

Abstract
Francisella tularensis is a facultative intracellular bacterium that causes the deadly disease tularemia. Most evidence suggests that Francisella is not well recognized by the innate immune system that normally leads to cytokine expression and cell death. In previous work, we identified new bacterial factors that were hyper-cytotoxic to macrophages. Four of the identified hyper-cytotoxic strains (lpcC, manB, manC, and kdtA) had an impaired lipopolysaccharide (LPS) synthesis and produced an exposed lipid A lacking the O-antigen. These mutants were not only hyper-cytotoxic but also were phagocytosed at much higher rates compared with the wild type parent strain. To elucidate the cellular signaling underlying this enhanced phagocytosis and cell death, we performed a large-scale comparative phosphoproteomic analysis of cells infected with wild-type and delta-lpcC F. novicida. Our data suggest that not only actin but also intermediate filaments and microtubules are important for F. novicida entry into the host cells. In addition, we observed differential phosphorylation of tristetraprolin, a key component of the mRNA-degrading machinery that controls the expression of a variety of genes including many cytokines. Infection with the delta-lpcC mutant induced the hyper-phosphorylation and inhibition of tristetraprolin, leading to the production of cytokines such as IL-1beta and TNF-alpha that may kill the host cells by triggering apoptosis. Together, our data provide new insights for Francisella invasion and a post-transcriptional mechanism that prevents the expression of host immune response factors that control infection by this pathogen.
Francisella tularensis is the Gram-negative facultative intracellular bacterium that causes the disease tularemia in humans and diverse animals. This bacterium is among the most virulent human pathogens known, with inhalation or inoculation of as few as 10 bacteria being sufficient to cause a fatal infection. Its low infectious dose and relative ease of airborne transmission led to the development of F. tularensis as a biological weapon (1). Because of potential threats to national security and public health, the U.S. Centers for Disease Control and Prevention (CDC) classifies F. tularensis as a Category A bioterrorism agent, and the Departments of Health and Human Services (HHS) and Agriculture (USDA) list this pathogen as a Tier 1 select agent. (http://www.bt.cdc.gov/agent/agentlist-category.asp; http://www.selectagents.gov/select%20agents%20and%20toxins%20list.html).
Francisella tularensis includes three subspecies: tularensis (abbreviated as F. tularensis), holarctica (abbreviated as F. holarctica), and mediasiatica (abbreviated as F. mediasiatica) (2). Of these, F. tularensis subsp. tularensis and subsp. holarctica are capable of causing disease in humans and must be manipulated in a Biosafety Level 3 environment. Francisella novicida (abbreviated as F. novicida) is a closely related species to F. tularensis and shares high genetic similarity to other F. tularensis subspecies (∼98% DNA sequence identity) (3, 4). Although some differences have been observed so far between F. novicida and F. tularensis, mainly in their lipopolysaccharide (LPS)1 O-antigen and the ability to activate the inflammasome, most of the mutations in orthologous genes generate similar phenotypes (5–7). Because of the avirulence in humans for the U112 isolate (8) and ability to cause a tularemia-like disease in animal models, F. novicida provides a safe and appropriate research alternative to virulent F. tularensis strains.
Although research efforts have elucidated many of the molecular mechanisms of Francisella pathogenesis over the past decade (9–12), the detailed mechanistic interplay between bacterial factors and the host immune response remains incomplete. One of the first steps during the bacteria-host interaction is the phagocytosis or entry of bacteria into the host cells. To date, there are five receptors described in the phagocytosis of Francisella by macrophages: scavenger receptor A (SRA), mannose receptor (MR), Fc-gamma receptor (FcγR), CR3 complement receptor (CR3) and surface-exposed nucleolin (SEN) (13–16). After being recognized by one of these receptors Francisella triggers a signaling cascade that culminates in the rearrangement of actin filaments and engulfment of the bacteria (17–19). This signaling cascade is in part regulated by phosphorylation and dependent on kinases such as Akt, Syk and Erk (20, 21). More recently, Francisella was also shown to be endocytosed by liver cells in a mechanism that involved cholesterol-rich domains on the plasma membrane and clathrin-coated vesicles (22). In addition to phagocytosis, the host inflammatory response through TLR signaling and the activation of the inflammasome are also essential factors during the early stages of the infection (9–11, 23).
Francisella has quite a distinct inflammatory response compared with other Gram-negative bacteria. Unlike E. coli or Salmonella, the lipopolysaccharide (LPS) from Francisella is not endotoxic and lacks the ability to stimulate toll-like receptor 4 (TLR4), the pro-inflammatory receptor that recognizes Gram-negative bacteria (24). The lack of TLR4 activation is related to hypoacylation and the addition of very long chain fatty acids to the diglucosamine backbone of the lipid A portion of LPS (24). Typically, TLR4 is activated by lipid A containing six short (12–14 carbon) fatty acids, whereas Francisella lipid A has four fatty acids with longer carbon chains (16–18 carbon) (11, 24). On the other hand, Francisella has been shown to activate TLR2 (25, 26) that recognizes lipoproteins, glycans, and glycophospholipids more frequently present in Gram-positive bacteria, fungi, and protozoan parasites, respectively (27). Francisella also activates another component of innate immune system—the inflammasome (10). Assembly of the inflammasome is activated by Francisella infection and is responsible for triggering a pro-inflammatory response through processing and maturing cytokines, such as IL-1β and IL-18 (28–32), in addition to triggering a pro-inflammatory response that leads to cell death by pyroptosis (30–33). More recently, Peng et al. showed that several Francisella mutants defective in membrane proteins were hyper-cytotoxic to host cells in a process that is dependent on the inflammasome (29).
It has been proposed that Francisella actively suppresses signaling by TLRs and the inflammasome to evade the host immune system and effectively establish an infection (11). To identify genes that are involved in host immune evasion, we recently screened a comprehensive mutant library containing over 3000 F. novicida strains for mutants that were hyper-cytotoxic (34). Among the identified strains four F. novicida genes were involved in LPS core synthesis and assembly. These mutants are unable to synthesize the LPS core and thus produce LPS with a very short glycan compared with the long polysaccharide chains of the wild-type (WT) (34). Interestingly, three mutants (ΔlpcC, ΔmanB, and ΔmanC) are hyper-cytotoxic to RAW 264.7 cells, a murine macrophage-like cell line that lacks the production of ASC (apoptosis-associated speck-like protein containing a caspase recruiting domain), a key component of the inflammasome signaling. This observation suggests that although ΔlpcC, ΔmanB, and ΔmanC are also defective in membrane components, the mechanism of cell killing may be different than the mutant strains studied by Peng et al. (29). Also surprising was that ΔlpcC, ΔmanB, and ΔmanC strains were phagocytosed at a much higher rate than WT F. novicida via a mechanism that does not require actin polymerization (34). Actin polymerization has thus far been demonstrated to be involved in internalization of Francisella by either phagocytic or endocytic pathways (21, 22, 35); therefore, the finding that ΔlpcC, ΔmanB, and ΔmanC mutant strains are able to invade host cells independent of actin filaments remains to be understood. Deletion of lpcC from F. tularensis Schu S4 also results in a similar phenotype, suggesting that this phenomenon acts broadly among the different Francisella species rather than specific to F. novicida (Tempel and Heffron, unpublished observations).
To better understand the mechanism of infection and cell killing by these hyper-cytotoxic F. novicida strains, we evaluated cell signaling stimulated by the F. novicida ΔlpcC strain in comparison to its parent WT strain. Here we carried out a comprehensive quantitative phosphoproteomic analysis using isobaric tags for relative and absolute quantification (iTRAQ), which allowed us to analyze WT and ΔlpcC F. novicida infection with multiple replicates in the same experiment. By combining network and function-enrichment analyses followed by immunological and cellular experiments we identified new mechanistic aspects of host phagocytosis and cell killing by F. novicida.
EXPERIMENTAL PROCEDURES
Ethics Statement
Mouse experiments were approved under the protocol of the Oregon Health and Science University Institutional Animal Care and Use Committee (IACUC B11220) and performed in accordance with the guidelines for the Care and Use of Laboratory Animals of the National Institutes of Health to minimize animal suffering.
Bacterial Culture
Wild type and ΔlpcC Francisella novicida were grown on cysteine heart agar (CHA) from −80 °C freezer stocks before culturing in liquid media. Bacteria were cultured overnight in tryptic soy broth + 0.1% cysteine (TSBC) with shaking (200 rpm), and the optical density at 600 nm (OD600) was measured to determine CFU/ml.
