

Abstract
Bacteria, particularly of the genus Bacillus, produce a wide variety of antifungal compounds. They act by affecting the lipid bilayers of fungal membranes, causing curvature-induced strain and eventual permeabilization. One class of these, known as fengycins, has been commercialized for treating agricultural infections and shows some promise as a possible antifungal pharmaceutical. Understanding the mechanism by which fengycins damage lipid bilayers could prove useful to the future development of related antifungal treatments. In this work, we present multi-microsecond-long simulations of fengycin interacting with different lipid bilayer systems. We see fengycin aggregation and uncover a clear aggregation pattern that is partially influenced by bilayer composition. We also quantify some local bilayer perturbations caused by fengycin binding, including curvature of the lipid bilayer and local electrostatic-driven reorganization.
Introduction
Fengycin, a class of lipopeptides originally isolated in Bacillus subtilis (1), has shown promise as an antifungal agent for use in controlling plant disease (2). Natural fengycins expressed from a variety of bacterial strains have been used to treat many agricultural infections: clubroot disease (Plasmodiophora brassicae) in cruciferous plants (3), maize rot (Fusarium moniliforme) (4), barley head blight (Fusarium graminearum) (5), and black bread mold (Rhizopus stolonifer) (6). The potential for fengycin use as a major agricultural tool has been enhanced by the development of protocols for maximizing fengycin synthesis in strains of Bacillus subtilis (7), and it has been commercialized as the green antifungal product Serenade (AgraQuest, Davis, CA). It also has hemolytic activity lower than other agents (8), making it a strong candidate as a novel pharmaceutical for treating fungal infections.
There are multiple possible mechanisms for fengycin action. It has been shown that similar peptides, particularly surfactins, stimulate natural plant responses to infection (9) and may have an effect on quorum-sensing (10). They also serve as powerful biosurfactants capable of inducing cell lysis (11) and membrane leakage (12).
The focus of this work will be on the membrane permeabilization mechanism. Fengycin has been shown to perturb DPPC monolayers in a dose-dependent manner (13), affect the mixing properties of monolayers (1), and alter the formation of lipid domains (14). However, experimental methods to date have not characterized the near-atomic interactions involved in these membrane perturbations.
Molecular dynamics simulation (MD) has long been used to address a variety of problems in membrane biophysics (15,16), including the substantial body of work simulating membrane proteins (17) like G protein-coupled receptors (18–20) and membrane transporters (21). Furthermore, a number of groups have used MD to quantify and analyze interactions between antimicrobial peptides and lipids in bilayers (22,23). Most of these studies used all-atom molecular dynamics, which explicitly represents every atom in the system. Although this approach has remarkable resolution in time and space, and arguably represents the gold standard for biomolecular simulation, it can be prohibitively expensive computationally to obtain adequate statistical sampling because of the large conformational space available to antimicrobial peptides, especially those that require the aggregation of multiple molecules and large patches of bilayer.
To overcome this challenge, coarse-grained models have been employed. In these models, subsets of atoms are abstracted into functional units and treated as a single bead that attempts to represent the underlying atoms. By reducing the quantity of degrees of freedom, there are fewer interactions to compute and it is possible to use a larger time step for integrating the equations of motion. One such model, the MARTINI force field (24), reduces computational costs by roughly two orders of magnitude relative to comparable all-atom models. As a result, it is one of the more common models for bilayer simulations and has been used to address a number of membrane problems, including vesicle fusion (25), phase behavior (26), and the formation of lipid domains (27). This model has also been used extensively in the study of antimicrobial peptides that interact with lipid bilayers and alter bilayer properties (28–32), including cyclic peptides (33).
In this study, we explore the structure and aggregation of fengycin interacting with different model membrane compositions using coarse-grained MD, and analyze its effects on bilayer structure to propose a mechanism for its function. Specifically, fengycin’s tendency to aggregate is sensitive to the composition of the bilayer, and this in turn modulates its ability to induce membrane curvature.
Methods
System construction
The coarse-grained force-field MARTINI, Ver. 2.1 (University of Groningen, Groningen, Netherlands), was used to describe the interactions in our system (24,34,35). We chose a fengycin structure that has been noted previously in Wu et al. (36) and has been recently characterized by mass spectrometry (37). With a glutamate at position 8, it differs from fengycin IX (38) and fengycin A2, which have a glutamine at that position. Because of this, our structure has a net negative charge, whereas these other fengycins tend to be neutral. Due to a lack of available PDB structures, we built a MARTINI model for this structure by hand, shown in Fig. 1. The ornithine residue was constructed by copying a lysine residue and shortening the bond length between the backbone bead and the first bead of the side chain (from 0.33 nm in lysine to 0.31 nm in our ornithine).
Figure 1.
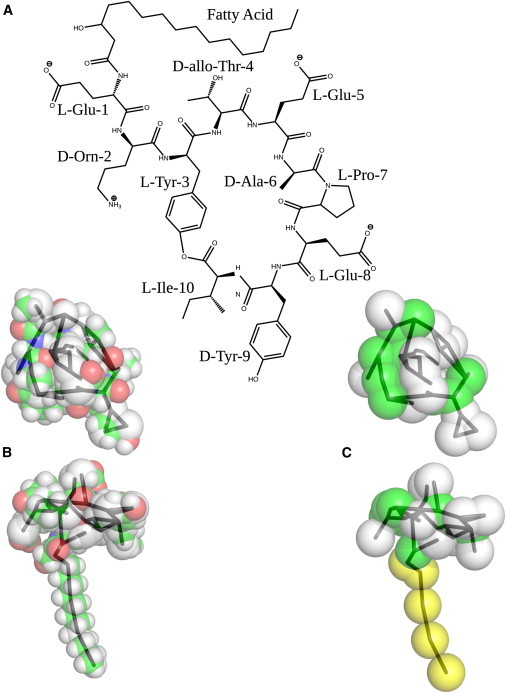
(A) Chemical structure of our fengycin of interest. Also shown are space-filling representations shown from the top and side of fengycin with (B) all-atom resolution and our (C) coarse-grained representation. (For the all-atom model, the atoms are colored using green for carbons, white for hydrogens, red for oxygens, and blue for nitrogens. In the coarse-grained model, yellow is used for tail beads, green for peptide backbone beads, and white for amino-acid side chains. In both representations, a black backbone trace of the coarse-grained model is shown for reference.)
To build the lipopeptide-free bilayer systems, we began with a 128-lipid dipalmitoyl phosphatidylcholine (DPPC) bilayer system available at the MARTINI website. We tessellated this system to create a 512-lipid system and equilibrated it for 400 ns. This bilayer was converted to a pure palmitoyl oleoyl phosphatidylcholine (POPC) bilayer by adding one bead and double bond character to a tail in each lipid, converting one chain from a palmitoyl to oleoyl. This bilayer was also converted to a 2:1 mixture of palmitoyl oleoyl phosphatidylethanolamine (POPE) and palmitoyl oleoyl phosphatidylglycerol (POPG). The conversion of palmitoyl to oleoyl was accomplished as before, although the headgroup change was accomplished by changing the identity of the positive headgroup bead (PC, PE, and PG all contain the same number of MARTINI beads). Lipids were randomly chosen to become PE or PG, such that both leaflets had the same 2:1 PE/PG composition. This process was performed again for each replicate of the system; the resulting snapshots were then equilibrated for 300 ns.
