

Abstract
A role of heat shock protein 27 (HSP27) as a potential biomarker has been reported in various tumour entities, but comprehensive studies in pancreatic cancer are lacking. Applying tissue microarray (TMA) analysis, we correlated HSP27 protein expression status with clinicopathologic parameters in pancreatic ductal adenocarcinoma specimens from 86 patients. Complementary, we established HSP27 overexpression and RNA-interference models to assess the impact of HSP27 on chemo- and radiosensitivity directly in pancreatic cancer cells. In the TMA study, HSP27 expression was found in 49% of tumour samples. Applying univariate analyses, a significant correlation was found between HSP27 expression and survival. In the multivariate Cox-regression model, HSP27 expression emerged as an independent prognostic factor. HSP27 expression also correlated inversely with nuclear p53 accumulation, indicating either protein interactions between HSP27 and p53 or TP53 mutation-dependent HSP27-regulation in pancreatic cancer. In the sensitivity studies, HSP27 overexpression rendered HSP27 low-expressing PL5 pancreatic cancer cells more susceptible towards treatment with gemcitabine. Vice versa, HSP27 protein depletion in HSP27 high-expressing AsPC-1 cells caused increased gemcitabine resistance. Importantly, HSP27 expression was inducible in pancreatic cancer cell lines as well as primary cells. Taken together, our study suggests a role for HSP27 as a prognostic and predictive marker in pancreatic cancer. Assessment of HSP27 expression could thus facilitate the identification of specific patient subpopulations that might benefit from individualized treatment options. Additional studies need to clarify whether modulation of HSP27 expression could represent an attractive concept to support the incorporation of hyperthermia in clinical treatment protocols for pancreatic cancer.
Keywords: HSP27, biomarker, gemcitabine, pancreatic cancer, TMA
Introduction
Pancreatic cancer is one of the most aggressive types of cancer, being the fourth leading cause of cancer-related deaths in the United States with an overall 5-year survival rate of less than 5% and a median survival of about 6 months [1, 2]. The poor prognosis has remained basically unchanged over the last decades despite advances in surgical, chemo- and radiotherapy [3]. Major contributing factors are the lack of early symptoms and the absence of reliable screening methods, leading to a late presentation of the disease and resulting in an advanced tumour stage and metastases in the majority of patients at the time of diagnosis [2]. Analyses of tissue microarrays (TMA), generated from surgical pancreatic cancer specimens, facilitate the comprehensive correlation of protein expression levels with clinicopathologic variables and represent a valuable tool for the identification of novel biomarkers for the disease [4].
HSP27 belongs to the family of heat shock proteins, a diverse group of highly conserved proteins, which are classified into five subgroups according to their molecular size [5], and which mainly act as molecular chaperones in cells exposed to different stresses, including heat shock, consecutively counteracting the formation of misfolded proteins and allowing correct protein refolding [6, 7]. In addition to these functions, HSP27 has been implicated in proteasome-mediated protein degradation as well as the regulation of apoptotic pathways [8].
Although HSP27 activity appears to be regulated mainly at the transcriptional level, HSP27 can also be post-translationally modified through rapid phosphorylation at three serine residues (15, 78 and 82) [9]. These phosphorylation events have been implicated in a wide variety of cellular processes, including especially the structural organization of HSP27 through oligomerization, but also survival, proliferation, apoptosis and cell differentiation. However, the exact functions of the different phosphorylated forms of Hsp27 remain insufficiently understood [10].
Expression of HSP27 is strongly induced by heat shock as well as various other physical and chemical stress stimuli, including irradiation, oxidative stress and diverse chemotherapeutics [5, 8]. In addition, HSP27 expression varies during development and differentiation in some tissues [8, 11]. Finally, HSP27 is constitutively expressed in the cytoplasm of many normal and malignant cell types in the absence of stress, with wide variations in regard to the basal level of expression among different tissues [5, 12].
In malignancy, the expression levels of HSP27 have been shown to be elevated in a wide spectrum of human cancers. Consecutively, potential diagnostic, prognostic, predictive or treatment implications of HSP27 overexpression have been investigated in multiple cancer types. These studies, comprehensively reviewed in Ref. [13], in summary suggest that HSP27 could represent a valuable prognostic biomarker in specific cancer types and predict the individual patient's response to certain chemotherapeutics, whereas its applicability as a diagnostic marker appears to be limited [13]. Notably, while much work has been devoted to the role of heat shock proteins including HSP27 during pancreatic inflammation, i.e. during the course of acute pancreatitis [14-16], only little and partly conflicting information is available on the significance of HSP27 expression in pancreatic malignancy. For example, protein expression profiling of nine patient samples showed a significantly higher HSP27 expression in normal pancreatic tissue as compared to pancreatic cancer in one study [17], whereas another study applying protein expression profiling and immunohistochemistry in nine samples showed that HSP27 expression was up-regulated in micro-dissected pancreatic cancer tissue as compared to normal pancreatic tissue [18]. Furthermore, HSP27 has been proposed as a potential serum marker for pancreatic cancer [18], but might not discriminate between chronic pancreatitis and pancreatic carcinoma, as serum HSP27 levels are elevated in both pancreatitis and pancreatic cancer [19]. Finally, proteomic studies support a role for HSP27 in modulating chemoresistance towards gemcitabine in pancreatic cancer [20-22].
The aim of our study was to comprehensively evaluate the significance of HSP27 expression in regard to its potential relevance as a diagnostic, prognostic or predictive marker in pancreatic cancer. In regard to a potential role for HSP27 as a diagnostic or prognostic marker, TMA analysis of 86 surgical pancreatic adenocarcinoma specimens was performed and HSP27 status correlated with multiple clinicopathologic parameters. In regard to a potential role for HSP27 as a predictive marker for therapeutic response, a well-controlled HSP27 overexpression model was generated in PL5 pancreatic cancer cells and consecutively, the influence of HSP27 expression on the radio- and chemotherapeutic response assessed. Complementary, RNA-interference methodology was used to validate the data obtained from the overexpression model.
Material and methods
Case identification, selection and patients’ follow-up
Eighty-nine consecutive patients (n= 89), with histologically confirmed well, moderately or poorly differentiated pancreatic ductal adenocarcinoma, who underwent surgery for pancreatic cancer (Whipple procedure, distal pancreatectomy, or total pancreatectomy) at the Department of Surgery at the Ludwig-Maximilians-University of Munich between January 31, 2003 and June 14, 2007, were chosen for TMA construction. None of these patients received neoadjuvant chemotherapy or irradiation. Patients who died from immediate postoperative complications (n= 3) were excluded. All patients selected for our study (n= 86) had a complete follow-up either until death (n= 67) or until their most recent contact (n= 19) on July 1, 2009. The shortest follow-up after surgery for patients still alive as of July 1, 2009 was 24.5 months, with a 2-year survival rate of 40% (34 of 86 patients). The longest follow-up for patients still alive was 68 months. The median patient age at the time of surgery was 65 years (range 32–81). All clinicopathologic data were collected from the database of the Munich Cancer Registry and the original patients’ charts.
TMA construction
Paraffin-embedded archived tissue material of tumour and surrounding normal pancreatic tissue was used for TMA construction. TMAs were prepared as published before [23]. In brief, the area of interest to be sampled was identified and marked on haematoxylin-eosin stained tissue slides. From the corresponding paraffin block (donor block), tissue core biopsies (each 0.6 mm in diameter) were taken out and then arrayed in a recipient TMA block using a manual arrayer (Beecher Instruments, Sun Prairie, WI, USA).
Each case was represented by three core biopsies from different parts of the pancreatic carcinoma and two core biopsies from corresponding normal pancreatic tissue to exclude artefacts due to heterogeneous antigen expression and to allow comparisons between normal exocrine pancreatic tissue and tumour tissue. Immunohistochemistry was performed on 2 μm sections of the TMA.
Histology and immunohistochemistry
Histological diagnosis of pancreatic adenocarcinoma and the tumour grading were blindly confirmed for each case by two pathologists (GA and TK). Immunohistochemistry was performed using an anti-pHSP27 rabbit monoclonal antibody (1:250; Epitomics, Burlingame, CA, USA), and an anti-HSP27 mouse monoclonal antibody (1:50; Zytomed Systems, Berlin, Germany), or an anti-p53 mouse monoclonal antibody (1:80; Dako, Hamburg, Germany), respectively, together with Vectastain Elite ABC Kits (Vector Laboratories, Burlingame, CA, USA), applying varying detection and antigen retrieval methods. Incubation time for the primary antibodies was 60 min. Pre-treatment was performed using either citrate for 30 min. (HSP27) or TRS for 30 min. (pHSP27). Slides were counterstained with haematoxylin. Omission of the primary antibody was used as negative control. Substitution of the primary antibody by non-immunogenic immunoglobulin of the same class was used as isotype control.
Evaluation of HSP27 and p53 labelling
The immunohistochemical scoring of pancreatic cancer tissue samples for pHSP27, HSP27 and p53 was performed by two pathologists (GA and TK) in blinded fashion without knowledge of the corresponding clinical data. High interobserver agreement was confirmed using Cohen's Kappa coefficient (κ= 0.91, P < 0.001). In case of interobserver differences, consensus was achieved through simultaneous reassessment of the respective specimen by both pathologists. For evaluation of immunohistochemical staining intensity of cytoplasmic HSP27 and pHSP27 a three-graded system was applied (negative, weakly positive, strongly positive). Samples were defined as positive when at least 5% of the tumour cells displayed HSP27 staining. Similarly, a two-graded system was applied to evaluate nuclear p53 accumulation, with no or minimal staining defined as negative, and moderate to strong staining defined as positive.
KRAS mutation analysis
The sequences of codons 12 and 13 of the KRAS oncogene were analysed using pyrosequencing. Isolated DNA from three punches of TMA tissue served as PCR template [24]. PCR was performed using HotStar DNA-polymerase (Qiagen, Hilden, Germany) and published primer sequences [25]. Subsequently, PCR products were sequenced applying the primer TGTGGTAGTTGGAGCT together with Pyro-Gold reagents (Qiagen) on a Q24 pyrosequencing device (Qiagen) with the injection list GNTGRCGTAGGCAA.
