

Background: How does the multiple sclerosis therapeutic antibody natalizumab bind to α4 integrins?
Results: Natalizumab binds the α4 β-propeller domain outside the ligand binding groove for domain 1 of vascular cell adhesion molecule (VCAM) and non-competitively antagonizes binding.
Conclusion: Natalizumab may push domain 2 of VCAM into a non-preferred orientation upon integrin binding.
Significance: Positioning of species-specific substitutions outside of ligand-binding sites leads to surprising antibody mechanisms of action.
Keywords: Adhesion, Cell Surface, Crystal Structure, Immunology, Integrins, Structure
Abstract
Natalizumab antibody to α4-integrins is used in therapy of multiple sclerosis and Crohn's disease. A crystal structure of the Fab bound to an α4 integrin β-propeller and thigh domain fragment shows that natalizumab recognizes human-mouse differences on the circumference of the β-propeller domain. The epitope is adjacent to but outside of a ligand-binding groove formed at the interface with the β-subunit βI domain and shows no difference in structure when bound to Fab. Competition between Fab and the ligand vascular cell adhesion molecule (VCAM) for binding to cell surface α4β1 shows noncompetitive antagonism. In agreement, VCAM docking models suggest that binding of domain 1 of VCAM to α4-integrins is unimpeded by the Fab, and that bound Fab requires a change in orientation between domains 1 and 2 of VCAM for binding to α4β1. Mapping of species-specific differences onto α4β1 and α4β7 shows that their ligand-binding sites are highly conserved. Skewing away from these conserved regions of the epitopes recognized by current therapeutic function-blocking antibodies has resulted in previously unanticipated mechanisms of action.
Introduction
Adhesion receptors of the immune system are required for all cell-cell interactions in innate and adaptive immune responses, as well as for leukocyte emigration from the bloodstream and migration within tissues (1). A new class of therapeutics termed selective adhesion molecule inhibitors target these receptors (2). A leading example is natalizumab, directed to the integrin α4 subunit that is present in both integrins α4β1 and α4β7 (1, 3, 4). Natalizumab is more effective than any other drug in preventing relapse in multiple sclerosis, where it has been used in over 100,000 patients (5, 6). Natalizumab also is efficacious in an inflammatory bowel disease, Crohn's disease (2, 7). However, little is known about the mechanism of adhesion blockade by natalizumab.
Integrin α4β1 binds to the immunoglobulin supergene family (IgSF)3 molecule, vascular cell adhesion molecule (VCAM) (1). VCAM is induced on venular endothelium in inflammation, including in brain in experimental autoimmune encephalitis. Antibody to α4β1 blocks lymphocyte emigration into brain, α4β1-dependent costimulation of immune responses, and experimental autoimmune encephalitis (8, 9).
Integrin α4β7 binds to the IgSF member mucosal addressin cell adhesion molecule (MAdCAM), which is expressed on vascular cells in mucosal tissues (1). α4β1 and not α4β7 is critical for experimental autoimmune encephalitis (10). Conversely, the efficacy of natalizumab in Crohn's disease appears to reflect blockade of α4β7-dependent interactions (2). Vedolizumab, an antibody specific for α4β7, shows promise in Crohn's disease (7).
We have previously reported the crystal structure of the integrin α4β7 headpiece bound to a small molecule antagonist, and to Fab of the mouse precursor of vedolizumab, Act-1 Fab (11). Because the complementarity-determining regions (CDRs) of Act-1 and vedolizumab are identical, we use these names interchangeably here. Vedolizumab bound on the β7 side of a long, wide ligand-binding groove at an extensive interface between the α4-subunit β-propeller domain and the β7-subunit βI domain.
Here, we report the crystal structure of natalizumab Fab bound to the integrin α4-subunit. Natalizumab binds on the opposite, α4 side of the ligand-binding groove. Because natalizumab did not appear to block binding of domain 1 of VCAM to the groove, we examined its mechanism of inhibition, which turns out to be noncompetitive. Apparently, natalizumab Fab imposes a change in orientation between D1 and D2 of VCAM bound to α4β1. Species-specific differences outside but near ligand-binding pockets appear to be targeted by many mouse anti-human therapeutic antibodies. Greater complexity exists in the mechanism of action of selective adhesion molecule inhibitors than was originally envisioned.
EXPERIMENTAL PROCEDURES
Protein Preparation and Purification
An α4β7 headpiece fragment containing α4 residues 1–587 and β7 residues 1–493, and C-terminal TEV cleavage sites, ACID-BASE coiled-coil, and purification tags secreted by CHO-Lec3.2.8.1 was prepared and purified as described (11). Pharmaceutical natalizumab (1 mg/ml) was digested with papain (0.01 mg/ml) in 10 mm cysteine in PBS, purified by Mono S eluting with a gradient of 0 to 0.4 m NaCl in MES, pH 6.0, and then by gel filtration with Superdex 75 in 20 mm Tris, pH 7.5, 0.15 m NaCl (TBS). The α4β7 headpiece (0.3 mg/ml) was mixed with natalizumab Fab (0.3 mg/ml) (a mole ratio of 1:3) and simultaneously digested by added TEV protease (0.1 mg/ml) and endo-β-N-acetylglucosaminidase H (Roche Applied Science) (0.1 mg/ml) at room temperature overnight. The mixture was passed through a nickel-nitrilotriacetic acid column and the Fab-α4β7 headpiece complex was further purified by gel filtration on Superdex 200 in TBS, 1 mm Ca2+, 1 mm Mg2+.
A cDNA encoding the VCAM D1D2 fragment (mature residues 1–202) was inserted into the in-house ET8 (ExpressTag-8) vector. ET8 is similar to ET1 (12), except it utilizes ligation-independent cloning and contains a HHHHHHA tag at the C terminus. The protein was stably expressed in HEK293S GnTI−/− cells (13) and purified by nickel-nitrilotriacetic acid-Sepharose and Superdex 75 gel filtration.
Crystallization and Diffraction
The α4β7-natalizumab Fab complex was concentrated to 4.5 mg/ml in 20 mm Tris, pH 7.5, 0.15 m NaCl, 1 mm CaCl2, 1 mm MgCl2 for crystallization. Using 0.1 μl of hanging drop vapor diffusion at 4 °C we obtained crystal-like solids from 10% PEG 20,000, 0.1 m Bicine, pH 9.0, 2% dioxane. We first optimized the concentration of PEG 20,000 and dioxane and the pH of Bicine, and then found NDSB-195 (non-detergent sulfobetaine, Hampton Research) by additive screening. Final needle crystals grew in 6% PEG 20,000, 0.1 m Bicine, pH 9.0, 5% dioxane, and 300 mm NDSB-195. Crystals were cryoprotected by direct transfer to 9% PEG 20000, 0.1 m Bicine, pH 9.0, 5% dioxane, 300 mm NDSB-195, 30% PEG 400 and flash-cooled in liquid nitrogen. Diffraction data were collected at APS GM/CA-CAT beamline and processed with HKL2000 (14) to 3.05 Å. Late in refinement, data were reprocessed with XDS (15). Higher completeness was obtained in higher resolution shells, and useful data were found to extend to 2.84 Å using the cross-correlation method (16).