We built single-fengycin systems by adding a fengycin molecule to each equilibrated lipid system and simulating until fengycin bound to the bilayer. We simulated fengycin bound in each bilayer for another 100 ns, chose 16 frames post-binding from each system trajectory, gave them new velocities, and restarted from these frames. The result was a set of 16 independent systems for each bilayer with one fengycin bound.
Fengycin’s amphipathicity suggested that it would likely aggregate in solution, sequestering the hydrocarbon chain away from solution. With this in mind, we built aggregates of fengycin by randomly placing fengycin molecules in solution and running for 100 ns, at which point a loose micelle had formed that persisted for some time. We excised this 13-fengycin aggregate from the system and placed it into the equilibrated POPE/POPG bilayer system with water and salt; in addition to neutralizing salt, we added enough NaCl to set the free salt concentration at ∼100 mM. A weak harmonic potential was applied to move the micelle toward the bilayer until they were in contact. Copies of this system were made and the POPE and POPG lipids were converted to POPC and DPPC lipids (using the same procedure described above) to give us all three lipid compositions with a fengycin micelle bound. These systems were energy-minimized and equilibrated for a nanosecond. Sixteen replicas of each system were given unique starting velocities to create 16 independent trajectories. In these simulations, the lipopeptides rapidly insert into the bilayer. Again, we focused on the behavior of lipopeptides after insertion, ignoring the binding process.
Simulation protocol
These simulations were run using Ver. 4.5.5 of the GROMACS simulation package (39,40). We employed a time step of 20 fs with the neighbor list updated every 5 steps. We held the temperature at 300 K for the POPC and POPE/POPG systems and 323 K for the DPPC systems using Nosé-Hoover temperature coupling (41,42). Pressure was treated semiisotropically using the Parrinello-Rahman barostat with a reference of 1 bar (43). Electrostatics were computed with a shift function with a cutoff of 12 Å. Shift was used for van der Waals, with a switch distance of 9 Å and a cutoff of 12 Å.
Each simulation with fengycin is >2 μs long and lipopeptide-free bilayer simulations are >1 μs long. There are nine distinct systems, each of which was simulated 16 times, for a total of >252 μs of simulation time (a complete list of systems and average simulation lengths can be found in Table S1 in the Supporting Material). This can be considered an effective total time of greater than a millisecond when we consider the faster kinetics in the MARTINI model (24); the times reported here are actual simulation times.
Simulation analysis
All analysis was performed with tools developed with the Lightweight Object Oriented Structure (LOOS) analysis library (44). LOOS is an object-oriented library implemented in C++ that provides functionality for creating new tools for the analysis of molecular dynamics simulations. (LOOS is available for download at http://loos.sourceforge.net, licensed under Ver. 3 of the GNU Project’s General Public License (GPL).) All analysis was done on the trajectories at 100-ps resolution. The first 100 ns were excluded from each trajectory as equilibration and to ensure that the lipopeptides were completely bound. Unless otherwise noted, error bars shown on plots reflect the standard error computed with each trajectory treated as a single independent data value.
Density distributions
The density distribution of lipopeptide residues along the membrane normal was computed by binning counts of each residue’s backbone bead using 0.25 Å bins. Each histogram was shifted relative to the phosphate density peak to make it easier to separate bilayer penetration from differences in bilayer thickness.
Aggregation
To quantify fengycin aggregation, we calculated the contacts between molecules and we defined clusters of fengycin molecules based on these criteria. Coarse-grained beads were considered in contact if they were spaced ≤6 Å apart. A pair of lipopeptides was considered members of the same cluster if they had two or more contacts.
Cluster contact map
We described the interactions between aggregated fengycins by employing a map of intermolecular residue-residue contacts. If two fengycin molecules were clustered based on the criteria we defined for aggregation, we checked every pairwise combination of intermolecular contacts for contact (beads spaced <6 Å). We report the likelihood that a given contact between beads exists when two lipopeptides are clustered; a value of 1 indicates that a given pair of atoms is always in contact when a pair of fengycins meets the criteria for aggregation.
Radial distribution function
We computed a variety of radial distribution functions (RDF) to quantify local enrichments in our system. For quantifying the lateral ordering of lipid species about lipopeptides, we computed the RDF in the plane of the bilayer only and treated each molecule as a single point, centered at the centroid. For pairwise interactions between headgroup beads, the RDF was computed in three dimensions.
Bilayer curvature
To quantify the curvature of the bilayer induced by fengycin, we created maps measuring the height of the lipid phosphate beads relative to the center of the lipid bilayer as a function of lateral distance from fengycin. We began by hand-curating a collection of clusters from the 13 fengycin in DPPC and POPC simulations. Details on the cluster curation process can be found in the Supporting Material.
Each cluster was centered in the plane of the bilayer, and the axis of the cluster (defined as the vector between the centroids of the terminal fengycins) was aligned along the X axis. This allows clusters of the same size from different simulations to be compared to one another. For each frame of the simulations, we binned the heights of the phosphates in a grid on the plane parallel to the membrane, with 1 Å2 bins. This calculation was performed for both the leaflet with lipopeptides bound (proximal) and the opposite leaflet (distal) of the bilayer. The heights were normalized compared to lipopeptide-free bilayers to make comparison between different lipid compositions possible. These data are presented as a heat map, with each bin presented as the average height for all the phosphates in that bin.
We also calculated the phosphate heights as a function of distance from each lipopeptide. Here we position each lipopeptide at the center of the bilayer plane and bin the phosphate heights by their distance from the lipopeptide centroid. This calculation was done for both leaflets to show whether the curvature is localized to one leaflet or is an effect across the membrane.
Principal component order parameters
Because the chains do not contain all of the carbons, we cannot compute order parameters that are directly comparable to the 2H quadrupolar splittings measured via solid-state nuclear magnetic resonance. However, we can quantify lipopeptide-induced changes to bilayer structure through a simplified order parameter. In this calculation, we compute the three principal axes for each lipid palmitoyl chain; the first principal axis lies in the direction of the chain, whereas the others define the plane perpendicular to the chain. The angle between these other axes and the membrane normal can be treated as mock carbon-hydrogen bonds to calculate the molecular order parameter,
| (1) |
where θ is the angle between the membrane normal and the second or third principal axis. We binned these order parameters based on distance in the plane of the bilayer from each lipopeptide in the same leaflet.
