;178:33–44. doi: 10.1016/j.neuroscience.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 346.Bown CD, Wang JF, Chen B, Young LT. Regulation of ER stress proteins by valproate: therapeutic implications. Bipolar Disorders. 2002;4(2):145–151. doi: 10.1034/j.1399-5618.2002.t01-1-40201.x. [DOI] [PubMed] [Google Scholar]
- 347.Hiroi T, Wei H, Hough C, Leeds P, Chuang DM. Protracted lithium treatment protects against the ER stress elicited by thapsigargin in rat PC12 cells: roles of intracellular calcium, GRP78 and Bcl-2. Pharmacogenomics Journal. 2005;5(2):102–111. doi: 10.1038/sj.tpj.6500296. [DOI] [PubMed] [Google Scholar]
- 348.Shao L, Sun X, Xu L, Young LT, Wang JF. Mood stabilizing drug lithium increases expression of endoplasmic reticulum stress proteins in primary cultured rat cerebral cortical cells. Life Sciences. 2006;78(12):1317–1323. doi: 10.1016/j.lfs.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 349.Inokuchi Y, Nakajima Y, Shimazawa M, et al. Effect of an inducer of BiP, a molecular chaperone, on endoplasmic reticulum (ER) stress-induced retinal cell death. Investigative Ophthalmology and Visual Science. 2009;50(1):334–344. doi: 10.1167/iovs.08-2123. [DOI] [PubMed] [Google Scholar]
- 350.Kudo T, Kanemoto S, Hara H, et al. A molecular chaperone inducer protects neurons from ER stress. Cell Death and Differentiation. 2008;15(2):364–375. doi: 10.1038/sj.cdd.4402276. [DOI] [PubMed] [Google Scholar]
- 351.Boyce M, Bryant KF, Jousse C, et al. A selective inhibitor of elF2α dephosphorylation protects cells from ER stress. Science. 2005;307(5711):935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 352.Fullwood MJ, Zhou W, Shenolikar S. Targeting phosphorylation of eukaryotic initiation factor-2alpha to treat human disease. Progress in Molecular Biology and Translational Science. 2012;106:75–106. doi: 10.1016/B978-0-12-396456-4.00005-5. [DOI] [PubMed] [Google Scholar]
- 353.Tsaytler P, Harding HP, Ron D, Bertolotti A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science. 2011;332(6025):91–94. doi: 10.1126/science.1201396. [DOI] [PubMed] [Google Scholar]
- 354.Appierto V, Tiberio P, Villani MG, Cavadini E, Formelli F. PLAB induction in fenretinide-induced apoptosis of ovarian cancer cells occurs via a ROS-dependent mechanism involving ER stress and JNK activation. Carcinogenesis. 2009;30(5):824–831. doi: 10.1093/carcin/bgp067. [DOI] [PubMed] [Google Scholar]
- 355.Choi JH, Choi AY, Yoon H, et al. Baicalein protects HT22 murine hippocampal neuronal cells against endoplasmic reticulum stress-induced apoptosis through inhibition of reactive oxygen species production and CHOP induction. Experimental and Molecular Medicine. 2010;42(12):811–822. doi: 10.3858/emm.2010.42.12.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Malhotra JD, Miao H, Zhang K, et al. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(47):18525–18530. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Takizawa S, Izuhara Y, Kitao Y, et al. A novel inhibitor of advanced glycation and endoplasmic reticulum stress reduces infarct volume in rat focal cerebral ischemia. Brain Research. 2007;1183(1):124–137. doi: 10.1016/j.brainres.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 358.Satoh T, Okamoto SI, Cui J, et al. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophillic phase II inducers. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(3):768–773. doi: 10.1073/pnas.0505723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. Journal of Neuroscience. 2004;24(5):1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. Journal of Neuroscience. 2005;25(44):10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Dinkova-Kostova AT, Liby KT, Stephenson KK, et al. Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(12):4584–4589. doi: 10.1073/pnas.0500815102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.A ELO, Han J, Ben Abdrabbah M, Isoda H. Down regulation effect of Rosmarinus officinalis polyphenols on cellular stress proteins in rat pheochromocytoma PC12 cells. Cytotechnology. 2012;64:231–240. doi: 10.1007/s10616-011-9352-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Satoh T, Rezaie T, Seki M, et al. Dual neuroprotective pathways of a proelectrophilic compound via HSF-1-activated heat-shock proteins and Nrf2-activated phase 2 antioxidant response enzymes. Journal of Neurochemistry. 2011;119:569–578. doi: 10.1111/j.1471-4159.2011.07449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Wang X, Ron D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP kinase. Science. 1996;272(5266):1347–1349. doi: 10.1126/science.272.5266.1347. [DOI] [PubMed] [Google Scholar]