Statistical models
Overall survival in months was defined as the interval from the date of surgery to death or to the most recent contact (for censored events) as of July 1, 2009. Categorical data were compared by chi-square or Fisher's exact tests, continuous data were compared by t-test. Overall median survival times were estimated using the Kaplan–Meier method. The log-rank test was used to test for homogeneity of the survival curves. Univariate and multivariate Cox proportional hazards models were used to assess the effects of variables on overall survival. Multivariate models were built by using backward elimination for variable selection relied on the likelihood ratio test. Follow-up maturity was validated by assessment of follow-up curves for the living patients to ensure comparable follow-up times of survival curves between the respective independent groups [26]. Statistical analyses were performed using Statistical Package for Sciences software (SPSS Inc., Chicago, IL, USA). Two-sided P values of less than 0.05 were considered statistically significant.
Cell lines and culture conditions
Cell lines PL5 and PL11 were kindly provided by S.E. Kern (Johns Hopkins University, Baltimore, MD, USA). All other cell lines used were purchased from the European Collection of Cell Cultures (Sigma-Aldrich, Munich, Germany) or the American Type Culture Collection (LGC Standards, Wesel, Germany), respectively. Primary human pancreatic cancer cells PPC-0039 were derived and propagated in our laboratory from a surgical specimen of pancreatic adenocarcinoma. Cells were grown in DMEM supplemented with 10% foetal calf serum, L-glutamine and penicillin/streptomycin (PAA, Coelbe, Germany).
Generation of PL5 cell clones stably overexpressing different HSP27 protein variants
PL5 cells were transfected with vector pcDNA3.1 (conferring neomycin resistance), which contained the complete coding sequence of either of three different HSP27 constructs under control of the human HSP27 promoter. The HSP27 constructs consisted either of wild-type human HSP27 (hu) or mutants with the serines 15, 78 and 82 substituted to alanines (3A) or aspartic acids (3D). The mutant 3A represents a non-phosphorylatable kinase-dead form of HSP27 while 3D imitates a permanently phosphorylated form through insertion of the three negatively charged residues [27, 28]. For the generation of a control cell line, PL5 cells were transfected with unaltered vector pcDNA3.1 (empty vector, EV). After transfection, the cells were maintained in DMEM containing 0.4 mg/ml G-418 (Roth, Karlsruhe, Germany). After 4 weeks of selection, single G-418-resistant cell clones were seeded and grown in 96-well plates, and consecutively screened by immunoblotting for high expression of HSP27 as compared to parental PL5 cells. The clones with the highest expression of HSP27 hu, 3A and 3D and control cells were labelled correspondingly (PL5/hu16, hu18, hu20, PL5/3A, PL5/3D and PL5/EV) and used for colony formation and cell proliferation assays.
siRNA-mediated depletion of HSP27 protein
AsPc1 cells at 40–60% confluence were transfected using Oligofectamine (Invitrogen) and siRNA directed against HSP27 (sense: GGACGAGCAUGGCUACAUCTT, antisense: GAUGUAGCCAUGCUCGUCCTT; Qiagen) at a final concentration of 100 nM. The transfection proceeded for 4 hrs before adding serum-containing medium. HSP27 protein depletion was quantified by immunoblotting at 48, 72, 96, 120, 144, 168 and 192 hrs after transfection. Gemcitabine was added to the cells at the time point of maximal HSP27 down-regulation (120–144 hrs), which was separately confirmed for each experiment.
Immunoblotting
Cells were lysed and protein extracts boiled and loaded on 10% polyacrylamide gels. After electrophoresis, proteins were transferred to PVDF membranes, which were blocked for 1 hr in TBS-0.1% Tween20/2% milk before the primary antibody was applied overnight at 4°C (1:1000; anti-HSP27 SPA-800 or SPA-803 or anti-phospho-Ser78 HSP27 SPA-523, all Stressgen/Enzo, Lörrach, Germany). Anti-#223;-ACTIN antibody (1:10000, AC-15; Sigma-Aldrich) served as loading control. The membranes were washed and stained with either anti-mouse or anti-rabbit HRP-conjugated antibody (1:10.000; GE Healthcare, Freiburg, Germany). Enhanced chemo-luminescence was elicited using SuperSignal West Pico chemo-luminescence substrate (Thermo Scientific, Schwerte, Germany) according to the manufacturer's instructions.
Cell proliferation assays
All assays were performed over a broad range of concentrations covering 100% to 0% survival. A total of 1000–1500 cells/well were plated in 96-well plates, allowed to adhere, and treated. Following incubation for 6 days, the cells were washed, lysed in 100 μl H2O, and 0.5% Picogreen (Molecular Probes, Invitrogen, Karlsruhe, Germany) was added. Fluorescence was measured (Cytofluor Series 4000, Applied Biosystems, Darmstadt, Germany) and growth inhibition calculated as compared to the untreated control samples. At least three independent experiments were performed per agent, with each data point reflecting triplicate wells. Error bars represent standard error of the mean (SEM) from three experiments. Prior to each set of experiments, HSP27 expression was quantitated in all utilized cell lines by immunoblotting.
Colony formation assays
Cells were plated in 6-well plates, allowed to adhere, and exposed to ionizing cesium-137 γ-radiation (IR). Each sample consisted of two different cell concentrations, each performed in duplicate (amount of cells plated was 20 and 100 cells at 0 Gy, 100 and 300 cells at 2 Gy, 2000 and 5000 cells at 4 Gy, 5000 and 15,000 cells at 6 Gy, 50,000 and 100,000 cells at 8 Gy). Cells were subsequently incubated for 12 days, fixed, and stained (PBS/10% formalin, 1:500 crystal violet). All macroscopically visible colonies were counted. Three to four experiments were performed per sample.
Heat shock in pancreatic cancer cells
Cells were plated in flasks, allowed to adhere and propagated until ∼80% confluent. Consecutively, heat shock was carried out for 1 hr at 39°C or 41°C using a water bath, followed by an incubation step for 3 hrs at 37°C. Cells incubated at identical conditions without heat shock were used as controls.
Results
Immunohistochemical TMA analysis of HSP27 in 86 pancreatic ductal adenocarcinoma and corresponding normal tissues
Expression of HSP27 and phosphorylated HSP27 (pHSP27) was assessed immunohistochemically in tissue specimens from 86 pancreatic cancer patients. HSP27 and pHSP27 staining was predominantly observed in the cytoplasm of malignant and normal (ductal epithelium and acinar) cells, while membranous staining was observed only sporadically. Nuclear staining was not detected. Cytoplasmic HSP27 and pHSP27 expression was defined as positive when at least 5% of cells displayed HSP27 staining and consecutively classified into three categories on the basis of staining intensity, i.e. negative, weakly positive or strongly positive, respectively (Fig. 1). Of the analysed 86 pancreatic ductal adenocarcinomas, approximately half (49%) were HSP27-positive, whereas 51% were HSP27-negative. Of the corresponding 86 normal pancreatic tissues, 71% were HSP27-positive, whereas 29% were HSP27-negative. Regarding pHSP27, approximately half of the pancreatic cancer tissues (48%) were pHSP27-positive, whereas 52% were pHSP27-negative. Of the corresponding 86 normal pancreatic tissues, 66% were pHSP27 positive, whereas 34% were pHSP27-negative. A strong correlation was observed between HSP27 and pHSP27 expression both in normal (P≤ 0.001) and tumour tissue (P= 0.001) (Table 1). Furthermore, significantly less tumour samples than normal tissue samples stained positive for HSP27 or pHSP27, respectively (McNemar's chi-square test for HSP27 P= 0.003, for pHSP27 P= 0.009).
Fig 1.
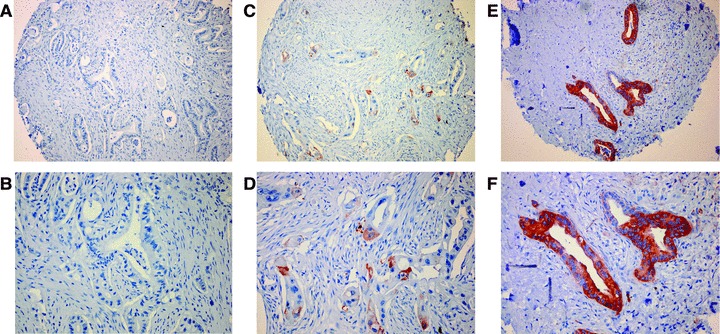
Immunohistochemical staining of HSP27 in pancreatic cancer. Representative microscopic pictures of pancreatic ductal adenocarcinomas considered negative (A and B), weakly positive (C and D) or strongly positive (E and F).
Table 1.
Summary of HSP27 staining and pHSP27 staining
| No. | Negative | Weak | Strong | Negative/positive (%) | |
|---|---|---|---|---|---|
| HSP27 | |||||
| Tumour tissue | 86 | 44 | 20 | 22 | 51/49 |
| Normal tissue | 86 | 25 | 36 | 25 | 29/71 |
| pHSP27 | |||||
| Tumour tissue | 86 | 45 | 21 | 20 | 52/48 |
| Normal tissue | 86 | 29 | 31 | 26 | 34/66 |
Correlations between HSP27 expression status and clinicopathologic features
The clinicopathologic characteristics assessed in our study population are listed (Table 2). No associations were observed between HSP27 expression status in tumours and either age, gender, tumour size, tumour differentiation, TNM classification or margin status, respectively. In contrast, the 2-year survival rate was significantly higher for patients with HSP27-positive tumours (52%) than for patients with HSP27-negative tumours (27%) (P= 0.017). In addition, a strong inverse correlation was observed between HSP27 expression and nuclear p53 accumulation in tumours (P < 0.001), while there was only a weak correlation between KRAS mutation status and HSP27 expression (P= 0.030). Regarding pHSP27 expression in tumours, no significant associations were observed for any of the above parameters (data not shown).
Table 2.