Cell Culture and Infection
RAW 264.7 or J774 murine macrophage-like cells (American Type Culture Collection, Manassas, VA) were cultured in DMEM/High Glucose (HyClone, Logan, UT) supplemented with 10% fetal bovine serum (FBS; Invitrogen, Rockville, MD) at 37°C in the presence of 5% CO2. For infection, bacteria were added to 50–80% confluent cells in tissue culture plates at a multiplicity of infection (MOI) of 100. The cells were centrifuged at 1000 × g for 5 min at room temperature and incubated at 37°C with 5% CO2. Thirty minutes to 1 h after infection, cells were washed three times with PBS and either prepared for analysis (time = 0) or overlaid with DMEM containing 100 μg/ml gentamicin to prevent growth of extracellular bacteria and incubated at 37°C with 5% CO2. After one hour, cells were washed three times with PBS and either prepared for analysis or overlaid with 10 μg/ml gentamicin and incubated at 37°C with 5% CO2 for the duration of the infection. Each infection assay is performed in triplicate unless otherwise stated.
Western Blot Analysis
RAW 264.7 or J774 murine macrophage-like cell lines were cultured as described in 6-well plates and either subjected to mock infections or acute 1 h infections with wild-type (U112) or ΔlpcC F. novicida strains, as indicated. Following infection, cells were washed three times in PBS and incubated with media containing 100 nm okadaic acid and 10 μg/ml gentamycin. Four hours after initial exposure to Francisella, cells were collected in PBS and pellets stored at −80°C for 10 min. Cell pellets were then lysed in the presence or absence of phosphatase inhibitors and EDTA depending on whether they were to undergo lambda phosphatase treatment (20 mm Tris pH7.4, 150 mm NaCl, 0.5% Nonidet P-40, 1 mm dithiothreitol (DTT), protease inhibitor mixture (Roche), ± 5 mm EDTA, ± 10 mm NaF, ± 10 mm Na3VO4, ± 10 mm p-Nitrophenyl phosphate, ± 100 nm Okadaic Acid, and ± 1× HALT phosphatase inhibitor mixture (Pierce, Rockford, IL)). Thirty μg of cleared whole cell lysates (quantified using the Bradford assay) was resolved on 10% Tris-Glycine SDS-PAGE. Manganese and 5 U of lambda phosphatase (New England Biolabs, Ipswich, MA) were added to whole cell lysates as indicated and the reaction was incubated at 30°C for 30 min; stopped with Laemmli protein loading buffer. Transferred proteins were subjected to Western blot analysis using anti-TTP 1:1000 (Millipore ABE285), anti-RNA Pol II Rpb1 1:1000 (SCBT sc-900), and anti-rabbit IgG-HRP secondary 1:10,000 (Promega, Madison, WI).
Cytokine Assay
RAW 264.7 cells were infected in triplicate with ΔlpcC, parent, mock, or untreated in 24-well plates as described above for 0, 2, or 6 h. At the end of each time point, 350 μl of supernatant was frozen at −80°C and submitted for analysis. The samples were tested on Quansys Biosciences' (Logan, UT) Q-Plex ArrayTM kits for expression of 21 unique cytokines. Q-PlexTM technology is a multiplex approach that involves the micro-spotting of individual groups of capture antibody in either a Cartesian or polar coordinate system on the bottom of a 96-well plate, each spot being its own micro ELISA. Each well was identically spotted. Standard ELISA procedures were applied notably initial sample incubation, washing, secondary antibody incubation, washing, incubation with the label, and measurement are involved. The label and reporting system used in a Q-Plex ArrayTM is chemiluminescent.
Apoptosis and Necrosis Assay
RAW 264.7 cells were infected for 18 h in triplicate in an opaque white 96-well tissue culture plate (BD, Billerica, MA) with ΔlpcC, WT U112, mock, or uninfected. 10 mm staurosporine or 100 mm ionomycin were added to triplicate wells as controls for apoptosis or necrosis, respectively. Thirty minutes before analysis, 5 μl of 10% saponin was added to triplicate control wells as a lysis control. The cells were analyzed for viability, cytotoxicity, and apoptosis using the ApoTox-GloTM Triplex Assay (Promega) according to manufacturer's instructions. The assay was read using a SpectraMax® GeminiTM XS plate reader (Molecular Devices, Sunnyvale, CA).
Cytotoxicity (Cell Invasion) Assay
J774 cells were infected in triplicate in 24-well tissue culture plates (BD, Billerica, MA) with ΔlpcC, WT, mock, or uninfected, ± cells invasion inhibitors (cytochalasin D, 5 μg/ml; nocodazole, 10 μg/ml; withaferin A, 5 μm; wortmannin, 0.1 μm). One hour after infection, the cells were washed and incubated with DMEM containing 100 μg/ml gentamicin, as above. After one hour of incubation with gentamicin, macrophages were washed and lysed with 0.5% saponin for 30 min at 37°C, and the lysates were serially diluted and plated on CHA plates to determine F. novicida CFU/ml.
Protein Extraction and Trypsin Digestion
In preparation for phosphorylation analyses, RAW 264.7 macrophage-like cells were infected with either wild-type or ΔlpcC Francisella strains as described above for 30 min, washed with PBS and incubated for an additional 0, 1, 2, and 4 h. The contents of each well of the 6-well plates were resuspended in 500 μl of lysis buffer (8 m urea in 100 mm NH4HCO3 pH 8.0 containing Sigma phosphatase inhibitor cocktails 1 and 3). The contents of 3 wells were combined and stored at −80°C. Proteins were then thawed and quantified by BCA assay (Pierce). Protein disulfide bonds were reduced by DTT (5 mm) for 1 h at 37°C, and the resulting free thiol groups were alkylated by iodoacetamide (20 mm) for 1 h at room temperature in the dark. After alkylation, samples were diluted 4-fold with 50 mm NH4HCO3 (pH 7.8) and digested with trypsin at ratio of 1:50 (trypsin/protein) for 4 h at 37°C, diluted by another 2-fold with the same buffer, and subjected to a second treatment with trypsin and incubated at room temperature overnight. The reaction was stopped by lowering the pH to 2.5 with trifluoroacetic acid. The resulting peptides were desalted using C18 SPE (Discovery DSC-18, Supelco) cartridges, concentrated down in SpeedVac SC250 Epress (ThermoSavant) and their concentrations were measured by BCA assay.
iTRAQ Labeling and Phosphopeptide Enrichment
Peptides (50 μg) from each sample were subjected to 8-plex iTRAQ (Applied Biosystems) labeling as the experimental design in Fig. 1, and according to the manufacturer recommendations. Each iTRAQ experiment had biological duplicate controls with mock infections and biological triplicates of RAW 264.7 cells infected with either wild-type or ΔlpcC F. novicida strains, all collected at the same time point. After labeling and mixing equal amounts of peptides, samples were desalted using C18 SPE cartridges, concentrated in a SpeedVac and submitted to quantification by BCA assay. The phosphopeptide enrichment was performed exactly as described by Nguyen et al., using Fe3+-nitriloacetic acid functionalized magnetic beads (36). After enrichment, phosphopeptides were concentrated by vacuum centrifugation before mass spectrometry analysis.
Fig. 1.
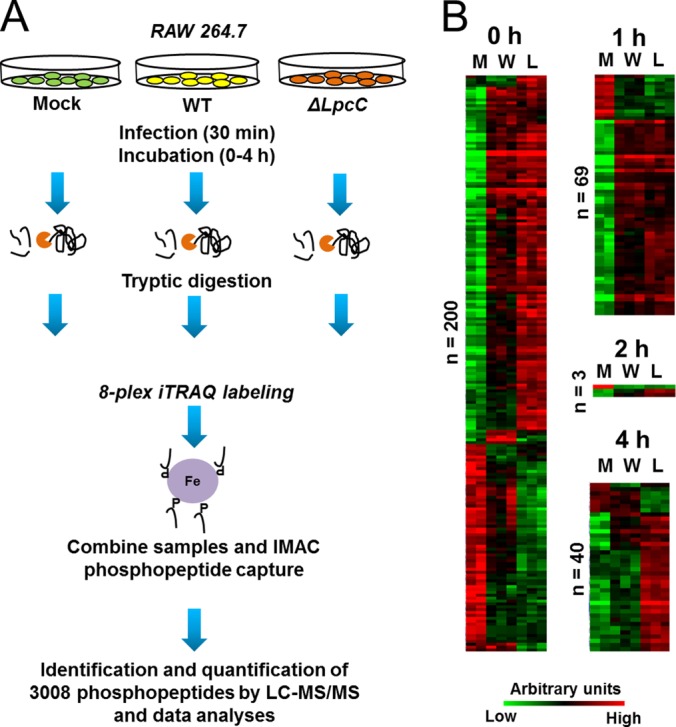
Phosphoproteome analysis of cells infected with F. novicida. A, Experimental design. RAW 264.7 macrophage-like cells were infected for 30 min with wild-type (WT) Francisella or with ΔlpcC, followed by incubation for 0–4 h before harvesting. A negative control was run in parallel using media alone (Mock). After infection, cells were harvested, digested with trypsin and the resulting peptides were labeled with iTRAQ. Equal parts of each iTRAQ sample were combined and phosphopeptides were captured by immobilized metal affinity chromatography (IMAC). Enriched phosphopeptide fractions were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) leading to the identification of 3008 phosphopeptides. B, Differentially abundant phosphopeptides from RAW 264.7 cells after Francisella infection. Abbreviations: L, ΔlpcC-infected; M, mock-infected; n, number of phosphopeptides; W, wild type-infected.