- 365.Tajiri S, Oyadomari S, Yano S, et al. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death and Differentiation. 2004;11(4):403–415. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- 366.Zhu H, Xiao S, Sun H, et al. Activation and crosstalk between the endoplasmic reticulum road and JNK pathway in ischemia-reperfusion brain injury. Acta Neurochirurgica. 2012;154:1197–1203. doi: 10.1007/s00701-012-1396-z. [DOI] [PubMed] [Google Scholar]
- 367.Park GB, Kim YS, Lee HK, et al. Endoplasmic reticulum stress-mediated apoptosis of EBV-transformed B cells by cross-linking of CD70 is dependent upon generation of reactive oxygen species and activation of p38 MAPK and JNK pathway. Journal of Immunology. 2010;185(12):7274–7284. doi: 10.4049/jimmunol.1001547. [DOI] [PubMed] [Google Scholar]
- 368.Christensen SB, Mondrup Skytte D, Denmeade SR, et al. A trojan horse in drug development: targeting of thapsigargins towards prostate cancer cells. Anti-Cancer Agents in Medicinal Chemistry. 2009;9(3):276–294. doi: 10.2174/1871520610909030276. [DOI] [PubMed] [Google Scholar]
- 369.Schönthal AH. Antitumor properties of dimethyl-celecoxib, a derivative of celecoxib that does not inhibit cyclooxygenase-2: implications for glioma therapy. Neurosurgical Focus. 2006;20(4):p. E21. doi: 10.3171/foc.2006.20.4.14. [DOI] [PubMed] [Google Scholar]
- 370.Criddle DN, Gerasimenko JV, Baumgartner HK, et al. Calcium signalling and pancreatic cell death: apoptosis or necrosis? Cell Death and Differentiation. 2007;14(7):1285–1294. doi: 10.1038/sj.cdd.4402150. [DOI] [PubMed] [Google Scholar]
- 371.Görlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxidants and Redox Signaling. 2006;8(9-10):1391–1418. doi: 10.1089/ars.2006.8.1391. [DOI] [PubMed] [Google Scholar]
- 372.Banerjee A, Lang JY, Hung MC, et al. Unfolded protein response is required in nu/nu mice microvasculature for treating breast tumor with tunicamycin. Journal of Biological Chemistry. 2011;286:29127–29138. doi: 10.1074/jbc.M110.169771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Elbein AD. The tunicamycins—useful tools for studies on glycoproteins. Trends in Biochemical Sciences. 1981;6:219–221. [Google Scholar]
- 374.Andresen L, Skovbakke SL, Persson G, et al. 2-deoxy D-glucose prevents cell surface expression of NKG2D ligands through inhibition of N-linked glycosylation. The Journal of Immunology. 2012;188:1847–1855. doi: 10.4049/jimmunol.1004085. [DOI] [PubMed] [Google Scholar]
- 375.Kishi S, Shimoke K, Nakatani Y, et al. Nerve growth factor attenuates 2-deoxy-d-glucose-triggered endoplasmic reticulum stress-mediated apoptosis via enhanced expression of GRP78. Neuroscience Research. 2010;66(1):14–21. doi: 10.1016/j.neures.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 376.Csala M, Margittai É, Bánhegyi G. Redox control of endoplasmic reticulum function. Antioxidants and Redox Signaling. 2010;13(1):77–108. doi: 10.1089/ars.2009.2529. [DOI] [PubMed] [Google Scholar]
- 377.Bánhegyi G, Mandl J, Csala M. Redox-based endoplasmic reticulum dysfunction in neurological diseases. Journal of Neurochemistry. 2008;107(1):20–34. doi: 10.1111/j.1471-4159.2008.05571.x. [DOI] [PubMed] [Google Scholar]
- 378.Gorska M, Popowska U, Sielicka-Dudzin A, et al. Geldanamycin and its derivatives as Hsp90 inhibitors. Frontiers in Bioscience. 2012;17:2269–2277. doi: 10.2741/4050. [DOI] [PubMed] [Google Scholar]
- 379.Lawson B, Brewer JW, Hendershot LM. Geldanamycin, an hsp90/GRP94-binding drug, induces increased transcription of endoplasmic reticulum (ER) chaperones via the ER stress pathway. Journal of Cellular Physiology. 1998;174:170–178. doi: 10.1002/(SICI)1097-4652(199802)174:2<170::AID-JCP4>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 380.Carew JS, Nawrocki ST, Krupnik YV, et al. Targeting endoplasmic reticulum protein transport: a novel strategy to kill malignant B cells and overcome fludarabine resistance in CLL. Blood. 2006;107(1):222–231. doi: 10.1182/blood-2005-05-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Casanova JE. Regulation of Arf activation: the Sec7 family of guanine nucleotide exchange factors. Traffic. 2007;8(11):1476–1485. doi: 10.1111/j.1600-0854.2007.00634.x. [DOI] [PubMed] [Google Scholar]
- 382.Fels DR, Ye J, Segan AT, et al. Preferential cytotoxicity of bortezomib toward hypoxic tumor cells via overactivation of endoplasmic reticulum stress pathways. Cancer Research. 2008;68(22):9323–9330. doi: 10.1158/0008-5472.CAN-08-2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Gills JJ, Lopiccolo J, Dennis PA. Nelfinavir, a new anti-cancer drug with pleiotropic effects and many paths to autophagy. Autophagy. 2008;4(1):107–109. doi: 10.4161/auto.5224. [DOI] [PubMed] [Google Scholar]
- 384.Nawrocki ST, Carew JS, Dunner K, Jr., et al. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Research. 2005;65(24):11510–11519. doi: 10.1158/0008-5472.CAN-05-2394. [DOI] [PubMed] [Google Scholar]