Results and Discussion
Lipopeptide binding and orientation in bilayers
All of the simulations started with fengycin close to the bilayer, and insertion always occurred within the 100-ns equilibration period. As a result, all of the analysis presented here is for the fully inserted state.
Fengycin’s orientation is largely the same in all three bilayer compositions, with the palmitoyl chain toward the interior of the bilayer, spanning one leaflet, and the peptide ring interacting with the lipid headgroups and extending into the solution. Fig. 2 shows normalized density distributions along the membrane normal for each of the 10 amino-acid residues of the fengycin chain in the DPPC system; for convenience, the distances are shifted such that z = 0 is the location of the peak of the lipid phosphate distribution. For comparison, the density for the hydrocarbon tail is shown as well, extending far below the phosphate peak and tapering off just outside the peak ∼z = 2 Å. Two hydrophobic residues have peaks inside the phosphate peak: the tyrosine at position 9 and the isoleucine at position 10. Most of the other amino acids, including the two amino acids connected directly to the hydrocarbon tail and not part of the ring (Glu1 and Orn2), have peaks within a window from 0 to 4 Å and interact primarily with the headgroups of the lipids. Two residues, the threonine at position 4 and the glutamate at position 5, are both above 4 Å, and are exposed to the water environment significantly more than the rest of the peptide.
Figure 2.
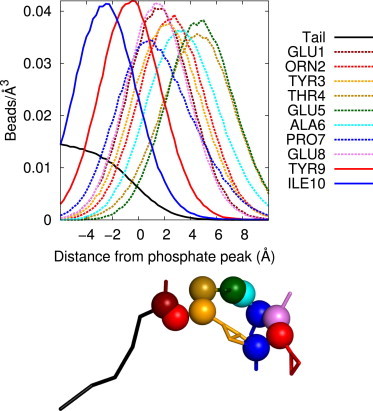
Normalized density of each fengycin residue relative to the phosphate density peak for the DPPC systems with one fengycin. (Solid and dashed lines represent residues with peaks below or above the phosphate peak, respectively.) Also shown is a snapshot from a simulation of the fengycin peptide that fits the distribution in the plot (backbone beads are shown as spheres; residues colored to match). Standard error was no larger than 1% at any point (omitted for clarity).
The entire lipopeptide is anchored by the hydrocarbon inserted into the hydrophobic bilayer interior. The ring of fengycin is tilted in the headgroup region to accommodate the insertion of Tyr9 and Ile10’s hydrophobic side chains into the bilayer. On the other side of the ring, Thr4 and Glu5, with polar side chains, are oriented toward the water outside the bilayer. The rest of the residues fall along a tilted plane between these two ends, resting primarily in the area of the lipid headgroups. A snapshot from a trajectory in which the fengycin ring fits this description and matches the density plots well, can be found in Fig. 2.
The relative spacing of the residues is nearly identical for the POPC and POPE/POPG systems (data not shown). However, there are subtle changes to fengycin’s overall positioning within the bilayer. It has been suggested that DPPC’s tight packing and lower fluidity may result in shallower lipopeptide penetration compared to bilayers with lipids with unsaturated hydrocarbons (DOPC, for instance) (14). In our simulations, we see a similar effect. This is quantified in Fig. S1 in the Supporting Material, using the normalized density distribution of the backbone beads for the fengycin ring, again shifted relative to the peak of the phosphate density. The peptide backbone of fengycin inserts most deeply into POPC, followed by POPE/POPG, with fengycin least deeply inserted into the DPPC bilayer.
Though the fengycin peptide density is shifted closer to the bilayer center in the POPC system, most of the density is still outside the phosphate peak. This implies that the peptide interacts mostly with the lipid headgroups and the aqueous environment, as opposed to the hydrophobic core of the bilayer. This makes sense chemically, given the preponderance of polar side chains.
Fengycin forms organized aggregates
Before building our bilayer systems, we began by simulating multiple fengycins in water. The fengycins readily formed micelles, which has been seen experimentally (45), leading to our decision to place a fengycin micelle at the surface of a lipid bilayer to explore the effect these micelles have on membranes.
Visual inspection of our micelle-bilayer simulations suggests that the preferred aggregation state of fengycin bound to membrane is sensitive to the headgroup composition. Fig. 3 quantifies this by showing the number of unique fengycin clusters present in the simulations as a function of time; because each simulation began with the 13 fengycins in a single cluster, we expect the value to increase over time simply for entropic reasons. Each curve is the average of 16 trajectories, with error bars representing the standard error of the mean to highlight the degree of variation between trajectories. Averaging the time series from independent trajectories obscures the details of individual simulations but highlights global trends. In this case, we see that the 13-fengycin micelle falls apart into smaller clusters in all of the systems. After 2 μs of simulation time the POPC and DPPC systems have leveled off with an average of ∼2 and 3 clusters per trajectory, respectively. This means that the equilibrated clusters tend to be ∼5–6 fengycins per cluster in POPC bilayers and four fengycins in DPPC bilayers. By contrast, the average number of clusters climbs to nearly six clusters per trajectory in the POPE/POPG system, and the absence of a pronounced plateau suggests that the true equilibrium value is higher. It is possible that the monomer is the most stable state when bound to the charged bacteria-like membrane, although significantly longer simulations would be necessary to prove this conclusively.
Figure 3.
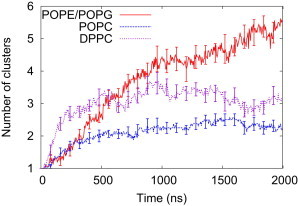
Average number of aggregates of fengycin over time for the systems with 13 fengycins stabilized. To see this figure in color, go online.
One interesting question is whether fengycin aggregation is driven by specific interactions, or merely nonspecific clustering. To answer it, we plot in Fig. 4 the average interresidue contact maps for clustered fengycins (see Methods for details). It is shown as a probability heat map, with bright spots corresponding to areas where two beads are often in contact when two lipopeptides are in contact. The maps for each of the different bilayer systems are qualitatively very similar, but the intensity of regions with high contact differs in key ways. The region of highest contact is Tyr3 (atoms 6–9). This is the tyrosine residue that connects to the C-terminus of the peptide portion to close the peptide ring. The map indicates that when fengycins are in contact, it most frequently involves contact of their Tyr3 residues. Panel D in Fig. 4 shows a sample cluster with the Tyr3 side-chain beads shown as spheres to highlight the stacking of Tyr3 residues and the linear aggregation of fengycin molecules. This tyrosine is conserved among fengycin and plipastatin variants (albeit with varying chirality). However, it is not present in surfactins and is positioned differently in iturins, all of which differ dramatically in their aggregation properties, which suggests that this tyrosine is a critical part of the unique aggregation mechanism of fengycins.
Figure 4.