Proteomic Analysis
Phosphopeptides were loaded to a trap column (150 μm i.d. × 4 cm; 5 μm C18 particles, Phenomenex) and the separation was achieved in a longer column (50 μm i.d. × 40 cm; 5 μm C18 particles, Phenomenex) using an isobaric gradient of acetonitrile over 200 min (37). Eluted peptides were directly analyzed by electrospray ionization - mass spectrometer (Orbitrap Velos equipped with ETD). Full scan - 400–200 m/z with resolution of 60,000 at 400 m/z. The five most intense ions were selected for HCD (40% normalized collision energy) followed by decision tree ETD (z = 3, m/z<650; z = 4, m/z<900; z = 5, m/z<950) or CID scans (z = 2; z = 3, m/z>650; z = 4, m/z>900; z = 5, m/z>950). MS/MS spectra were converted to peak lists using DeconMSn (version 2.2.2.2, http://omics.pnl.gov/software/DeconMSn.php) (38) using default parameters and database searched using SEQUEST v27 against murine protein sequences from Uniprot (downloaded on August 22nd, 2011) and porcine trypsin both in forward and reverse orientations (32,780 total sequences). As searching parameters, precursor ion mass tolerance was 50 ppm, and fragmentation tolerances for CID, ETD, and HCD were 0.5, 0.5, and 0.05 Da, respectively. Only fully tryptic peptides were considered, with two missed cleavages allowed. Phosphorylation (+79.9663 Da) of serine, threonine, or tyrosine was searched as a dynamic modification. Cysteine carbamidomethylation (+57.0215 Da) and lysine and N terminus labeling with iTRAQ (+304.2022 Da) were searched as static modifications. The maximum number of variable modifications was set to 3. Peptides were then filtered using a cutoff of MSGF (39) ≤ 1.0e-9, which resulted in ∼1% false-discovery rate. When peptides matched multiple sequences only the first in the database was reported. Ascore (40) was used to estimate the confidence of phosphorylation modification site assignment, Sites with an A-Score ≥ 19 were considered as unambiguous sites (≥ 99% confidence). Sites with −1 Ascore indicate these sites are the only choices for modification on the peptide identified.
Quantitative Data Analysis
iTRAQ reporter ion intensities were extracted with MASIC (MS/MS Automated Selected Ion Chromatogram Generator, version v2.5.3923, http://omics.pnl.gov/software/MASIC.php) (41) and the phosphopeptides fragmented multiple times were summed together to remove redundancy in quantification and improve signal to noise ratio followed by log transformation. To convert log2 (ion intensities) to relative abundance measures we subtracted the mean log intensity values for each individual peptide. The normalization step was based on assumption that the majority of phosphopeptides do not change in one direction, and the median relative abundance should be equal between samples. This type of normalization allows removing biases arising from pipetting error or labeling efficiency. The missing data was imputed using K-NN approach similar to described previously (42), using Spearman correlation as a distance measure. Because of the low number of replicates (2–3), for testing of statistical significance of differential abundance of phosphopeptides we applied a moderated ANOVA test available in the “limma” BioConductor package (43). Pairwise differences were inferred by applying post-hoc contrasts analysis (Mock-WT, Mock-ΔlpcC, WT-ΔlpcC). The significance threshold was set at 0.05 of Benjamin-Hochberg adjusted p value. All of the time points (0, 1, 2 and 4 h) were analyzed independently.
Sequence Analysis
Motif analysis for specific kinases was performed online using motif-x (http://motif-x.med.harvard.edu/motif-x.html) (44). For this analysis only confident phosphorylation sites with Ascore ≥ 19.0 or –1 were used, and results were filtered with an enrichment significance ≤ 0.01. The number of occurrences was adjusted to 5 or 10 depending on the number of queried phosphosites. The protein-protein interaction network was built based on the information deposited at InnateDB (innatebd.org, updated February 16th, 2012) (45). Proteins from experimentally verified interactions were retrieved from InnateDB and the network was built in Cytoscape v2.8.3 (cytoscape.org) (46). After building the network, only proteins that directly interact with at least 2 phosphoproteins were kept. The function-enrichment analysis was performed online using the Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.7 (http://david.abcc.ncifcrf.gov/home.jsp) (47).
Murine Infection
Four- to six-week-old female C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, Maine) and allowed to acclimatize for 1 week before infection. Groups of three mice were infected intraperitoneally (intraperitoneal) with 2 × 104 CFU of WT or ΔlpcC F. novicida, mock infected with PBS, or left untreated. At 48 h postinfection, whole spleens were harvested, cut into 3 pieces and placed in RNAlater (Ambion) at 4°C overnight before storing at −80°C. All procedures and protocols were performed at OHSU in compliance with Institutional Animal Care and Usage Committee guidelines.
Generation of Probes for Microarray Experiments
DNA probes for microarray experiments were generated by adding 2 μg of total RNA in a mixture containing 6 μg of random hexamers (Invitrogen), 0.01 m dithiothreitol, an aminoallyl-deoxynucleoside triphosphate mixture containing 25 mm each dATP, dCTP, and dGTP, 15 mm dTTP, and 10 mm amino-allyl-dUTP (aa-dUTP) (Sigma), reaction buffer, and 400 units of SuperScript III reverse transcriptase (Invitrogen) and incubating at 42°C overnight. The RNA template then was hydrolyzed by adding NaOH and EDTA to a final concentration of 0.2 and 0.1 m, respectively, and incubating at 70°C for 15 min. Unincorporated aa-dUTP was removed with a QIAquick column (Qiagen). The probe was eluted with phosphate elution buffer (4 mm KH2PO4, pH 8.5, in ultrapure water), dried, and resuspended in 0.1 m sodium carbonate buffer (pH 9.0). To couple the amino-allyl cDNA with fluorescent labels, Cy3 or Cy5 (Amersham Biosciences) was added for 1h. Uncoupled label was removed using the Qiagen QIAquick PCR purification kit (Valencia, CA).
Microarray Hybridization, Scanning, Image Analysis, Normalization, and Analysis
Agilent 4 × 44K mouse gene expression arrays were used. Hybridization protocol was performed by manufacturer's suggestion. Equal volumes of the appropriate Cy3- and Cy5-labeled probes were combined, dried, and then resuspended in the hybridization buffer and were heated to 95°C before hybridization. The probe mixture then was added to the microarray slide and allowed to hybridize overnight at 42°C with rotation in the Agilent hybridization oven. Hybridized slides were washed sequentially in Agilent wash buffer #1 and wash buffer #2. Individual TIFF images from each channel were captured and analyzed with Agilent Feature Extractor. Microarray data were normalized by LOWESS in TIGR MIDAS software (available at http://www.jcvi.org/software/tm4) and data visualized in TMEV.
mRNA Stability Assay
RAW 264.7 macrophage-like cells were infected with either wild-type or ΔlpcC Francisella strains in biological triplicate for 1 h, followed by washes and addition of gentamicin as described above. Cells were then treated with Actinomycin D (5 μg/ml) and indicated time points were harvested directly in Trizol (Invitrogen). From each replicate and time point, 500 ng of purified total RNA was used for cDNA synthesis using oligoDT primers. Quantitative PCR was performed using gene-specific primers for rRNA 18S, Gapdh, IL-1β, and RANTES transcripts. (IL-1β: TTGACAGTGATGAGAATGACCTG, GAGATTTGAAGCTGGATGCTCT; RANTES: TTGCAGTCGTGTTTGTCACTC, TTCTTGAACCCACTTCTTCTCTG). Values were normalized to rRNA 18S transcript as well as relative to their baseline abundance at time 0.