Structure Determination and Refinement
The structure was solved by molecular replacement using Phaser (17) first searching with the α4 β-propeller domain, then with different Fab, and then the α4 thigh domain (11). Models were iteratively improved by building with COOT (18), refinement with PHENIX (19), and validation with MOLPROBITY (20).
Radioligand Binding Assay
Natalizumab Fab was labeled with 125I (PerkinElmer Life Sciences) using IODO-GEN to a specific activity of 134 Ci/mmol as described (21), and concentrated to 1.5 mg/ml. Jurkat cells (1 × 107/ml) in HBS (20 mm HEPES, pH 7.4, 137 mm NaCl, 5 mm KCl, 5.5 mm glucose, 10 mg/ml of BSA) were treated with 4 mm Mn2+ and 0.4 mm Ca2+ (with or without VCAM D1D2) for 30 min at 37 °C. Cells were aliquoted into 0.6-ml centrifugal tubes (10 μl) and put on ice. Various concentrations of 125I-natalizumab Fab in 10 μl of HBS were added to the cells to give final cation concentrations of 2 mm Mn2+ and 0.2 mm Ca2+. After 2 h, bound ligand was quantified as described (21). Nonspecific binding was determined in the presence of 100 μg/ml of cold natalizumab IgG for Fab concentrations of 10 nm or lower. At higher Fab concentrations, the IgG concentration was increased proportionally. Specific binding was calculated by subtracting nonspecific Fab binding from total binding.
Competition with increasing concentrations of non-labeled natalizumab Fab or IgG was performed similarly with 20 nm 125I-natalizumab Fab. Protein concentrations were determined by measuring A280. Extinction coefficients for natalizumab Fab and IgG were calculated from amino acid sequences for the constant regions of human immunoglobulin γ4 and κ chains (Uniprot) and the variable regions of natalizumab (22). Values were 76,250 m−1 for Fab fragment and 224,320 m−1 for IgG.
Nonlinear Curve Fitting
Specific natalizumab Fab saturation binding data were fitted to Equation 1 with Prism version 5.04 (GraphPad Software, San Diego, CA), which uses the Levenberg-Marquardt method for performing nonlinear regression.
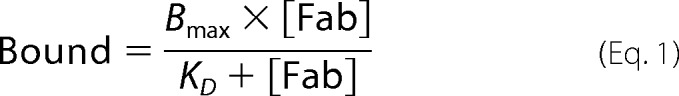 |
A series of natalizumab Fab saturation curves that were obtained in the presence of increasing concentrations of VCAM were fitted globally with Prism to a modified Gaddum/Schild equation (23) (Equation 2).
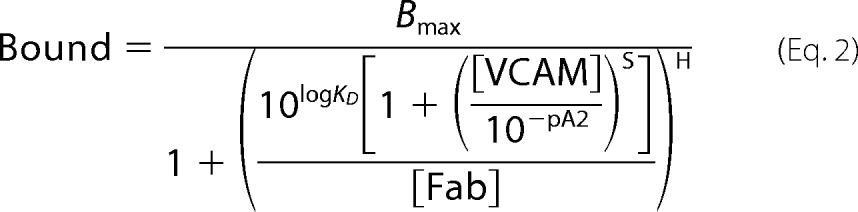 |
Bmax is the number of binding sites for natalizumab Fab, KD is the dissociation constant for natalizumab Fab binding in the absence of VCAM, [Fab] and [VCAM] are the concentrations of natalizumab Fab and VCAM (D1D2), respectively, pA2 is the negative logarithm of the antagonist concentration required for shifting the dose-response curve by a factor of 2, H is the Hill slope, and S is the Schild slope, which is 1 in case of competitive antagonism. Data fitting revealed that H did not significantly deviate from 1 (F-test, p = 0.82) and was therefore constrained to H = 1.
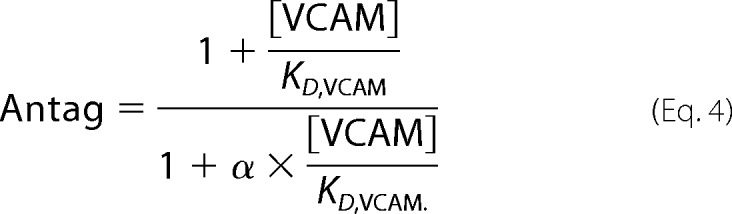
To calculate ternary complex model constants, the same data set was fitted to the allosteric EC50 shift model built into the Prism software (Equations 3 and 4) (24). Noncompetitive antagonism is described by the ternary complex model, in which the KD for a ternary complex equals KD/α for the binary complex (24). α, the ternary complex constant, describes how an allosteric modulator affects ligand binding, and KD,VCAM is the equilibrium dissociation constant for VCAM binding.
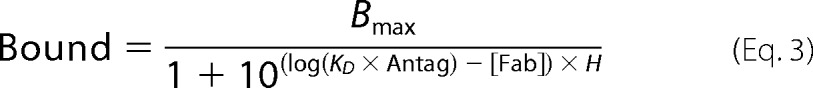 |
From Equation 3,
 |
Similar to the modified Gaddum/Schild model, the Hill coefficient was not significantly different from 1 (F-test, p = 0.74) and was constrained to H = 1.
To derive KI values for nonlabeled natalizumab Fab or IgG, competition binding data were fitted as described in Ref. 21 to a one site binding model. Radioligand concentration and binding affinity were [125I-Fab] = 20 nm and KD,Fab = 19.6 nm, respectively.
Accession Code
The coordinates and the structure factors of α4β7-natalizumab Fab complex have been deposited to the RCSB Protein Data Bank with the accession code 4IRZ.
RESULTS
Natalizumab Fab co-crystallized with the integrin α4-subunit from an α4β7 headpiece preparation (Figs. 1A and 2) with one complex in the asymmetric unit. Resolution could be extended (16) to 2.84 Å (Table 1). The β7 headpiece fragment dissociated during crystallization; only the Fab and α4 headpiece fragment are present in crystals (Figs. 1A and 2B). Dissociation was unrelated to natalizumab Fab binding, because natalizumab actually stabilizes association between α4 and β7 (Fig. 2, A and B), as previously demonstrated with vedolizumab (11). Furthermore, previous electron microscopy (EM) shows natalizumab binding to the α4β7 headpiece with excellent density for the β7 subunit, and the same orientation between the α4 subunit and natalizumab Fab as seen in the crystal structure (Fig. 1D) (11). The high pH of 9.0 and 5% organic solvent dioxane in crystallization may have contributed to α4 and β7 dissociation. The structure of the α4 β-propeller and thigh domains are essentially identical to those in the vedolizumab Fab complex, except for a difference in orientation of 8 to 24° at the flexible interface between the β-propeller and thigh domains (Fig. 1, A and B).
FIGURE 1.

Overall natalizumab complex structure. A, natalizumab Fab bound to the α4 headpiece. B, vedolizumab and a small molecule antagonist (shown in stick with pink carbons) bound to the α4β7 headpiece (11), in identical orientation. Residues Tyr-187 and Trp-188 mutationally important in VCAM binding (27) are shown in stick. C, the natalizumab binding site with a view down the β-propeller pseudosymmetry axis. The α4 β-propeller domain is shown in rainbow colors, from N (blue) to C (red). α4 side chains that contact natalizumab are shown in stick. D, EM class averages of natalizumab Fab bound to the α4β7 headpiece from Fig. 7C of Ref. 11. From left to right are representative averages with a closed headpiece, intermediate headpiece, and open headpiece. The orientation is similar to that of the crystal structure in A.