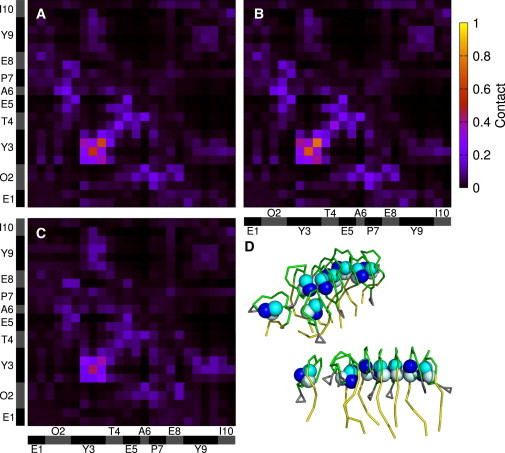
Bead-bead contact maps between clustered fengycins for (A) DPPC, (B) POPC, and (C) POPE/POPG bilayers. Residue sequences are shown as axes, with the complete coarse-grained bead list for the peptide portion of fengycin to the right. (D) Also shown are two different views for a representative cluster (palmitoyl chains in yellow, backbones in green, side-chain tyrosine in gray, and the three beads that make up the cyclic-linking tyrosine in blue shades to show the orientation of packing).
Cryo-transmission electron microscopy data indicate that fengycin forms a variety of aggregate structures in POPC systems, including crystals, amorphous aggregates, and fibers (46), leading us to suggest that the organized pattern of aggregation in our simulations may be an early nucleation structure. From visual inspection of the aggregates we can see an alternating pattern within aggregates, where neighboring lipopeptides in the chain are often rotated relative to one another, with the opposite face of the ring interacting. At fengycin concentrations slightly higher than the one in our system (fengycin/lipid ratio of 0.026:1 compared to our 0.025:1), fengycin has been observed experimentally beginning to form larger aggregates in DPPC bilayers (14). The experiments show that aggregation increases with higher concentration, and eventually higher-order structures are stabilized.
The fengycin-packing pattern in POPE/POPG is similar to that in DPPC and POPC, but less pronounced in the Tyr3 region. In addition, areas outside of this region are brighter in POPE/POPG. This leads us to believe that whereas clusters tend to follow the described packing pattern in the POPE/POPG system, fengycin contacts that qualify as clusters by our criteria frequently involve nonstandard interactions that are likely transient, as the clusters (which are larger in number) move about in the lipid bilayer.
Preferential interactions with POPG lipids
Fig. 5 shows a radial distribution function (RDF) in the plane of the bilayer for each lipid component, tracking their distribution about fengycin. The DPPC and POPC curves are not especially informative, because there is no other lipid component for comparison in these pure bilayers. However, the absence of a contact peak suggests the absence of any strong specific structuring of lipid headgroups in the vicinity of the peptide.
Figure 5.
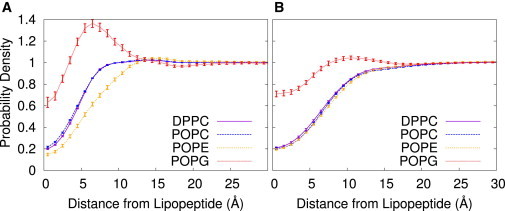
Lateral radial distribution of lipids as a function of distance from lipopeptides in the bound leaflet for systems with (A) one fengycin and (B) 13 fengycins. To see this figure in color, go online.
By contrast, the results for the POPE/POPG system reveal striking (and surprising) effects on lateral ordering. In both the single fengycin and the micelle systems, we see a preference for POPG lipids at short range. In the single fengycin system, this preference is substantial and leads to the occlusion of POPE at short range. In the 13-fengycin system, the POPG preference is far less dramatic, and the POPE RDF is not noticeably different from the DPPC and POPC curves. The presence of other lipopeptides in that system and the weak aggregates of 2–3 lipopeptides is probably washing out the effect. The alternating pattern of fengycins within clusters means that if there are preferential POPG interactions sites on a single fengycin, the aggregate as a whole likely has alternating interaction sites as well, convoluting the RDF calculation for individual lipopeptides within this context. Fengycin’s inability to exist in persistent clusters within the POPE/POPG system is explained to some degree by fengycin’s strong interactions with POPG lipids—by dissolving the aggregates, each fengycin can maximize contacts with these lipids.
The preferential interaction between POPG and fengycin is at first very surprising, because a naive electrostatic interpretation would suggest that the fengycin’s net −2 charge would repel the anionic headgroup. To rationalize this phenomenon, we computed three-dimensional RDFs between the headgroups and specific components of the fengycin, to figure out which interactions were stabilizing these interactions. The headgroup beads of interest were the anionic phosphate bead for POPE, the cationic amine for POPE (which gives it zwitterionic character), the anionic phosphate bead for POPG, and the neutral glycerol for POPG.
Fig. 6 shows the RDFs, separated by the side chain used as the center. For the ornithine, the RDFs appear exactly as we would expect. There are favorable interactions between the positively charged ornithine bead and both beads of the PG headgroup; these are presumably driven by favorable electrostatics between the phosphate and the ornithine (the covalent bond between the phosphate and glycerol beads accounts for the apparent interaction between ornithine and glycerol). By contrast, the interaction with POPE is very weak, likely because POPE tends to be displaced by POPG.
Figure 6.
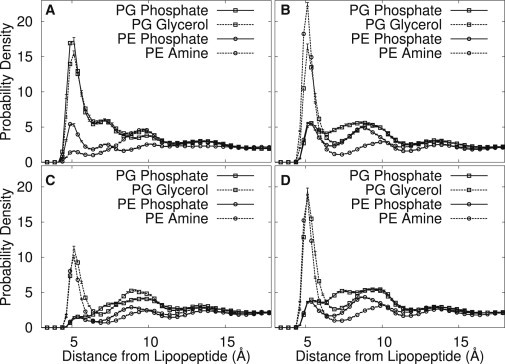
Radial distribution of the headgroup beads of PG and PE lipids relative to the charged beads of (A) Orn2, (B) Glu1, (C) Glu5, and (D) Glu8 in the POPE/POPG system with one fengycin.
The plots for Glu1 and Glu8 are similar to one another and provide insight into fengycin’s overall POPG preference. As expected, because of repulsive electrostatics, we see little interaction with the phosphate groups from both POPE and POPG. We do see favorable contact peaks for both the POPE amine group (due to complementary electrostatics) and the POPG glycerol—a strong polar interaction. For the glutamates, each lipid has both one preferential interaction and one repulsive. By contrast, the ornithine residue has only attractive interactions with PG headgroups, and this is sufficient to cause a net attraction to PG. Visual inspection of the trajectories indicates that Glu1 and Glu8 are constantly close to Orn2, with the POPG lipids frequently oriented in a way to maximize preferential interactions between all involved residues.
The curves for Glu5 are qualitatively similar to the other glutamates, but the peaks are significantly smaller. This occurs because the layout of the fengycin ring places Glu5 farther from the membrane (see Fig. 2), where it interacts mostly with water rather than the lipid headgroups.