RESULTS AND DISCUSSION
Changes in Host Cell Phosphoproteome Induced by F. novicida Infection
In an effort to identify pathways that are activated during cell invasion by Francisella we performed a quantitative phosphoproteomic analysis of RAW 264.7 macrophage-like cells infected with either wild-type or ΔlpcC Francisella strains. A total of 3008 phosphopeptides from 1256 phosphoproteins were identified with a false-discovery rate of ∼1% in phosphopeptide level (Fig. 1A, supplemental Table S1).
The quantitative analysis revealed 200, 69, 3, and 40 phosphopeptides that were significantly differentially abundant comparing infected and control (mock) cells at 0, 1, 2, or 4 h, respectively (Fig. 1B and supplemental Fig. S1, supplemental Tables S2–S5). To provide insights on the kinase families activated or repressed during Francisella infection, specific kinase motifs were searched. Although protein kinase motifs are often redundant, it is possible to assign these sequences to enzyme families, thus providing insights about classes of kinases differentially activated during the cell invasion process (48). At the early time point (0 h postinfection) two motifs (RxxS and SP, x being any amino acid residue) were found to be enriched among the up-regulated phosphorylation sites, and one motif (SP) was identified among down-regulated phosphorylation sites (Fig. 2). At 1 h postinfection, both RxxS and SP motifs were overrepresented among the up-regulated phosphorylation sites, but no overrepresented motif was found for the down-regulated sites, likely because of the low number of down-regulated phosphopeptides. At the last time point, only the RxxS motif was found to be overrepresented in the up-regulated sites (Fig. 2). The motif RxxS is consistent with either calmodulin kinase 2 (CaMK-II), PKA, PBK/AKT, or PKC, whereas SP is consistent with ERK/MAPK, CDK, and CDK-like motifs, thus suggesting that enzymes from these families might be regulated during Francisella infection. In agreement with these observations, Francisella infection is known to activate TLR2, which triggers a cascade of MAPKs (26).
Fig. 2.
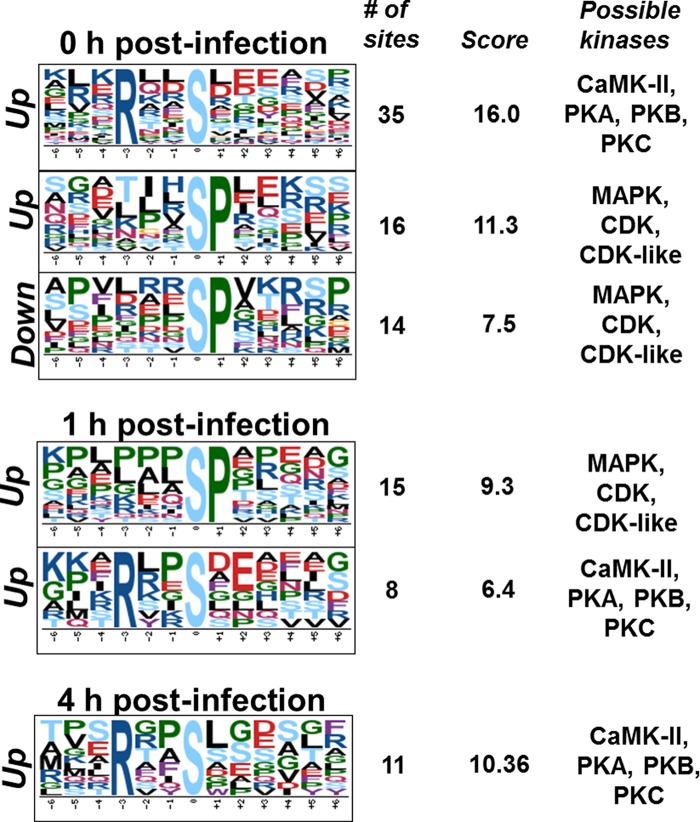
Analysis of phosphorylation motifs differentially abundant in cells infected with F. novicida. Enrichment of phosphorylation site motifs was performed using Motif-x (http://motif-x.med.harvard.edu/) and only the significantly different phosphopeptides in the course of infection. Abbreviations: Up, up-regulated phosphorylation sites; Down, down-regulated phosphorylation sites.
To investigate potential pathways activated during Francisella infection, we built a protein–protein interaction (PPI) network and performed a function-enrichment analysis. For this analysis we combined differentially phosphorylated proteins found in all time points because proteins from the same pathway are not necessarily simultaneously phosphorylated. The PPI network was based on experimentally identified interactions deposited in InnateDB. To have a more insightful network, only proteins with significantly changing phosphorylation sites were used, resulting in a network of 222 proteins (nodes) and 383 connections (edges). The network showed a great number of proteins involved in transcription and translation regulation (RNA processing and post-transcription regulation), as well as degradation of polypeptides (proteasome) (Fig. 3A). In support of the network analysis, these functions, besides protein degradation, were found to be significantly overrepresented by a DAVID analysis (Fig. 3B). Interestingly, regulators of cytoskeleton organization and phagocytosis were also enriched among the differentially phosphorylated proteins (Figs. 3A and 3B), which may suggest the utilization of these pathways in the host cell entry process by the bacteria. The protein network and function-enrichment analysis were also consistent with motifs found to be overrepresented in our analysis. For instance, the motif analyses suggested that CDKs might be regulated during the infection process and, in accordance with these data, cell cycle function was suggested by the network and functional enrichment analysis to be altered during infection (Figs. 1B, 3A, and 3B).
Fig. 3.

Analysis of functions overrepresented among differentially phosphorylated proteins in F. novicida infection. A, Network of differentially phosphorylated proteins in RAW 264.7 cells during Francisella infection. A protein–protein interaction map of the differentially phosphorylated proteins was retrieved from InnateDB and the network was built using Cytoscape. Circles (nodes) represent each protein and lines indicate physical interactions. Nodes were colored according to the phosphorylation abundance in infected cells compared with the mock infection: red, up-regulated; green, down-regulated; blue, either unchanged or not identified by LC-MS/MS. B, Enriched functions among the differentially phosphorylated proteins in RAW 264.7 cells during Francisella infection. Overrepresented functions were annotated using Database for Annotation, Visualization and Integrated Discovery (DAVID).
Comparative Analysis of Wild-Type Versus ΔlpcC Infected Cells
To better understand the mechanism of host cell invasion and killing by the ΔlpcC mutant, we investigated the differences in phosphorylation patterns between RAW 264.7 cells infected with WT or ΔlpcC F. novicida strains. At the earliest time point (0 h postinfection) phosphorylation signals, seemed to be stronger, but not significant, in ΔlpcC-infected cells compared with WT-infected cells (Fig. 1B). At 1 and 2 h postinfection the levels of protein phosphorylation in both ΔlpcC- and WT-infected cells were almost identical (Fig. 1B). The largest differences were observed at 4 h postinfection (Fig. 1B). We speculate that the signaling observed 4 h postinfection is dependent on bacterial escape from the phagosome into the host cell cytosol. Supporting this notion, the escape from the phagosome occurs from 30 min to 4 h postinfection (11). Furthermore, after the bacteria gain access to the host cell cytosol some signaling occurs, such as inflammasome activation by AIM2 (29).
To determine the significantly different phosphopeptides from WT- and ΔlpcC-infected cells a pairwise analysis was done using the Benjamin-Hochberg test. A total of 36 phosphopeptides were significantly different in ΔlpcC infection compared with WT infection and were only observed at 4 h postinfection (Fig. 4A, supplemental Table S6). Among the enriched motifs, the RxxS motif was enriched within the up-regulated phosphorylation sites (Fig. 4B), which suggests that kinases with preference to basic motifs, such as PKB/Akt, are active at this time point.
Fig. 4.

Differentially phosphorylated proteins in cells infected with wild-type (W) or ΔlpcC (L) F. novicida. A, Significantly different phosphopeptides in WT or ΔlpcC infection. Assigned phosphorylation sites are in parentheses and sites that were not confidently assigned to a single amino acid residue (Ascore < 19.0 or −1) are represented by the number of phosphogroups and range of potential modification sites. B, Enriched motif among the phosphosites that were differentially abundant comparing WT and ΔlpcC F. novicida infection. C, Enriched functions among the differentially phosphorylated proteins in RAW 264.7 cells comparing infection with WT and ΔlpcC F. novicida strains.