Association between HSP27 expression and clinicopathologic features
| Feature | No./Avg. | HSP27 status | P | |
|---|---|---|---|---|
| No expression (n= 44) | Expression (n= 42) | |||
| Mean age at the time of surgery (year) | 65.7 ± 9.3 | 65.1 ± 9.9 (32.2–81.9) | 66.4 ± 8.7 (44.9–78.9) | 0.667 |
| Gender | ||||
| Female | 44 | 26 | 18 | 0.132 |
| Male | 42 | 18 | 24 | |
| Tumour differentiation | ||||
| Well and moderate | 28 | 14 | 14 | 0.881 |
| Poor | 58 | 30 | 28 | |
| Tumour pathologic stage | ||||
| T1 | 2 | 1 | 1 | 0.892 |
| T2 | 9 | 4 | 5 | |
| T3 | 72 | 38 | 34 | |
| T4 | 3 | 1 | 2 | |
| Mean tumour size (cm) | 3.66 ± 1.27 | 3.56 ± 1.33 (0.9–8.0) | 3.76 ± 1.21 (1.5–7.0) | 0.483 |
| Lymph node status | ||||
| N0 | 37 | 17 | 20 | 0.400 |
| N1 | 49 | 27 | 22 | |
| Mean n of positive nodes, N0/N1 | 1.79 ± 2.90 | 1.80 ± 2.46 (0–19) | 1.79 ± 3.34 (0–12) | 0.988 |
| Metastasis status | ||||
| M0 | 75 | 37 | 38 | 0.522 |
| M1 | 11 | 7 | 4 | |
| Margin status | ||||
| Negative | 44 | 22 | 22 | |
| Positive | 42 | 22 | 20 | 0.825 |
| Postoperative radiotherapy | ||||
| No | 35 | 19 | 16 | |
| Yes | 51 | 25 | 26 | 0.631 |
| Postoperative chemotherapy | ||||
| No | 13 | 8 | 5 | |
| Yes | 73 | 36 | 37 | 0.417 |
| Nuclear p53 accumulation* | ||||
| Negative | 52 | 20 | 32 | |
| Positive | 30 | 24 | 6 | <0.001 |
| KRAS status* | ||||
| Wild-type | 36 | 14 | 22 | |
| Mutant | 46 | 29 | 17 | 0.030 |
| 2-year survival rate (%)† | 40 | 27.3 | 52.4 | 0.017 |
4/86 samples were not informative in regard to p53 or KRAS status, respectively (results from n= 82 are shown). †The shortest follow-up was 24.5 months for patients still alive as of July 1, 2009.
Univariate analyses of overall survival for HSP27 expression status and other clinicopathologic features
Postoperative overall survival was correlated to HSP27 expression in tumours as well as clinicopathologic parameters in univariate analyses applying the Cox proportional hazards model and Kaplan–Meier survival curves. Follow-up maturity was calculated for all assessed groups and no significant differences were observed. Data were censored for 19 patients that were still alive at the latest follow-up as of July 1, 2009. In the univariate Cox models, we found no significant correlation between survival and either gender, age, tumour differentiation, margin status or nuclear p53 accumulation. In contrast, a significant correlation was observed between survival and HSP27 expression in tumours [negative versus positive staining hazard ratio (HR) 0.51, P= 0.006], as well as between survival and KRAS mutation status (wild-type versus mutant HR 1.78, P= 0.026), lymph node status (N0 versus N1 HR 1.87, P= 0.014), metastasis status (M0 versus M1 HR 2.65, P= 0.006) and tumour size (median, <3.5 cm versus≥3.5cm HR 2.11, P= 0.004), respectively. Subgroup analyses showed that patients with tumours having strong HSP27 staining had a significant better outcome than those with HSP27-negative tumours (HR 0.48, P= 0.018), whereas data comparing patients with tumours having weak HSP27 staining versus those with no staining reached borderline statistical significance (HR 0.53, P= 0.050) (Table 3). Kaplan–Meier curves of survival are shown for absolute (positive versus negative) HSP27 expression (Fig. 2A) as well as for relative (staining intensity) HSP27 expression (Fig. 2B). Survival was also correlated to pHSP27 expression in tumours, but in contrast to HSP27, no significant associations were observed (data not shown).
Table 3.
Univariate analyses of survival for HSP27 expression and clinicopathologic features
| Variables | No. | Survival* | HR (95% CI) | P |
|---|---|---|---|---|
| HSP27 expression | ||||
| Negative | 44 | 13.5 | 1.00 | |
| Positive | 42 | 25.9 | 0.51 (0.31–0.83) | 0.006 |
| HSP27 staining intensity | ||||
| Negative | 44 | 13.5 | 1.00 | |
| Weak | 20 | 20.0 | 0.53 (0.28–1.00) | 0.050 |
| Strong | 22 | 28.4 | 0.48 (0.26–0.88) | 0.018 |
| Median age (year) | ||||
| ≤65 | 40 | 19.3 | 1.00 | |
| >65 | 46 | 16.6 | 1.01 (0.62–1.64) | 0.975 |
| Gender | ||||
| Female | 44 | 13.1 | 1.00 | |
| Male | 42 | 19.6 | 0.72 (0.44−1.17) | 0.183 |
| Tumour differentiation | ||||
| Well to moderate | 28 | 28.7 | 1.00 | |
| Poor | 58 | 14.9 | 1.29 (0.77−2.17) | 0.339 |
| Median tumour size (cm) | ||||
| <3.5 | 34 | 28.4 | 1.00 | |
| ≥3.5 | 52 | 13.1 | 2.11 (1.26−3.52) | 0.004 |
| Lymph node status | ||||
| N0 | 37 | 28.6 | 1.00 | |
| N1 | 49 | 13.4 | 1.87 (1.13−3.07) | 0.014 |
| Metastasis status | ||||
| M0 | 75 | 19.9 | 1.00 | |
| M1 | 11 | 10.2 | 2.65 (1.32−5.33) | 0.006 |
| Margin status | ||||
| Negative | 44 | 19.3 | 1.00 | |
| Positive | 42 | 16.1 | 1.19 (0.73−1.92) | 0.486 |
| Nuclear p53 accumulation† | ||||
| Negative | 52 | 17.1 | 1.00 | |
| Positive | 30 | 16.1 | 1.16 (0.69−1.93) | 0.562 |
| KRAS status† | ||||
| Wild-type | 36 | 25.9 | 1.00 | |
| Mutant | 46 | 13.6 | 1.78 (1.07−2.96) | 0.026 |
HR: hazard ratio; CI: confidence interval. *Median survival time in months from date of surgery to death or to most recent contact. †4/86 samples were not informative in regard to p53 or KRAS status, respectively (results from n= 82 are shown).
Fig 2.
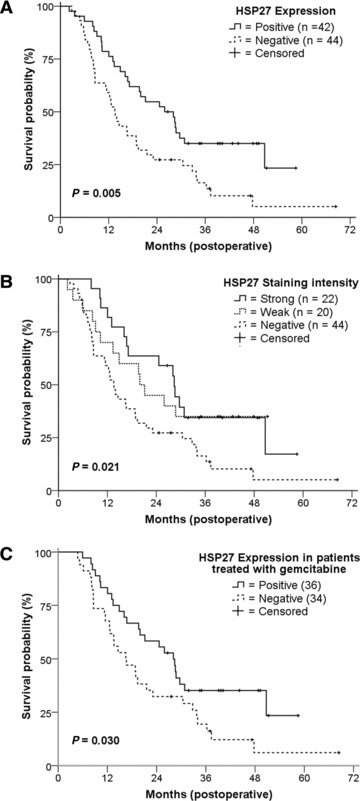
Kaplan–Meier plots of overall survival for HSP27 expression of all patients and of the gemcitabine-treated patient subpopulation. (A) Patients whose tumours showed expression of HSP27 had a longer median overall survival than those whose tumour did not (P= 0.005, median survival 13.5 versus 25.9 months). (B) Patients whose tumours showed weak or strong HSP27 staining intensity had a longer median overall survival than patients whose tumours showed no expression (P= 0.021, median survival 13.5 versus 20.0 versus 28.4 months). (C) Gemcitabine-treated patients whose tumours showed expression of HSP27 had a longer median overall survival than those whose tumours did not (P= 0.030, median survival 16.4 versus 28.1 months).
Multivariate analyses of overall survival for HSP27 expression status and selected clinicopathologic features
In order to establish whether HSP27 expression was an independent prognostic factor for postoperative overall survival, we performed multivariate analyses using the Cox proportional hazards model adjusting HSP27 expression status with age, gender, differentiation, size, number of positive nodes and margin status (Table 4). In these analyses, HSP27 expression (both absolute and relative) emerged as an independent marker for survival. Of note, HSP27 expression remained an independent survival marker after applying backward elimination (negative versus positive staining HR 0.47, P= 0.002, weak versus no staining HR 0.53, P= 0.046, strong versus no staining HR 0.42, P= 0.006). In addition to HSP27 expression, tumour size and number of positive lymph nodes were prognostic factors for survival independent of patient's age, gender, tumour differentiation and margin status. In contrast, KRAS mutation status correlated with survival only in the univariate but not in the multivariate analyses (wild-type versus mutant HR = 1.45, P= 0.230).
Table 4.