Curvature and perturbation of lipid bilayers
Fengycin’s size relative to common phospholipids suggests that it would induce positive curvature in the lipid bilayer and promote micelle formation (14). To quantify possible fengycin-induced curvature, we computed the heights of the lipid phosphate beads relative to the center of the bilayer along the bilayer normal. It is shown in Fig. 7 as a heat map for the bound and lipopeptide-free leaflets. We curated clusters of six fengycins and nine fengycins for both the DPPC and POPC systems. We could not do the same thing for POPE/POPG because we could not find stable, persistent clusters that were large enough.
Figure 7.
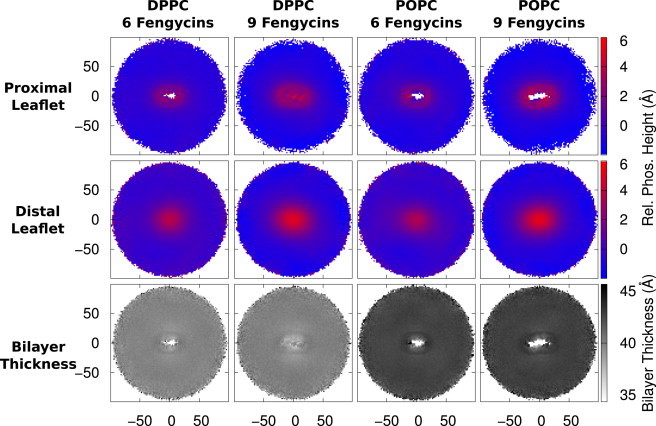
Bilayer curvature indicated by heat maps of phosphate heights relative to neat bilayers and bilayer thickness for clusters of six fengycins and nine fengycins bound to DPPC and POPC bilayers. The X and Y axes are the distance from the centered fengycin cluster (measured in Ångstroms). Extreme values near the origin reflect the fact that there is some occlusion of lipids by fengycin in one or both leaflets. To see this figure in color, go online.
We can see shifts in the phosphate heights for both leaflets in all the systems shown. The shifts are positive increases in both leaflets, toward the lipopeptides, suggesting positive curvature in the bilayer. In Fig. 7, we also show an indicator of the thickness of the bilayer, calculated by the difference between the leaflet maps, to see if the fengycins are inducing bilayer-thinning. Fengycin appears to promote curvature of both leaflets while slightly thinning the bilayer. (Sample trajectory snapshots of fengycin clusters inducing bilayer curvature are found in Fig. 8.)
Figure 8.
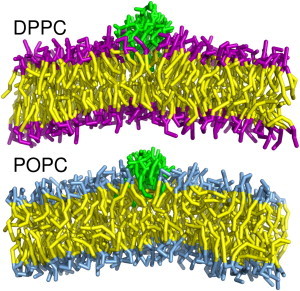
Sample images of curvature induced by nine fengycin clusters in both DPPC and POPC bilayers. (Fengycins are green, lipid tails are yellow, and headgroups for DPPC and POPC are purple and blue, respectively.)
These maps can be striking visual evidence for induced curvature, but are difficult to quantify. As a followup, we also calculated the phosphate heights as a function of distance from each lipopeptide. This allows us to include clusters of every size in the micelle simulations to see if the clustering has a general effect on curvature. The single fengycin systems show very little bilayer perturbation, with the largest differences between the phosphate peak and the plateau of the curve measuring <1 Å (data not shown). Fig. 9 shows the same quantity for both leaflets in the 13-fengycin systems. The first microsecond of simulation was omitted from the calculation so that we can compare the effects after the clusters have equilibrated (this corresponds with the plateaus in the DPPC and POPC aggregation curves). The fengycin clusters have a much more dramatic effect on the bilayer curvature. The phosphate peaks for the POPC system show the greatest curvature. The peak-to-plateau differences for the bound leaflet and the unbound leaflet are 3.25 and 3.87 Å, respectively. For DPPC, the differences are 2.26 and 3.14 Å and for POPE/POPG, they are 2.05 and 2.34 Å. It is clear that the curvature effect is dependent on cluster size, because the mixed POPE/POPG systems show the smallest effect on the bilayer. These values also allow us to quantify the weak bilayer thinning effect that these clusters induce. By taking the difference between the peak-to-plateau curvature changes between the two leaflets, we calculate that the membrane is thinned by <1 Å in all of our systems.
Figure 9.
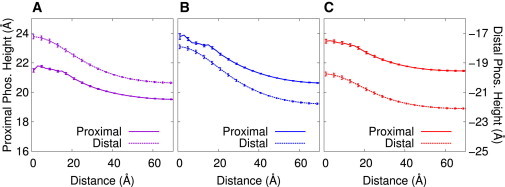
Height of phosphates above the center of the bilayer as a function of distance from the centroid of each fengycin, plotted for both the proximal and distal leaflets (left and right axes, respectively) for the (A) DPPC, (B) POPC, and (C) POPE/POPG systems with 13 fengycins. To see this figure in color, go online.
Fengycin also perturbs bilayer structure by affecting the order within the lipid bilayer. Our analysis of principal component-based order parameters calculated as a function of distance from the lipopeptides shows this disordering effect (seen in Fig. 10). The molecular order parameter effectively measures the tilt of the lipid palmitoyl chains on a scale comparable to standard deuterium order parameters. At distances far from the lipopeptides the curves match the calculated molecular order parameters from bilayers without any lipopeptides, indicating the simulated systems are large enough to recover bulk-like structure at long range. Near the peptide, the bilayer is significantly perturbed. The effect is enhanced in the presence of fengycin clusters, both in strength and range. However, the effect decays relatively rapidly, and is nearly invisible beyond 40 Å from the largest lipopeptide clusters. Experimental work shows that fengycin has little effect on bulk bilayer order, but has a disrupting effect locally (47). One model suggests this heterogeneous perturbation is the result of fengycin’s poor bilayer mixing, which leads to detergent-rich clusters that cause short-range order effects (47).
Figure 10.
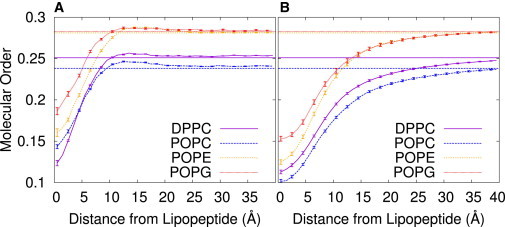
Principal component-based order parameters for the palmitoyl chains in the three systems, with POPE and POPG shown separately, for the systems with (A) one fengycin and those with (B) 13 fengycins. (Dashed lines indicate the value calculated from equivalent fengycin-free systems.) To see this figure in color, go online.
DPPC and POPC, which differ only in one lipid tail, are affected differently by fengycin, at least for the systems with only one lipopeptide. The order parameter for DPPC in those systems drops much more dramatically. This might be expected, because fengycin has been shown to affect DPPC compressibility in monolayers and has a fluidizing effect in DPPC bilayers, while reducing the formation of condensed domains (13) (this effect is similar to that for cholesterol (48)).
