

Abstract
Morphine is a potent analgesic, but the molecular mechanism for tolerance formation after repeated use is not fully understood. Binding immunoglobulin protein (BiP) is an endoplasmic reticulum (ER) chaperone that is central to ER function. We examined knock-in mice expressing a mutant BiP with the retrieval sequence deleted in order to elucidate physiological processes that are sensitive to BiP functions. We tested the thermal antinociceptive effect of morphine in heterozygous mutant BiP mice in a hot plate test. Paw withdrawal latencies before and after a single administration of morphine were not significantly different between the wild-type and mutant BiP mice. Repeated morphine administration caused the development of morphine tolerance in the wild-type mice. The activation of glycogen synthase kinase 3b (GSK-3b) was associated with morphine tolerance, because an inhibitor of GSK-3β prevented it. On the other hand, the mutant BiP mice showed less morphine tolerance, and the activation of GSK-3b was suppressed in their brain. These results suggest that BiP may play an important role in the development of morphine tolerance. Furthermore, we found that a chemical chaperone which improves ER protein folding capacity also attenuated the development of morphine tolerance in wild-type mice, suggesting a possible clinical application of chemical chaperones in preventing morphine tolerance.
Keywords: opioid tolerance, morphine, mu opioid receptor, endoplasmic reticulum, chaperone, ER stress, glycogen synthase kinase
Introduction
Opioids are potent analgesics that are widely used to control acute and chronic pain [1]. Although repeated administration of opioids, particularly morphine, induces tolerance that reduces the effectiveness of the analgesic, the precise molecular mechanism for the development of tolerance remains uncertain.
Opioids bind to the μ opioid receptor (MOR) to activate various signalling molecules through heterotrimeric guanine nucleotide-binding proteins (G proteins), leading to a decrease in neuronal excitability by the inhibition of voltage-dependent calcium channels and the activation of inwardly rectifying potassium channels [2]. Activation of MOR also induces the phosphorylation of MOR by G-protein-coupled receptor kinases [3, 4]. Phosphorylated MOR is recognized by arrestins [5], and internalized by clathrin-coated vesicles. The transient uncoupling of MOR from signalling pathways due to the phosphorylation and intracellular trafficking of MOR causes opioid desensitization. Most of the internalized MORs return to the cell surface, resulting in resensitization [6–8].
Chronic morphine tolerance may be derived from adaptations in the intracellular signal transduction of post-MOR activation, as morphine does not induce effective MOR phosphorylation and internalization [9]. Persistent MOR activation may alter signal transduction, including changes in MOR-coupled G proteins from Gia to Gsa [10], increased activity of protein kinase C [11], and the up-regulation of N-methyl-D-aspartate receptor signalling [12]. These changes may contribute to the development of morphine tolerance. Chronic morphine treatment also activates the cyclin-dependent kinase 5 and glycogen synthase kinase 3β (GSK-3β) signalling pathway, whereas the inhibition of them diminishes morphine tolerance and restores analgesia in rats [13]. GSK-3β is expressed ubiquitously and is one of the central molecules in intracellular signal transduction [14]. It may play an important role in diverse physiological and pathological states [15].
Proteins destined for secretory pathways, such as cell surface receptors including MOR, are inserted into the endoplasmic reticulum (ER), where their folding or degradation intermediaries interact with molecular chaperones like binding immunoglobulin protein (BiP/GRP78) [16]. Many physiological and pathological conditions, such as secretory demands, ischemia, hypoxia and genetic mutations, can cause aberrant protein folding and the accumulation of misfolded proteins in the ER. These insults lead to ER stress and initiate the unfolded protein response (UPR) [17, 18], which increases the capacity for ER quality control by reducing general protein synthesis, increasing the level of ER chaperones and promoting ER-associated protein degradation. Recent studies have suggested that chronic ER stress might modulate intracellular signalling pathways, resulting in several chronic disorders, such as type II diabetes [19] and interstitial pneumonia [20].
