

Abstract
Metastasis is the major cause of death for cancer patients with solid tumours, due mainly to the ineffectiveness of current therapies once metastases begin to form. Further insight into the biology of metastasis is therefore essential in order to gain a greater understanding of this process and ultimately to develop better cancer therapies. Metastasis is an inefficient process, such that very few cells that leave a tumour successfully form macrometastases in distant sites. This suggests that only a small subset of cells can successfully navigate the metastatic cascade and eventually re-initiate tumour growth to form life-threatening metastases. Recently, there has been growing support for the cancer stem cell (CSC) hypothesis which stipulates that primary tumours are initiated and maintained by a small subpopulation of cancer cells that possess “stem-like” characteristics. Classical properties of normal stem cells are strikingly reminiscent of the observed experimental and clinical behaviour of metastatic cancer cells, including an unlimited capacity for self renewal; the requirement for a specific ‘niche’or microenvironment to grow; use of the stromal cell-derived factor 1 (SDF-1)/chemokine receptor 4 (CXCR4) axis for migration; enhanced resistance to apoptosis and an increased capacity for drug resistance. Therefore, in addition to playing a role in primary tumour formation, we believe that CSCs are also key players in the metastatic process. We will review the current evidence supporting this idea and discuss the potential implications of the CSC hypothesis with regards to experimental investigation and treatment of metastatic disease.
Keywords: metastasis, metastatic ineffiiency, stem cell, cancer stem cell (CSC), CD44, CD133, metastatic niche, SDF-1/CXCR4 axis, resistance to therapy
Introduction
It is estimated that North Americans have an approximately 40% risk of developing cancer in their lifetimes [1, 2]. With many cancers, early detection and timely treatment with existing therapies can successfully reduce morbidity and mortality, such that a little less than half of patients diagnosed with cancer will actually die of the disease [1]. Depending on the type and severity of the cancer, current therapeutic options include surgery, radiation therapy (RT), and systemic therapies such as cytotoxic chemotherapy and/or hormonal therapy [3, 4]. More recently, several promising molecular targeted agents have been approved for use in the clinic, including targeting of Her-2 with Herceptin® (trastuzumab;breast cancer) [5], targeting of vascular endothelial growth factor (VEGF) with Avastin® (bevacizumab; colorectal and lung cancer) [6, 7] and targeting of the epidermal growth factor (EGFR) with Iressa® (gefitinib; lung cancer) [8].
Despite these promising advances, the majority (if not all) of these therapies fail in the metastatic setting. For example, even after complete remission in response to therapy, less than 20% of patients with metastatic breast cancer will remain disease-free for more than 5 years [9]. Therefore, there is an essential need to better understand the biology of metastasis in order to develop new therapeutic approaches that will be effective for combating this deadly process. The emergence of the cancer stem cell (CSC) hypothesis, predicting that a small subpopulation of ‘stem-like’ cells are responsible for initiating and maintaining cancer growth, may hold promise in terms of new approaches to cancer therapy in the metastatic setting. This review will discuss the metastatic process and the CSC hypothesis; describe the striking similarities between CSCs, normal stem cells and metastatic cells; and speculate about the implications that these ideas may have for the progression and treatment of metastatic disease.
The metastatic process
Metastasis is a sequential, multi-step process that requires cancer cells to escape from the primary tumour, intravasate into the circulation or lymphatic system, migrate through the body, adhere at a secondary site, extravasate from the circulation and into the secondary tissue, form micrometastases, develop a blood supply, and finally form macroscopic, clinically relevant metastases (Fig. 1) [10, 11].
1.
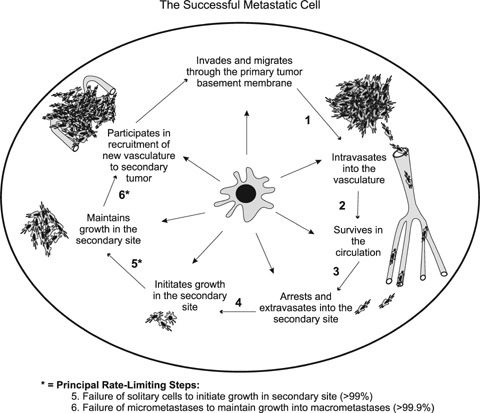
The successful metastatic cell must carry out a number of sequential steps in order to form clinically relevant metastases. Based on the complexity of the metastatic process, it seems unlikely that all cancer cells would be able to successfully complete all the steps necessary to form macrometastases. Indeed, it is known that metastasis is a highly inefficient process, and that not all the steps of the metastatic process are equally inefficient. The principal rate limiting steps are (5) initiation of growth into micrometastases; and (6) maintenance of growth into macrometastases, whereby less than ∼2% of solitary cells are able to initiate growth and less than ∼0.02% of cells are able to maintain growth into clinically relevant metastases [10, 12, 13].
Metastatic inefficiency
Based on the complexity of the metastatic process, it seems unlikely that all cancer cells would be able to successfully complete all the steps necessary to form clinically relevant metastases. Indeed, it is known that metastasis is a highly inefficient process, and interestingly, that not all the steps of the metastatic process are equally inefficient [10, 12, 13]. Previous studies by Luzzi et al. (1998) used in vivo video microscopy to follow the fate of metastatic cells. They observed that cancer cells were very proficient at intravasating, migrating through the circulation, adhering at the secondary site and extravasating back out of the vasculature. In fact, almost 90% of metastatic cells could successfully complete these early steps in the metastatic process. However, only ∼2% of disseminated cells were able to form micrometastases, and only ∼0.02% of cells were able to develop into vascularized macrometastases [13]. These results suggest that only a subset of tumour cells are capable of successfully traversing the entire metastatic cascade, and that the initiation and maintenance of cancer cell growth at a secondary site is what limits metastasis (Fig. 1).
Organ-specific metastasis: seed and soil
This regulation of growth at the secondary site also differs depending on which organ the tumour cells metastasize to, and many cancers show an organ-specific pattern of metastasis. For example, colorectal cancers preferentially metastasize to the liver, prostate cancer often metastasizes to bone and breast cancer favours metastasis to regional lymph nodes, bone, liver, brain and lungs [10, 11]. It was originally believed that many metastases could be explained purely by circulatory patterns [14]. According to this mechanical hypothesis, since cancer cells are much larger than blood cells, cancer cells would be forced to arrest in the capillary bed of the first organ they encounter in the circulation, and thus form metastases wherever they are stopped [10, 14].
However, several theories have challenged this idea by proposing that there are additional, molecularlevel mechanisms which explain why and how cancer cells can arrest and grow in ‘favourite’metastatic sites. The most central of these theories is the ‘seed and soil’ theory of metastasis, first proposed in 1889 by Stephen Paget [15]. Paget predicted that cancer cells (the ‘seed’) can survive and proliferate only in secondary sites (the ‘soil’) that produce growth factors appropriate to that type of cell, and this theory has largely withstood the test of time [16]. In a meta-analysis of published autopsy study data [17], Weiss showed that, in many cases, metastases detected at autopsy were in proportion to the blood flow from the primary tumour site to the secondary organ. However, in some cases, more metastases (notably breast cancer metastasis to bone) or fewer metastases were detected than would be expected by blood flow alone, indicating that the ‘soil’ or microenvironment in the secondary organ is likely very important [15–17]. Another concept, often called the homing theory, proposes that different organs produce chemotactic factors (i.e. chemokines, such as stromal cell-derived factor 1 [SDF-1]) which can attract specific types of tumour cells to ‘home’ to and arrest in a particular organ [18–20]. It is likely that all these theories could be correct simultaneously. Since metastasis is such an inefficient process, and since the inefficiency lies in the growth of cancer cells in the secondary tissue, it is possible that the primary method of dissemination is mechanical (i.e. blood flow patterns) and/or dependent on chemotactic factors, and whether or not a tumour will form depends on if the metastatic microenvironment is suitable to sustain tumour growth.