The order of POPE and POPG lipids is affected differently as well, despite the fact that they show the same order in a well-mixed system. Given what we have already seen about fengycin’s ability to separate POPG and POPE, with a preference for POPG, it is not surprising that the values between the two would be different. The effect on POPE is stronger, suggesting another reason for the preference for POPG near the lipid surface: to avoid unfavorable lipid-peptide interactions, the lipid must undergo presumably unfavorable contortions. However, the thermodynamic significance of this effect is unclear.
Limitations of this work and future goals
Improvements to the MARTINI force field might improve the accuracy of these simulations. For instance, MARTINI Ver. 2.2 includes changes to proline residues, which have been shown to be too hydrophobic (49). This would have some effect, given the importance of amphipathicity to fengycin’s binding and positioning in the bilayer. Also, there are concerns about the accuracy of electrostatics in our chosen model. Because of the lack of polarity in water and the absence of partial charges in the system, a screening constant is employed to simulate distance-dependent dielectric screening. However, this screening is the same strength no matter the environment, resulting in weak attractions between oppositely charged residues in apolar mediums (like the center of the lipid bilayer). This could have an effect on the lipid-fengycin interactions in our system, especially in the case of fengycin in the POPE/POPG system where a huge number of charged moieties exist near the bilayer. One way to improve this, at the expense of computational speed, would be to utilize a polarizable water model (50).
There is a large variety of fengycins with differences in primary sequence, particularly substitutions of glutamine for the charged Glu8. But even among those with identical primary sequence, there are differences in chirality of the side chains, specifically Tyr3 and Tyr9 (51). Exploring the role of chirality in the mechanism of fengycin is beyond the resolution of the MARTINI model, which has too few interaction sites in each amino acid to effectively consider stereoisomers; going forward, we plan to perform all-atom simulations to address these questions.
It has also been shown that the ionic environment has an effect on the structure of fengycin. In particular, calcium causes changes to fengycin that alters the environment around the tyrosines as detected by nuclear magnetic resonance (52). We have only explicitly considered NaCl in our systems, as is commonly done to model physiological conditions. It would be worth considering the role of ionic strength on fengycin structure and fengycin binding to bilayers (especially the negatively charged POPE/POPG bilayer system). However, treating the solvation behavior for divalent cations is challenging even for all-atom force fields that explicitly treat polarization (53), let alone simpler models such as the one used here.
As a result, future work will definitely include all-atom simulations of fengycin. These will be challenging, because membrane binding and lateral reorganization are relatively slow phenomena requiring long trajectories (preferably multiple independent ones) of relatively large systems. However, we could speed the process by choosing snapshots from a number of different system states in our coarse-grained trajectories and using them to construct equivalent all-atom systems that can be used to seed new runs. Comparison of these all-atom runs to the coarse-grained results would help confirm the validity of the models and provide more detailed information about the interactions between fengycin and lipids.
We also intend to simulate the same bilayer systems with dispersed, bound lipopeptides, rather than beginning from a micelle structure. This would allow us to see if the lipopeptides will cluster and form equilibrated aggregates of the sizes we see from our aggregate dispersion data. Furthermore, we plan to simulate larger bilayer systems to try to further capture the bilayer curvature effects of the fengycin molecules. Because of the use of periodic boundary conditions, any curvature in the bilayer is limited to what is able to fit within the box dimensions. Simulating a box that is much larger would allow us to see if the curvature is even greater than what is reported here.
Future simulations must also include more variety in fengycin structures. The fengycin structure chosen for this work is atypical and the literature on it is sparse. For comparison, simulations of fengycin IX (38), a much more common structure with a deeper literature, would be useful for more validation with experimental results. Considering the importance we place on Tyr3 in the aggregation properties of fengycin, we suggest mutating this residue to a hydrophobic, noncyclic residue experimentally to see if this affects aggregation. Followup simulations will include this test case, as well as include alanine to valine substitutions at position 6, a variation among known fengycins. Because this change is minor, we would not expect a major effect, but it might result in subtle changes to aggregation and insertion properties.
Conclusions
According to our coarse-grained simulations, fengycin aggregates in solution and remains in clusters, even during long timescale simulations. They readily partition into bilayers, existing as partially inserted and partially solvent-exposed. It has been theorized that membranes do not readily accommodate the large peptide head of fengycin, leading to partial insertion and a preference for clusters (46).
The predicted clusters form and bind to membranes, regardless of the headgroup composition, and remain stable as the peptides insert into zwitterionic PC membranes. By contrast, in anionic-mixed POPE/POPG systems, the clusters do not persist, but fall apart into a mixture of dimers and monomers. Further simulation may lead to complete disintegration of the clusters in those bilayers. Because the 2:1 POPE/POPG bilayer is an effective (if simplified) model for bacteria-like membranes, which have high concentrations of PG headgroups and a net negative charge (54), this suggests a mechanism for how fengycin could avoid damaging bacterial membranes while attacking those of fungi. If lipopeptide clustering at higher concentrations plays a major role in fengycin’s function—our simulations suggest that fengycin oligomers have much larger effects on bilayer structure—then this inability to form clusters in the bacteria-like bilayer might explain fengycin’s specificity for fungi as opposed to bacteria (1). The fact that the clustering occurs via specific structural motifs further suggests that this is part of its evolved mechanism.
Fengycin has multiple effects on bilayer systems. We have shown that fengycin demixes POPE/POPG bilayers, surprisingly attracting the anionic POPG lipids. We have quantified pairwise interactions between lipids and residues that explain this contact despite an anionic peptide and an anionic lipid. Fengycin also promotes positive curvature in all of our tested bilayer systems. This effect seems to be much more dramatic when the fengycins are clustered. Finally, fengycins also perturb the intrinsic order of the bilayer locally.
All of this evidence suggests a more complete model for fengycin-bilayer interactions that is in agreement with experimental evidence. Fengycin readily aggregates and binds to lipid bilayers, increasing bilayer fluidity and promoting curvature that could lead to micelle formation. The concentration of fengycin in our systems is too low to expect to see major bilayer deformations, but the curvature effects do appear to be concentration-dependent. At very high concentrations, fengycin likely acts as a detergent, but at lower concentrations that are still above the effective threshold, fengycin might form oligomeric structures that cause bilayer perturbation, bending, and micelle formation that might damage the membrane (13). This process has been reported for surfactins, which are very similar structurally to fengycins (55,56).
We have presented a number of long-timescale coarse-grained simulations that explore the effects of fengycin on a variety of lipid bilayer systems. We have identified a number of fengycin-induced bilayer perturbations that are theorized and described in the experimental literature, allowing us to contribute to the model of activity for fengycin.
Acknowledgments
We thank the Center for Integrated Research Computing at the University of Rochester for providing computational resources, including access to the BlueGene/Q.