In this study, we examined whether ER function may attenuate the MOR signalling pathway, which might cause the development of morphine tolerance. GSK-3β is one possible candidate molecule that may play key roles in both the UPR and MOR signalling pathways. ER stress induces the activation of GSK-3β[21, 22]. GSK-3β may also be involved in the MOR signalling pathway, as stimulation of MOR by morphine may lead to the activation of phosphatidylinositol 3-kinase (PI3K)/Akt signalling pathway [23]. The kinase Akt inactivates GSK-3β by phosphorylation of Ser9.
BiP is central to ER function [24]. We have previously produced knock-in mice expressing a mutant BiP in order to elucidate the physiological processes that are sensitive to BiP function in adulthood [25]. The mutant BiP protein lacks the retrieval carboxyl-terminal Lys-Asp-Glu-Leu (KDEL) sequence [26, 27], that normally functions to return BiP to the ER from the secretory pathway by the KDEL receptor in the Golgi complex. This mutant allows us to examine the effects of a defect in ER function without completely eliminating BiP function. The homozygous mutant BiP mice have defects in some professional secretory cells. They survive only several hours after birth due to impaired pulmonary surfactant biosynthesis by alveolar type II epithelial cells and respiratory failure [25]. They also have cortical dysplasia due to impaired synthesis of reelin by Cajal-Retzius cells [28]. Heterozygous mutant BiP mice produce pulmonary surfactant and reelin, and grow to be apparently normal adults.
We evaluated the thermal antinociceptive effect of morphine in heterozygous mutant BiP mice. Although the BiP mutant mice had normal sensory transmission and analgesia, they were much less likely to develop morphine tolerance after repeated use. The activation of GSK-3β was associated with morphine tolerance in wild-type mice. On the other hand, GSK-3β activation was attenuated in the mutant BiP mice. Furthermore, we found that a chemical chaperone that improves ER protein folding capacity and suppresses the expression of BiP [29, 30] attenuated the development of morphine tolerance in wild-type mice. These results suggest that BiP, an ER chaperone, may play an important role in the development of morphine tolerance.
Materials and methods
Animals
All animal experimental procedures were in accordance with a protocol approved by the Institutional Animal Care Committee of Chiba University, Chiba, Japan. We used homologous recombination to establish knock-in mice expressing a mutant BiP lacking the carboxyl-terminal KDEL sequence [25]. The missing KDEL sequence was replaced by a hemagglutinin tag. The heterozygous mutant BiP mice were maintained over 10 generations with crossing to C57BL/6 mice. Mutant BiP male mice, their wild-type male littermates and C57BL/6 male mice (20–25 g body weight, 10–15 weeks old) were used. All mice were provided with food and water ad libitum before the experiment.
Cells and reagents
Mouse embryonic fibroblasts (MEFs) were prepared from 13.5-day-old embryos [25]. MEFs were grown in a complete medium that consisted of Dulbecco’s modified Eagle’s medium (DMEM; Sigma Chemical Co., Irvine, UK) with 10% foetal bovine serum, 2 mM glutamine, 50 mg/ml streptomycin and 50 U/ml penicillin G at 37°C in a 5% CO2 incubator.
The following antibodies were used: rabbit polyclonal antibody against MOR-1 (Chemicon, Temecula, CA, USA), rabbit polyclonal antibody against phospho-GSK3β (Ser9), rabbit polyclonal antibody against GSK3β, rabbit polyclonal antibody against phospho Akt1/2/3 (Ser473) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit polyclonal antibody against Akt (Cell Signaling Technology, Beverly, MA, USA), mouse monoclonal antibody (mAb) against phospho-GSK3β (Tyr279/Tyr216) (Upstate Biotechnology, Chicago, IL, USA), mouse mAβ 9E10 against the myc epitope (ATCC, Manassas, VA, USA), mouse mAb 15E6 against the hemagglutinin epitope (a kind gift from VW Hsu, Harvard Medical School, Boston, MA, USA), mouse mAb against γ-tubulin (Sigma Chemical Co.), mouse mAb SPA-827 against BiP (KDEL sequence) (Stressgen, Victoria, Canada), Cy-2- or Cy-3-conjugated donkey antibody against rabbit IgG, and Cy-2- or Cy-3-conjugated donkey antibody against mouse IgG (Jackson Immunoresearch Laboratories, West Grove, PA, USA). The following reagents were used: [D-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO), thapsigargin (Sigma Chemical Co.), morphine hydrochloride (Takeda Pharmaceutical Co., Tokyo, Japan), Hoechst 33258 (Invitrogen, Carlsbad, CA, USA), SB216763 (Biomol International, Plymouth Meeting, PA, USA) and tauroursodeoxycholic acid (TUDCA, Calbiochem, San Diego, CA, USA).