Epithelial-mesenchymal transition (EMT)
Another theory that has gained favour in recent years is the idea that epithelial-mesenchymal transition (EMT) can contribute to the metastatic process. First identified in embryonic development, EMT involves conversion of epithelial cells to a mesenchymal phenotype via loss of polarity and cell–cell contacts and dramatic cytoskeletal remodelling [21]. Cells undergoing EMT also acquire expression of mesenchymal proteins and develop an enhanced ability to migrate, thus assisting in cell distribution throughout the embryo and organ development.
In cancer, it is believed that epithelial tumour cells may be able to somehow activate this primitive developmental program, thus converting differentiated epithelial cancer cells into de-differentiated cells that possess more mesenchymal characteristics [22]. The EMT phenotype in cancer has been associated with a decrease in tumour growth, increased resistance to apoptosis, increased motility and invasiveness, and enhanced metastatic ability [23]. These phenotypic transitions are reversible, and it is hypothesized that once tumour cells have reached their destination, they may transform back into an epithelial phenotype in order to facilitate tumour growth in the secondary site [24]. EMT is characterized by the expression of various factors responsible for mediating this process at the molecular level. The growth factor transforming growth factor β (TGF-β) has been shown to induce reversible EMT, along with Wnt pathway proteins (in particular β-catenin), Notch, and Hedgehog signalling pathways, which often act in a sequential manner to induce EMT [24–28]. Additionally, transcription factors such as Snail and NF-κB have been shown to confer apoptotic resistance to tumour cells undergoing EMT [29, 30]. Given the developmental origin of EMT, it is interesting but not surprising that many of these molecules have also been associated with stem cell maintenance [22]. In fact, metastatic cells share many cellular and molecular characteristics of cells important to early development, and growing evidence suggests that many cancers may contain small subsets of ‘stem-like’ cells or ‘CSCs’ that are responsible for tumour initiation and progression.
The CSC hypothesis
There are currently two conflicting views in the literature that attempt to explain tumour formation. The Stochastic Model suggests that every cell within a tumour is a potential tumour-initiator, but that entry into the cell cycle is governed by a low probability of stochastic events. According to this model, it would be impossible to tell which cell would initiate the potential tumour since each cell should have an equal ability to be tumourigenic. In contrast, the Hierarchy Theory proposes that only a small subset of cells in a tumour are capable of initiating tumour growth, but that these cells all do so at a high frequency. According to this theory, it should be possible to identify the cells responsible for new tumour formation because not all the cells have the same phenotypic and functional characteristics [31]. This idea paved the way for the CSC hypothesis, which postulates that cancer arises from a subpopulation of tumour-initiating cells, or CSCs.
The CSC hypothesis is not a new idea. In fact, in 1855, Rudolph Virchow proposed that cancer arises from embryonic-like cells [32]. However, the existing technology at the time was not sensitive enough to properly test this hypothesis. The recent emergence of new techniques allowing accurate identification, isolation, and characterization of single cells within a population (such as normal stem cells and stem-like cells in tumours) has led to the identification of embryonic-like stem cells in some adult tissues [33] and a resurgence of interest and debate about the CSC hypothesis.
Evidence supporting the CSC hypothesis
In 1994, the first solid evidence supporting the CSC hypothesis was provided when a population of stem-like cells was found in acutemyeloid leukaemia (AML) [34]. The cell surface phenotype CD34+CD38−was associated with cells displaying stem-like properties, and this was confirmed by their ability to engraft non-obese diabetic (NOD)/ severe combined immunodeficiency (SCID) mice and to produce large numbers of colony-forming progenitors. Another pivotal study by Hope et al. (2004) demonstrated for the first time that AML originated from a cellular hierarchy, with only the most primitive of cells being able to initiate and sustain the leukaemia [35]. Further to these findings in leukaemia, tremendous research has been directed towards identifying stem-like cells in solid tumours, and there is strong evidence supporting the existence of stem cell-like populations in brain, colon, breast, prostate, and pancreatic tumours [36–42].
Stem-like cells have been identified in brain, colon, and pancreatic tumours based on CD133+ marker expression [36, 37, 42]. When CD133+ expressing cells were injected into immunocompromised mice, as few as one hundred stem-like cells were sufficient to cause tumour formation in mice, whereas injections of ten thousand CD133− cancer cells consistently failed to form tumours. In breast cancer, stem-like cells were identified based on CD44+CD24−/low cell surface phenotype, and isolated from primary tumours or pleural effusions from breast cancer patients [38]. These cells were then injected into the mammary fat pad of immunocompromised mice to measure the cells’ability to form new breast tumours. Again, as few as one hundred of these cells were sufficient to cause tumour formation in the mice. In contrast, even injections of tens of thousands of cancer cells that did not express the CD44+CD24−/low phenotype failed to cause tumour formation. Interestingly, Notch 3 (which plays a role in stem cell self-renewal, cell fate, apoptosis, and proliferation) has been found to be up-regulated in CD44+ populations of normal and breast cancer cells [43]. This suggests that Notch expression could be increased in the CD44+CD24−(CSC) population of breast cancer cells. It is known that the amplification of Notch receptors and the presence of ligands (i.e. Jagged-1) is correlated with a more aggressive phenotype [28, 43, 44], and this supports the idea that the CSCs are the aggressive cancer cells within a tumour.
Populations of CD44+ stem-like prostate cancer cells have also been identified, and these cells express higher mRNA levels of several ‘stemness’ genes, such as Smoothened (SMO) and Oct ¾ when compared to CD44− cells. Additionally, CD44+ prostate cancer cells can generate CD44− cells in vitro and in vivo, and also appear to undergo asymetrical division in clonal analysis [39, 40]. Small populations of cancer cells within pancreatic tumours were also identified based on CD44+CD24+ESA+ cell surface marker expression, and these cells demonstrated a 100-fold increase in tumourigenic potential compared to the rest of the cells in the tumour [41]. Furthermore, tumours that formed from CD44+CD24+ESA+ cells were indistinguishable from the human tumours in which they originated, indicating that the tumour-initiating cells were stem cell-like in their ability to self-renew and give rise to a heterogeneous cell population.
Defining the CSC
Given the intense research interest in the CSC hypothesis, it is important at this stage to more clearly define what makes a cancer cell a ‘CSC’ so that researchers working in different model systems are able to compare cells exhibiting a common set of properties. Recently, a consensus definition was proposed by Clarke et al. (2006) such that a CSC is ‘a cell within a tumour that possesses the capacity to self-renew and to cause the heterogeneous lineages of cancer cells that comprise the tumour’[45]. It should be emphasized that self-renewal is not tantamount to proliferation. Normal stem cells can self-renew and give rise to either two identical daughter stem cells (symmetric division) or give rise to one daughter stem cell and one more differentiated progenitor cell (asymmetric division), thus generating a heterogeneous population [46]. There is evidence that CSCs can also self-renew to form heterogeneous cell populations, since injection of isolated brain, colon, breast, prostate or pancreatic cancer stem-like cells into immunocompromised mice results in the formation of tumours that are phenotypically identical to the original tumour and contain both stem-like cells and non-stem-like cells [36–42, 46, 47].
Identifying and isolating CSCs
In order to study CSCs, is important that these cells are accurately identified and prospectively isolated from the rest of the tumour cell population in a consistent manner. This task has proven to be challenging because of the lack of universality in morphologic characteristics and marker expression between cancer types. Nonetheless, as described above, certain markers have recently emerged as possible CSC identifiers, including CD133 and CD44 [36–42, 47]. Interestingly, CD133 is a marker expressed by many types of normal stem cells, including neural and haematopoietic stem cells [36, 37]. CD44 is a cell surface receptor for hyaluronic acid, and is involved in cell migration and adhesion when interacting with other common ligands including osteo-pontin (OPN), collagens and matrix metalloproteinases (MMPs) [47, 48] Furthermore, CD44 is commonly implicated in metastasis [47], supporting the idea that these stem-like, tumour-initiating cells may also be the cells that survive the metastatic process and continue on to form clinically relevant metastases.