FIGURE 2.
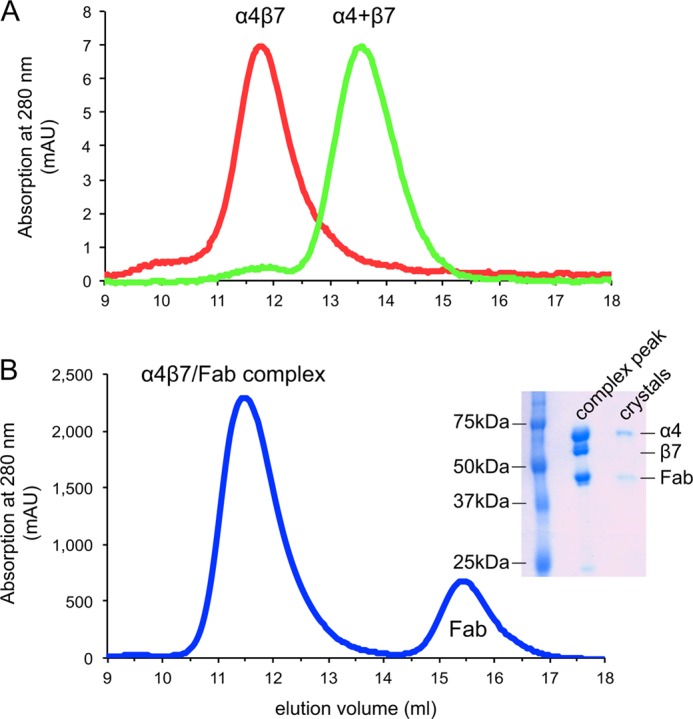
Integrin subunit association or dissociation. A, gel filtration of α4β7 headpiece before (red curve) and after TEV protease treatment (green curve). B, gel filtration of the α4β7 headpiece treated with TEV in the presence of natalizumab Fab. The α4β7 headpiece was incubated with TEV at 3:1 mass ratio at room temperature for 12 h and then separated by Superdex 200 in TBS with 1 mm Ca2+/Mg2+. The α4 and β7 subunits dissociated in the absence of natalizumab (green curve in A) and remained associated in the presence of natalizumab Fab (blue curve in B). The inset in B shows SDS-PAGE of protein samples from the α4β7-Fab complex peak in gel filtration and from crystals.
TABLE 1.
Diffraction data and structure refinement statistics
Numbers in parentheses correspond to the last resolution shell.
| Diffraction Data | |
| Wavelength (Å) | 1.000 |
| Space group | P212121 |
| Cell parameters | |
| a, b, c (Å) | 73.6, 77.9, 217.7 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution range (Å) | 50.0–2.84 (3.01–2.84) |
| Observed reflections | 221,831 |
| Unique reflections | 30,300 |
| Redundancy | 7.3 (7.1) |
| I/σI | 6.32 (0.6) |
| Completeness (%) | 99.7 (98.2) |
| Wilson B factors (Å2) | 60.3 |
| Rmerge (%)a | 34 (307.6) |
| CC½b | 98.6 (25.2) |
| Refinement | |
| Rworkc | 0.237 |
| Rfreed | 0.287 |
| Root mean square deviation | |
| Bond (Å) | 0.004 |
| Angle (°) | 0.588 |
| Ramachandran plot (%)e | 92.8, 7.1, 0.1 |
a Rmerge = ΣhklΣi|Ii(hkl)i − 〈I(hkl)〉|ΣhklΣiIi(hkl).
b Pearson's correlation coefficient between average intensities of random half-datasets of the measurements for each unique reflection (16).
c Rwork = Σhkl‖Fobs(hkl)| − |Fcalc(hkl)‖/Σhkl|Fobs(hkl)|.
d Rfree is the R value obtained for a test of reflections.
e Residues in favored, allowed, and outlier regions calculated with the MolProbity server (20).
A previously undescribed (11) interaction between the thigh FG′ loop and the β-propeller domain is conserved in all five examples of α4β7 and α4 in crystal lattices. When superposition is based on the β-propeller domain, residues 563–569 in the highly extended thigh FG′ loop vary little in position (Fig. 3A). In contrast, when superposition is based on the thigh domain, their position varies markedly (Fig. 3B). Stronger coupling of the thigh FG′ loop to the β-propeller domain than to the thigh domain is consistent with extensive structural interactions.
FIGURE 3.
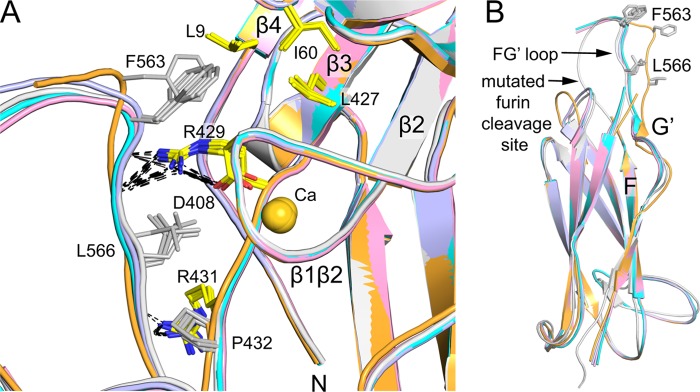
Interaction between the thigh FG′ loop and β-propeller domain. Views are after superposition of five examples of α4 structures (here and Ref. 11) on the β-propeller (A) and thigh domain (B). Important side chains are shown in stick and hydrogen bonds are dashed. The calcium ion in blade W7 is shown as a sphere.
Before describing this interaction, let us briefly review the integrin β-propeller structure (25). Each β-sheet, also termed a propeller blade or “W,” has four anti-parallel β-strands arranged like legs of the letter W. The blades assemble around a 7-fold pseudosymmetry axis, with β-strand 1 lining a solvent-filled central cavity and β-strand 4 forming the circumference of the propeller (Fig. 1C). The last three blades in α4 have Ca2+-binding sites in their β1-β2 loops (26). The sequence wraps around the propeller so blades 1 and 7 are adjacent; furthermore, in blade 7, β1-β3 and β4 are from C-terminal and N-terminal portions of the β-propeller sequence, respectively (Fig. 1C).
The thigh FG′ loop stabilizes the region of the β-propeller where its N and C termini are knit together in W7 (Figs. 1C and 3A). Thigh residue Phe-563 binds in a hydrophobic pocket formed by Leu-9 at the beginning of β-strand 4 and Leu-427 at the end of β-strand 3 in blade 7, and Ile-60 in the β3-β4 loop of blade 6 (Fig. 3A). Phe-563 also packs against the aliphatic portion of Asp-408 that coordinates Ca2+ in the β1-β2 loop of blade 7. Moreover, Arg-429 in the loop following the β3-strand of blade 7 forms a π-cation bond to Phe-563, and hydrogen bonds to both Asp-408 and the backbone following Phe-563 (Fig. 3A). Leu-566 contributes a hydrophobic environment that stabilizes these polar interactions, and also interacts with Pro-432 and the aliphatic portion of Arg-431, which hydrogen bonds to the backbone of thigh residue 567. These interactions with the thigh domain may help the β-propeller domain to fold, and are consistent with findings for a large number of different integrins that heterodimer fragments containing α-subunits truncated after the thigh domain, but not after the β-propeller domain, are well expressed.