Multivariate analyses of survival for HSP27 expression and staining intensity
| Variables | No. | HR (95% CI) | P |
|---|---|---|---|
| HSP27 expression | |||
| Adjusted for all | |||
| HSP27 expression | |||
| Negative | 44 | 1.00 | |
| Positive | 42 | 0.49 (0.28−0.84) | 0.010 |
| Age (year) | |||
| Continuous | 86 | 1.01 (0.98−1.04) | 0.454 |
| Gender | |||
| Female | 44 | 1.00 | |
| Male | 42 | 0.94 (0.54−1.62) | 0.816 |
| Tumour differentiation | |||
| Well to moderate | 28 | 1.00 | |
| Poor | 58 | 1.29 (0.75−2.24) | 0.362 |
| Tumour size [cm] | |||
| Continuous | 86 | 1.61 (1.30−1.99) | <0.001 |
| Number of positive nodes | |||
| Continuous | 86 | 1.13 (1.02−1.25) | 0.016 |
| Margin status | |||
| Negative | 44 | 1.00 | |
| Positive | 42 | 0.87 (0.52−1.44) | 0.867 |
| Backward elimination | |||
| HSP27 expression | |||
| Negative | 44 | 1.00 | |
| Positive | 42 | 0.47 (0.29−0.77) | 0.002 |
| Tumour size (cm) | |||
| Continuous | 86 | 1.57 (1.29−1.91) | <0.001 |
| Number of positive nodes | |||
| Continuous | 86 | 1.13 (1.02−1.24) | 0.013 |
| HSP27 staining intensity* | |||
| Adjusted for all | |||
| HSP27 staining intensity | |||
| Negative | 44 | 1.00 | |
| Weak | 20 | 0.56 (0.28−1.09) | 0.088 |
| Strong | 22 | 0.44 (0.23−0.84) | 0.013 |
| Tumour size (cm) | |||
| Continuous | 86 | 1.62 (1.31−2.01) | <0.001 |
| Number of positive nodes | |||
| Continuous | 86 | 1.13 (1.02−1.25) | 0.020 |
| Backward elimination | |||
| HSP27 staining intensity | |||
| Negative | 44 | 1.00 | |
| Weak | 20 | 0.53 (0.28−0.99) | 0.046 |
| Strong | 22 | 0.42 (0.23−0.78) | 0.006 |
| Tumour size (cm) | |||
| Continuous | 86 | 1.58 (1.29−1.92) | <0.001 |
| Number of positive nodes | |||
| Continuous | 86 | 1.12 (1.02−1.24) | 0.017 |
Only variables that were significant in multivariate analyses are shown.
Overall survival according to HSP27 expression in patients treated with gemcitabine
In our study, the majority of patients (70 of 86, 81%) received adjuvant therapy including gemcitabine, either as monotherapy (n= 17) or in combination with radiotherapy (n= 13) and/or other agents (n= 40), including 5-fluorouracil, oxaliplatin and cisplatin. In this gemcitabine-treated subpopulation, patients with HSP27 expression had a significant longer survival as compared to those without HSP27 expression in both univariate (negative versus positive staining HR 0.55, P= 0.033) and multivariate analyses using backward elimination (negative versus positive staining HR 0.46, P= 0.006) (Table 5 and Fig. 2C). Consistent with our analyses of the total patient population, HSP27 expression in patients of the gemcitabine-treated subgroup was not associated with other clinicopathologic features (data not shown). Vice versa, HSP27 expression had no significant influence on survival in the complementary subgroup of 16 patients not having received gemcitabine (negative versus positive staining HR = 1.91, P= 0.297).
Table 5.
Uni-/multivariate analyses for HSP27 expression in gemcitabine-treated patients
| Variables | No. | HR (95% CI) | P |
|---|---|---|---|
| Univariate analyses* | |||
| HSP27 expression | |||
| Negative | 34 | 1.00 | |
| Positive | 36 | 0.55 (0.32–0.95) | 0.033 |
| Multivariate analyses* | |||
| Adjusted for all† | |||
| HSP27 expression | |||
| Negative | 34 | 1.00 | |
| Positive | 36 | 0.49 (0.27–0.90) | 0.021 |
| Backward elimination | |||
| HSP27 expression | |||
| Negative | 34 | 1.00 | |
| Positive | 36 | 0.46 (0.27–0.79) | 0.006 |
Only data for HSP27 are shown. †Adjusted for age, gender, tumour differentiation, tumour size, number of positive nodes and margin status.
HSP27 expression patterns in pancreatic cancer cell lines
HSP27 expression patterns were assessed in ten established pancreatic cancer cell lines using immunoblotting and immunofluorescence. HSP27 was constitutively expressed at varying degrees in all cell lines, with six lines displaying a strong expression (AsPC-1, BxPC3, MIA Paca-2, Capan-1, Capan-2, PANC-1) and four lines displaying a comparatively weaker expression (CFPAC-1, PL11, PL5 and Su.86.86). The phosphorylated form of HSP27 (pHSP27) was constitutively expressed only in Capan-1 and Capan-2 cells, while all other cell lines did not show constitutive HSP27 phosphorylation (Fig. 3A). As expected, HSP27 expression was restricted mainly to the cytoplasm in all tested cell lines (Fig. 3B).
Fig 3.
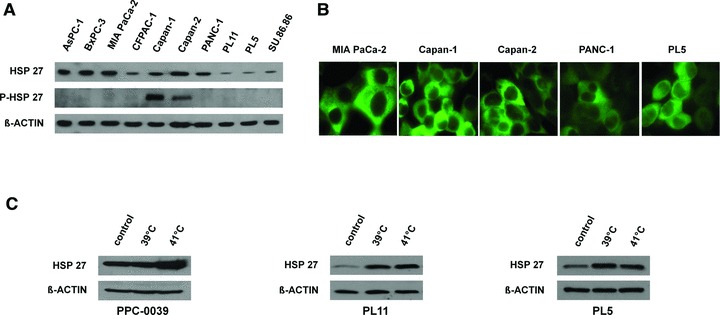
HSP27 expression patterns and heat-shock inducible HSP27 expression in pancreatic cancer cells. (A) Immunoblotting to assess constitutive HSP27 and phospho-HSP27 expression in 10 pancreatic cancer cell lines. β-ACTIN served as loading control. (B) Immunofluorescence to assess spatial HSP27 expression patterns in 5 pancreatic cancer cell lines. (C) Immunoblotting to assess inducible HSP27 expression upon heat shock (39°C and 41°C) in two established pancreatic cancer cell lines (PL5, PL11) and patient-derived short-term passaged primary pancreatic cancer cells (PPC-0039). β-ACTIN served as loading control.
Heat shock-inducible HSP27 expression in pancreatic cancer cell lines and primary cells
We next tested whether HSP27 was inducible upon heat shock in pancreatic cancer cells. Therefore, two cell lines displaying low constitutive HSP27 expression (PL5, PL11) as well as early-passage, patient-derived primary pancreatic cancer cells (PPC-0039) were assessed. Heat shock-inducible expression of HSP27 was demonstrated at varying degrees in all samples (Fig. 3C).
Influence of HSP27 protein overexpression on chemosensitivity in pancreatic cancer cells
To investigate potential influences of HSP27 or pHSP27 expression on chemo- and radiosensitivity in pancreatic cancer cells, we first established an isogenic (p-)HSP27-overexpression model, using a pancreatic cancer cell line displaying low constitutive HSP27 expression: PL5 cells were stably transfected with either the complete coding sequence of unmodified human HSP27 (hu), or a constitutive pseudo-phosphorylated variant with all three serine residues (15, 78, 82) changed to aspartic acid, or a non- phosphorylatable kinase-dead variant with all serine residues changed to alanine. Single-cell diluted PL5 clones stably overexpressing these HSP27 isoforms upon transfection were identified by immunoblotting (Fig. 4A) and subsequently used for sensitivity studies. Stably HSP27-transfected PL5 clones did not significantly differ from parental control cells in survival upon treatment with gamma-irradiation (Fig. 4B). Likewise, no relevant proliferation differences were observed upon treatment with cisplatin, paclitaxel or 5-fluorouracil (5-FU). In contrast, HSP27-overexpressing PL5 clones were significantly more sensitive to gemcitabine than parental control cells (Fig. 4C). Increased gemcitabine sensitivity was observed in all HSP27-overexpressing PL5 clones (hu16, 3A, 3D), indicating that this effect was attributable to the total amount of HSP27 protein, but independent of HSP27 phosphorylation status. Artefacts due to clonal variability or variability among the non-clonal parental cell population were excluded by using an empty vector-transfected PL5 cell clone (PL5/ev), which displayed gemcitabine sensitivity comparable to that of parental cells and by using multiple HSP27-overexpressing cell clones, which all displayed increased gemcitabine sensitivity (Fig. 4D and data not shown). In addition, HSP27-transfected PL5 cell clones, which in the absence of continuous neomycin selection lost HSP27 overexpression during passaging (PL5/3D-lost), also lost the increased sensitivity towards gemcitabine (Fig. 4E), further supporting that HSP27 expression but not clonal variability was responsible for the increased gemcitabine sensitivity in the utilized clones.
Fig 4.