However, because of the heterogeneous nature and inherent genomic instability of solid cancers, caution should be exercised when basing CSC identity on marker expression alone [45, 47]. Assays that measure functional characteristics of normal stem cells may therefore prove useful in helping to identify CSCs in a way that is complementary to marker analysis, but avoids the problem of gene expression instability that commonly occurs in cancer cells. An example of these functional assays is ‘side-population’ analysis, which identifies a subset of cells within a population that can more efficiently efflux dyes or drugs [49–51]. Normal stem cells have a high ability to efflux drugs and dyes due to the high expression of ATP binding cassette (ABC) transporters, which actively pump drugs out of the cell [49]. Hirschman-Jax et al. (2004) found that there was a small SP in neuroblastomas, as well as in breast cancer, lung cancer and glioblas-toma cell lines that could efflux the Hoechst 33342 dye at a comparable rate to normal stem cells [50]. Similarly, measurement of the activity of aldehyde dehydrogenase (ALDH) (another enzyme implicated in drug resistance) is a side population assay that has been used to identify normal and CSCs [51]. Other potentially useful functional assays for identifying CSCs include serial colony-forming unit (CFU) assays and sphere formation assays (i.e. neu-rospheres, mammospheres). Regardless of the approach used for prospective isolation of stem cells, the ‘gold standard’ for identifying stem cells (including CSCs) is in vivo reconstitution ability [45].
Once CSCs can be reliably identified and isolated from different tumour types, they can be compared to non-CSCs to assess differences in malignant behav-iour, such as growth, resistance to apoptosis, adhesion, migration, invasion, metastasis and drug resistance. Since the emergence of the first evidence supporting the CSC hypothesis, CSCs have become hot topic in cancer biology. However, one of the most under-investigated areas of study in the CSC field is the relationship between stem cells, CSCs, and metastasis.
Parallels between stem cell behaviour and metastatic behaviour
As discussed earlier, it is well known that metastasis in an inefficient process. At present, this inefficiency is explained in terms of the need for cancer cells to find the proper microenvironment, since initiation and maintenance of tumour growth in secondary organs are the most inefficient steps of the metastatic process. We and others believe that there may be an alternative explanation for this inefficiency, one related to the CSC hypothesis [52–56].
Rare cells drive metastatic progression, a fact illustrated by the observation that less than 1% of disseminated cancer cells are able to form clinically relevant macrometastases [13]. Application of the CSC hypothesis to metastasis therefore suggests that this rare subset of cells within a primary tumour that are capable of re-initiating growth to form metastases in distant sites may in fact be CSCs. It also suggests that the inefficiency of metastasis may be due both to the rarity of CSCs and the unsuitable habitats in different microenvironments. This is supported by the observation that metastatic cells and stem cells share a number of key properties, including an unlimited capacity for self-renewal (described earlier); the requirement for a specific ‘niche’ or microenvironment to grow (and, juxtapositional with that, a self-protective ability to grow in harsh environments);use of the SDF-1/CXCR4 axis for migration; enhanced resistance to apoptosis;and an increased capacity for drug resistance (Table 1).
1.
Stem cell properties displayed by metastatic cells could be potentially exploited for therapeutic targeting of cancer stem cells (CSCs).
| Stem cell property | Potential molecular factors involved | Proposed therapeutic strategy | ||
|---|---|---|---|---|
| Requirement for specific ‘niche’or microenvironment: | • TGF-β | Differentiation therapy | ||
| • Other factors? | ||||
| • growth versus differentiation | ||||
| • maintenance of the CSC pool | ||||
| Use of the SDF-1/CXCR-4 axis: | • SDF-1 | Treatment with agents that target CXCR4 | ||
| • homing to secondary sites | • CXCR4 | (i.e. TN14003, AMD3100) | ||
| • adhesion, migration, invasion | • Other factors? | |||
| Resistance to apoptosis and protection from cellular insult: | • TGF-β | Treatment with agents that target: | ||
| • Hedgehog (HH) | • HH signalling | |||
| • maintenance of the CSC pool | • Bmi-1 | (i.e. cyclopamine) | ||
| • drug resistance | • Bcl-2 | • Bmi-1 | ||
| • resistance to DNA damage | • Notch-1 | • Bcl-2 | ||
| • ABC transporters (i.e. ABCC1, ABCB1, ABCG2) | • Notch-1 | |||
| • DNA checkpoint proteins (i.e. Rad17, Chk1, Chk2) | • DNA checkpoint proteins | |||
| • Other factors? | • ABC transporters | |||
References [18, 19, 48, 68, 69, 72–74, 76–79, 81, 91, 92, 96, 97, 101, 102].
The metastatic niche
Normal stem cells require a specific niche or microenvironment in order to grow and survive [57–59]. The stem cell niche is an anatomically defined space that has been identified in many different tissue types, and it serves to regulate stem cell number and function as well as to modulate stem cells under conditions of physiologic change. The niche cells and the microenvironment they create allow the niche to maintain the stem cell pool and prevent its differentiation, while at the same time also directing tissue growth and renewal through more differentiated daughter progenitor cells [57]. Furthermore, the niche provides protection to stem cells. In those tissues where the niche is well defined, the stem cell may be practically enveloped by differentiated cells which work to house and interact with the stem cells. For example, in the mammary gland, putative stem and progenitor cells are sequestered from both the basement membrane and the lumen in structurally specialized spaces. The presence of a niche within the epithelium can provide nourishment, yet exclusion from molecules that may cause differentiation or mutation [58]. Besides protective effects, the stem cell niche can also play a role in determining cell fate. The physical orientation of a stem cell in its niche can affect the symmetry of cell division, and niche-forming cells can be stimulated by growth factors to produce ligands that act on stem cell receptors such as Notch to initiate stem cell mitosis or specify differentiation [58, 59].
Metastatic cells, like normal stem cells, require a particular niche to grow. This was elegantly demonstrated by Kaplan et al. (2005), who showed that bone marrow-derived VEGFR1+ haematopoietic cells (HPC) can home to tumour-specific pre-metastatic sites and form cellular clusters before the arrival of metastatic tumour cells in mice [60]. At these sites, the bone marrow-derived cells express several haematopoietic markers, such as CD34, CD116, c-kit and Sca-1, which help in maintaining their progenitor cell status within the tissue parenchyma in the pre-metastatic niche. These VEGFR1+ HPCs (along with fibronectin) alter the local microenvironment, which leads to activation of integrins and chemokines (such as SDF-1) that promote attachment, survival, and growth of tumour cells. When treated with an anti-VEGFR1+ antibody, the supportive pre-metastatic cell clusters were abolished and metastasis was prevented, indicating that these clusters play an important role in the metastatic process [60–63].
Additional studies have shown that other niche cells, such as bone marrow-derived mesenchymal stem cells have been shown to localize to breast carcinomas, and seem to be involved in cancer metastasis [64]. When mesenchymal stem cells were mixed with non-metastatic MCF-7/Ras human breast cancer cells, their metastatic potency increased in immunocompromised mice. Furthermore, breast cancer cells stimulate chemokine secretion from the mesenchy-mal cells, which acts in a paracrine fashion on cancer cells to enhance their motility, invasion and metastasis.
Lysyl oxidase (LOX) and its regulator, hypoxia inducible factor (HIF), may also be factors that influence the tumour microenvironment or niche to favour metastasis. It is known that patients with high LOX-expressing tumours (i.e. increased hypoxia in tumours) have decreased survival due to more aggressive metastasis. Secreted LOX is thought to be responsible for the invasive properties of hypoxic human cancer cells through FAK activity and cell-matrix adhesion [65]. Interestingly, when LOX was inhibited (using β-aminoproprionitrile [BAPN]. or an antibody targeting LOX), metastasis was eliminated in mice, but there was no effect on the primary tumour. This suggests that hypoxic environments and the induced expression of LOX and HIF are important in the metastatic setting. Normal stem cells also do well in hypoxic environments, typically maintaining an undifferentiated state that is important to preservation of the stem cell pool [66].