The Natalizumab Binding Site
Natalizumab binds to α4 β-propeller blades 2 to 4 (Fig. 1C). The ligand binding groove in α4β7 is formed at the interface between the α4 β-propeller and β7 βI domains (Fig. 1B) (11). α4 forms one wall of this groove and part of its lower surface. Natalizumab binds beside this groove (Fig. 1A). Long loops in the β-propeller that help form this groove separate natalizumab from the groove (Fig. 1, A and B). Among a large number of individual α4 β-propeller amino acid substitutions tested, only mutations of Tyr-187, Trp-188, and Gly-190 decreased binding to VCAM (27). Tyr-187 forms part of the α4 wall, Trp-188 forms part of the bottom of the groove (Fig. 1A) and each contacts a small molecule α4 antagonist (Fig. 1B). Notably, Tyr-187 and Trp-188 are distal from the natalizumab footprint (Fig. 1A). Gly-190 is in a highly conserved, buried position in the seven FG-GAP sequence repeats that form integrin β-propellers. The Ala side chain in the G190A mutation clashes with the backbone of Pro-183 and is expected to alter the conformation of the loop bearing Tyr-187 and Trp-188.
The circumference of integrin β-propellers is formed by the β4 strands; the ligand binding and βI domain-binding face is formed by the β4-β1 and β2-β3 loops, and the opposite face is formed by the β1-β2 loops (some of which bind cations) and β3-β4 loops. Natalizumab Fab binds to the circumference of the β-propeller, with its footprint extending all the way to the face bearing the β3-β4 loops, but missing by 15 to 20 Å the face of the β-propeller that binds the βI domain and ligand and bears Tyr-187 and Trp-188 (Fig. 1A and Fig. 4). The epitope includes the β4-β1 loop preceding W3, the β4-strand of W3, and the β3-β4 loops of W3 and W4 (Fig. 4). The footprint extends to the tips of the β3-β4 loops of W3 and W4 on the divalent cation binding β face, but includes only tip-distal portions of the W3 β4-β1 loop on the ligand binding face.
FIGURE 4.

The natalizumab binding site. A, the epitope on α4 from the point of view of Fab. The α4 β-propeller from the natalizumab complex is shown in rainbow colors, and a superimposed α4 β-propeller domain from the vedolizumab complex is shown in gray, together with a bound small molecule antagonist in stick with gold carbons as a marker of the position of the ligand-binding site. All side chains (and one N-acetylglucosamine residue) within 4 Å of natalizumab are shown in stick, and backbone atoms that hydrogen bond to antibody are shown as spheres. B, a different view of the epitope that includes the Fab. Carbons of α4 and natalizumab H and L chains are in rainbow colors, gray, and light blue, respectively. All natalizumab-α4 hydrogen bonds are shown as dashes. All side chains with contacts of less than 3.9 Å across the interface are shown. Backbone atoms that participate in hydrogen bonds are shown as spheres.
Natalizumab induces no significant change in α4 conformation. The β-sheets and loops of the α4 β-propeller domain have the same conformations as when natalizumab is not bound and the β7 βI domain is bound (Fig. 4A). The side chains differ no more in rotamer than would be seen for the same protein in different crystal lattices.
Natalizumab buries a total solvent accessible surface area of 2060 Å2, with the heavy and light chains burying close to 75 and 25% of this area, respectively. The contact is dominated by the H chain CDR3 loop, which extends into a cleft on the cylindrical β-propeller surface formed between W2 and W3 (Fig. 4B). The central portion of the cleft is lined by invariant α4 hydrophobic residues that include Tyr-202 in β4 of W2, Pro-149 near the end of β4 of W2, and Leu-206 and Phe-162 of the β1–4 loops following W2 and W3, respectively (Fig. 4A). The long CDR H3 loop extends deep into this crevice, enabling its Tyr-101 and Tyr-108 residues both to bury hydrophobic residues and form hydrogen bonds to main chain atoms in β4 of W3 and in the segment following β4 of W2 (Fig. 4B). Contact even reaches deeper to Phe-193 in the β3-strand of W3 (Fig. 4A). β-Propeller loops that form and surround the hydrophobic pocket also contribute contacts, including the β4-β1 loops following W1, W2, and W3 and the β3-β4 loops of W3 and W4. Lys-201 in the W3 β4-strand and Lys-256 in the W4 β3-β4 loop, each more peripheral than the central hydrophobic region yet still well within the antibody-binding interface, make important, charged hydrogen bonds to the antibody and insert in negatively charged antibody pockets. The Gln-152 side chain forms a hydrogen bond to the CDR H3 backbone (Fig. 4B).
Natalizumab recognizes an α4 region with significant sequence variation among human and mammals commonly used in disease models and toxicology (Fig. 5). By contrast, the long narrow binding groove for small molecule antagonists and the biological ligands MAdCAM and VCAM is highly conserved in α4β7 (Fig. 5A) and invariant in α4β1 (Fig. 5C). Domain 1 of VCAM can be docked in this groove with some precision (Fig. 5D) by placing the Asp side chain of its rigid, integrin-binding loop in the same βI domain MIDAS Mg2+-coordinating position as the carboxyl group of a co-crystallized antagonist (11) (Fig. 5A). This model is well supported by the excellent fit of D1 of VCAM into the α4β1 or α4β7 ligand-binding groove with the long axis of D1 aligned with the groove (11). Furthermore, this model places VCAM in contact with α4 residues Tyr-187 and Trp-188 (Figs. 1A and 5), shown mutationally to be important in binding VCAM (27). The orientation between D1 and D2 of VCAM-1 is variable among crystal structures (28, 29) (Fig. 6). D1 of docked VCAM does not clash with natalizumab Fab; however, where D2 of VCAM emerges from the groove, it clashes in some orientations (Fig. 6, A and B). Our docking model thus predicts a noncompetitive mechanism of antagonism where natalizumab would lower affinity of α4β1 by limiting the number of VCAM conformations accessible for binding (Fig. 6B).
FIGURE 5.

Species-specific differences around the integrin α4β7 and α4β1 ligand-binding sites. Conservation on the solvent accessible surface is displayed from invariant (1, green or blue) to low (0.1, red) for A, C, and D using the species shown in panel E. In B, mouse-human sequence is shown as invariant (green or blue) or different (red). Invariant α and β subunit residues are shown in green and blue, respectively, to visualize the subunit interface. Additionally, invariant VCAM-binding α4 residues Tyr-187 and Trp-188 are in yellow and β subunit MIDAS and ADMIDAS metal ions are shown as green spheres. Antibody footprints are outlined in yellow dashes (N for natalizumab) and key antigenic residues are labeled. A small molecule antagonist bound to α4β7 is shown in stick with orange carbons (11). VCAM is shown in ribbon representation in the orientation found previously in docking to α4β7 (11). A–D are in identical orientations. The α4β1 model was made by superimposing the β1 βI domain from α5β1 (44) onto α4β7 (11). The location of the ligand binding groove is marked with dashed lines. Sequence conservation was calculated by AL2CO (51) with species equally weighted, using the sum of pairs measure with the BLOSUM62 matrix with normalization of the scoring matrix. There was no further normalization, so results for α4, β1, and β7 all use the same scale. E, sequence variation in the natalizumab epitope. Sequences are of species found to be positive (+) or negative (−) for natalizumab reactivity in European Medicines Agency filings. Residues in the epitope are numbered. Residues most important for species reactivity are in bold. Residues that differ from human are red.