This work was supported by grant No. GM095496 (to A.G.) and grant No. GM068411 from the Institutional Ruth L. Kirchstein National Research Service Award (to J.N.H.).
Supporting Material
References
- 1.Vanittanakom N., Loeffler W., Jung G. Fengycin—a novel antifungal lipopeptide antibiotic produced by Bacillus subtilis f-29–3. J. Antibiot. (Tokyo) 1986;39:888–901. doi: 10.7164/antibiotics.39.888. [DOI] [PubMed] [Google Scholar]
- 2.Ongena M., Jacques P. Bacillus lipopeptides: versatile weapons for plant disease biocontrol. Trends Microbiol. 2008;16:115–125. doi: 10.1016/j.tim.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 3.Li X.-Y., Mao Z.-C., Long C.-L. Diversity and active mechanism of fengycin-type cyclopeptides from Bacillus subtilis xf-1 against Plasmodiophora brassicae. J. Microbiol. Biotechnol. 2013;23:313–321. doi: 10.4014/jmb.1208.08065. [DOI] [PubMed] [Google Scholar]
- 4.Hu L.B., Shi Z.Q., Yang Z.M. Fengycin antibiotics isolated from B-FS01 culture inhibit the growth of Fusarium moniliforme Sheldon ATCC 38932. FEMS Microbiol. Lett. 2007;272:91–98. doi: 10.1111/j.1574-6968.2007.00743.x. [DOI] [PubMed] [Google Scholar]
- 5.Chan Y.-K., Savard M., Seguin C. Identification of lipopeptide antibiotics of a Bacillus subtilis isolate and their control of Fusarium graminearum diseases in maize and wheat. BioControl. 2009;54:567–574. [Google Scholar]
- 6.Tao Y., Bie X.M., Lu Z.X. Antifungal activity and mechanism of fengycin in the presence and absence of commercial surfactin against Rhizopus stolonifer. J. Microbiol. 2011;49:146–150. doi: 10.1007/s12275-011-0171-9. [DOI] [PubMed] [Google Scholar]
- 7.Jacques P., Hbid C., Thonart P. Optimization of biosurfactant lipopeptide production from Bacillus subtilis S499 by Plackett-Burman design. Appl. Biochem. Biotechnol. 1999;77:223–233. [Google Scholar]
- 8.Loeffler W., Tschen J.S.-M., Wu T.-G. Antifungal effects of bacilysin and fengymycin from Bacillus subtilis F-29–3: a comparison with activities of other Bacillus antibiotics. J. Phytopathol. 1986;115:204–213. [Google Scholar]
- 9.Ongena M., Jourdan E., Thonart P. Surfactin and fengycin lipopeptides of Bacillus subtilis as elicitors of induced systemic resistance in plants. Environ. Microbiol. 2007;9:1084–1090. doi: 10.1111/j.1462-2920.2006.01202.x. [DOI] [PubMed] [Google Scholar]
- 10.Jourdan E., Henry G., Ongena M. Insights into the defense-related events occurring in plant cells following perception of surfactin-type lipopeptide from Bacillus subtilis. Mol. Plant Microbe Interact. 2009;22:456–468. doi: 10.1094/MPMI-22-4-0456. [DOI] [PubMed] [Google Scholar]
- 11.Vollenbroich D., Pauli G., Vater J. Antimycoplasma properties and application in cell culture of surfactin, a lipopeptide antibiotic from Bacillus subtilis. Appl. Environ. Microbiol. 1997;63:44–49. doi: 10.1128/aem.63.1.44-49.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heerklotz H., Seelig J. Leakage and lysis of lipid membranes induced by the lipopeptide surfactin. Eur. Biophys. J. 2007;36:305–314. doi: 10.1007/s00249-006-0091-5. [DOI] [PubMed] [Google Scholar]
- 13.Deleu M., Paquot M., Nylander T. Fengycin interaction with lipid monolayers at the air-aqueous interface-implications for the effect of fengycin on biological membranes. J. Colloid Interface Sci. 2005;283:358–365. doi: 10.1016/j.jcis.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 14.Deleu M., Paquot M., Nylander T. Effect of fengycin, a lipopeptide produced by bacillus subtilis, on model biomembranes. Biophys. J. 2008;94:2667–2679. doi: 10.1529/biophysj.107.114090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heller H., Schaefer M., Schulten K. Molecular dynamics simulation of a bilayer of 200 lipids in the gel and in the liquid-crystal phases. J. Phys. Chem. 1993;97:8343–8360. [Google Scholar]
- 16.Pastor R.W., Feller S.E. Time scales of lipid dynamics and molecular dynamics. In: Merz K.M. Jr., Roux B., editors. Biological Membranes: A Molecular Perspective from Computation and Experiment. Birkhauser; Basel, Switzerland: 1996. pp. 3–30. [Google Scholar]
- 17.Lindahl E., Sansom M.S. Membrane proteins: molecular dynamics simulations. Curr. Opin. Struct. Biol. 2008;18:425–431. doi: 10.1016/j.sbi.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 18.Dror R.O., Arlow D.H., Shaw D.E. Identification of two distinct inactive conformations of the β2-adrenergic receptor reconciles structural and biochemical observations. Proc. Natl. Acad. Sci. USA. 2009;106:4689–4694. doi: 10.1073/pnas.0811065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grossfield A. Recent progress in the study of G protein-coupled receptors with molecular dynamics computer simulations. Biochim. Biophys, Acta Biomembr. 2011;1808:1868–1878. doi: 10.1016/j.bbamem.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 20.Mertz B., Struts A.V., Brown M.F. Molecular simulations and solid-state NMR investigate dynamical structure in rhodopsin activation. Biochim. Biophys, Acta Biomembr. 2012;1818:241–251. doi: 10.1016/j.bbamem.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaikh S.A., Li J., Tajkhorshid E. Visualizing functional motions of membrane transporters with molecular dynamics simulations. Biochemistry. 2013;52:569–587. doi: 10.1021/bi301086x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolintineanu D.S., Kaznessis Y.N. Computational studies of protegrin antimicrobial peptides: a review. Peptides. 2011;32:188–201. doi: 10.1016/j.peptides.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mátyus E., Kandt C., Tieleman D.P. Computer simulation of antimicrobial peptides. Curr. Med. Chem. 2007;14:2789–2798. doi: 10.2174/092986707782360105. [DOI] [PubMed] [Google Scholar]
- 24.Marrink S.J., Risselada H.J., de Vries A.H. The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B. 2007;111:7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- 25.Baoukina S., Tieleman D.P. Direct simulation of protein-mediated vesicle fusion: lung surfactant protein B. Biophys. J. 2010;99:2134–2142. doi: 10.1016/j.bpj.2010.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodgers J.M., Srensen J., Smit B. Understanding the phase behavior of coarse-grained model lipid bilayers through computational calorimetry. J. Phys. Chem. B. 2012;116:1551–1569. doi: 10.1021/jp207837v. [DOI] [PubMed] [Google Scholar]