Immunohistochemistry
Mice were deeply anesthetized with pentobarbital (Dainippon Sumitomo Pharma, Osaka, Japan) and were fixed by transcardiac perfusion with 4% paraformaldehyde in phosphate-buffered saline (PBS). The brains were further immersion-fixed for 12 hrs in 4% paraformaldehyde at 4°C. After fixation, they were dehydrated in increasing concentrations of ethanol and embedded in paraffin wax. For immunofluorescence, sections (8 μm) were incubated with 10% normal goat or bovine serum in PBS for 30 min. to block non-specific antibody binding, and then incubated with a primary antibody in PBS for 1 hr at room temperature. The sections were rinsed with PBS and then incubated with a mixture of Cy2-conjugated anti-rabbit IgG and Cy3-conjugated anti-mouse IgG in PBS for 1 hr at room temperature. Then, the sections were rinsed with PBS and mounted on glass slides with Perma Fluor (Immunon, Pittsburgh, PA, USA). Immunolocalization was observed with a fluorescence microscope using FITC/rhodamine filters and a Plan-Neofluar 20× and 40× NA 0.75 objective (Axiovert 200M, Carl Zeiss, Oberkochen, Germany). The brightness and contrast were optimized by AxioVision 4.4 software (Carl Zeiss), and immunofluorescence images were captured with a digital camera (AxioCam MRm, Carl Zeiss). The mean grey values of the cells with the background subtraction were used for densitometry.
Transfection and confocal laser scanning microscopy
A cDNA-encoding rat MOR was obtained from wild-type rat brain mRNA using the following primers: 5′-cggtaccaagcaccatggacagcagc-3′and 5′-cggtaccaagggcaatggagcagtttc-3′. The cDNA was subcloned into a pcDNA3.1 myc-His vector (Invitrogen). The DNA sequence was verified using the Applied Biosystems ABI Prism 310 genetic analyser (Applied Biosystems, Foster City, CA, USA). Transfection was performed with Fugene 6 (Roche Applied Science, Indianapolis, IN, USA). Forty hours after transfection, cells on cover slips were fixed in methanol at –20°C for 1 hr and then processed as described [31]. The labelled cells were examined by a confocal laser scanning microscope (Axiovert 100M, LSM510, Ver. 3.2, Carl Zeiss) fitted with krypton and argon lasers using a Plan-Apochromat 100× NA 1.40 oil objective.
Western blotting
In order to obtain embryonic brains, the pregnant mice were deeply anesthetized by pentobarbital, and embryos (E18.5) were removed by caesarean section. To obtain adult brains, mice were deeply anesthetized with pentobarbital and were perfused with ice-cold PBS by transcardiac cannulation. Brain stems were removed for Western blotting. The brains were homogenized by supersonic wave (UR-20P, TOMY, Tokyo, Japan) in a buffer containing 0.4% (w/v) Nonidet P-40, 0.2% N-lauroylsarcosine, 30 mM Tris/HCl pH 8.0, 1 mM ethylenediaminetetraacetic acid, 10 mg/ml aprotinin, 10 μg/ml leupeptin, 30 μg/ml N-acetyl-l-leucinal-l-lecinal-l-norleucinal (ALLN, Sigma Chemical Co.). Cultured cells were washed twice with ice-cold PBS and then homogenized in the same buffer. The lysates were centrifuged, and the supernatants in SDS-PAGE sample buffer were separated by SDS-PAGE under reducing conditions. The proteins were transferred from the gels to polyvinylidene fluoride membranes (Immobilon-P, Millipore Corp., Billerica, MA, USA), and Western blotting was done as previously described [32]. Imaging was obtained by LAS-1000 and Image Gauge software (Fuji Photo Film Co. Ltd., Tokyo, Japan). Densitometry was performed with LAS-1000 and ImageJ software (Wayne Rasband, NIH, Bethesda, MD, USA).