FIGURE 6.

Model of VCAM binding to a natalizumab Fab-α4β1 complex. Transparent solvent accessible surfaces are shown of the α4 β-propeller domain (wheat), β1 βI domain (light blue), and natalizumab Fab (gray). Two examples of VCAM D1D2 crystal structures (chain A from Ref. 28 and chain B from Ref. 29) that differ the most in D1-D2 orientation are shown in ribbon representation, docked as described (11). The model of α4β1 is described in the legend to Fig. 5. Strong clashes show regions where the schematic is largely obscured by the transparent surface.
Mechanism of Inhibition by Natalizumab
We set up an assay of competition between VCAM and 125I-natalizumab Fab. Affinities were measured on Jurkat T lymphoblastoid cells in buffer containing 2 mm Mn2+ and 0.2 mm Ca2+ to activate integrin α4β1; Jurkat cells express α4β1 and lack α4β7. Binding was maximal after 2 h (Fig. 7A) and was saturable with 86,000 ± 6,000 Fab bound per cell with a KD = 19.6 ± 4.6 nm (Fig. 7B). Given the importance of tyrosines in the CDR loops of natalizumab, we expected that their iodination might lower affinity. Non-labeled natalizumab Fab inhibited radiolabeled Fab binding with KI = 6.4 nm (Fig. 7C). The difference in affinity between labeled and unlabeled Fab is significant and important for appreciating the affinity of the unlabeled Fab; however, it does not affect the measurements of competition with VCAM described in the following section. Inhibition of 125I-natalizumab Fab by unlabeled natalizumab IgG occurs with an apparent KI = 0.28 nm (Fig. 7C). The 20-fold higher avidity of the IgG than the affinity of the Fab demonstrates bivalent binding of natalizumab IgG to cell surface α4β1.
FIGURE 7.

Binding of 125I-labeled natalizumab Fab to T lymphoblastoid cells. A, time course of binding. B, saturation binding. C, inhibition of binding by unlabeled natalizumab Fab and IgG. D and E, binding of 125I-Fab at different concentrations of input Fab and VCAM D1D2 fragment fit to an allosteric ternary complex model (D) or the Gaddum-Schild equation (E). Bars show S.E. for each data point in two or three triplicate experiments. F, different plot of same data as in D and E. Lines in B-D show fit to equations. Fit values are shown together with S.E. unless 95% confidence intervals (CI) are otherwise shown. α4β1 integrins on Jurkat T lymphoblastoid cells were activated with Mn2+ for 30 min at 37 °C, and equilibrium 125I-Fab binding was measured after 2 h on ice as described under ”Experimental Procedures.“
125I-Natalizumab Fab binding was competed with a VCAM fragment containing domains 1 and 2 (D1D2) (Fig. 7D). The range of VCAM concentrations from 0.1 to 100 μm spanned its KD for α4β1 as shown below (Fig. 7D). Furthermore, the range of 125I-Fab concentrations from 0.1 to 100 nm spanned the KD values of Fab for α4β1 and extended to the concentration giving saturation binding in the absence of VCAM. All data were fit globally to an allosteric ternary complex model (24), which gave the lines shown in Fig. 7D and to a Gaddum-Schild equation, which gave the lines shown in Fig. 7E. The two methods yielded nearly identical estimates of the KD of VCAM (1.9 or 1.6 μm) and the KD of 125I-natalizumab Fab (24.7 or 22.9 nm). The Schild slope was S = 0.595 ± 0.084, with a 95% confidence interval of 0.429 to 0.760 (Fig. 7E). The data fit noncompetitive antagonism significantly better than competitive antagonism with S = 1 (F-test, p = 0.0003). The fit to the allosteric model yielded a ternary complex constant α = 0.083 (Fig. 7D). The α value is significantly less than 1, showing that natalizumab is an allosteric inhibitor (24).
The allosteric coupling constant is the factor by which the affinity constant is modified by binding of the allosteric modulator (noncompetitive antagonist). Thus the KD of VCAM for Mn2+-activated α4β1 changes from 1.9 μm in the absence of natalizumab to 23 μm in the presence of natalizumab Fab. The corresponding free energy penalty for binding in the presence of Fab is 1.35 kcal/mol.
DISCUSSION
Our study shows how a clinically important antibody binds to the β-propeller domain of α4 integrins, noncompetitively blocks binding of VCAM to α4β1, and illuminates a large number of previous studies on α4 integrin structure and function. Integrin α4 antibodies were classified by effect on function and cross-competition as binding to epitopes A, B, or C (30). Antibodies to epitope B block binding to VCAM and fibronectin, whereas antibodies to epitopes A and C partially inhibit binding only to fibronectin or are non-inhibitory, respectively. The mouse anti-human precursor to natalizumab (31), TY21/6, binds the B epitope (32).
Among the α4 residues that natalizumab contacts, only four differ between mouse and human (Fig. 5, B and E). Previous chimera work showed that replacing the human α4 sequence with mouse segments of 50 to 100 residues between residues 108 and 268, i.e. β-propeller blades 2 to 4, was sufficient to abolish or diminish reactivity of all tested B epitope antibodies (33, 34). Because the smallest regions studied encompassed an entire β-propeller blade, whether antibodies mapped to the propeller circumference bearing the β4-strand, to the βI-proximal β4-β1 or β2-β3 loops, or to the cation-loop proximal β1-β2 and β3-β4 loops, could not be defined. Furthermore, because none of the studies demonstrated that a reciprocal swap would reconstitute the epitope, species-specific residues required for the epitope might have extended beyond the minimal mouse region required to eliminate binding, and indeed this was suggested by some swaps that did not completely eliminate reactivity. Therefore, little could be said previously about where B epitope antibodies bound, except that their epitopes included portions of β-propeller blades 2–4.
However, based on our structure, previous mapping data, and the finding that all B epitope antibodies cross-block one another, it now seems likely that all B epitope antibodies bind to a site that overlaps with natalizumab, and also do not overlap with the ligand-binding site. Thus, reactivity of HP1/2, HP2/1, HP2/4, L25, and P4C2 is abolished or decreased ≥80% by mouse residues 152–203 (34), which replace four human residues including two in the natalizumab epitope, Gln-152 and Lys-201 (Table 2). P4C2 and Z0E4 reactivity is abolished by mouse residues 108–182 (33), which alter nine human residues including Gln-152 (Table 2). HP2/1 and SG/73 reactivity is abolished by mouse residues 195–268 (33), which replace four residues including Lys-201, Lys-208, and Lys-256 in the natalizumab epitope (Table 2). In summary, there is substantial overlap between the species-specific residues recognized by natalizumab and the regions to which reactivity has been mapped for all previously characterized, function-blocking α4 integrin antibodies.
TABLE 2.