There has been some controversy over whether or not CSCs require specific niches to grow. However, in leukaemia, studies have shown that leukaemia stem cells actually occupy a similar region as normal haematopoietic stem cells (the perioendosteal region), suggesting that CSCs may in fact require a niche to protect and maintain the tumour initiating/tumour sustaining CSCs [57]. In the situation of metastasis, we might consider that the tumour microenvironment in a secondary organ is in fact a ‘metastatic niche’. Initially, it was believed that the microenvironment where a metastatic cell ended up either passively supported tumour development and facilitated tumour formation, or it did not support growth, and no tumour was able to form. As discussed above, there is now evidence to suggest that the tumour microenvironment actively contributes to the growth and invasion of metastatic tumours, and that non-CSCs may actually contribute to the creation of the CSC niche [31, 57, 59, 60, 67, 68]. In metastasis, the niche could be supporting the establishment and expansion of CSCs either through normal or dysregulated signalling. For example, TGF-β has been shown to inhibit the proliferation of quiescent haematopoietic stem cells [69], so perhaps non-CSCs in leukaemia control TGF-β secretion to dys-regulate CSC division, thus influencing the balance between CSC proliferation, self-renewal, differentiation, and senescence. In solid cancers, it could be hypothesized that CSCs in metastatic sites are similarly regulated by inappropriate signalling from the metastatic niche [70, 71]. Currently, the similarities between stem cell niches in different tissues remains poorly understood, in particular with regards to whether tissue-specific stem cells can be regulated by stem cell niches in other organs. This knowledge will have important implications for understanding metastatic growth in secondary sites, including the possibility that some CSCs (i.e. breast, prostate) may favour growth in the bone marrow because it provides a particularly rich stem cell niche [52].
The SDF-1/CXCR4 axis
Another similarity between metastatic cells and stem cells is their use of chemokine pathways for migration. The chemokine SDF-1 is believed to play a critical role in stem cell migration in cooperation with its receptor CXCR4 [19]. SDF-1 is an ideal candidate for aiding in metastasis because its major biological effects are related to the ability of this chemokine to induce motility, chemotactic responses, adhesion, secretion of MMPs and secretion of angiopoietic factors such as VEGF in cells that express CXCR4. SDF-1 also increases adhesion of cells to VCAM-1, fibronectin and fibrinogen by activating/modulating the function of several cell surface integrins [18]. In cancer development, fibroblast expression of SDF-1 and tumour cell expression of CXCR4 is often increased within hypoxic areas of the tumour, subsequently triggering tumour cell growth, motility and invasiveness. Furthermore, many CXCR4+ metastatic cells use the SDF-1/CXCR4 axis to migrate through the body according to an SDF-1 gradient [18, 72]. In support of this, studies have demonstrated that breast cancer cells treated with a CXCR4 inhibitor show significantly inhibited metastatic ability [73], and intracranial glioblastoma and medulloblastoma xenografts treated with a CXCR4 antagonist (AMD3100) show reduced cell growth and increased tumour cell apoptosis [74]. This axis may also help to explain the organ-specific nature of metastatic growth, since CXCR4-expressing cancer cells may home to organs that express high levels of SDF-1. For example, breast cancer has been shown to metastasize experimentally using the SDF-1/CXCR4 axis, with CXCR4-expressing breast cancer cells preferentially metastasizing to SDF-1-expressing organs such as lymph nodes, liver, and bones [10, 11, 75]. Many cancers are positive for CXCR4 expression [18, 72, 75], and it seems, at least in breast cancer, that CXCR4 expression correlates with the CSC content, and thus the aggressiveness, of cancer cell lines. For example, relative to non-metastatic MCF-7 breast cancer cells, highly metastatic MDA-MB-231 cells have a larger proportion of CSCs and express higher levels of CXCR4 [72, and our unpublished data]. Furthermore, in pancreatic cancer, it was recently shown that a CD133+ CSC population was heterogeneous in nature with regards to CXCR4 expression, and that only the CD133+CXCR4+ CSCs were able to metastasize [42].
Resistance to apoptosis and protection from cellular insult
Resistance to apoptosis is another key property shared by both normal stem cells and highly malignant tumour cells. Stem cells can resist apoptosis by a number of mechanisms, including via TGF-β signalling or activation of the Hedgehog (HH) pathway [76]. Additionally, stem cells also express higher levels of anti-apoptotic proteins than differentiated cells, including members of the Bcl-2 family [77]. Stem cells must also resist early senescence in order to maintain the stem cell pool, a process facilitated by Bmi-1 expression. Interestingly, Bmi-1, Bcl-2, TGF-β and HH pathway components have all been shown to be up-regulated in cancer cells [48, 76–79]. Furthermore, despite their limitless self-renewal capacity, normal stem cells are relatively quiescent and divide infrequently unless activated [80–82]. Similarly, CSCs may cycle through long periods of quiescence and short bursts of proliferation, and since most chemotherapeutic anti-cancer agents are designed to target rapidly dividing cells, this may be one mechanism by which CSCs escape cytoxicity from these drugs.
From an evolutionary point of view, normal stem cells have a number of unique properties that help protect them from cellular insult and ensure their long lifespan. For instance, normal stem cells express high levels of ABC transporters that facilitate rapid efflux of toxins and drugs, but these genes get turned off in committed progenitors and mature cells [81]. These transporters include ABCB1, which encodes P-glycoprotein, and ABCG2, which encodes a protein called breast cancer resistance protein (BCRP). These two transporter proteins can help a cell efflux a large number of different chemotherapeutic agents, including doxorubicin and paclitaxel. Along with ABCC1, ABCB1 and ABCG2 represent the three principal multi-drug resistance (MDR) genes overexpressed in tumour cells [80, 83–85]. This drug resistance could be an inherent feature of CSCs, and could help explain the high level of drug resistance in metastatic disease [52, 81].
Finally, stem cells are also thought to be more resistant to DNA damaging agents than differentiated cells because of their ability to undergo asynchronous DNA synthesis and their enhanced capacity for DNA repair. During asynchronous DNA synthesis, the parental ‘immortal’ DNA strand always segregates with the new stem cell rather than with the differentiating progeny, thus helping to protect the stem cell population from DNA damage [86–90]. Similarly, CSCs are believed to be resistant to radiation therapy by preferentially up-regulating their DNA proofreading mechanisms in order to avoid cellular death due to DNA damage [91–92]. Studies have shown that treating a tumour with radiation can deplete the non-CSC population and increase the CSC population by 3–5 fold, thus rendering the tumour even more aggressive and resistant to treatment [92]. It is also possible that CSCs may tend to be located in the hypoxic regions of tumours, which would affect their sensitivity to radiation via the oxygen enhancement ratio. It is more likely that radioresistance is a general property of CSCs, due to their ability to more efficiently repair their DNA than non-CSCs [93].
Origin of the CSC: normal stem cell gone bad?
Given the many parallels between stem cells and metastatic cancer cells, one of the most elusive and highly debated research questions surrounding the CSC hypothesis is regarding the definitive cellular origin of the CSC [45, 53]. If these cells arise from normal stem cells, then cancer cells could requisition the existing stem cell regulatory pathways for self-renew-al. On the other hand, if these cells arise from mature, differentiated cells, oncogenic mutations would be required to drive de-differentiation and self-renewal.
The fact that multiple mutations are necessary for a cell to become tumourigenic and metastatic has implications for the cellular origin of cancer cells. It can be argued that mature cells have a very limited lifespan, and thus it is unlikely that all the necessary mutations could occur during the relatively short life of these cells. In contrast, the infinite self-renewal capacity of normal stem cells means these cells may be the only cells that are around long enough to accumulate the necessary mutations [52]. It has been suggested that stem or progenitor cells must be the initial targets for malignant transformation since the CSCs must be able to self-renew, and it would be more difficult for a differentiated cell to regain self-renewal properties through mutations. However, since the presence of stem cells in adult tissue is extremely rare, progenitor cells (which retain partial self-renewal capacity but are more abundant in adult tissue) may be the cells from which CSCs are derived [53, 56, 94]. Alternatively, it has been demonstrated that the actual number of mutations required to form a pluripotent stem cell from an adult fibroblast is surprisingly small. Forced expression of Oct-4, c-Myc, Sox2 and klf4 in adult fibroblasts results in cells that are both morphologically similar to embryonic stem cells, and have stem cell-like replicative potential [66].
Therefore, although it is presently unclear whether tumours initiate from a stem or progenitor cell that has accumulated genetic mutations towards a more cancerous phenotype, or whether a cancerous cell somehow de-differentiates to become more stem cell-like, what is clear is that this stem-like population in tumours is important for the initiation and maintenance of tumour growth. Focusing research attention on these populations would thus be very beneficial for understanding cancer biology and potentially discovering new therapeutic targets to combat cancer.