To identify pathways that were affected by proteins differentially phosphorylated between WT and ΔlpcC infections, we searched for overrepresented functions. Only four gene ontology categories were enriched: cell leading edge, anchoring junction, negative regulation of gene expression and RNA processing (Fig. 4C). Indeed cell leading edge and anchoring junction functions shared several proteins, thus we believe that indeed they reflect the same pathway of cytoskeleton rearrangement. The same was observed for negative regulation of gene expression and RNA processing, which we interpret as down-regulation of gene expression by RNA degradation. Accordingly, we next performed a more complete analysis of the proteins involved in these pathways and attempted to correlate them with ΔlpcC phenotypes.
Phosphorylation of Phagocytic and Cytoskeleton Proteins
To identify phosphorylation events that could be involved in the host cell invasion by ΔlpcC, we examined the phosphorylation quantification of proteins known to be involved in Francisella entry in the host cells. Francisella entry into host cells occurs mainly by two general mechanisms: endocytosis and phagocytosis (11, 22). In the endocytic process, Francisella enters host cells through cholesterol-rich domains in the plasma membrane. This process depends on clathrin and two adaptor proteins, Eps15 and AP2 (22). In the phosphoproteomic data, phosphorylation of the heavy chain of clathrin (Cltc) was shown to be reduced at the earliest time point after infection (0 h) but unchanged at the later time points (Fig. 5A). Cltc was also slightly less phosphorylated during the infection with the WT compared with ΔlpcC, but this difference was not significant (supplemental Table S2). Eps15 phosphorylation levels were unchanged over the two time points when a peptide from this protein was detected, and no phosphopeptides derived from AP2 were found. These observations suggest that the endocytic process might not be involved in the enhanced infectivity by ΔlpcC or that this process is not controlled by phosphorylation.
Fig. 5.

Phosphorylation on endocytic, phagocytic, and cytoskeleton proteins, and the mechanism of cell invasion. A, Phosphorylation on endocytic, phagocytic and cytoskeleton proteins. The endocytic, phagocytic and cytoskeleton rearrangement pathways were built based on literature search (see text for references) and KEGG database. B, Contribution of cytoskeleton filaments to host cell entry of WT and ΔlpcC F. novicida strains. J774 cells were left untreated (Unt) or treated with 5 μg/ml cytochalasin D (CytD or C), 10 μg/ml nocodazole (Noc or N), or 5 μm withaferin A (WFA or W) before infection with WT and ΔlpcC F. novicida strains. Data points significantly different to the untreated control (t test, p ≤ 0.05) are marked the asterisks.
The phagocytic process of Francisella by host cells involves at least five receptors: MR, SRA, FcγR, CR3, and SEN (17–19). Of these known Francisella receptors, only phosphopeptides from FcγR were identified in our phosphoproteomic dataset, and no changes in the phosphorylation levels were observed (Fig. 5A). It is possible that ΔlpcC is more easily recognized by one or more of these receptors because of the lack of LPS polysaccharides on the cell surface. Indeed, previous work has shown the importance of LPS O antigen polysaccharide on the regulation of complement deposition on Francisella surface (49). The deposition of the complement protein C3 on Francisella surface enables the recognition of the bacteria by CR3 promoting phagocytosis by opsonization (13, 50); however, we inactivated complement proteins in our experimental conditions.
After recognizing bacteria, the phagocytic receptors trigger cell signaling that results in the rearrangement of cytoskeleton and engulfment of the particle. In the case of Francisella, two pathways have been reported to be activated during the phagocytosis process: phosphoinositol 3-kinase (PI3K)/Akt and the tyrosine kinase Syk/mitogen-activated protein kinase (MAPK) Erk2 pathways (20, 21). For the PI3K/Akt pathway, phosphopeptides for PI3K and ribosomal protein S6 kinase beta-1 (Rps6kb1) were identified. Although the phosphorylation on PI3K remained unchanged, Rps6kb1 phosphorylation decreased in both WT and ΔlpcC infection in early time points (Fig. 5A). The lack of phosphorylation up-regulation suggests that this pathway was not activated during the infection. Indeed, another report has provided contradictory results regarding the role of PI3K/Akt pathway during Francisella phagocytosis (20), which could be potentially explained by their use of different bacterial strains. On the other hand, two proteins from the Syk/Erk2 pathway, kinase Erk2 and phospholipase A2 (PLA2), were shown to be more phosphorylated in both WT and ΔlpcC Francisella infection at 0 h postinfection (Fig. 5A). That only the last two enzymes of the cascade were found to be overphosphorylated may indicate that the upstream signaling had already faded. Interestingly, phosphorylation of these proteins was up-regulated in ΔlpcC-infected cells as compared with WT infection at 4 h (Fig. 5A), which suggests that this pathway could also be involved in the phagocytosis of ΔlpcC.
Next, we analyzed cytoskeletal proteins that can be involved in the engulfment of bacteria. Although more proteins involved in actin filament organization were found to be phosphorylated, only three were previously described to have a role in phagocytic or endocytic pathways: mDia2, and formin-like proteins 1 (Fmnl1) and 3 (Fmnl3) (51). Among these proteins, only phosphorylation on mDia2 was found to be up-regulated at the earliest infection time point, and no difference between WT and ΔlpcC infection was observed (Fig. 5A). Among the microtubule proteins, tubulins were found to be phosphorylated, but their modification levels did not change with the F. novicida infection (Fig. 5A). On the other hand, the intermediate filament protein vimentin was phosphorylated in multiple sites (Table S1). Vimentin phosphorylation was higher in the early time point in both WT and ΔlpcC infections, but was up-regulated only in ΔlpcC at 4 h postinfection in a pattern that was consistent with Erk2 and PLA2 phosphorylation (Fig. 5A). Indeed, vimentin has previously been shown to interact with PLA2 (52); therefore, it is reasonable to believe that they could be involved in similar cellular signaling pathways. Furthermore, previous studies indicate that vimentin filaments are regulated by phosphorylation (53).
To test whether intermediate filaments and microtubules have a function in Francisella entry into host cells, J774 cells were treated with cytochalasin D, nocodazole and withaferin A, which are inhibitors of actin, microtubules, and intermediate (vimentin) filaments, respectively. As previously shown (34), ΔlpcC F. novicida infected at a higher rate compared with WT, and the inhibition of host cell actin polymerization with cytochalasin D decreased the infectivity of the WT bacteria (more than 2 logs) to a much greater extent than in the ΔlpcC infection (Fig. 5B). Blocking microtubule polymerization with nocodazole also led to a similar phenotype of decreasing bacterial entry in a greater extent in the WT (∼1 log) compared with the ΔlpcC (not significant) infection (Fig. 5B). Inhibition of vimentin polymerization with withaferin A by itself had only a small effect on the entry of WT Francisella into the host cells, but in combination with nocodazole and cytochalasin D it significantly reduced the number of internalized bacteria (Fig. 5B). The individual treatments with cytochalasin D, nocodazole and withaferin A had no effect on the entry of ΔlpcC into host cells, but in combination these inhibitors significantly reduced the number of internalized bacteria (up to ∼1 log) (Fig. 5B). It should be noted that the reduction in the number of internalized bacteria was smaller in ΔlpcC infection compared with WT for all the treatments, suggesting that whereas all these 3 cytoskeleton filaments play a role during Francisella infection an additional factor may be required for entry of ΔlpcC F. novicida uptake via the endocytic pathway.
Phosphorylation of RNA-processing Proteins, Post-transcription Regulation, and Mechanism of Cell Killing by ΔlpcC
We next performed an in-depth analysis of the proteins involved in RNA degradation, as this group was overrepresented among the differentially abundant phosphoproteins in cells infected with ΔlpcC compared with WT Francisella. The phosphoprotein tristetraprolin (TTP), product of the gene zfp36, drew our interest in particular because it contained two phosphopeptides with the most pronounced difference between ΔlpcC versus WT Francisella infection (Fig. 4A). Thus, we decided to validate TTP and investigate possible functions for differential phosphorylation during Francisella infection. Fig. 6A shows the MS/MS spectrum of a peptide corresponding to the phosphorylation at serine 178, and the magnified region details the signal intensity of the iTRAQ report ions used for the quantitative analysis clearly shows a difference between WT and ΔlpcC infections. The quantification at the 4 h time point exhibited 2.5- and 12.6-fold increases in WT and ΔlpcC Francisella infection compared with the mock, respectively (Fig. 6B). This observation was further validated by Western blot analysis. In independent experiments using both J774 and RAW 264.7 macrophages, we observed a dramatic increase in the phosphorylation status of endogenous TTP, indicated by the appearance of multiple higher molecular weight bands, particularly following infection with ΔlpcC Francisella compared with WT (Fig. 6C). Importantly, these higher molecular weight bands were diminished on treatment with λ-phosphatase, consistent with a shift in electrophoretic mobility because of phosphorylation (Fig. 6C). The Western blot against subunit 1 of RNA Polymerase II (Rbp1) served as a loading control, as well as a positive control for the phosphatase treatment (Fig. 6C).