Species-specific differences in α4 β-propeller blades 2–4
Residues in the natalizumab epitope are in bold.
| Region | Residue | Human | Mouse |
|---|---|---|---|
| W2, β2-β3 | 121 | Ile | Met |
| W2, β2-β3 | 123 | Asn | Ser |
| W2, β2-β3 | 124 | Glu | Asp |
| W2, β3 | 131 | Gly | Ile |
| W2, β3 | 134 | Gly | Val |
| W2, β3 | 135 | Val | Met |
| W2, β3-β4 | 137 | Pro | Ser |
| W2, β4 | 147 | Ile | Met |
| W3, β4-β1 | 152 | Gln | Lys |
| W3, β4-β1 | 155 | Val | Thr |
| W3, β1-β2 | 175 | Lys | Gln |
| W3, β3-β4 | 201 | Lys | Gln |
| W4, β4-β1 | 208 | Lys | Arg |
| W4, β1-β2 | 230 | Gln | Pro |
| W4, β3-β4 | 256 | Lys | Asn |
During development of natalizumab, it was tested on a range of species in disease model and toxicology studies. The species variation in reactivity with natalizumab (Fig. 5E) is in excellent agreement with our structural analysis in demonstrating the importance of residues Gln-152, Lys-201, and Lys-256. When any one of these differs from human, reactivity with natalizumab is lost (Fig. 5E). By contrast, Lys-208, on the edge of the epitope (Fig. 4A), is not required for reactivity (Fig. 5E).
The KD of natalizumab Fab for cell surface α4β1 is 6.4 nm. The KD of its IgG is 0.28 nm, in agreement with previous measurements (22). The 20-fold higher affinity of the IgG strongly suggests that it binds bivalently to cell surface α4 integrins.
Although natalizumab starts out bivalent, after administration much of it becomes functionally monovalent. Like other IgG4 antibodies, its hinge region cysteines are partially reduced, and half-molecules containing a heavy and light chain pair exchange with other IgG4 molecules. Thus a substantial fraction of natalizumab becomes monovalent (35). Therefore in patients natalizumab is both bivalent and monovalent, and the monovalent IgG4 is expected to bind with a KD of 6.4 nm as measured here with Fab.
The VCAM D1D2 fragment has a KD of 2 μm for Mn2+-activated, cell surface integrin α4β1. The binding site centered in D1 includes a portion of D2, but there is no evidence it extends beyond D2 into D3 (36, 37). MAdCAM, which contains only two IgSF domains, binds to integrin α4β7 similarly to the docking model with VCAM, with much of D2 not in contact with the integrin; D3 if present would locate far from the integrin (11). VCAM is alternatively spliced, with IgSF domain 4 (D4) present in an isoform with 7 IgSF domains (7D) and absent in the isoform with 6 IgSF domains (6D) in the ectodomain. Domain 4 shows high sequence identity to domain 1, an identical integrin-binding motif, and binds to α4β1. The 7D isoform with D1 mutationally removed binds similarly to the native 6D isoform (38–40).
The D1D2 fragment of VCAM used here ensures measurement of true, i.e. monovalent, affinity. Previous careful estimates of the affinity of the 7D fragment of VCAM for cell surface α4β1 in Mn2+ range from 9 to 33 nm (41). The much higher affinity estimates for 7D than D1D2 VCAM fragment strongly suggest that the 7D fragment binds bivalently to cell surface α4β1. In general, adhesion molecules have low affinity for their ligands compared with receptors for soluble ligands. A full-length five IgSF ectodomain fragment of ICAM-1 binds to Mn2+-activated cell surface LFA-1 (integrin αLβ2) with a KD of 9 μm (21), similar to the KD of 2 μm measured here for binding of α4β1 to VCAM D1D2.
The noncompetitive inhibition mechanism described here is a departure from previous concepts on how antibodies block function, particularly for integrins, which are known to undergo conformational change. Natalizumab clearly binds to a different site on α4β1 than VCAM, making the noncompetitive mechanism conceptually easy to appreciate. A change in the conformational space accessible to D2 of VCAM that is imposed by natalizumab is the simplest explanation of the structural and ligand competition data. Crystal structures show multiple orientations between domains 1 and 2 of VCAM that differ by up to 35° (Fig. 6A). No doubt, greater variation in the D1-D2 orientation is possible than has yet been sampled by crystallography. D2 of VCAM has a role in binding integrin α4β1 (37), and another interpretation of our results is that natalizumab would not only affect the D1-D2 orientation but also disrupt some of the interactions through D2 while leaving intact the more important interactions with D1. The net result is a 12-fold loss in affinity of α4β1 for VCAM imposed by natalizumab. In contrast to these results, structural studies with several inhibitory antibodies to integrin β-subunits have directly demonstrated truly allosteric effects. These antibodies bind too far from the ligand-binding site to sterically hinder ligand binding, and stabilize the low-affinity, closed integrin headpiece relative to the high-affinity, open integrin headpiece (42–44).
Other B epitope antibodies inhibit VCAM binding to α4β1 by a similar noncompetitive mechanism (32). The measurements were less quantitative, because they used bivalent IgG and ELISA with wash steps that disrupt equilibrium binding. Nonetheless, plots of single-reciprocal antibody binding were hyperbolic with increasing VCAM concentration (Fig. 7E shows this type of plot), and suggested an allosteric or noncompetitive mechanism of action. These data were interpreted as an allosteric change induced by VCAM in the integrin α4-subunit (32).
Formally, neither our structural or competition data can rule out this alternative mechanism of a VCAM-induced change in the conformation of the α4 β-propeller domain that lowers the affinity of natalizumab for its epitope. The epitope does not change conformation in α4 structures to date, i.e. when bound to natalizumab, when α4 is bound to the β7 subunit, which in turn is bound to vedolizumab, or when additionally a small molecule antagonist binds to α4 and β7 in the ligand-binding groove (11). As is the case for most integrin antagonists (45), this antagonist causes reshaping of the integrin β-subunit βI domain but not the α-subunit β-propeller domain (11). Whether VCAM induces a conformational change in α4 must await a co-crystal structure.
It is on structural grounds that we believe it is unlikely that VCAM binding would transmit conformational change through the β-propeller domain to the natalizumab epitope. β-Propellers are exceptionally large domains that are formed from unusually closely packed β-sheets (46). β-Sheets are rigid structural elements, and whereas most domains are two or three layers thick, β-propellers are eight layers thick across their diameter. Thus β-propellers are predicted to be unusually rigid. Indeed, their function in trimeric G proteins and integrins may be to serve as rigid platforms to stabilize their allosteric signaling β/α domain partners. Integrin βI domains, with their three layer β/α structures, transmit conformational change through motions of α-helices (45, 47).