Therapeutic implications
Consideration of the CSC hypothesis in the context of metastasis has far-reaching implications to the way that we not only study cancer, but also how we treat it. Although some early stage cancers can be successfully treated by surgery, radiation and/or systemic cytotoxic therapy, the majority of current therapies fail in the metastatic setting. Metastatic cells are often highly resistant to therapy, and this is reflected by the high mortality rate once a primary tumour has metastasized. One of the major questions raised by CSC hypothesis is whether or not current therapies are in fact targeting the right cells. As discussed above, it has been speculated that CSCs have the ability to avoid or resist current cancer therapies, although this has yet to be definitively proven in the clinical setting. It has also been hypothesized that the proportion of CSCs within a tumour may correlate with the severity of the cancer [92, 93, 95, and our unpublished data]. Therefore, less aggressive cancers may be comprised of mostly therapy-sensitive non-CSCs, which may make early stage tumours more susceptible to successful treatment with both chemotherapy and radiation. In contrast, more aggressive metastatic tumours may be mostly populated with therapy-resistant CSCs, making them extremely difficult to treat. In this section, we consider potential therapeutic approaches for targeting CSCs (Table 1).
Radiation therapy and CSCs
Experimental studies in breast and brain cancer model systems have provided some interesting data with regards to CSCs and radiation resistance. A serious clinical problem associated with fractionated RT is accelerated re-population, or the increase in rate of growth as a result of time between treatments. During accelerated repopulation, each day of a treatment gap reduces the efficacy of RT by about 0.6 Gy, making it one of the major reasons for local failure of RT [91, 93]. In breast cancer model systems, CD44+/CD24−/low CSCs from MCF-7 and MDA-MB-231 cancer cell lines were isolated and subjected to a single dose of radiation [91]. The CSCs were observed to be more radioresistant, had fewer or no double stranded DNA breaks (or they were quickly repaired), and had a 50% lower dose-dependent formation of reactive oxygen species (ROS) in response to the radiation. In addition, the increase in the CSC population was associated with the activation of Notch-1 (important in specifying cell fate during development), so it is possible that CSCs activate this developmental pathway in response to radiation [68, 91].
Another elegant study by Bao et al. (2006) demonstrated that glioma cells expressing CD133 (CSCs) showed preferential survival following radiation treatment as compared to CD133- cells (non-CSCs) [92]. Interestingly, even after radiation of up to 5Gy, the CD133+ CSCs retained a similar tumour formation ability and multi-lineage differentiation potential as the un-irradiated CSCs. The CSCs also demonstrated reduced apoptosis relative to non-CSCs, and this was supported by a decrease in caspase-3 activation and increased activation of the DNA checkpoint proteins Rad17, Chk1 and Chk2 in response to DNA damage by radiation.
Thus, in the face of radiotherapy, CSCs appear to survive better, repair their DNA more efficiently, begin to self-renew in order to increase the CSC population within the tumour and ultimately this may allow the tumour to become even more radioresistant. It may be reasonable to suggest that targeted therapies, such as blockage of Notch-1 expression within tumours could be beneficial in preventing the expansion of the CSC pool following radiation. Similarly, therapies targeting DNA checkpoint proteins may sensitize CSCs to radiation, resulting in a cancer that is potentially less resistant to radiation because the cells will no longer be able to proofread their DNA at such a superior rate.
Cytotoxic/targeted chemotherapy and CSCs
As discussed earlier, one of the intriguing parallels between normal stem cells and metastatic cancer cells is increased drug resistance via up-regulation of drug transporter proteins. ABCB1, ABCC1 and ABCG2 represent three of the most common multi-drug resistant genes that have been identified in stem cells and CSCs [81–85]. It may be possible to sensitize CSCs to chemotherapy by blocking the function of one or more of these ABC transporters such that CSCs would be unable to efflux the cytotoxic agents as efficiently, resulting in enhanced cell death.
In addition to a decreased sensitivity to chemotherapeutic agents due to a high expression of drug resistance genes, expression of both HH and Bmi-1 is activated in breast CSCs [48]. In many types of normal stem cells, HH signalling is essential for promoting stem cell self-renewal and proliferation. HH signalling also increases Bmi-1 expression, and Bmi-1 has been shown to play an important role in the regulation of self-renewal of haematopoietic stem cells and neuronal stem cells [79]. The cytotoxic agent cyclopamine exerts its effect on cancer cells by binding to and inhibiting SMO, which inhibits the growth of tumours with activated HH signalling [76, 78]. Studies have demonstrated that xenograft tumours resulting from injection of mice with DU-145 and PC-3 prostate cancer cells can be virtually eliminated by treatment with cyclopamine [96, 97]. It is possible that this is a result of the drug being able to inhibit CSC self-renewal, and hence the overall growth of the tumour. In addition to regulating stem cell self-renewal, Bmi-1 expression is also important for prevention of stem cell senescence, which is vital for maintaining organ homeostasis throughout life [79]. Bmi-1 expression has been shown to extent replicative lifespan in mouse embryonic fibroblasts (MEFs). MEFs usually reach replicative senescence after 7 passages in culture, but MEFs from Bmi-1 deficient mice show a premature senescence phenotype after the third passage [98]. Since Bmi-1 is over-expressed in many cancers, including breast cancer [48], it may be useful to target CSCs via targeting of Bmi-1. If Bmi-1 is responsible for delaying senescence in CSCs, then blocking its activity would drastically shorten the lifespan of CSCs and thereby decrease the overall CSC population within the tumour.
Additional studies have shown encouraging results with regards to targeting specific molecules known to be associated with CSCs. For example, TN14003 is a synthetic polypeptide that specifically targets CXCR4 to inhibit its activity. In breast cancer, TN14003 was successful in limiting metastasis in SCID mice [73]. In brain cancer, intracranial glioblastoma and medulloblastoma xenografts treated with another CXCR4 antagonist (AMD3100) showed reduced cell growth and increased tumour cell apoptosis [74]. Furthermore, Jin et al. (2006) were able to show that by targeting CD44, leukaemic stem cells can be eliminated in an AML model. The authors hypothesized that this result was due to interference with transport to stem cell-supportive microenvironmental niches and/or alteration of CSC fate towards differentiation [99]. Other studies have shown that treatment of prostate and breast cancer cell lines with an siRNA against CD44 can decrease cancer cell adhesion to bone marrow endothelial cells [100]. This could reduce cellular ability to migrate and invade tissues, and further supports the idea of using a CD44 blocker to target CSCs in cancer therapy.
Differentiation therapy and CSCs
It has also been speculated that CSCs may be targeted using therapies designed to modify cell differentiation. Although differentiation therapy does not kill cancer cells, it does have the potential to restrain their self-renewal capacity and perhaps increase the efficacy of more conventional therapies (such as chemotherapy) which are often most effective on differentiated cells. Furthermore, differentiation agents often have less toxicity than conventional cancer treatments [101, 102]. This type of therapeutic strategy has had the greatest impact in haematologic malignancies such as leukaemia, where the cancer-initiating cell and the cellular differentiation hierarchy is well-characterized [35, 103, 104]. The first differentiation agent found to be successful in the clinic was all-trans-retinoic acid (ATRA) in the treatment of acute promyelocytic leukaemia (APL) [103–105]. It remains to be seen whether this type of approach would also be therapeutically advantageous for eliminating CSCs in solid cancers, either via direct targeting of CSCs or by targeting the metastat-ic niche in order to induce CSC differentiation.