- 27.Rosetti C., Pastorino C. Comparison of ternary bilayer mixtures with asymmetric or symmetric unsaturated phosphatidylcholine lipids by coarse grained molecular dynamics simulations. J. Phys. Chem. B. 2012;116:3525–3537. doi: 10.1021/jp212406u. [DOI] [PubMed] [Google Scholar]
- 28.Rzepiela A.J., Sengupta D., Marrink S.J. Membrane poration by antimicrobial peptides combining atomistic and coarse-grained descriptions. Faraday Discuss. 2010;144:431–443. doi: 10.1039/b901615e. [DOI] [PubMed] [Google Scholar]
- 29.Polyansky A.A., Ramaswamy R., Efremov R.G. Antimicrobial peptides induce growth of phosphatidylglycerol domains in a model bacterial membrane. J. Phys. Chem. Lett. 2010;1:3108–3111. [Google Scholar]
- 30.Woo H.-J., Wallqvist A. Spontaneous buckling of lipid bilayer and vesicle budding induced by antimicrobial peptide magainin 2: a coarse-grained simulation study. J. Phys. Chem. B. 2011;115:8122–8129. doi: 10.1021/jp2023023. [DOI] [PubMed] [Google Scholar]
- 31.Horn J.N., Sengillo J.D., Grossfield A. Characterization of a potent antimicrobial lipopeptide via coarse-grained molecular dynamics. Biochim. Biophys, Acta Biomembr. 2012;1818:212–218. doi: 10.1016/j.bbamem.2011.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santo K.P., Berkowitz M.L. Difference between magainin-2 and melittin assemblies in phosphatidylcholine bilayers: results from coarse-grained simulations. J. Phys. Chem. B. 2012;116:3021–3030. doi: 10.1021/jp212018f. [DOI] [PubMed] [Google Scholar]
- 33.Khalfa A., Tarek M. On the antibacterial action of cyclic peptides: insights from coarse-grained MD simulations. J. Phys. Chem. B. 2010;114:2676–2684. doi: 10.1021/jp9064196. [DOI] [PubMed] [Google Scholar]
- 34.Periole X., Marrink S.-J. The MARTINI coarse-grained force field. In: Monticelli L., Salonen E., editors. Biomolecular Simulations. Vol. 924. Humana Press; Totowa, NJ: 2013. pp. 533–565. (Methods in Molecular Biology). [DOI] [PubMed] [Google Scholar]
- 35.MARTINI. 2012. Biomolecular force field for coarse-grained simulations. Accessed Nov. 1, 2012. http://md.chem.rug.nl/cgmartini/.
- 36.Wu C.-Y., Chen C.-L., Liu S.-T. Nonribosomal synthesis of fengycin on an enzyme complex formed by fengycin synthetases. J. Biol. Chem. 2007;282:5608–5616. doi: 10.1074/jbc.M609726200. [DOI] [PubMed] [Google Scholar]
- 37.Pathak K., Keharia H., Balaram P. Lipopeptides from the banyan endophyte, Bacillus subtilis k1: mass spectrometric characterization of a library of fengycins. J. Am. Soc. Mass Spectrom. 2012;23:1716–1728. doi: 10.1007/s13361-012-0437-4. [DOI] [PubMed] [Google Scholar]
- 38.Honma M., Tanaka K., Hashimoto M. Termination of the structural confusion between plipastatin A1 and fengycin IX. Bioorg. Med. Chem. 2012;20:3793–3798. doi: 10.1016/j.bmc.2012.04.040. [DOI] [PubMed] [Google Scholar]
- 39.van der Spoel D., Lindahl E., Berendsen H.J.C. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 40.Hess B., Kutzner C., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 41.Nosé S., Klein M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983;50:1055–1076. [Google Scholar]
- 42.Hoover W.G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 43.Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. [Google Scholar]
- 44.Romo T.D., Grossfield A. LOOS: an extensible platform for the structural analysis of simulations. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2009;2009:2332–2335. doi: 10.1109/IEMBS.2009.5335065. [DOI] [PubMed] [Google Scholar]
- 45.Deleu M., Razafindralambo H., Paquot M. Interfacial and emulsifying properties of lipopeptides from Bacillus subtilis. Colloid Surface A. 1999;152:3–10. [Google Scholar]
- 46.Patel H., Tscheka C., Heerklotz H. All-or-none membrane permeabilization by fengycin-type lipopeptides from Bacillus subtilis QST713. Biochim. Biophys, Acta Biomembr. 2011;1808:2000–2008. doi: 10.1016/j.bbamem.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 47.Nazari M., Kurdi M., Heerklotz H. Classifying surfactants with respect to their effect on lipid membrane order. Biophys. J. 2012;102:498–506. doi: 10.1016/j.bpj.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gong K., Feng S.-S., Soew P.H. Effects of pH on the stability and compressibility of DPPC/cholesterol monolayers at the air-water interface. Colloids Surf. A Physicochem. Eng. Asp. 2002;207:113–125. [Google Scholar]
- 49.de Jong D.H., Singh G., Marrink S.J. Improved parameters for the MARTINI coarse-grained protein force field. J. Chem. Theory Comput. 2013;9:687–697. doi: 10.1021/ct300646g. [DOI] [PubMed] [Google Scholar]
- 50.Yesylevskyy S.O., Schäfer L.V., Marrink S.J. Polarizable water model for the coarse-grained MARTINI force field. PLOS Comput. Biol. 2010;6:e1000810. doi: 10.1371/journal.pcbi.1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jacques P. Surfactin and other lipopeptides from Bacillus spp. In: Sobern-Chavez G., editor. Biosurfactants. Vol. 20. Springer; Berlin, Germany: 2011. pp. 57–91. (Microbiology Monographs). [Google Scholar]
- 52.Nasir M.N., Laurent P., Deleu M. Analysis of calcium-induced effects on the conformation of fengycin. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2013;110:450–457. doi: 10.1016/j.saa.2013.03.063. [DOI] [PubMed] [Google Scholar]
- 53.Jiao D., King C., Ren P. Simulation of Ca2+ and Mg2+ solvation using polarizable atomic multipole potential. J. Phys. Chem. B. 2006;110:18553–18559. doi: 10.1021/jp062230r. [DOI] [PubMed] [Google Scholar]
- 54.Wadhwani P., Epand R., Epand R. Membrane-active peptides and the clustering of anionic lipids. Biophys. J. 2012;103:265–274. doi: 10.1016/j.bpj.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heerklotz H., Seelig J. Detergent-like action of the antibiotic peptide surfactin on lipid membranes. Biophys. J. 2001;81:1547–1554. doi: 10.1016/S0006-3495(01)75808-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bouffioux O., Berquand A., Deleu M. Molecular organization of surfactin-phospholipid monolayers: effect of phospholipid chain length and polar head. Biochim. Biophys, Acta Biomembr. 2007;1768:1758–1768. doi: 10.1016/j.bbamem.2007.04.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.