Our findings demonstrate that antibodies to integrins that inhibit by a noncompetitive mechanism can nonetheless be highly effective therapeutics. Strong blockade of adhesion in vitro is also not required. Consistent with noncompetitive inhibition, it is difficult to inhibit cell adhesion to VCAM with natalizumab in Mn2+, particularly at high VCAM density.4
Because ligand-binding sites are well conserved across species (Fig. 4), the finding here that a function-blocking, species-specific antibody binds beside, rather than in, a ligand-binding site may be more common than not. Another species-specific therapeutic antibody, vedolizumab, also binds to the edge of the ligand-binding site, but on the opposite, β7 side (11) (Fig. 1B). Vedolizumab inhibits binding of MAdCAM, but not VCAM, to integrin α4β7 (48). This unexpected result appears to be a consequence of binding close enough to the ligand-binding site to inhibit binding of MAdCAM, but not quite close enough to inhibit binding of VCAM. Similarly, efalizumab to LFA-1 binds to the αL αI domain outside the binding site for D1 of ICAM-1, in a position where it would clash with or require bending away of D2 of ICAM-1 (49). The greater presence of species-specific differences outside of the ligand-binding site has thus skewed the current generation of therapeutic antibodies toward previously unanticipated mechanisms of action, which include noncompetitive antagonism and inhibition of binding of some and not other ligands. Currently, technology is maturing to generate synthetic antibody libraries that are devoid of tolerance to self (50). It will be interesting to see whether next generation antibodies recognize a broader range of epitopes including highly conserved ligand-binding epitopes, and reveal even further surprises.
Acknowledgment
We gratefully acknowledge Eric Fedyk of Millennium Pharmaceuticals, a Takeda Company (Cambridge, MA).
This work was supported, in whole or in part, by National Institutes of Health Grant HL-103526.
The atomic coordinates and structure factors (code 4IRZ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
T. Yednock, Elan Pharmaceuticals, personal communication.
- IgSF
- immunoglobulin supergene family
- VCAM
- vascular cell adhesion molecule
- MAdCAM
- mucosal addressin cell adhesion molecule
- CDR
- complementarity-determining region
- TEV
- tobacco etch virus
- Bicine
- N,N-bis(2-hydroxyethyl)glycine
- ICAM-1
- intercellular adhesion molecule 1.
REFERENCES
- 1. Springer T. A. (1994) Traffic signals for lymphocyte recirculation and leukocyte emigration. The multi-step paradigm. Cell 76, 301–314 [DOI] [PubMed] [Google Scholar]
- 2. Thomas S., Baumgart D. C. (2012) Targeting leukocyte migration and adhesion in Crohn's disease and ulcerative colitis. Inflammopharmacology 20, 1–18 [DOI] [PubMed] [Google Scholar]
- 3. von Andrian U. H., Engelhardt B. (2003) α4 integrins as therapeutic targets in autoimmune disease. New Engl. J. Med. 348, 68–72 [DOI] [PubMed] [Google Scholar]
- 4. Polman C. H., O'Connor P. W., Havrdova E., Hutchinson M., Kappos L., Miller D. H., Phillips J. T., Lublin F. D., Giovannoni G., Wajgt A., Toal M., Lynn F., Panzara M. A., Sandrock A. W. (2006) A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. New Engl. J. Med. 354, 899–910 [DOI] [PubMed] [Google Scholar]
- 5. Bakhshai J., Bleu-Lainé R., Jung M., Lim J., Reyes C., Sun L., Rochester C., Shaya F. T. (2010) The cost effectiveness and budget impact of natalizumab for formulary inclusion. J. Med. Econ. 13, 63–69 [DOI] [PubMed] [Google Scholar]
- 6. O'Day K., Meyer K., Miller R. M., Agarwal S., Franklin M. (2011) Cost-effectiveness of natalizumab versus fingolimod for the treatment of relapsing multiple sclerosis. J. Med. Econ. 14, 617–627 [DOI] [PubMed] [Google Scholar]
- 7. Targan S. R., Feagan B. G., Fedorak R. N., Lashner B. A., Panaccione R., Present D. H., Spehlmann M. E., Rutgeerts P. J., Tulassay Z., Volfova M., Wolf D. C., Hernandez C., Bornstein J., Sandborn W. J. (2007) Natalizumab for the treatment of active Crohn's disease. Results of the ENCORE Trial. Gastroenterology 132, 1672–1683 [DOI] [PubMed] [Google Scholar]
- 8. Yednock T. A., Cannon C., Fritz L. C., Sanchez-Madrid F., Steinman L., Karin N. (1992) Prevention of experimental autoimmune encephalomyelitis by antibodies against α4β1 integrin. Nature 356, 63–66 [DOI] [PubMed] [Google Scholar]
- 9. Davis L. S., Oppenheimer-Marks N., Bednarczyk J. L., McIntyre B. W., Lipsky P. E. (1990) Fibronectin promotes proliferation of naive and memory T cells by signaling through both the VLA-4 and VLA-5 integrin molecules. J. Immunol. 145, 785–793 [PubMed] [Google Scholar]
- 10. Bauer M., Brakebusch C., Coisne C., Sixt M., Wekerle H., Engelhardt B., Fässler R. (2009) β1 integrins differentially control extravasation of inflammatory cell subsets into the CNS during autoimmunity. Proc. Natl. Acad. Sci. U.S.A. 106, 1920–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yu Y., Zhu J., Mi L. Z., Walz T., Sun H., Chen J.-F., Springer T. A. (2012) Structural specializations of α4β7, an integrin that mediates rolling adhesion. J. Cell Biol. 196, 131–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mi L. Z., Grey M. J., Nishida N., Walz T., Lu C., Springer T. A. (2008) Functional and structural stability of the epidermal growth factor receptor in detergent micelles and phospholipid nanodiscs. Biochemistry 47, 10314–10323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Reeves P. J., Callewaert N., Contreras R., Khorana H. G. (2002) Structure and function in rhodopsin. High-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyl transferase I-negative HEK293S stable mammalian cell line. Proc. Natl. Acad. Sci. U.S.A. 99, 13419–13424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 15. Kabsch W. (2001) in International Tables for Crystallography. F, Crystallography of Biological Macromolecules (Rossmann M. G., Arnold E., eds) pp. 730–734, Kluwer Academic Publishers, Dordrecht [Google Scholar]
- 16. Karplus P. A., Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 19. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) PHENIX. Building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 20. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) MolProbity. All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schürpf T., Springer T. A. (2011) Regulation of integrin affinity on cell surfaces. EMBO J. 30, 4712–4727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Léger O. J., Yednock T. A., Tanner L., Horner H. C., Hines D. K., Keen S., Saldanha J., Jones S. T., Fritz L. C., Bendig M. M. (1997) Humanization of a mouse antibody against human α4 integrin. A potential therapeutic for the treatment of multiple sclerosis. Hum. Antibodies 8, 3–16 [PubMed] [Google Scholar]
- 23. Lazareno S., Birdsall N. J. (1993) Estimation of competitive antagonist affinity from functional inhibition curves using the Gaddum, Schild and Cheng-Prusoff equations. Br. J. Pharmacol. 109, 1110–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Christopoulos A., Kenakin T. (2002) G protein-coupled receptor allosterism and complexing. Pharmacol. Rev. 54, 323–374 [DOI] [PubMed] [Google Scholar]