Prognostic implications
Finally, the use of gene expression arrays for identification of novel prognostic markers and/or new therapeutic targets for cancer has generally failed to account for the cellular heterogeneity present within tumours. Since the expression patterns of normal stem cells can vary significantly from their differentiated counterparts [106], then it is probable that CSCs also have different expression characteristics than the bulk of the non-tumourigenic cell population. Thus, expression analysis of isolated, enriched populations of CSCs could potentially uncover better prognostic markers and more effective therapeutic targets than the current practice of analyzing mixed populations of cells present in tumour tissue. This idea is supported by a recent study by Liu et al. (2007), in which CD44+CD24low‘tumourigenic’ cells from breast tumour tissue were isolated and compared for gene expression differences relative to that of normal breast epithelium. Differentially expressed genes were used to generate a 186-gene ‘invasiveness’ gene signature which could be correlated with reduced overall survival and reduced metastasis-free survival in patients with breast or other types of cancer [107]. However, the large body of work from Joan Massagué's group (reviewed in [108]) and additional ‘CSC’ subpopulation gene expression studies by Shipitsin et al. (2007) [47] indicate that further work is clearly needed to confirm that the cells that are being isolated and analysed are in fact CSCs, and also, most importantly, that these cells are the cells that predict metastatic progression, treatment response, and clinical outcome.
Conclusions and future directions
Metastasis is a lethal yet entirely inefficient process. There are several theories to explain this inefficiency, including the CSC hypothesis. If CSCs are the only cells within a tumour that can initiate and sustain primary tumour growth, then it is very feasible to hypothesize that that CSCs are also the cells responsible for initiating and sustaining metastatic growth. Furthermore, metastatic cells share many similarities with normal stem cells, including an unlimited capacity for self renewal; the requirement for a specific ‘niche’ or microenvironment to grow; use of the SDF-1/CXCR4 axis for migration; enhanced resistance to apoptosis; and an increased capacity for drug resistance. Therapeutically, it could be advantageous if the important cells to target in cancer are similar to normal stem cells, because we can use much of the knowledge gained about normal stem cells to design new strategies against CSCs (Table 1). However, the potential therapeutic approaches discussed above need to be taken with a cautionary grain of salt. Most of these ideas centre around attacking the stem-like properties of CSCs, and are risky because of the fact that normal stem cells may also be targeted in the process. For example, blocking ABC transporters, such as ABCG2 and ABCB1 may make CSCs more sensitive to chemotherapy, but may also cause the body's normal stem cells to become sensitive to drugs and die prematurely. Furthermore, ABCG2 and ABCB1 are important for maintaining the blood–brain barrier (BBB), so blocking these transporters also carries the risk of jeopardizing the BBB96]. Similarly, blocking DNA checkpoint proteins, HH signalling and Bmi-1 expression is also risky because normal stem cells may become unable to self-renew or may senesce prematurely. More work is clearly needed with regards to delineating the similarities and differences between normal stem cells, CSCs and metasta-tic CSCs in order to identify possible therapeutic strategies that would effectively eradicate CSCs while leaving normal stem cells unharmed. Although much experimental investigation remains to be undertaken in order to definitively prove that metastatic CSCs are similar to CSCs and that these cells are responsible for driving metastasis, the behavioural parallels between normal stem cells, CSCs, and metastatic cells are too intriguing to ignore.
Acknowledgments
We thank members of our laboratory and our collaborators for their research work and helpful discussions. The authors' research on CSCs and metastasis is supported by a grant from the London Regional Cancer Program.A.K.C. is the recipient of a Canadian Institute of Health Research-Strategic Training Program Scholarship and a Translational Breast Cancer Scholarship through the London Regional Cancer Program. A.L.A. is supported by the Imperial Oil Foundation.
References
- 1.Canadian Cancer Statistics. 2007. http://www.cancer.ca.
- 2.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 3.Clarke M, Collins R, Darby S, Davies C, Elphinstone P, Evans E, Godwin J, Gray R, Hicks C, James S, MacKinnon E, McGale P, McHugh T, Peto R, Taylor C, Wang Y. Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;366:2087–106. doi: 10.1016/S0140-6736(05)67887-7. [DOI] [PubMed] [Google Scholar]
- 4.Cole BF, Gelber RD, Gelber S, Coates AS, Goldhirsch A. Polychemotherapy for early breast cancer: an overview of the randomised clinical trials with quality-adjusted survival analysis. Lancet. 2001;358:277–86. doi: 10.1016/S0140-6736(01)05483-6. [DOI] [PubMed] [Google Scholar]
- 5.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpress-es HER2. N Engl J Med. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 6.Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, Johnson DH. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–50. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 7.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 8.Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, Ciuleanu T, Von Pawel J, Thongprasert S, Tan EH, Pemberton K, Archer V, Carroll K. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomized placebo-controlled, multi-centre study (Iressa Survival Evaluation in Lung Cancer) Lancet. 2005;366:1527–37. doi: 10.1016/S0140-6736(05)67625-8. [DOI] [PubMed] [Google Scholar]
- 9.Pusztai L, Hortobagyi GN. High-dose chemotherapy: how resistant is breast cancer? Drug Resist Updat. 1998;1:62–72. doi: 10.1016/s1368-7646(98)80216-1. [DOI] [PubMed] [Google Scholar]
- 10.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–72. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 11.Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2004;4:448–56. doi: 10.1038/nrc1370. [DOI] [PubMed] [Google Scholar]
- 12.Weiss L. Metastatic inefficiency. Adv. Cancer Res. 1990;54:159–211. doi: 10.1016/s0065-230x(08)60811-8. [DOI] [PubMed] [Google Scholar]
- 13.Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, Groom AC. Multistep nature of metastatic inefficiency:dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998;153:865–73. doi: 10.1016/S0002-9440(10)65628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ewing J. Neoplastic Diseases. London: W.B. Saunders Co; 1928. A treatise on tumors; pp. 77–89. [Google Scholar]
- 15.Paget S. The distribution of secondary growths in cancer of the breast (re-publication of the original 1889 Lancet article) Cancer Met Rev. 1989;8:98–101. [PubMed] [Google Scholar]
- 16.Fidler IJ. Seed and soil revisited: contribution of the organ microenvironment to cancer metastasis. Surg Oncol Clin N Am. 2001;10:257–69. vii–viii. [PubMed] [Google Scholar]
- 17.Weiss L. Comments on hematogenous metastatic patterns in humans as revealed by autopsy. Clin Exp Metastasis. 1992;10:191–9. doi: 10.1007/BF00132751. [DOI] [PubMed] [Google Scholar]
- 18.Ratajczak MZ, Zuba-Surma E, Kucia M, Reca R, Wojakowski W, Ratajczak J. The pleiotropic effects of the SDF-1-CXCR4 axis in organogenesis, regeneration and tumorigenesis. Leukemia. 2006;20:1915–24. doi: 10.1038/sj.leu.2404357. [DOI] [PubMed] [Google Scholar]
- 19.Kucia M, Reca R, Miekus K, Wanzeck J, Wojakowski W, Janowska-Wieczorek A, Ratajczak J, Ratajczak MZ. Trafficking of normal stem cells and metastasis of cancer stem cells involve similar mechanisms:pivotal role of the SDF-1-CXCR4 axis. Stem Cells. 2005;23:879–94. doi: 10.1634/stemcells.2004-0342. [DOI] [PubMed] [Google Scholar]
- 20.Smith MCP, Luker EK, Garbow JR, Prior JL, Jackson E, Piwnica-Worms D, Luker GD. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004;64:8604–12. doi: 10.1158/0008-5472.CAN-04-1844. [DOI] [PubMed] [Google Scholar]