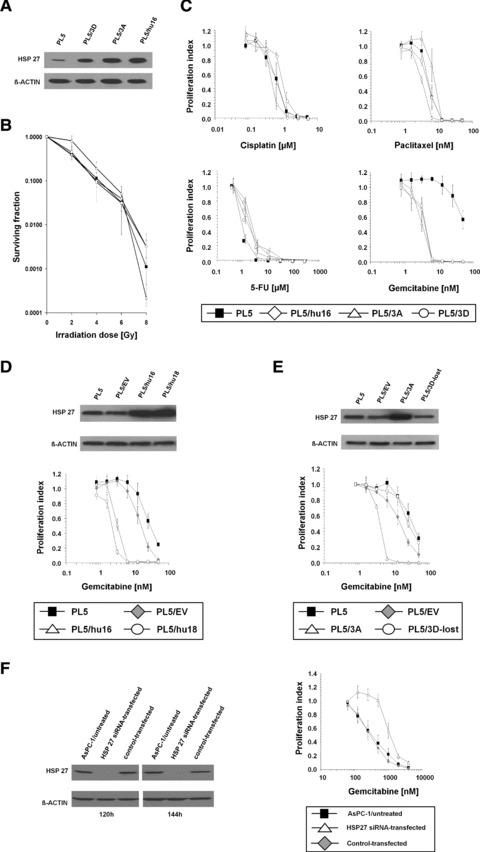
Impact of HSP27 expression levels on radio- and chemosensitivity in pancreatic cancer cells. (A) Immunoblotting to confirm exogenous HSP27 overexpression in selected PL5 cell clones upon stable transfection with three different HSP27 constructs, i.e. wild-type human HSP27 (labelled PL5/hu16), or a HSP27 mutant form having three serine residues changed to alanine (labelled PL5/3A), or a HSP27 mutant form having three serine residues changed to aspartic acid (labelled PL5/3D) prior to the set of experiments shown in (B) and (C). β-ACTIN served as loading control. (B) Colony formation assays to assess radiosensitivity of parental PL5 cells as compared to the HSP27-overexpressing cell clones PL5/hu16, PL5/3A, PL5/3D upon treatment with γ-irradiation (0–8 Gy). Error bars represent SEM of three independent experiments. (C) Cell proliferation assays to assess chemosensitivity of parental PL5 cells as compared to the HSP27-overexpressing cell clones PL5/hu16, PL5/3A, PL5/3D upon treatment with the indicated chemotherapeutic agents. Error bars represent SEM of at least three independent experiments. (D) Gemcitabine sensitivity of parental (PL5) and empty vector-transfected (PL5/ev) control PL5 cells as compared to two different HSP27-overexpressing PL5/hu cell clones (hu16, hu18) to exclude artefacts due to clonal variability. Error bars represent SEM of three independent experiments. Corresponding HSP27 expression of the utilized cell clones as assessed by immunoblotting directly prior to the set of experiments is provided. (E) Gemcitabine sensitivity of parental and empty-vector transfected control PL5 cells as compared to HSP27 overexpressing PL5/3A cells and PL5/3D cells that lost HSP27 overexpression during cell culture in the absence of continuous selection (PL5/3D-lost). Corresponding HSP27 expression as assessed by immunoblotting directly prior to the set of experiments is provided. (F) Gemcitabine sensitivity of untreated and control-transfected AsPC-1 cells as compared to HSP27-siRNA-transfected AsPC-1 cells. Error bars represent SEM of four independent experiments. HSP27 expression upon siRNA treatment from one representative experiment is provided.
Influence of HSP27 protein depletion on chemosensitivity in pancreatic cancer cells
We additionally used RNA-interference in a pancreatic cancer cell line exhibiting constitutively high HSP27 expression to validate the experiments applying HSP27 overexpression and to exclude cell line-specific or methodology-dependent artefacts. Gemcitabine sensitivity was assessed upon siRNA-mediated HSP27 down-regulation in AsPC-1 cells. HSP27 protein depletion efficiency was validated by immunoblotting 48–192 hrs after siRNA transfection and the time points of maximal HSP27 down-regulation were used for gemcitabine treatment. A significantly decreased sensitivity of HSP27 siRNA-treated AsPC-1 cells, expressing virtually no detectable HSP27 protein at 120–144 hrs after siRNA transfection as compared to HSP27-expressing untreated or control-transfected cells, was observed upon treatment with gemcitabine at 120–144 hrs (Fig. 4F).
Discussion
In this study, we correlated HSP27 expression status with a variety of clinicopathologic parameters in a large number of surgical pancreatic cancer specimens in an effort to determine the significance of HSP27 as a diagnostic or prognostic biomarker in pancreatic cancer. Complementary, we evaluated the impact of HSP27 on radio- and chemotherapy in HSP27-overexpression and RNA-interference models to determine its significance as a predictive marker for therapeutic response.
Only about half of the pancreatic cancer specimens expressed detectable amounts of HSP27 in our study. A general overexpression of HSP27 in pancreatic cancer, which has previously been indicated in a small series of nine tissue samples [18], could thus not be confirmed in our study [18]. On the contrary, HSP27 expression was even more prevalent in normal cells (ductal epithelium and acinar cells) than in malignant cells. Consequently, our data suggest that HSP27 detection cannot serve as an immunohistochemical diagnostic tumour marker in pancreatic cancer, a conclusion, which also applies to most other tumour entities [13].
When correlating HSP27 expression status with clinicopathologic parameters, we found no significant association of HSP27 expression status with age, gender, tumour size, tumour differentiation, TNM classification or margin status, respectively. In contrast, patients whose tumours exhibited HSP27 expression demonstrated a significantly longer postoperative overall survival than patients with HSP27 non-expressing tumours. More specifically, survival correlated with the staining intensity for HSP27, with patients, whose tumours exhibited strong staining having the longest median survival. After adjusting for age, gender, tumour differentiation, tumour size, number of positive nodes and margin status in subsequent multivariate analyses and consecutive stepwise backward elimination, HSP27 expression emerged as an independent prognostic factor for overall survival in pancreatic cancer. Other parameters affecting survival to a similar extent in regard to comparable hazard ratios were expectedly lymph node status, metastasis status and tumour size, whereas gender, age, tumour differentiation and margin status had no discernible influence. Tumour size was the most significant clinicopathologic factor deductible from our univariate survival analyses. Taken together, these data well reflect many of the typical clinical features of pancreatic adenocarcinoma and illustrate the representativeness of our TMA [29].
Since HSP27 expression levels might be related to the genetic changes that occur during pancreatic carcinogenesis [13] and since KRAS as well as TP53 are both frequently genetically mutated in pancreatic cancer [30-32], we additionally analysed KRAS mutation status and p53 expression in our cohort. Immunohistochemical accumulation of nuclear p53 expression was chosen as a surrogate marker for TP53 mutations due to its feasibility on TMA tissues at the expense of a slightly decreased sensitivity (30–50%versus 50–75% detection rate) [33]. While only a weak association was found between KRAS mutation status and HSP27 expression (P= 0.030), a strong inverse correlation was observed between nuclear p53 accumulation and HSP27 expression (P < 0.001). This phenomenon might be attributable to several molecular mechanisms that have previously been described for HSP27 and p53 in other tumour entities. On the one hand, TP53 mutation status might have a direct influence on inducible HSP27 expression. Consistent with this hypothesis and our results, it has been shown that exogenous expression of only wild-type but not mutant p53 induced HSP27 expression in prostate cancer cells [34]. Likewise, it has been reported that several HSP promoters, including the HSP27 promoter, include consensus sequence motifs for p53 binding [35, 36] and that the induction of other small HSPs is regulated by p53-dependent mechanisms [35]. On the other hand, HSP27 expression could vice versa modulate p53 expression in pancreatic cancer cells. This hypothesis is corroborated by studies in breast and colon cancer cells, which show that HSP27 accelerated proteasomal degradation of p53 and consistently, that HSP27 overexpression caused p53 depletion, while HSP27 down-regulation stabilized p53, even in the absence of genotoxic stress [36, 37]. However, it should be noted that HSP27 overexpression appears not to correlate with p53 status in several other tumour types (comprehensively reviewed in Ref. [13]), and the strong correlation between HSP27 expression and nuclear p53 accumulation in pancreatic cancer cells observed in our study might therefore constitute a tissue-specific phenomenon. Regarding potential correlations between survival and KRAS or p53 status, respectively, in our cohort, an association was found only for KRAS but not for p53. This is consistent with previous studies on the roles of KRAS and p53 as prognostic markers in pancreatic cancer [38] and importantly, excludes p53 status as a confounding variable for the observed correlation between HSP27 expression and survival in our study. Likewise, only HSP27 expression but not KRAS status was confirmed as an independent marker for survival in the multivariate analyses.
Expression levels of HSP27 appear to be elevated in a wide spectrum of human cancers, and accordingly, HSP27 expression and correlation studies have been performed in a plethora of cancer entities other than pancreatic cancer [13], in summary suggesting that HSP27 could on the one hand represent a prognostic biomarker in specific cancer types and on the other hand even predict the individual patient's response to certain chemotherapeutics, while its applicability as a diagnostic marker appears to be rather limited.
Regarding the role of HSP27 as a prognostic marker, different conclusions were reached depending on the tumour type under investigation. For example, HSP27 expression has been associated with poor prognosis in osteosarcoma [39], hepatocellular carcinoma [40] and prostate cancer [41, 42], whereas HSP27 expression has been associated with good prognosis in endometrial adenocarcinoma [43], oesophageal squamous cell carcinoma [44] and malignant fibrous histiocytoma [45]. In contrast, HSP27 expression has been reported to have no effect on prognosis in head and neck squamous cell carcinoma [46], bladder cancer [47] or renal cell carcinoma [48]. Data are inconclusive in oral squamous cell carcinoma [49-52], gastric cancer [53-55] and ovarian cancer [56-61]. Studies in pancreatic cancer were still lacking. Our study now suggests that HSP27 expression strongly correlates with good prognosis in pancreatic cancer, both complementing and adding a new aspect to the prior literature. Taken together, HSP27 expression might serve as a useful prognostic marker in certain but not all cancer types. Furthermore, the implications of HSP27 expression on prognosis appear to be highly variable depending on the tumour entity under investigation, suggesting that the influence of HSP27 expression could be highly dependent on the unique molecular context of each tumour type [13], e.g. tumour milieu, genetic and epigenetic changes, mutation signature, and protein expression profiles (e.g. receptor status).
In addition to its role as a prognostic marker, HSP27 may also predict the response towards radio- and/or chemotherapy in certain tumours of individual patients [7, 13], including bladder cancer [62], breast cancer [63, 64], oesophageal cancer [65, 66], ovarian cancer [56, 59] and prostate cancer [67]. However, studies exist that report no predictive value of HSP27 in other as well as in some of the above mentioned tumour types [45, 68-73]. Studies investigating the role of HSP27 as predictive marker for chemosensitivity in large numbers of pancreatic cancers are lacking. However, indirect evidence for an influence of HSP27 on chemosensitivity, especially towards treatment with gemcitabine, came from three recent studies: Proteomic analyses found increased HSP27 expression and HSP27 phosphorylation in the gemcitabine-resistant pancreatic cancer cell line KLM1-R when compared to gemcitabine-sensitive KLM1 control cells [20, 22]. Additionally, interferon-γ induced HSP27 down-regulation and concomitantly increased cytotoxicity upon treatment with gemcitabine [21]. In our TMA study, 81% of the patients (70 of 86) received adjuvant therapy including gemcitabine, either alone or in combination with radiotherapy and/or other chemotherapeutic agents, prompting us to investigate whether this particular subgroup of gemcitabine-treated patients also had a significantly different outcome dependent on HSP27 expression. We found that HSP27 remained an independent prognostic marker in this subpopulation, indicating that the better survival of patients with HSP27-positive tumours might partly be attributable to a better response towards gemcitabine. Vice versa, HSP27 expression would be expected not to influence survival in patients not having received gemcitabine. Even though this was confirmed in our study, this specific subgroup consisting of 16 patients might have been too small to allow significant conclusions. Therefore, future studies are required specifically investigating HSP27 as prognostic marker in a large cohort of patients stratified for adjuvant treatment with gemcitabine.