- 25. Springer T. A. (1997) Folding of the N-terminal, ligand-binding region of integrin α-subunits into a β-propeller domain. Proc. Natl. Acad. Sci. U.S.A. 94, 65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Springer T. A., Jing H., Takagi J. (2000) A novel Ca2+-binding β-hairpin loop better resembles integrin sequence motifs than the EF-hand. Cell 102, 275–277 [DOI] [PubMed] [Google Scholar]
- 27. Irie A., Kamata T., Puzon-McLaughlin W., Takada Y. (1995) Critical amino acid residues for ligand binding are clustered in a predicted β-turn of the third N-terminal repeat in the integrin α4 and α5 subunits. EMBO J. 14, 5550–5556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jones E. Y., Harlos K., Bottomley M. J., Robinson R. C., Driscoll P. C., Edwards R. M., Clements J. M., Dudgeon T. J., Stuart D. I. (1995) Crystal structure of an integrin-binding fragment of vascular cell adhesion molecule-1 at 1.8-Å resolution. Nature 373, 539–544 [DOI] [PubMed] [Google Scholar]
- 29. Wang J.-h., Stehle T., Pepinsky B., Liu J.-h., Karpusas M., Osborn L. (1996) Structure of a functional fragment of VCAM-1 refined at 1.9-Å resolution. Acta Crystallogr. D Biol. Crystallogr. 52, 369–379 [DOI] [PubMed] [Google Scholar]
- 30. Pulido R., Elices M. J., Campanero M. R., Osborn L., Schiffer S., García-Pardo A., Lobb R., Hemler M. E., Sánchez-Madrid F. (1991) Functional evidence for three distinct and independently inhibitable adhesion activities mediated by the human integrin VLA-4. Correlation with distinct α4 epitopes. J. Biol. Chem. 266, 10241–10245 [PubMed] [Google Scholar]
- 31. Kent S. J., Karlik S. J., Cannon C., Hines D. K., Yednock T. A., Fritz L. C., Horner H. C. (1995) A monoclonal antibody to α4 integrin suppresses and reverses active experimental allergic encephalomyelitis. J. Neuroimmunol. 58, 1–10 [DOI] [PubMed] [Google Scholar]
- 32. Newham P., Craig S. E., Clark K., Mould A. P., Humphries M. J. (1998) Analysis of ligand-induced and ligand-attenuated epitopes on the leukocyte integrin α4β1. VCAM-1, mucosal addressin cell adhesion molecule-1, and fibronectin induce distinct conformational changes. J. Immunol. 160, 4508–4517 [PubMed] [Google Scholar]
- 33. Kamata T., Puzon W., Takada Y. (1995) Identification of putative ligand-binding sites of the integrin α4β1 (VLA-4, CD49d/CD29). Biochem. J. 305, 945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schiffer S. G., Hemler M. E., Lobb R. R., Tizard R., Osborn L. (1995) Molecular mapping of functional antibody binding sites of α4 integrin. J. Biol. Chem. 270, 14270–14273 [DOI] [PubMed] [Google Scholar]
- 35. Shapiro R. I., Plavina T., Schlain B. R., Pepinsky R. B., Garber E. A., Jarpe M., Hochman P. S., Wehner N. G., Bard F., Motter R., Yednock T. A., Taylor F. R. (2011) Development and validation of immunoassays to quantify the half-antibody exchange of an IgG4 antibody, natalizumab (Tysabri(R)) with endogenous IgG4. J. Pharm. Biomed. Anal. 55, 168–175 [DOI] [PubMed] [Google Scholar]
- 36. Newham P., Craig S. E., Seddon G. N., Schofield N. R., Rees A., Edwards R. M., Jones E. Y., Humphries M. J. (1997) α4 integrin binding interfaces on VCAM-1 and MAdCAM-1. Integrin binding footprints identify accessory binding sites that play a role in integrin specificity. J. Biol. Chem. 272, 19429–19440 [DOI] [PubMed] [Google Scholar]
- 37. Chiu H. H., Crowe D. T., Renz M. E., Presta L. G., Jones S., Weissman I. L., Fong S. (1995) Similar but nonidentical amino acid residues on vascular cell adhesion molecule-1 are involved in the interaction with α4β1 and α4β7 under different activity states. J. Immunol. 155, 5257–5267 [PubMed] [Google Scholar]
- 38. Vonderheide R. H., Springer T. A. (1992) Lymphocyte adhesion through VLA-4. Evidence for a novel binding site in the alternatively spliced domain of VCAM-1 and an additional α4 integrin counter-receptor on stimulated endothelium. J. Exp. Med. 175, 1433–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vonderheide R. H., Tedder T. F., Springer T. A., Staunton D. E. (1994) Residues within a conserved amino acid motif of domains 1 and 4 of VCAM-1 are required for binding to VLA-4. J. Cell Biol. 125, 215–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chuluyan H. E., Osborn L., Lobb R., Issekutz A. C. (1995) Domains 1 and 4 of vascular cell adhesion molecule-1 (CD106) both support very late activation antigen-4 (CD49d/CD29)-dependent monocyte transendothelial migration. J. Immunol. 155, 3135–3144 [PubMed] [Google Scholar]
- 41. Chigaev A., Zwartz G., Graves S. W., Dwyer D. C., Tsuji H., Foutz T. D., Edwards B. S., Prossnitz E. R., Larson R. S., Sklar L. A. (2003) α4β1 integrin affinity changes govern cell adhesion. J. Biol. Chem. 278, 38174–38182 [DOI] [PubMed] [Google Scholar]
- 42. Luo B.-H., Strokovich K., Walz T., Springer T. A., Takagi J. (2004) Allosteric β1 integrin antibodies that stabilize the low affinity state by preventing the swing-out of the hybrid domain. J. Biol. Chem. 279, 27466–27471 [DOI] [PubMed] [Google Scholar]
- 43. Chen X., Xie C., Nishida N., Li Z., Walz T., Springer T. A. (2010) Requirement of open headpiece conformation for activation of leukocyte integrin αXβ2. Proc. Natl. Acad. Sci. U.S.A. 107, 14727–14732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nagae M., Re S., Mihara E., Nogi T., Sugita Y., Takagi J. (2012) Crystal structure of α5β1 integrin ectodomain. Atomic details of the fibronectin receptor. J. Cell Biol. 197, 131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Springer T. A., Dustin M. L. (2012) Integrin inside-out signaling and the immunological synapse. Curr. Opin. Cell Biol. 24, 107–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Murzin A. G. (1992) Structural principles for the propeller assembly of β-sheets. The preference for seven-fold symmetry. Proteins 14, 191–201 [DOI] [PubMed] [Google Scholar]
- 47. Xiao T., Takagi J., Wang J.-H., Coller B. S., Springer T. A. (2004) Structural basis for allostery in integrins and binding of fibrinogen-mimetic therapeutics. Nature 432, 59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Soler D., Chapman T., Yang L. L., Wyant T., Egan R., Fedyk E. R. (2009) The binding specificity and selective antagonism of vedolizumab, an anti-α4β7 integrin therapeutic antibody in development for inflammatory bowel diseases. J. Pharmacol. Exp. Ther. 330, 864–875 [DOI] [PubMed] [Google Scholar]
- 49. Li S., Wang H., Peng B., Zhang M., Zhang D., Hou S., Guo Y., Ding J. (2009) Efalizumab binding to the LFA-1 αL I domain blocks ICAM-1 binding via steric hindrance. Proc. Natl. Acad. Sci. U.S.A. 106, 4349–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sidhu S. S. (2012) Antibodies for all. The case for genome-wide affinity reagents. FEBS Lett. 586, 2778–2779 [DOI] [PubMed] [Google Scholar]
- 51. Pei J., Grishin N. V. (2001) AL2CO. Calculation of positional conservation in a protein sequence alignment. Bioinformatics 17, 700–712 [DOI] [PubMed] [Google Scholar]