- 21.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 22.Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. Lancet. 2007;369:1742–57. doi: 10.1016/S0140-6736(07)60781-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang Y, Massague J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004;118:277–9. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 24.Stover DG, Bierie B, Moses HL. A delicate balance: TGF-β and the tumor microenvironment. J Cell Biochem. 2007;101:851–61. doi: 10.1002/jcb.21149. [DOI] [PubMed] [Google Scholar]
- 25.Zavadil J, Cermak L, Soto-Nieves N, Bottinger E. Integration of TGF-β/Smad and Jagged1/Notch signaling in epithelial-to-mesenchymal transition. EMBO J. 2004;23:1155–65. doi: 10.1038/sj.emboj.7600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moustakas A, Heldin C-H. Signaling networks guiding epithelial-mesenchyml transitions during embryo-genesis and cancer progression. Cancer Sci. 2007;98:1512–20. doi: 10.1111/j.1349-7006.2007.00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muller T, Bain G, Wang X, Papkoff J. Regulation of epithelial cell migration and tumor formation by β-catenin signaling. Exp Cell Res. 2002;280:119–33. doi: 10.1006/excr.2002.5630. [DOI] [PubMed] [Google Scholar]
- 28.Dreesen O, Brivanlou AH. Signaling pathways in cancer and embryonic stem cells. Stem Cell Rev. 2007;3:7–17. doi: 10.1007/s12015-007-0004-8. [DOI] [PubMed] [Google Scholar]
- 29.Derksen PWB, Liu X, Saridin F, Van Der Gulden H, Zevenhoven J, Evers B, Van Beijnum JR, Griffioen AW, Vink J, Krimpenfort P, Peterse JL, Cardiff RD, Berns A, Jonkers J. Somatic inactivation of E-cad-herin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell. 2006;10:437–49. doi: 10.1016/j.ccr.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 30.Rennebeck G, Martelli M, Kyprianou N. Anoikis and survival connections in the tumor microenvironment: is there a role in prostate cancer metastasis? Cancer Res. 2005;65:11230–5. doi: 10.1158/0008-5472.CAN-05-2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 32.Virchow R. Editorial. Virchows Arch Pathol Anat Physiol Klin Med. 1855;3:23. [Google Scholar]
- 33.Kucia M, Reca R, Campbell FR, Zuba-Surma E, Majka, Ratajczak J, Ratajczak MZ. A population of very small embryonic-like (VSEL) CXCR4+SSEA-1+ Oct-4+ stem cells identified in adult bone marrow. Leukemia. 2006;20:857–69. doi: 10.1038/sj.leu.2404171. [DOI] [PubMed] [Google Scholar]
- 34.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuris MA, Dick JE. A cell initiating human acute myeloid leukemia after transplantation into SCID mice. Nature. 1994;367:645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 35.Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol. 2004;5:738–43. doi: 10.1038/ni1080. [DOI] [PubMed] [Google Scholar]
- 36.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 37.O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon caner cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–10. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 38.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, Reilly JG, Chandra D, Zhou J, Claypool K, Coghlan L, Tang DG. Highly purified CD44+ prostate cancer cells from xenografts human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 2006;25:1696–708. doi: 10.1038/sj.onc.1209327. [DOI] [PubMed] [Google Scholar]
- 40.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland MJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946–51. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 41.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 42.Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–23. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 43.Farnie G, Clarke RB. Mammary stem cells and breast cancer – role of notch signaling. Stem Cell Rev. 2007;3:169–75. doi: 10.1007/s12015-007-0023-5. [DOI] [PubMed] [Google Scholar]
- 44.Bailey JM, Singh PK, Hollingsworth MA. Cancer metastasis facilitated by developmental pathways: Sonic hedgehog, Notch, and bone morphogenic proteins. J Cell Biochem. 2007;102:829–39. doi: 10.1002/jcb.21509. [DOI] [PubMed] [Google Scholar]
- 45.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer Stem Cells–Perspectives on Current Status and Future Directions: AACR Workshop on Cancer Stem Cells. Cancer Res. 2006;66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 46.Al-Hajj M, Clarke MF. Self-renewal and solid tumor stem cells. Oncogene. 2004;23:7274–82. doi: 10.1038/sj.onc.1207947. [DOI] [PubMed] [Google Scholar]
- 47.Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebrylskaya T, Beroukhim R, Hu M, Halushka MK, Sukumar S, Parker LM, Anderson KS, Harris LN, Garber JE, Richardson AL, Schnitt SJ, Nikolsky Y, Gelman RS, Polyak K. Molecular definition of breast cancer heterogeneity. Cancer Cell. 2007;11:259–73. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 48.Liu S, Dontu G, Mantle ID, Patel S, Ahn N-S, Jackson KW, Suri P, Wicha MS. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–71. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hadnagy A, Gaboury L, Beaulieu R, Balicki D. SP analysis may be used to identify cancer stem cell populations. Exp Cell Res. 2006;312:3701–10. doi: 10.1016/j.yexcr.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 50.Hirschmann-Jax C, Fodter AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, Goodell MA, Brenner M. A distinct “side population”of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA. 2004;101:14228–33. doi: 10.1073/pnas.0400067101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corti S, Locatelli F, Papadimitriou D, Donadoni C, Salani S, Del Bo R, Strazzer S, Bresolini N, Comi GP. Identification of a primitive brain-derived neual stem cell population based on aldehyde dehydroge-nase activity. Stem Cells. 2006;24:975–85. doi: 10.1634/stemcells.2005-0217. [DOI] [PubMed] [Google Scholar]
- 52.Allan AL, Vantyghem SA, Tuck AB, Chambers AF. Tumor dormancy and cancer stem cells: implications for the biology and treatment of breast cancer metastasis. Breast Dis. 2007;26:87–98. doi: 10.3233/bd-2007-26108. [DOI] [PubMed] [Google Scholar]
- 53.Li F, Tiede B, Massagué J, Kang Y. Beyond tumori-genesis: cancer stem cells in metastasis. Cell Res. 2007;17:3–14. doi: 10.1038/sj.cr.7310118. [DOI] [PubMed] [Google Scholar]
- 54.Spillane JB, Henderson MA. Cancer stem cells: a review. ANZ J Surg. 2007;77:464–8. doi: 10.1111/j.1445-2197.2007.04096.x. [DOI] [PubMed] [Google Scholar]
- 55.Vaidya JS. An alternative model of cancer cell growth and metastasis. Int J Surg. 2007;5:73–5. doi: 10.1016/j.ijsu.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 56.Kucia M, Ratajczak MZ. Stem cells as a two edged sword – from regeneration to tumor formation. J Physiol Pharmacol. 2006;57:5–16. [PubMed] [Google Scholar]
- 57.Scadden DT. The stem cell niche in health and leukemic disease. Clin Hematol. 2007;20:19–27. doi: 10.1016/j.beha.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chepko G, Dickson RB. Ultrastructure of the putative stem cell niche in rat mammary epithelium. Tissue Cell. 2003;35:83–93. doi: 10.1016/s0040-8166(02)00107-6. [DOI] [PubMed] [Google Scholar]
- 59.Hendrix MJC, Seftor EA, Seftor REB, Kasemeier-Kulesa J, Kulesa PM, Postovit L-M. Reprogramming metastatic tumor cells with embryonic microenvironments. Nature. 2007;7:246–55. doi: 10.1038/nrc2108. [DOI] [PubMed] [Google Scholar]
- 60.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, Zhu Z, Hicklin D, Wu Y, Port JL, Altorki N, Port ER, Ruggero D, Shmelkov SV, Jensen KK, Rafil S, Lyden D. VEGFR1-positive haematopietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–6. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaplan RN, Rafii S, Lyden D. Preparing the “soil”: the premetastatic niche. Cancer Res. 2006;66:11089–93. doi: 10.1158/0008-5472.CAN-06-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Psaila B, Kaplan RN, Port ER, Lyden D. Priming the ‘soil’ for breast cancer metastasis:the pre-metastatic niche. Breast Dis. 20062007;26:65–74. doi: 10.3233/bd-2007-26106. [DOI] [PubMed] [Google Scholar]