Hot plate test
The hot plate test was carried out to assess the effects of an agent on the thermal nociceptive threshold of the mice. Mice were placed on a 54.5°C hot plate (Socrel hot-plate model DS37, Ugo Basile, Italy). The response latency to either a hind paw lick or a jump was recorded. In the absence of a response, the animals were removed from the hot plate at 60 sec. to avoid tissue injury, and a 60 sec. latency was assigned as the response. The agents were administered intraperitoneally twice a day for five consecutive days. The hot plate test was performed after the first administration of the drug on day 1 and the tenth administration of the drug on day 5. The hot plate latency was measured at 5, 15, 30, 45 and 60 min. after the drug injection. Before the drug administration, the hot plate latency was measured three times, and the average of the second and the third measurements was obtained as the pre-drug response latency at time 0 min. To obtain control data, the vehicle (saline) was injected intraperitoneally.
To analyse the effects of the drugs on animal performance in the hot plate test, the percentage maximum possible effect (MPE) was calculated, where percentage MPE = ([post-drug maximum response latency – pre-drug response latency]/[cut-off time (60 sec.) – pre-drug response latency]) × 100. The post-drug maximum response latency was defined as the single longest response latency during the entire time course of the hot plate test.
The experimental protocol for the dose–response study has been reported [5]. The hot plate test was performed on both mutant BiP mice and wild-type mice for a cumulative dose of 0 mg/kg. Then, both groups of mice were injected intraperitoneally with 5 mg/kg morphine for a cumulative dose of 5 mg/kg. Antinociception was assessed 45 min. after the injection using the hot plate test. These mice were again injected with 5 mg/kg morphine to yield a cumulative dose of 10 mg/kg, and the hot plate test was performed again. In this way, we reached cumulative doses of 20, 40 and 80 mg/kg. Finally, the percentage MPE of each hot plate test was obtained to determine the dose–response curve. Dose–response studies were performed before and after the intraperitoneal administration of morphine (20 mg/kg) twice a day for five consecutive days.
Statistical analysis
To compare the hot plate percentage MPE, latencies and other values between groups, one-way or two-way ANOVA was used, and to compare the mean values of percentage MPE or other values between two groups, the Mann-Whitney U-test was used (GraphPad Prism 4.0, GraphPad Software, San Diego, CA, USA).
Results
Morphine tolerance is attenuated in mice expressing a mutant BiP
We examined whether BiP might affect morphine analgesia using heterozygous mutant BiP mice. We evaluated morphine-induced antinociception by measuring response latencies in a hot plate test. Morphine tolerance was induced by intraperitoneal morphine injection (20 mg/kg) twice a day for five consecutive days. We performed hot plate tests at the first and the tenth morphine treatments (Fig. 1A). The response latencies of the mutant BiP mice and their wild-type littermates before morphine treatment on day 1 were not significantly different (Fig. 1A, at time 0). The time courses of response latencies of the both groups at the first morphine treatment on day 1 were almost similar, and both latencies reached the 60 sec. cut-off point 30 min. after injection (Fig. 1A). Even just before the tenth morphine treatment on day 5, the response latencies of the both groups were not significantly different (Fig. 1A, at time 0). These results indicate that the mutant BiP mice have normal sensory transmission and analgesia. After the tenth morphine treatment on day 5, the response latencies of the wild-type mice were significantly reduced, indicating that morphine tolerance had developed (Fig. 1A). However, the response latencies of the mutant BiP mice after the tenth morphine treatment were significantly longer than those of their wild-type littermates at 30, 45 and 60 min. after injection. The distribution of the percentage MPE after repetitive morphine treatment is shown in Fig. 1B. The percentage MPE of mutant heterozygous BiP mice was somewhat heterogeneous. One-half of the mutant mice remained sensitive to morphine treatment and did not develop morphine tolerance. The mean percentage MPE of the mutant BiP mice (72.24%) was significantly greater than that of their wild-type littermates (29.30%). The morphine dose–response curve of the wild-type littermates, but not that of the mutant BiP mice, obtained after repetitive administration of morphine for 5 days shifted to higher morphine doses from that obtained on day 1 (Fig. 1C). The effective concentrations of 50% MPE (EC50s) of pre- and post-repeated morphine treatment of wild-type mice were 12.0 and 34.6 mg/kg, respectively, and those of the mutant BiP mice were 16.7 and 17.0 mg/kg, respectively. These results show that the mutant BiP mice have normal sensory transmission and analgesia, but that they are impaired in the development of morphine tolerance.