To further clarify the discrepancy between our hypothesis, suggesting increased gemcitabine sensitivity in HSP27-expressing pancreatic cancers, and the above mentioned studies, suggesting increased gemcitabine resistance in HSP27-expressing pancreatic cancers, we additionally validated our hypothesis in vitro applying HSP27 overexpression and RNA-interference models in pancreatic cancer cell lines. Consistent with our hypothesis, HSP27 low-expressing PL5 pancreatic cancer cells, engineered to stably overexpress exogenous HSP27, were distinctly more sensitive towards treatment with gemcitabine than control PL5 cells, but not towards other common chemotherapeutics or radiation. Of note, gemcitabine sensitivity was increased independent of HSP27 phosphorylation status, excluding HSP27 phosphorylation as a major mechanism for HSP27-dependent modulation of gemcitabine sensitivity. Potential artefacts of the overexpression experiments due to clonal variability were excluded using multiple PL5 cell clones, including clones that lost HSP27 overexpression during passaging in the absence of continuous antibiotic selection. Vice versa, HSP27 high-expressing AsPC-1 pancreatic cancer cells exhibited a significantly increased resistance towards gemcitabine upon siRNA-mediated HSP27 protein depletion, additionally substantiating our results from the PL5 overexpression model and further excluding cell line-specific phenomena or artefacts due to clonal variability. Nevertheless, future studies, expanding these models to a large panel of pancreatic cell lines, are desirable to validate a further generalizability of our data.
Our hypothesis of increased gemcitabine sensitivity in HSP27 overexpressing pancreatic cancers has already been indirectly anticipated by preclinical and clinical studies. A recent in vitro study using the pancreatic cancer cell lines AsPC-1 and MIAPaCa-2 demonstrated increased cytotoxicity when gemcitabine was combined with heat treatment [74]. As a potential mechanism, the levels of heat-inducible HSP70 were shown to be increased in these lines upon heat treatment. We now demonstrate that heat treatment increases also HSP27 expression in pancreatic cancer cell lines as well as short-term cultivated primary pancreatic cancer cells, indicating HSP27 as a potential contributing factor for heat shock-induced gemcitabine sensitivity. Similar evidence came from two recent clinical studies. Ohguri et al. demonstrated that progression-free and overall survival was significantly better in patients with locally advanced pancreatic cancer when regional hyperthermia was added to radiochemotherapy with gemcitabine [75]. Furthermore, Tschoep et al. showed that regional hyperthermia combined with gemcitabine and cisplatin as a second line treatment exhibited significant antitumour activity even in patients with gemcitabine-refractory pancreatic cancer [76].
Given the similar function and regulation of many heat shock proteins in cancer cells, it has to be considered whether the observed association between HSP27 and survival could partly be attributable to similar expression patterns of HSP27 and other HSPs in our TMA study. This might be especially true for HSP70, as HSP70 and HSP27 share high similarities in regard to their function and regulation [8], and both are elevated in a wide spectrum of human cancers [77]. However, despite an association between HSP27 and HSP70 expression in some tumours [69, 78], overexpression of HSP70 appears to occur more uniformly than overexpression of HSP27 in pancreatic cancer, with robust overexpression of HSP70 having been demonstrated on both the RNA and protein level in the vast majority of pancreatic cancer specimens as well as pancreatic cancer cell lines [79-81]. Analogous to the partly conflicting literature on the predictive ability and therapeutic implications of HSP27 expression in pancreatic cancer, the role of HSP70 also remains controversial in this tumour type. On the one hand, as has similarly been demonstrated for HSP27 in our study, expression of HSP70 in pancreatic cancer was reported to represent an independent prognostic factor for better survival in a small study assessing a cohort of 36 patients [81] and hyperthermia along with concomitant HSP70 induction has been shown to enhance the cytotoxicity of gemcitabine in pancreatic cancer cell lines [74]. On the other hand however, there exists considerable experimental-mechanistic evidence that HSP70 plays in fact a rather deleterious role in pancreatic cancer. In accordance with its tumour-specific overexpression mediating resistance to apoptotic cell death [82], inhibition of HSP70 expression through pharmacological approaches or RNA-interference has been demonstrated to cause caspase-dependent apoptotic cell death in pancreatic cancer cells in vitro and in vivo[79, 83] mediated by attenuation of cytosolic calcium and lysosome stabilization [84]. Taken together, despite HSP70 overexpression correlating with a better prognosis in pancreatic cancer patients [81], inhibition of HSP70 might paradoxically represent a highly promising therapeutic strategy in these tumours [82].
In conclusion, HSP27 expression significantly correlated with better patient survival in pancreatic cancer in our TMA study, suggesting that HSP27 could serve as a prognostic marker in this tumour type. Furthermore, exogenous overexpression of HSP27 in pancreatic cancer cells conferred increased sensitivity to gemcitabine, whereas depletion of HSP27 protein conferred increased resistance, indicating that HSP27 might serve as a predictive marker for therapeutic response in pancreatic cancer patients, and potentially explaining the impact of HSP27 expression on better survival in gemcitabine-treated patients in our study. Prospective studies comprising larger patient numbers and chemotherapy-stratified subpopulations are required to validate our data and to clarify the potential association between HSP27 expression and chemotherapeutic response, especially in regard to gemcitabine and p53 status. As HSP27 is inducible upon heat shock in pancreatic cancer cells, these studies could finally entail direct clinical implications, for example the incorporation of hyperthermia in clinical treatment protocols for pancreatic cancer, a rationale already supported by recent clinical trials [75, 76].
Acknowledgments
The authors thank A. Sendelhofert and M. Brand for excellent technical assistance and R. Issels for critical review of the manuscript. This work is part of the M.D. theses of D. Bader and D. Buchner. This work was supported by grants to C.S. (DFG Sch766/3–4, Friedrich-Baur-Foundation) and to E.G. (DFG Ga762/3-1, Bavarian Academy of Sciences and Arts, Foerderprogramm fuer Forschung und Lehre, Friedrich-Baur-Fund, K.L. Weigand-Fund, Curt-Bohnewand-Fund, MMW-Fund).
Authors’ contributions
CS: Conception and design, financial support, data analysis and interpretation, manuscript drafting; HS: Conception and design, collection and assembly of data, manuscript drafting; DBa: Collection and assembly of data, data analysis and interpretation, manuscript drafting; GA: Collection and assembly of data, data analysis and interpretation; DBu: Collection and assembly of data, data analysis and interpretation; YG: Collection and assembly of data, data analysis and interpretation; AZ: Collection and assembly of data, data analysis and interpretation; AP: Collection and assembly of data, data analysis and interpretation; SO: Collection and assembly of data, data analysis and interpretation; RPL: Data analysis and interpretation; AJ: Collection and assembly of data, data analysis and interpretation; ENT: Collection and assembly of data, data analysis and interpretation; TK: Conception and design, data analysis and interpretation; BG: Conception and design, financial support, data analysis and interpretation; CB: Conception and design, financial support, data analysis and interpretation; EG: Conception and design, financial support, collection and assembly of data, data analysis and interpretation, writing of the final manuscript.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Li D, Xie K, Wolff R, et al. Pancreatic cancer. Lancet. 2004;363:1049–57. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 3.Garcea G, Dennison AR, Pattenden CJ, et al. Survival following curative resection for pancreatic ductal adenocarcinoma. A systematic review of the literature. JOP. 2008;9:99–132. [PubMed] [Google Scholar]
- 4.Camp RL, Neumeister V, Rimm DL. A decade of tissue microarrays: progress in the discovery and validation of cancer biomarkers. J Clin Oncol. 2008;26:5630–7. doi: 10.1200/JCO.2008.17.3567. [DOI] [PubMed] [Google Scholar]
- 5.Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–77. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 6.Georgopoulos C, Welch WJ. Role of the major heat shock proteins as molecular chaperones. Annu Rev Cell Biol. 1993;9:601–34. doi: 10.1146/annurev.cb.09.110193.003125. [DOI] [PubMed] [Google Scholar]
- 7.Arrigo AP, Simon S, Gibert B, et al. Hsp27 (HspB1) and alphaB-crystallin (HspB5) as therapeutic targets. FEBS Lett. 2007;581:3665–74. doi: 10.1016/j.febslet.2007.04.033. [DOI] [PubMed] [Google Scholar]