- 63.Kaplan RN, Psaila B, Lyden D. Niche-to-niche migration of bone-marrow-derived cells. Trends Mol Med. 2006;13:72–81. doi: 10.1016/j.molmed.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 64.Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–65. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 65.Erler JT, Bennewith KL, Nciolau M, Dornhofer N, Kong C, Le Q-T, Chi J-TA, Jeffrey SS, Giaccia AJ. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–6. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 66.Barnhart BC, Simon MC. Metastasis and stem cell pathways. Cancer Metastasis Rev. 2007;26:261–71. doi: 10.1007/s10555-007-9053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang W, Eddy R, Condeelis J. The cofilin pathway in breast cancer invasion and metastasis. Nature. 2007;7:429–40. doi: 10.1038/nrc2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Politi K, Feirt N, Kitajewski J. Notch in mammary gland development and breast cancer. Semin Cancer Biol. 2004;14:341–7. doi: 10.1016/j.semcancer.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 69.Isufi I, Seetharam M, Zhou L, Sohal D, Opalinska J, Pahanish P, Verma A. Transforming growth factor-β signaling in normal and malignant hematopoiesis. J Interferon Cytokine Res. 2007;27:543–52. doi: 10.1089/jir.2007.0009. [DOI] [PubMed] [Google Scholar]
- 70.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea – a paradigm shift. Cancer Res. 2006;66:1883–90. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 71.Li L, Neaves WB. Normal stem cells and cancer stem cells: the niche matters. Cancer Res. 2006;66:4553–7. doi: 10.1158/0008-5472.CAN-05-3986. [DOI] [PubMed] [Google Scholar]
- 72.Dewan MZ, Ahmed S, Iwasaki Y, Ohba K, Toi M, Yamamoto N. Stromal cell-derived factor-1 and CXCR4 receptor interaction in tumor growth and metastasis of breast cancer. Biomed Pharmacother. 2006;60:273–6. doi: 10.1016/j.biopha.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 73.Liang Z, Wu T, Lou H, Yu X, Taichman RS, Lau SK, Nie S, Umbreit J, Shim H. Inhibition of breast cancer metastasis by selective synthetic polypeptide against CXCR4. Cancer Res. 2004;64:4302–8. doi: 10.1158/0008-5472.CAN-03-3958. [DOI] [PubMed] [Google Scholar]
- 74.Rubin JB, Kung AL, Klein RS, Chan JA, Sun Y, Schmidt K, Kieran MW, Luster AD, Segal RA. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc Natl Acad Sci USA. 2003;100:13513–8. doi: 10.1073/pnas.2235846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verástegui E, Zlotnik A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 76.Thayer SP, Pasca di Magliano M, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernandez-del Castillo C, Yajnik V, Antoniu B, McMahon M, Warshaw AL, Hebrok M. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–6. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang S, Yang D, Lippman ME. Targeting Bcl-2 and Bcl-XL with nonpeptidic small-molecule antagonists. Semin Oncol. 2003;30:133–42. doi: 10.1053/j.seminoncol.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 78.Berman DM, Karhadkar SS, Maltra A, Montes de Oca R, Gerstenblith MR, Briggs K, Parker AR, Shimada Y, Eshieman JR, Watkins DN, Beachy PA. Widespread requirement for hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425:846–51. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 79.Park I-K, Morrison SJ, Clarke MF. Bmi1, stem cells, and senescence regulation. J Clin Invest. 2004;113:175–9. doi: 10.1172/JCI20800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 81.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–84. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 82.Dean M. Cancer Stem Cells: Redefining the Paradigm of Cancer Treatment Strategies. Mol Interv. 2006;6:140–8. doi: 10.1124/mi.6.3.5. [DOI] [PubMed] [Google Scholar]
- 83.Scharenberg CW, Harkey MA, Torok-Storb B. The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood. 2002;99:507–12. doi: 10.1182/blood.v99.2.507. [DOI] [PubMed] [Google Scholar]
- 84.Kim M, Turnquist H, Jackson J, Sgagias M, Yan Y, Gong M, Dean M, Sharp JG, Cowan K. The mul-tidrug resistance transporter ABCG2 (breast cancer resistance protein 1) effluxes Hoechst 33342 and is overexpressed in hematopoietic stem cells. Clin Cancer Res. 2002;8:22–8. [PubMed] [Google Scholar]
- 85.Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, Ross DD. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci USA. 1998;95:15665–70. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cairns J. The cancer problem. Sci Am. 1975;233:64–72. doi: 10.1038/scientificamerican1175-64. [DOI] [PubMed] [Google Scholar]
- 87.Cairns J. Somatic stem cells and the kinetics of mutagenesis and carcinogenesis. Proc Natl Acad Sci USA. 2002;99:10567–70. doi: 10.1073/pnas.162369899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Potten CS, Owen G, Booth D. Intestinal stem cells protect their genome by selective segregation of template DNA strands. J Cell Sci. 2002;115:2381–8. doi: 10.1242/jcs.115.11.2381. [DOI] [PubMed] [Google Scholar]
- 89.Cai J, Weiss ML, Rao MS. In search of “stemness”. Exp Hematol. 2004;32:585–98. doi: 10.1016/j.exphem.2004.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Park Y, Gerson SL. DNA repair defects in stem cell function and aging. Annu Rev Med. 2005;56:495–508. doi: 10.1146/annurev.med.56.082103.104546. [DOI] [PubMed] [Google Scholar]
- 91.Phillips TM, McBride WH, Pajonk F. The response of CD24−/lowCD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777–85. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 92.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 93.Diehn M, Clarke MF. Cancer stem cells and radiotherapy: new insights into tumor radioresistance. J Natl Cancer Inst. 2006;98:1755–7. doi: 10.1093/jnci/djj505. [DOI] [PubMed] [Google Scholar]
- 94.Stingl J, Caldas C. Molecular heterogeneity of breast carcinomas and the cancer stem cell hypothesis. Nat Rev Cancer. 2007;7:791–8. doi: 10.1038/nrc2212. [DOI] [PubMed] [Google Scholar]
- 95.Smalley M, Ashworth A. Stem cells and breast cancer: A field in transit. Nat Rev Cancer. 2003;3:832–44. doi: 10.1038/nrc1212. [DOI] [PubMed] [Google Scholar]
- 96.Lou H, Dean M. Targeted therapy for cancer stem cells: the patched pathway and ABC transporters. Oncogene. 2007;26:1357–60. doi: 10.1038/sj.onc.1210200. [DOI] [PubMed] [Google Scholar]
- 97.Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, Isaacs JT, Berman DM, Beachy PA. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431:707–12. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 98.Jacobs JJ, Kieboom K, Marino S, DePinho RA, Van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–8. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 99.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–74. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 100.Draffin JE, McFarlane S, Hill A, Johnston PG, Waugh DJJ. CD44 potentiates the adherence of metastatic prostate and breast cancer cells to bone marrow endothelial cells. Cancer Res. 2004;64:5702–11. doi: 10.1158/0008-5472.CAN-04-0389. [DOI] [PubMed] [Google Scholar]
- 101.Leszczyniecka M, Roberts T, Dent P, Grant S, Fisher PB. Differentiation therapy of human cancer: basic science and clinical applications. Pharmacol Ther. 2001;90:105–56. doi: 10.1016/s0163-7258(01)00132-2. [DOI] [PubMed] [Google Scholar]
- 102.Sell S. Cancer stem cells and differentiation therapy. Tumour Biol. 2006;27:59–70. doi: 10.1159/000092323. [DOI] [PubMed] [Google Scholar]
- 103.Warrell RP, Jr, De Thé H, Wang ZY, Degos L. Acute promyelocytic leukemia. N Engl J Med. 1993;329:177–89. doi: 10.1056/NEJM199307153290307. [DOI] [PubMed] [Google Scholar]
- 104.Lo Coco F, Nervi C, Avvisati G, Mandelli F. Acute promyelocytic leukemia: a curable disease. Leukemia. 1998;12:1866–80. doi: 10.1038/sj.leu.2401230. [DOI] [PubMed] [Google Scholar]
- 105.Slack JL, Willman CL, Andersen JW, Li YP, Viswanatha DS, Bloomfield CD, Tallman MS, Gallagher RE. Molecular analysis and clinical outcome of adult APL patients with the type V PML-RARalpha isoform: results from intergroup protocol 0129. Blood. 2000;95:398–403. [PubMed] [Google Scholar]
- 106.Akashi K, He X, Chen J, Iwasaki H, Niu C, Steenhard B, Zhang J, Haug J, Li L. Transcriptional accessibility for genes of multiple tissues and hematopoietic lineages is hierarchically controlled during early hematopoiesis. Blood. 2003;101:383–9. doi: 10.1182/blood-2002-06-1780. [DOI] [PubMed] [Google Scholar]
- 107.Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T, Sherlock G, Lewicki J, Shedden K, Clarke MF. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med. 2007;356:217–26. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]
- 108.Nguyen DX, Massagué J. Genetic determinants of cancer metastasis. Nat Rev Genet. 2007;8:341–52. doi: 10.1038/nrg2101. [DOI] [PubMed] [Google Scholar]