Fig. 6.

Dynamics of TTP phosphorylation in RAW 264. 7 cells during Francisella infection. A, Tandem mass spectrum illustrating the identification and quantification of the phosphopeptide QSISpFSGLPSGR (corresponding to phosphorylation at serine 178) from tristetraprolin (Zfp36 or TTP) at 4 h postinfection. The expanded region highlights the iTRAQ report ions used for quantification. B, Time course quantification of TTP phosphorylation at serine 178. C, Western blot analysis of J774 and RAW 264.7 cell lines 4 h post infection with WT or ΔlpcC F. novicida. The presence of phosphorylation on TTP was determined by shift of electrophoretic mobility on treatment with λ-phosphatase (λPP).
With this support for hyper-phosphorylation of TTP by phosphoproteomic and Western blot analyses, we next investigated if this phenomenon could have consequences for in vivo murine infections. TTP is a well-known component of the mRNA-destabilizing machinery that binds to AU-rich elements (54, 55). TTP is also known to be hyper-phosphorylated by p38 MAPK, which is associated with a decrease in the RNA-binding capacity of this protein and results in the subsequent inhibition of RNA degradation (55, 56). In addition, as a negative feedback mechanism TTP phosphorylation was shown to be regulated by protein phosphatase 2A (57). Because TTP hyper-phosphorylation was observed with a much higher intensity in the ΔlpcC infection (p < 0.01), we hypothesized that Francisella could be down regulating TTP phosphorylation as a mechanism to shut down the translation of important factors that control its infection. To test this hypothesis we performed transcriptomic analyses using spleens of mice infected with either ΔlpcC or WT Francisella. We measured the abundance of ∼30,000 genes by microarray, which showed a more drastic change in expression profiles in mice infected with WT compared with ΔlpcC Francisella (supplemental Fig. S2A, supplemental Table S7). Next, we investigated the levels of transcripts that were recently identified as TTP substrates (54). Out of 310 mRNAs identified as targets of TTP, 245 were present in our dataset (supplemental Figs. S2B and S2C, supplemental Table S8). Although the TTP-regulated RNAs were on average slightly more abundant in ΔlpcC-infected samples compared with mock infections, they were slightly reduced in WT infections (supplemental Figs. S2B and S2C). To test if this difference in transcript abundance among the TTP-regulated RNAs was significant, we compared the transcripts found during ΔlpcC infections to those observed in WT infections (t test, p ≤ 0.05). Although 25% of the transcripts were more abundant in ΔlpcC compared with WT infections, this proportion was increased to 45% when only the TTP-regulated RNAs were considered, showing a clear enrichment (Chi-square contingency test, p = 0.0001). These data suggest that hyper-phosphorylation on TTP has an impact on the transcript levels during Francisella infection. These results may help explain the down-regulation of many immune response genes observed in monocytes infected with either F. novicida or F. tularensis Schu S4 (58).
TTP has been shown to be a key player on the regulation of cytokine production (54, 55, 59). Thus, we speculate that Francisella may inhibit TTP phosphorylation and consequently targeting the mRNA of selected cytokines to degradation and preventing their translation. To test this hypothesis, we infected RAW 264.7 cells and measured the cytokines by a multiplex ELISA array 6 h after infection. Six cytokines were shown to be produced in higher amounts during infection with ΔlpcC compared with WT: IL-1β, MDC, TARC, MCP-1, TNF-α and RANTES (Fig. 7A). Out of these six, IL-1β, MDC, MCP-1 and TNF-α, have been previously shown to be regulated by TTP. These results suggest that Francisella impairs the production of cytokines, likely by a mechanism that involves TTP-mediated mRNA degradation. To directly investigate this mechanism in RAW 264.7 cells, we used qPCR to monitor mRNA stability of IL-1β and RANTES cytokines after infection and in the presence of actinomycin D (Fig. 7B). IL-1β and RANTES were selected as representative pro-inflammatory cytokines and chemokines, respectively. With IL-1β, we observed a deficiency in its mRNA degradation following infection with ΔlpcC compared with WT; the most dramatic delineation appeared within the first hour. With RANTES, there was a trending delay in mRNA degradation with ΔlpcC within the first 2 h but it caught up with WT by 4 h postinfection. Importantly, there were no statistically significant differences in control Gapdh mRNA stability between WT and ΔlpcC infection. Together, our data suggest that Francisella can regulate the mRNA stability of particular cytokines, such as IL-1β, but for others it may only represent a contributing mechanism.
Fig. 7.

Cytokine profile and killing mechanism of cells infected with F. novicida. A, Cytokine profile. Cytokines were measured in culture supernatants 6 h postinfection using the Q-Plex ArrayTM kit. B, mRNA stability for IL-1β and RANTES was determined with qPCR following infection and Actinomycin D treatment. (n = 3, ±S.E., 2-way ANOVA, p < 0.001 for IL-1β, Bonferroni post tests, *p < 0.05, **p < 0.01) Viability (C), cytotoxicity (D), and apoptosis/necrosis (E) assays. Cells were infected with WT and ΔlpcC F. novicida strains and assayed for viability, cytotoxicity and apoptosis/necrosis using the ApoTox-GloTM Triplex Assay kit. As positive controls for lysis, necrosis and apoptosis, cells were treated with saponin, ionomycin (Ionom.), staurosporine (Stauros.), respectively.
Considering that several cytokines have cytotoxic effects, we analyzed the relationship of cytokine production and the hyper-cytotoxic phenotype of the ΔlpcC mutant. Because some of these cytokines, such as IL-1β and TNF-α, kill cells by triggering apoptosis (60, 61), we aimed to identify the mechanism of host cell death by ΔlpcC using the ApoTox-Glo™ triplex assay, which analyzes viability, necrosis, and apoptosis with the same assay well. Consistent with previous results (34), ΔlpcC is more cytotoxic to host cells than WT (Fig. 7C). Furthermore, ΔlpcC infection led to an increase in apoptosis, similar to the positive control with the apoptotic inducer staurosporine (Figs. 7D and 7E). These results suggest that Francisella avoids triggering the host apoptotic pathway potentially by inhibiting the production of select cytokines. In support of our results, Francisella has been shown to inhibit apoptotic signaling and to prolong the lifespan of infected neutrophils (62). Indeed, Francisella targeting of IL-1β- and TNF-α-mediated signaling has been proposed as potential mechanisms for anti-apoptotic activity in infected neutrophils (63).
CONCLUSIONS
We performed a comparative phosphoproteomic analysis of murine cells infected with F. novicida. Our data showed the participation of different cytoskeleton filaments in the process of bacterial entry into the host cells. Furthermore, by a combination of phosphoproteomics, transcriptomics, and cellular and immunological assays, we showed a deregulation of the signaling that controls TTP activity, a key component of post-transcriptional regulation in innate immune response (54, 57, 59). These findings suggest that Francisella targets host post-transcriptional regulatory machinery to repress the expression of innate immune factors that controls the infection.
Supplementary Material
Acknowledgments
We thank our colleagues Charles Ansong, Therese Clauss, Ron Moore, Penny Colton, and Brooke Kaiser for insightful comments, input, and suggestions.
Footnotes
* This work was supported by the National Institute of Allergy and Infectious Diseases (NIH/DHHS through interagency agreement Y1-AI-4894-01; project website www.SysBEP.org) and the National Institute for General Medicine (GM094623). This work used instrumentation and capabilities developed with support from the NIH grant 5P41RR018522-10, the National Institute of General Medical Sciences grant 8 P41 GM103493-10, and the U. S. Department of Energy Office of Biological and Environmental Research (DOE/BER). Significant portions of this work were performed in the EMSL, a DOE/BER national scientific user facility located at Pacific Northwest National Laboratory. The Pacific Northwest National Laboratory is operated for the DOE by Battelle under Contract DE-AC05-76RLO1830. This work was also supported by the NIH award NS076094 to X.A.C.
 This article contains supplemental Figs. S1 and S2 and Tables S1 to S8.
This article contains supplemental Figs. S1 and S2 and Tables S1 to S8.