Fig 1.
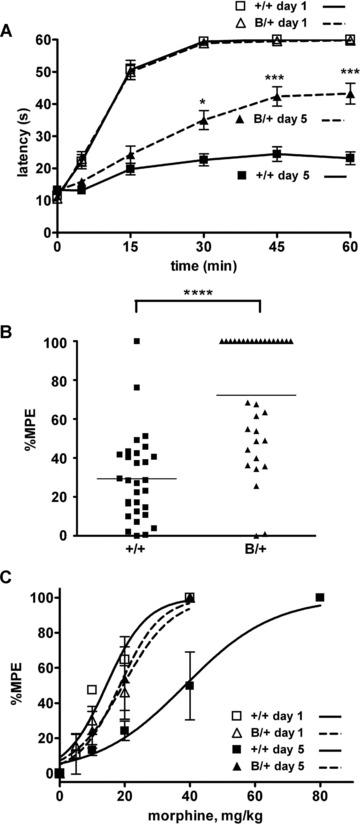
Morphine tolerance is attenuated in mice expressing a mutant BiP. (A) Heterozygous mutant BiP mice (B/+) and littermate wild-type mice (+/+) were injected intraperitoneally with 20 mg/kg morphine twice a day for 5 days, and the hot plate test was performed to evaluate analgesia at the first injection on day 1 and the tenth injection on day 5. The graph represents the response latencies (0–60 sec.) of wild-type mice on day 1, mutant BiP mice on day 1, wild-type mice on day 5 and mutant BiP mice on day 5. N = 27 (wild-type mice) and 29 (mutant BiP mice). The response latencies of the mutant BiP mice after the tenth injection on day 5 were significantly longer than those of their wild-type littermates at 30, 45 and 60 min. *P < 0.05, ***P < 0.001, two-way ANOVA with the Bonferroni post hoc test. (B) The distribution of percentage MPE of wild-type mice (N = 30) and mutant BiP mice (N = 33) after the repeated morphine treatment. The mean value of percentage MPE of the mutant BiP mice (72.2%) on day 5 was significantly larger than that of the wild-type mice (29.3%). ****P < 0.0001, the Mann-Whitney U-test. (C) The morphine dose–response curve of wild-type mice and mutant BiP mice obtained before and after repeated administration of morphine for 5 days. Wild-type mice on day 1, mutant BiP mice on day 1, wild-type mice on day 5, mutant BiP mice on day 5. N= 5 in each group.
Cell surface expression of MOR is preserved in the presence of mutant BiP
Because alterations in the intracellular trafficking of MOR might affect opioid analgesia, we examined the effect of the mutant BiP with the KDEL sequence deleted on the surface expression of MOR. MEFs from the wild-type and homozygous mutant embryos [25] were transfected with a myc-tagged MOR (Fig. 2). DAMGO, a selective peptidergic MOR ligand induces the internalization of MOR but morphine does not internalize MOR. We found the surface expression of MOR in both wild-type and mutant MEFs by confocal laser microscopy. Although DAMGO induced the internalization of MOR in both types of MEFs, MOR remained on the cell surface upon morphine treatment, suggesting that the mutant BiP did not affect the transport of MOR.
Fig 2.
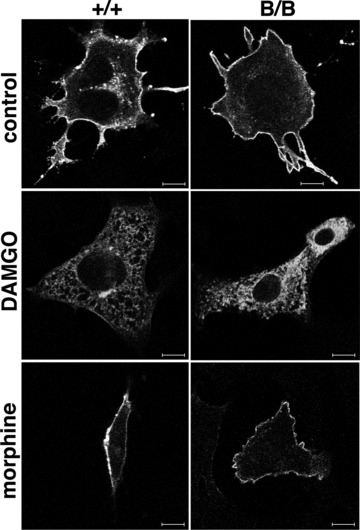
MOR is expressed on the cell surface of the mutant BiP MEF. MEFs from wild-type (+/+, left panels) and homozygous mutant (B/B, right panels) embryos were transfected with myc-tagged MOR. Thirty-eight hours after transfection, the cells were incubated with DAMGO (5 μM, 2 hrs) or morphine (1 μM, 2 hrs). Then, they were fixed, stained with monoclonal anti-myc antibody, and analysed by confocal laser microscopy. Scale bars represent 10 μm.