- 8.Garrido C, Brunet M, Didelot C, et al. Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell Cycle. 2006;5:2592–601. doi: 10.4161/cc.5.22.3448. [DOI] [PubMed] [Google Scholar]
- 9.Theriault JR, Lambert H, Chavez-Zobel AT, et al. Essential role of the NH2-terminal WD/EPF motif in the phosphorylation-activated protective function of mammalian Hsp27. J Biol Chem. 2004;279:23463–71. doi: 10.1074/jbc.M402325200. [DOI] [PubMed] [Google Scholar]
- 10.Kostenko S, Moens U. Heat shock protein 27 phosphorylation: kinases, phosphatases, functions and pathology. Cell Mol Life Sci. 2009;66:3289–307. doi: 10.1007/s00018-009-0086-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciocca DR, Adams DJ, Edwards DP, et al. Distribution of an estrogen-induced protein with a molecular weight of 24,000 in normal and malignant human tissues and cells. Cancer Res. 1983;43:1204–10. [PubMed] [Google Scholar]
- 12.Soldes OS, Kuick RD, Thompson IA, 2nd, et al. Differential expression of Hsp27 in normal oesophagus, Barrett's metaplasia and oesophageal adenocarcinomas. Br J Cancer. 1999;79:595–603. doi: 10.1038/sj.bjc.6690094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ciocca DR, Calderwood SK. Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones. 2005;10:86–103. doi: 10.1379/CSC-99r.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schafer C, Williams JA. Stress kinases and heat shock proteins in the pancreas: possible roles in normal function and disease. J Gastroenterol. 2000;35:1–9. doi: 10.1080/003655200750024443. [DOI] [PubMed] [Google Scholar]
- 15.Rakonczay Z, Jr, Takacs T, Boros I, et al. Heat shock proteins and the pancreas. J Cell Physiol. 2003;195:383–91. doi: 10.1002/jcp.10268. [DOI] [PubMed] [Google Scholar]
- 16.Kubisch C, Dimagno MJ, Tietz AB, et al. Overexpression of heat shock protein Hsp27 protects against cerulein-induced pancreatitis. Gastroenterology. 2004;127:275–86. doi: 10.1053/j.gastro.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Lu Z, Hu L, Evers S, et al. Differential expression profiling of human pancreatic adenocarcinoma and healthy pancreatic tissue. Proteomics. 2004;4:3975–88. doi: 10.1002/pmic.200300863. [DOI] [PubMed] [Google Scholar]
- 18.Melle C, Ernst G, Escher N, et al. Protein profiling of microdissected pancreas carcinoma and identification of HSP27 as a potential serum marker. Clin Chem. 2007;53:629–35. doi: 10.1373/clinchem.2006.079194. [DOI] [PubMed] [Google Scholar]
- 19.Liao WC, Wu MS, Wang HP, et al. Serum heat shock protein 27 is increased in chronic pancreatitis and pancreatic carcinoma. Pancreas. 2009;38:422–6. doi: 10.1097/MPA.0b013e318198281d. [DOI] [PubMed] [Google Scholar]
- 20.Mori-Iwamoto S, Kuramitsu Y, Ryozawa S, et al. Proteomics finding heat shock protein 27 as a biomarker for resistance of pancreatic cancer cells to gemcitabine. Int J Oncol. 2007;31:1345–50. [PubMed] [Google Scholar]
- 21.Mori-Iwamoto S, Taba K, Kuramitsu Y, et al. Interferon-gamma down-regulates heat shock protein 27 of pancreatic cancer cells and helps in the cytotoxic effect of gemcitabine. Pancreas. 2009;38:224–6. doi: 10.1097/MPA.0b013e3181773970. [DOI] [PubMed] [Google Scholar]
- 22.Taba K, Kuramitsu Y, Ryozawa S, et al. Heat-shock protein 27 is phosphorylated in gemcitabine-resistant pancreatic cancer cells. Anticancer Res. 2010;30:2539–43. [PubMed] [Google Scholar]
- 23.Kononen J, Bubendorf L, Kallioniemi A, et al. Tissue microarrays for high-throughput molecular profiling of tumour specimens. Nat Med. 1998;4:844–7. doi: 10.1038/nm0798-844. [DOI] [PubMed] [Google Scholar]
- 24.Neumann J, Zeindl-Eberhart E, Kirchner T, et al. Frequency and type of KRAS mutations in routine diagnostic analysis of metastatic colorectal cancer. Pathol Res Pract. 2009;205:858–62. doi: 10.1016/j.prp.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 25.Ogino S, Kawasaki T, Brahmandam M, et al. Sensitive sequencing method for KRAS mutation detection by Pyrosequencing. J Mol Diagn. 2005;7:413–21. doi: 10.1016/S1525-1578(10)60571-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parmar M, Machin D. Survival analysis – a practical approach. New York: Wiley & Sons; 1996. [Google Scholar]
- 27.Schafer C, Clapp P, Welsh MJ, et al. HSP27 expression regulates CCK-induced changes of the actin cytoskeleton in CHO-CCK-A cells. Am J Physiol. 1999;277:C1032–43. doi: 10.1152/ajpcell.1999.277.6.C1032. [DOI] [PubMed] [Google Scholar]
- 28.Mehlen P, Hickey E, Weber LA, et al. Large unphosphorylated aggregates as the active form of hsp27 which controls intracellular reactive oxygen species and glutathione levels and generates a protection against TNFalpha in NIH-3T3-ras cells. Biochem Biophys Res Commun. 1997;241:187–92. doi: 10.1006/bbrc.1997.7635. [DOI] [PubMed] [Google Scholar]
- 29.Katz MH, Hwang R, Fleming JB, et al. Tumour-node-metastasis staging of pancreatic adenocarcinoma. CA Cancer J Clin. 2008;58:111–25. doi: 10.3322/CA.2007.0012. [DOI] [PubMed] [Google Scholar]
- 30.Rozenblum E, Schutte M, Goggins M, et al. Tumour-suppressive pathways in pancreatic carcinoma. Cancer Res. 1997;57:1731–4. [PubMed] [Google Scholar]
- 31.Redston MS, Caldas C, Seymour AB, et al. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res. 1994;54:3025–33. [PubMed] [Google Scholar]
- 32.Almoguera C, Shibata D, Forrester K, et al. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–54. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 33.Maitra A, Adsay NV, Argani P, et al. Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Mod Pathol. 2003;16:902–12. doi: 10.1097/01.MP.0000086072.56290.FB. [DOI] [PubMed] [Google Scholar]
- 34.Gao C, Zou Z, Xu L, et al. p53-dependent induction of heat shock protein 27 (HSP27) expression. Int J Cancer. 2000;88:191–4. [PubMed] [Google Scholar]
- 35.Evans JR, Bosman JD, Brown-Endres L, et al. Induction of the small heat shock protein alphaB-crystallin by genotoxic stress is mediated by p53 and p73. Breast Cancer Res Treat. 2010;122:159–68. doi: 10.1007/s10549-009-0542-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanagasabai R, Krishnamurthy K, Druhan LJ, et al. Forced expression of Hsp27 reverses P-glycoprotein (ABCB1) mediated drug efflux and MDR1 gene expression in adriamycin resistant human breast cancer cells. J Biol Chem. 2011 doi: 10.1074/jbc.M111.249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Callaghan-Sunol C, Gabai VL, Sherman MY. Hsp27 modulates p53 signaling and suppresses cellular senescence. Cancer Res. 2007;67:11779–88. doi: 10.1158/0008-5472.CAN-07-2441. [DOI] [PubMed] [Google Scholar]
- 38.Garcea G, Neal CP, Pattenden CJ, et al. Molecular prognostic markers in pancreatic cancer: a systematic review. Eur J Cancer. 2005;41:2213–36. doi: 10.1016/j.ejca.2005.04.044. [DOI] [PubMed] [Google Scholar]
- 39.Uozaki H, Ishida T, Kakiuchi C, et al. Expression of heat shock proteins in osteosarcoma and its relationship to prognosis. Pathol Res Pract. 2000;196:665–73. doi: 10.1016/S0344-0338(00)80118-1. [DOI] [PubMed] [Google Scholar]
- 40.King KL, Li AF, Chau GY, et al. Prognostic significance of heat shock protein-27 expression in hepatocellular carcinoma and its relation to histologic grading and survival. Cancer. 2000;88:2464–70. doi: 10.1002/1097-0142(20000601)88:11<2464::aid-cncr6>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 41.Cornford PA, Dodson AR, Parsons KF, et al. Heat shock protein expression independently predicts clinical outcome in prostate cancer. Cancer Res. 2000;60:7099–105. [PubMed] [Google Scholar]
- 42.Foster CS, Dodson AR, Ambroisine L, et al. Hsp-27 expression at diagnosis predicts poor clinical outcome in prostate cancer independent of ETS-gene rearrangement. Br J Cancer. 2009;101:1137–44. doi: 10.1038/sj.bjc.6605227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geisler JP, Geisler HE, Tammela J, et al. A study of heat shock protein 27 in endometrial carcinoma. Gynecol Oncol. 1999;72:347–50. doi: 10.1006/gyno.1998.5283. [DOI] [PubMed] [Google Scholar]
- 44.Kawanishi K, Shiozaki H, Doki Y, et al. Prognostic significance of heat shock proteins 27 and 70 in patients with squamous cell carcinoma of the esophagus. Cancer. 1999;85:1649–57. doi: 10.1002/(sici)1097-0142(19990415)85:8<1649::aid-cncr2>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 45.Tetu B, Lacasse B, Bouchard HL, et al. Prognostic influence of HSP-27 expression in malignant fibrous histiocytoma: a clinicopathological and immunohistochemical study. Cancer Res. 1992;52:2325–8. [PubMed] [Google Scholar]
- 46.Gandour-Edwards R, Trock BJ, Gumerlock P, et al. Heat shock protein and p53 expression in head and neck squamous cell carcinoma. Otolaryngol Head Neck Surg. 1998;118:610–5. doi: 10.1177/019459989811800508. [DOI] [PubMed] [Google Scholar]
- 47.Storm FK, Mahvi DM, Gilchrist KW. Hsp-27 has no diagnostic or prognostic significance in prostate or bladder cancers. Urology. 1993;42:379–82. doi: 10.1016/0090-4295(93)90361-d. [DOI] [PubMed] [Google Scholar]