Data availability: The LC-MS/MS runs and annotated spectra are available at PeptideAtlas under the accession number PASS00297.
1 The abbreviations used are:
- LPS
- lipopolysaccharide
- ASC
- apoptosis-associated speck-like protein containing a caspase recruitment domain
- CDC
- Centers for Disease Control and Prevention
- CR3
- CR3 complement receptor
- CytD
- cytochalasin D
- (FcγR)
- Fc-gamma receptor
- iTRAQ
- isobaric tags for relative and absolute quantification
- MAPK
- mitogen-activated protein kinase
- MR
- mannose receptor
- SEN
- surface-exposed nucleolin
- SRA
- scavenger receptor A
- TLR
- toll-like receptor
- TTP
- tristetraprolin
- WT
- wild-type.
REFERENCES
- 1. Sjostedt A. (2007) Tularemia: history, epidemiology, pathogen physiology, and clinical manifestations. Ann. N. Y. Acad. Sci. 1105, 1–29 [DOI] [PubMed] [Google Scholar]
- 2. Sjostedt A. (2005) Family III. Francisellaceae. in Bergey's Manual® of Systematic Bacteriology (Brenner D. J., Krieg N. R., Staley J. T., Garrity G. M. eds.), Springer, New York, NY: pp 199–209 [Google Scholar]
- 3. Champion M. D., Zeng Q., Nix E. B., Nano F. E., Keim P., Kodira C. D., Borowsky M., Young S., Koehrsen M., Engels R., Pearson M., Howarth C., Larson L., White J., Alvarado L., Forsman M., Bearden S. W., Sjostedt A., Titball R., Michell S. L., Birren B., Galagan J. (2009) Comparative genomic characterization of Francisella tularensis strains belonging to low and high virulence subspecies. PLoS Pathogens 5, e1000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rohmer L., Fong C., Abmayr S., Wasnick M., Larson Freeman T. J., Radey M., Guina T., Svensson K., Hayden H. S., Jacobs M., Gallagher L. A., Manoil C., Ernst R. K., Drees B., Buckley D., Haugen E., Bovee D., Zhou Y., Chang J., Levy R., Lim R., Gillett W., Guenthener D., Kang A., Shaffer S. A., Taylor G., Chen J., Gallis B., D'Argenio D. A., Forsman M., Olson M. V., Goodlett D. R., Kaul R., Miller S. I., Brittnacher M. J. (2007) Comparison of Francisella tularensis genomes reveals evolutionary events associated with the emergence of human pathogenic strains. Genome Biol. 8, R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thomas R. M., Titball R. W., Oyston P. C., Griffin K., Waters E., Hitchen P. G., Michell S. L., Grice I. D., Wilson J. C., Prior J. L. (2007) The immunologically distinct O antigens from Francisella tularensis subspecies tularensis and Francisella novicida are both virulence determinants and protective antigens. Infection Immunity 75, 371–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Atianand M. K., Duffy E. B., Shah A., Kar S., Malik M., Harton J. A. (2011) Francisella tularensis reveals a disparity between human and mouse NLRP3 inflammasome activation. J. Biol. Chem. 286, 39033–39042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dotson R. J., Rabadi S. M., Westcott E. L., Bradley S., Catlett S. V., Banik S., Harton J. A., Bakshi C. S., Malik M. (2013) Repression of inflammasome by Francisella tularensis during early stages of infection. J. Biol. Chem. in press 288, 23844–23857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shen H., Chen W., Conlan J. W. (2004) Mice sublethally infected with Francisella novicida U112 develop only marginal protective immunity against systemic or aerosol challenge with virulent type A or B strains of F. tularensis. Microb. Pathogenesis 37, 107–110 [DOI] [PubMed] [Google Scholar]
- 9. Asare R., Kwaik Y. A. (2010) Exploitation of host cell biology and evasion of immunity by francisella tularensis. Front. Microbiol. 1, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Henry T., Monack D. M. (2007) Activation of the inflammasome upon Francisella tularensis infection: interplay of innate immune pathways and virulence factors. Cell. Microbiol. 9, 2543–2551 [DOI] [PubMed] [Google Scholar]
- 11. Jones C. L., Napier B. A., Sampson T. R., Llewellyn A. C., Schroeder M. R., Weiss D. S. (2012) Subversion of host recognition and defense systems by Francisella spp. Microbiol. Mol. Biol. Rev. 76, 383–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bosio C. M. (2011) The subversion of the immune system by francisella tularensis. Front. Microbiol. 2, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Balagopal A., MacFarlane A. S., Mohapatra N., Soni S., Gunn J. S., Schlesinger L. S. (2006) Characterization of the receptor-ligand pathways important for entry and survival of Francisella tularensis in human macrophages. Infection Immunity 74, 5114–5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barel M., Hovanessian A. G., Meibom K., Briand J. P., Dupuis M., Charbit A. (2008) A novel receptor - ligand pathway for entry of Francisella tularensis in monocyte-like THP-1 cells: interaction between surface nucleolin and bacterial elongation factor Tu. BMC Microbiol. 8, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Geier H., Celli J. (2011) Phagocytic receptors dictate phagosomal escape and intracellular proliferation of Francisella tularensis. Infection Immunity 79, 2204–2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schulert G. S., Allen L. A. (2006) Differential infection of mononuclear phagocytes by Francisella tularensis: role of the macrophage mannose receptor. J. Leukocyte Biol. 80, 563–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clemens D. L., Lee B. Y., Horwitz M. A. (2005) Francisella tularensis enters macrophages via a novel process involving pseudopod loops. Infection Immunity 73, 5892–5902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lai X. H., Golovliov I., Sjostedt A. (2001) Francisella tularensis induces cytopathogenicity and apoptosis in murine macrophages via a mechanism that requires intracellular bacterial multiplication. Infection Immunity 69, 4691–4694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lindemann S. R., McLendon M. K., Apicella M. A., Jones B. D. (2007) An in vitro model system used to study adherence and invasion of Francisella tularensis live vaccine strain in nonphagocytic cells. Infection Immunity 75, 3178–3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parsa K. V., Butchar J. P., Rajaram M. V., Cremer T. J., Tridandapani S. (2008) The tyrosine kinase Syk promotes phagocytosis of Francisella through the activation of Erk. Mol. Immunol. 45, 3012–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tamilselvam B., Daefler S. (2008) Francisella targets cholesterol-rich host cell membrane domains for entry into macrophages. J. Immunol. 180, 8262–8271 [DOI] [PubMed] [Google Scholar]
- 22. Law H. T., Lin A. E. J., Kim Y., Quach B., Nano F. E., Guttman J. A. (2011) Francisella tularensis uses cholesterol and clathrin-based endocytic mechanisms to invade hepatocytes. Sci Rep-Uk 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cremer T. J., Butchar J. P., Tridandapani S. (2011) Francisella subverts innate immune signaling: Focus On PI3K/Akt. Front. Microbiol. 5, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hajjar A. M., Harvey M. D., Shaffer S. A., Goodlett D. R., Sjostedt A., Edebro H., Forsman M., Bystrom M., Pelletier M., Wilson C. B., Miller S. I., Skerrett S. J., Ernst R. K. (2006) Lack of in vitro and in vivo recognition of Francisella tularensis subspecies lipopolysaccharide by Toll-like receptors. Infection Immunity 74, 6730–6738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Katz J., Zhang P., Martin M., Vogel S. N., Michalek S. M. (2006) Toll-like receptor 2 is required for inflammatory responses to Francisella tularensis LVS. Infection Immunity 74, 2809–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cole L. E., Shirey K. A., Barry E., Santiago A., Rallabhandi P., Elkins K. L., Puche A. C., Michalek S. M., Vogel S. N. (2007) Toll-like receptor 2-mediated signaling requirements for Francisella tularensis live vaccine strain infection of murine macrophages. Infection Immunity 75, 4127–4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kawai T., Akira S. (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650 [DOI] [PubMed] [Google Scholar]