Inhibition of GSK-3β signalling is associated with the prevention of morphine tolerance
Then, we speculated that the UPR signalling may attenuate the MOR signalling, which may cause the development of morphine tolerance. GSK-3b is one possible candidate molecule that may play key roles in both the UPR and MOR signalling pathways. Recently the inhibition of GSK-3b by the specific inhibitors SB216763 and (2′Z, 3′E)-6-bromoindirubin-3′-oxime was shown to diminish the development of morphine tolerance in rats after chronic intrathecal morphine treatment [13]. We evaluated the effect of inhibition of GSK-3β on the development of morphine tolerance in our mouse model through intraperitoneal injection, which might affect both spinal and supraspinal analgesia. We administered 0.4 mg/kg SB216763 and 20 mg/kg morphine twice a day for 5 days in wild-type mice, and hot plate tests were performed at the first and tenth treatments. The response latencies of the mice following co-administration of SB216763 and morphine were significantly longer on day 5 than those of control mice with morphine alone at 15, 30, 45 and 60 min. after morphine injection (Fig. 3A). The mean percentage MPE of the mice receiving both SB216763 and morphine was significantly greater than that of mice treated with morphine alone (Fig. 3B). Thus, the inhibition of GSK-3β prevented the development of morphine tolerance in mice.
Fig 3.
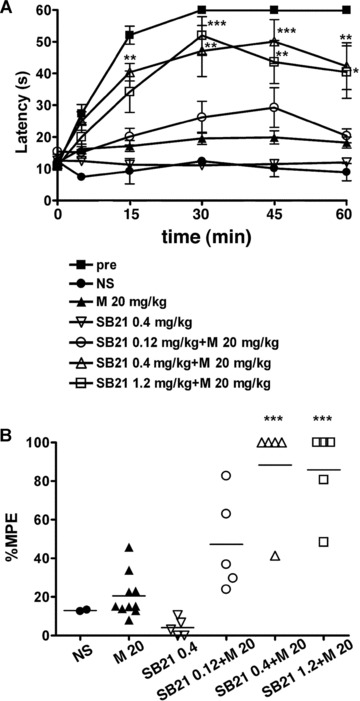
Inhibition of GSK3β is associated with prevention of morphine tolerance. Wild-type mice were injected intraperitoneally with SB216763 (SB21), a potent selective inhibitor of GSK3β, together with 20 mg/kg morphine twice a day for 5 days, and the hot plate test was performed to evaluate analgesia at the tenth injection on day 5. (A) The graph represents the response latencies (0–60 sec.) of the mice with saline control (NS, N = 2), morphine alone (M, N = 10), 0.4 mg/kg SB21 alone (SB21, N = 5), 0.12 mg/kg SB21 and morphine (N = 5), 0.4 mg/kg SB21 and morphine (N = 5), and 1.2 mg/kg SB21 and morphine (N = 5). ‘Pre’ represents control mice evaluated by the hot plate test after the first morphine injection on day 1 (N = 25). The response latencies of the mice following co-administration of SB216763 and morphine were significantly longer on day 5 than those of control mice with morphine alone at 15, 30, 45 and 60 min. after morphine injection. *P < 0.05, **P < 0.01, ***P < 0.001, two-way ANOVA with the Bonferroni post hoc test. (B) The distribution of percentage MPE after the treatment of wild-type mice. The mean percentage MPEs of the mice receiving both SB216763 and morphine were significantly larger than those of mice treated with morphine alone. ***P < 0.001, one-way ANOVA with the Bonferroni post hoc test.
GSK-3β is attenuated in the mutant BiP mice
The kinase activity of GSK-3b is regulated by its phosphorylation status. Phosphorylation of residue Ser9 inactivates the activity, whereas dephosphorylation of Ser9 and phosphorylation of Tyr216 enhance the activity [14]. We evaluated the phosphorylation status of GSK-3b in the brain stems of wild-type and heterozygous mutant BiP mice using specific antibodies against phosphorylated Tyr216 GSK-3b and phosphorylated Ser9 GSK-3β.