- 48.Erkizan O, Kirkali G, Yorukoglu K, et al. Significance of heat shock protein-27 expression in patients with renal cell carcinoma. Urology. 2004;64:474–8. doi: 10.1016/j.urology.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 49.Ito T, Kawabe R, Kurasono Y, et al. Expression of heat shock proteins in squamous cell carcinoma of the tongue: an immunohistochemical study. J Oral Pathol Med. 1998;27:18–22. doi: 10.1111/j.1600-0714.1998.tb02085.x. [DOI] [PubMed] [Google Scholar]
- 50.Mese H, Sasaki A, Nakayama S, et al. Prognostic significance of heat shock protein 27 (HSP27) in patients with oral squamous cell carcinoma. Oncol Rep. 2002;9:341–4. [PubMed] [Google Scholar]
- 51.Wang A, Liu X, Sheng S, et al. Dysregulation of heat shock protein 27 expression in oral tongue squamous cell carcinoma. BMC Cancer. 2009;9:167. doi: 10.1186/1471-2407-9-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lo Muzio L, Campisi G, Farina A, et al. Prognostic value of HSP27 in head and neck squamous cell carcinoma: a retrospective analysis of 57 tumours. Anticancer Res. 2006;26:1343–9. [PubMed] [Google Scholar]
- 53.Kapranos N, Kominea A, Konstantinopoulos PA, et al. Expression of the 27-kDa heat shock protein (HSP27) in gastric carcinomas and adjacent normal, metaplastic, and dysplastic gastric mucosa, and its prognostic significance. J Cancer Res Clin Oncol. 2002;128:426–32. doi: 10.1007/s00432-002-0357-y. [DOI] [PubMed] [Google Scholar]
- 54.Takeno S, Noguchi T, Kikuchi R, et al. Analysis of the survival period in resectable stage IV gastric cancer. Ann Surg Oncol. 2001;8:215–21. doi: 10.1007/s10434-001-0215-1. [DOI] [PubMed] [Google Scholar]
- 55.Giaginis C, Daskalopoulou SS, Vgenopoulou S, et al. Heat Shock Protein-27, −60 and −90 expression in gastric cancer: association with clinicopathological variables and patient survival. BMC Gastroenterol. 2009;9:14. doi: 10.1186/1471-230X-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langdon SP, Rabiasz GJ, Hirst GL, et al. Expression of the heat shock protein HSP27 in human ovarian cancer. Clin Cancer Res. 1995;1:1603–9. [PubMed] [Google Scholar]
- 57.Geisler JP, Geisler HE, Tammela J, et al. Heat shock protein 27: an independent prognostic indicator of survival in patients with epithelial ovarian carcinoma. Gynecol Oncol. 1998;69:14–6. doi: 10.1006/gyno.1998.4961. [DOI] [PubMed] [Google Scholar]
- 58.Schneider J, Jimenez E, Marenbach K, et al. Co-expression of the MDR1 gene and HSP27 in human ovarian cancer. Anticancer Res. 1998;18:2967–71. [PubMed] [Google Scholar]
- 59.Arts HJ, Hollema H, Lemstra W, et al. Heat-shock-protein-27 (hsp27) expression in ovarian carcinoma: relation in response to chemotherapy and prognosis. Int J Cancer. 1999;84:234–8. doi: 10.1002/(sici)1097-0215(19990621)84:3<234::aid-ijc6>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 60.Elpek GO, Karaveli S, Simsek T, et al. Expression of heat-shock proteins hsp27, hsp70 and hsp90 in malignant epithelial tumour of the ovaries. APMIS. 2003;111:523–30. doi: 10.1034/j.1600-0463.2003.1110411.x. [DOI] [PubMed] [Google Scholar]
- 61.Elstrand MB, Kleinberg L, Kohn EC, et al. Expression and clinical role of antiapoptotic proteins of the bag, heat shock, and Bcl-2 families in effusions, primary tumours, and solid metastases in ovarian carcinoma. Int J Gynecol Pathol. 2009;28:211–21. doi: 10.1097/PGP.0b013e31818b0f5e. [DOI] [PubMed] [Google Scholar]
- 62.Kassem H, Sangar V, Cowan R, et al. A potential role of heat shock proteins and nicotinamide N-methyl transferase in predicting response to radiation in bladder cancer. Int J Cancer. 2002;101:454–60. doi: 10.1002/ijc.10631. [DOI] [PubMed] [Google Scholar]
- 63.Seymour L, Bezwoda WR, Meyer K, et al. Detection of P24 protein in human breast cancer: influence of receptor status and oestrogen exposure. Br J Cancer. 1990;61:886–90. doi: 10.1038/bjc.1990.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vargas-Roig LM, Gago FE, Tello O, et al. Heat shock protein expression and drug resistance in breast cancer patients treated with induction chemotherapy. Int J Cancer. 1998;79:468–75. doi: 10.1002/(sici)1097-0215(19981023)79:5<468::aid-ijc4>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 65.Takeno S, Noguchi T, Takahashi Y, et al. Immunohistochemical and clinicopathologic analysis of response to neoadjuvant therapy for esophageal squamous cell carcinoma. Dis Esophagus. 2001;14:149–54. doi: 10.1046/j.1442-2050.2001.00174.x. [DOI] [PubMed] [Google Scholar]
- 66.Langer R, Ott K, Specht K, et al. Protein expression profiling in esophageal adenocarcinoma patients indicates association of heat-shock protein 27 expression and chemotherapy response. Clin Cancer Res. 2008;14:8279–87. doi: 10.1158/1078-0432.CCR-08-0679. [DOI] [PubMed] [Google Scholar]
- 67.Bubendorf L, Kolmer M, Kononen J, et al. Hormone therapy failure in human prostate cancer: analysis by complementary DNA and tissue microarrays. J Natl Cancer Inst. 1999;91:1758–64. doi: 10.1093/jnci/91.20.1758. [DOI] [PubMed] [Google Scholar]
- 68.Damstrup L, Andersen J, Kufe DW, et al. Immunocytochemical determination of the estrogen-regulated proteins Mr 24,000, Mr 52,000 and DF3 breast cancer associated antigen: clinical value in advanced breast cancer and correlation with estrogen receptor. Ann Oncol. 1992;3:71–7. doi: 10.1093/oxfordjournals.annonc.a058078. [DOI] [PubMed] [Google Scholar]
- 69.Ciocca DR, Green S, Elledge RM, et al. Heat shock proteins hsp27 and hsp70: lack of correlation with response to tamoxifen and clinical course of disease in estrogen receptor-positive metastatic breast cancer (a Southwest Oncology Group Study) Clin Cancer Res. 1998;4:1263–6. [PubMed] [Google Scholar]
- 70.Germain I, Tetu B, Brisson J, et al. Markers of chemoresistance in ovarian carcinomas: an immunohistochemical study of 86 cases. Int J Gynecol Pathol. 1996;15:54–62. doi: 10.1097/00004347-199601000-00009. [DOI] [PubMed] [Google Scholar]
- 71.Vargas Roig LM, Lotfi H, Olcese JE, et al. Effects of short-term tamoxifen administration in patients with invasive cervical carcinoma. Anticancer Res. 1993;13:2457–63. [PubMed] [Google Scholar]
- 72.Fortin A, Raybaud-Diogene H, Tetu B, et al. Overexpression of the 27 KDa heat shock protein is associated with thermoresistance and chemoresistance but not with radioresistance. Int J Radiat Oncol Biol Phys. 2000;46:1259–66. doi: 10.1016/s0360-3016(99)00410-1. [DOI] [PubMed] [Google Scholar]
- 73.Rau B, Gaestel M, Wust P, et al. Preoperative treatment of rectal cancer with radiation, chemotherapy and hyperthermia: analysis of treatment efficacy and heat-shock response. Radiat Res. 1999;151:479–88. [PubMed] [Google Scholar]
- 74.Adachi S, Kokura S, Okayama T, et al. Effect of hyperthermia combined with gemcitabine on apoptotic cell death in cultured human pancreatic cancer cell lines. Int J Hyperthermia. 2009;25:210–9. doi: 10.1080/02656730802657036. [DOI] [PubMed] [Google Scholar]
- 75.Ohguri T, Imada H, Yahara K, et al. Concurrent chemoradiotherapy with gemcitabine plus regional hyperthermia for locally advanced pancreatic carcinoma: initial experience. Radiat Med. 2008;26:587–96. doi: 10.1007/s11604-008-0279-y. [DOI] [PubMed] [Google Scholar]
- 76.Tschoep KE, Boeck S, Berger F, et al. Regional hyperthermia (RHT) combined with gemcitabine (GEM) + cisplatin (CIS) in patients with GEM-refractory advanced pancreatic cancer: results of the ESHO phase II trial. J Clin Oncol (Meeting Abstracts) 2008;26:4635. [Google Scholar]
- 77.Calderwood SK, Khaleque MA, Sawyer DB, et al. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci. 2006;31:164–72. doi: 10.1016/j.tibs.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 78.Stammler G, Volm M. Expression of heat shock proteins, glutathione peroxidase and catalase in childhood acute lymphoblastic leukemia and nephroblastoma. Cancer Lett. 1996;99:35–42. doi: 10.1016/0304-3835(95)04035-8. [DOI] [PubMed] [Google Scholar]
- 79.Aghdassi A, Phillips P, Dudeja V, et al. Heat shock protein 70 increases tumorigenicity and inhibits apoptosis in pancreatic adenocarcinoma. Cancer Res. 2007;67:616–25. doi: 10.1158/0008-5472.CAN-06-1567. [DOI] [PubMed] [Google Scholar]
- 80.Gress TM, Muller-Pillasch F, Weber C, et al. Differential expression of heat shock proteins in pancreatic carcinoma. Cancer Res. 1994;54:547–51. [PubMed] [Google Scholar]
- 81.Sagol O, Tuna B, Coker A, et al. Immunohistochemical detection of pS2 protein and heat shock protein-70 in pancreatic adenocarcinomas. Relationship with disease extent and patient survival. Pathol Res Pract. 2002;198:77–84. doi: 10.1078/0344-0338-00190. [DOI] [PubMed] [Google Scholar]
- 82.Saluja A, Dudeja V. Heat shock proteins in pancreatic diseases. J Gastroenterol Hepatol. 2008;23(Suppl 1):S42–5. doi: 10.1111/j.1440-1746.2007.05272.x. [DOI] [PubMed] [Google Scholar]
- 83.Phillips PA, Dudeja V, McCarroll JA, et al. Triptolide induces pancreatic cancer cell death via inhibition of heat shock protein 70. Cancer Res. 2007;67:9407–16. doi: 10.1158/0008-5472.CAN-07-1077. [DOI] [PubMed] [Google Scholar]
- 84.Dudeja V, Mujumdar N, Phillips P, et al. Heat shock protein 70 inhibits apoptosis in cancer cells through simultaneous and independent mechanisms. Gastroenterology. 2009;136:1772–82. doi: 10.1053/j.gastro.2009.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]