- 28. Belhocine K., Monack D. M. (2012) Francisella infection triggers activation of the AIM2 inflammasome in murine dendritic cells. Cell. Microbiol. 14, 71–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peng K., Broz P., Jones J., Joubert L. M., Monack D. (2011) Elevated AIM2-mediated pyroptosis triggered by hypercytotoxic Francisella mutant strains is attributed to increased intracellular bacteriolysis. Cell. Microbiol. 13, 1586–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jones J. W., Kayagaki N., Broz P., Henry T., Newton K., O'Rourke K., Chan S., Dong J., Qu Y., Roose-Girma M., Dixit V. M., Monack D. M. (2010) Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. U. S. A.107, 9771–9776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fernandes-Alnemri T., Yu J. W., Juliana C., Solorzano L., Kang S., Wu J., Datta P., McCormick M., Huang L., McDermott E., Eisenlohr L., Landel C. P., Alnemri E. S. (2010) The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rathinam V. A., Jiang Z., Waggoner S. N., Sharma S., Cole L. E., Waggoner L., Vanaja S. K., Monks B. G., Ganesan S., Latz E., Hornung V., Vogel S. N., Szomolanyi-Tsuda E., Fitzgerald K. A. (2010) The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Broz P., von Moltke J., Jones J. W., Vance R. E., Monack D. M. (2010) Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8, 471–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lai X. H., Shirley R. L., Crosa L., Kanistanon D., Tempel R., Ernst R. K., Gallagher L. A., Manoil C., Heffron F. (2010) Mutations of Francisella novicida that alter the mechanism of its phagocytosis by murine macrophages. PloS One 5, e11857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Craven R. R., Hall J. D., Fuller J. R., Taft-Benz S., Kawula T. H. (2008) Francisella tularensis invasion of lung epithelial cells. Infection Immunity 76, 2833–2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nguyen T. H., Brechenmacher L., Aldrich J. T., Clauss T. R., Gritsenko M. A., Hixson K. K., Libault M., Tanaka K., Yang F., Yao Q., Pasa-Tolic L., Xu D., Nguyen H. T., Stacey G. (2012) Quantitative Phosphoproteomic Analysis of Soybean Root Hairs Inoculated with Bradyrhizobium japonicum. Mol. Cell. Proteomics 11, 1140–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao R., Ding S. J., Shen Y., Camp D. G., 2nd, Livesay E. A., Udseth H., Smith R. D. (2009) Automated metal-free multiple-column nanoLC for improved phosphopeptide analysis sensitivity and throughput. J. Chromatog. 877, 663–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mayampurath A. M., Jaitly N., Purvine S. O., Monroe M. E., Auberry K. J., Adkins J. N., Smith R. D. (2008) DeconMSn: a software tool for accurate parent ion monoisotopic mass determination for tandem mass spectra. Bioinformatics 24, 1021–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim S., Gupta N., Pevzner P. A. (2008) Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. J. Proteome Res. 7, 3354–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Beausoleil S. A., Villen J., Gerber S. A., Rush J., Gygi S. P. (2006) A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 41. Monroe M. E., Shaw J. L., Daly D. S., Adkins J. N., Smith R. D. (2008) MASIC: a software program for fast quantitation and flexible visualization of chromatographic profiles from detected LC-MS(/MS) features. Comput. Biol. Chem. 32, 215–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Troyanskaya O., Cantor M., Sherlock G., Brown P., Hastie T., Tibshirani R., Botstein D., Altman R. B. (2001) Missing value estimation methods for DNA microarrays. Bioinformatics 17, 520–525 [DOI] [PubMed] [Google Scholar]
- 43. Smyth G. K. (2005) Limma: linear models for microarray data. in Bioinformatics and Computational Biology Solutions using R and Bioconductor (Gentleman R., Carey V., Dudoit S., Irizarry R., Huber W. eds.), Springer, New York, NY: pp 397–420 [Google Scholar]
- 44. Schwartz D., Gygi S. P. (2005) An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 23, 1391–1398 [DOI] [PubMed] [Google Scholar]
- 45. Lynn D. J., Winsor G. L., Chan C., Richard N., Laird M. R., Barsky A., Gardy J. L., Roche F. M., Chan T. H., Shah N., Lo R., Naseer M., Que J., Yau M., Acab M., Tulpan D., Whiteside M. D., Chikatamarla A., Mah B., Munzner T., Hokamp K., Hancock R. E., Brinkman F. S. (2008) InnateDB: facilitating systems-level analyses of the mammalian innate immune response. Mol. Sys. Biol. 4, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smoot M. E., Ono K., Ruscheinski J., Wang P. L., Ideker T. (2011) Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 48. Trost M., English L., Lemieux S., Courcelles M., Desjardins M., Thibault P. (2009) The phagosomal proteome in interferon-gamma-activated macrophages. Immunity 30, 143–154 [DOI] [PubMed] [Google Scholar]
- 49. Clay C. D., Soni S., Gunn J. S., Schlesinger L. S. (2008) Evasion of complement-mediated lysis and complement C3 deposition are regulated by Francisella tularensis lipopolysaccharide O antigen. Journal of immunology 181, 5568–5578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schwartz J. T., Barker J. H., Long M. E., Kaufman J., McCracken J., Allen L. A. (2012) Natural IgM mediates complement-dependent uptake of Francisella tularensis by human neutrophils via complement receptors 1 and 3 in nonimmune serum. J. Immunol. 189, 3064–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Campellone K. G., Welch M. D. (2010) A nucleator arms race: cellular control of actin assembly. Nat. Rev. 11, 237–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kuwata H., Yamamoto S., Miyazaki Y., Shimbara S., Nakatani Y., Suzuki H., Ueda N., Yamamoto S., Murakami M., Kudo I. (2000) Studies on a mechanism by which cytosolic phospholipase A2 regulates the expression and function of type IIA secretory phospholipase A2. J. Immunol. 165, 4024–4031 [DOI] [PubMed] [Google Scholar]
- 53. Eriksson J. E., He T., Trejo-Skalli A. V., Harmala-Brasken A. S., Hellman J., Chou Y. H., Goldman R. D. (2004) Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J. Cell Sci. 117, 919–932 [DOI] [PubMed] [Google Scholar]
- 54. Kratochvill F., Machacek C., Vogl C., Ebner F., Sedlyarov V., Gruber A. R., Hartweger H., Vielnascher R., Karaghiosoff M., Rulicke T., Muller M., Hofacker I., Lang R., Kovarik P. (2011) Tristetraprolin-driven regulatory circuit controls quality and timing of mRNA decay in inflammation. Mol. Sys. Biol. 7, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ronkina N., Menon M. B., Schwermann J., Tiedje C., Hitti E., Kotlyarov A., Gaestel M. (2010) MAPKAP kinases MK2 and MK3 in inflammation: complex regulation of TNF biosynthesis via expression and phosphorylation of tristetraprolin. Biochem. Pharmacol. 80, 1915–1920 [DOI] [PubMed] [Google Scholar]
- 56. Brooks S. A., Blackshear P. J. (2013) Tristetraprolin (TTP): interactions with mRNA and proteins, and current thoughts on mechanisms of action. Biochim. Biophys. Acta 1829, 666–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sun L., Stoecklin G., Van Way S., Hinkovska-Galcheva V., Guo R. F., Anderson P., Shanley T. P. (2007) Tristetraprolin (TTP)-14-3-3 complex formation protects TTP from dephosphorylation by protein phosphatase 2a and stabilizes tumor necrosis factor-alpha mRNA. J. Biol. Chem. 282, 3766–3777 [DOI] [PubMed] [Google Scholar]
- 58. Butchar J. P., Cremer T. J., Clay C. D., Gavrilin M. A., Wewers M. D., Marsh C. B., Schlesinger L. S., Tridandapani S. (2008) Microarray analysis of human monocytes infected with Francisella tularensis identifies new targets of host response subversion. PloS One 3, e2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Qiu L. Q., Stumpo D. J., Blackshear P. J. (2012) Myeloid-specific tristetraprolin deficiency in mice results in extreme lipopolysaccharide sensitivity in an otherwise minimal phenotype. J. Immunol. 188, 5150–5159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hogquist K. A., Nett M. A., Unanue E. R., Chaplin D. D. (1991) Interleukin 1 is processed and released during apoptosis. Proc. Natl. Acad. Sci. U. S. A. 88, 8485–8489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rath P. C., Aggarwal B. B. (1999) TNF-induced signaling in apoptosis. J. Clin. Immunol. 19, 350–364 [DOI] [PubMed] [Google Scholar]
- 62. Schwartz J. T., Barker J. H., Kaufman J., Fayram D. C., McCracken J. M., Allen L. A. (2012) Francisella tularensis inhibits the intrinsic and extrinsic pathways to delay constitutive apoptosis and prolong human neutrophil lifespan. J. Immunol. 188, 3351–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schwartz J. T., Bandyopadhyay S., Kobayashi S. D., McCracken J., Whitney A. R., Deleo F. R., Allen L. A. (2013) Francisella tularensis alters human neutrophil gene expression: insights into the molecular basis of delayed neutrophil apoptosis. J. Innate Immun. 5, 124–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.