After chronic morphine injection intraperitoneally for 5 days, the wild-type mice developed morphine tolerance, whereas the mutant BiP mice remained less tolerant to morphine. Because we injected morphine intraperitoneally, both spinal and supraspinal neurons were supposed to be affected. Neurons with MOR expression in the periaqueductal grey (PAG) matter contribute to morphine tolerance [33–35]. With repeated morphine treatment, the mutant BiP brain stems showed low levels of phosphorylation of Tyr216 in GSK-3β, in contrast to the prominent phosphorylation in wild-type mice by Western blotting (Fig. 4).
Fig 4.
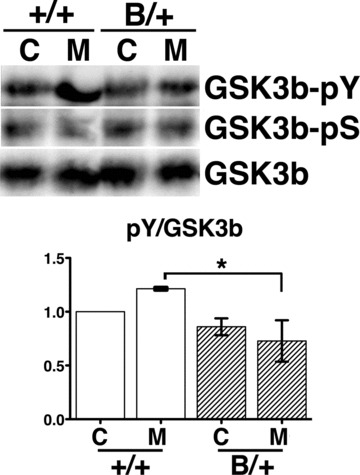
GSK3β is less activated in the mutant BiP mice with morphine treatment. The heterozygous mutant BiP mice (B/+) and wild-type mice (+/+) were injected with 20 mg/kg morphine intraperitoneally twice a day for 5 days (M) or with saline (C). The expression of phospho-tyrosine216 GSK3β (GSK3β-pY), phospho-serine9 GSK3β (GSK3β-pS) and GSK3β in brain stems from both types of mice were evaluated by Western blotting. The activation of GSK3β was assessed by comparing the relative density (arbitrary units) of the band corresponding to phospho-tyrosine216 GSK3β to the GSK3β band (lower panel, pY/GSK3β). Mean ± S.D. Three experiments. The pY/GSK3β values represent the relative values compared to those in control of the wild-type mice standardized as 1.0 in each experiment. The pY/GSK3β value of the wild-type mice were significantly larger than those of the mutant mice after morphine treatment. *P < 0.05, one-way ANOVA with the Bonferroni post hoc test.
After chronic morphine injection intraperitoneally for 5 days in both types of mice, the brains were sectioned and double immunostained with antibodies raised against MOR and tyrosine-phosphorylated GSK-3β. MOR-immunopositive neurons in the PAG region of wild-type brains showed more enhanced expression of tyrosine-phosphorylated GSK-3β significantly than those in the mutant BiP brains (Fig. 5).
Fig 5.
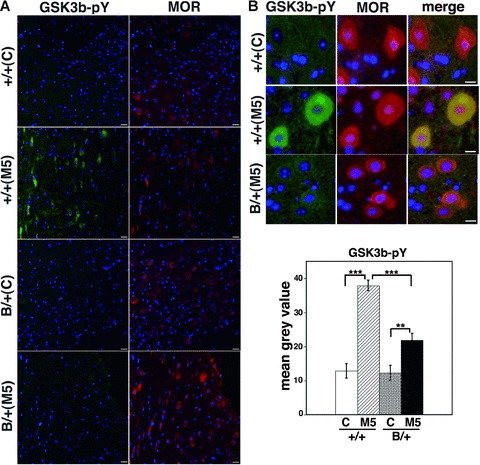
GSK3β is less activated in the mutant BiP mice with morphine treatment. The heterozygous mutant BiP mice (B/+) and wild-type mice (+/+) were injected with 20 mg/kg morphine intraperitoneally twice a day for 5 days (M5) or with saline (C). The brains were sectioned and double immunostained with anti-MOR (right panels) and anti-phospho-GSK3β (Tyr216, left panels). The nuclei were stained with Hoechst 33258. Scale bars represent 10 μm. (A) Low magnification and (B) upper panel; high magnification. MOR-immunopositive neurons in the periaqueductal grey matter of wild-type brains showed more enhanced expression of tyrosine-phosphorylated GSK3β after chronic morphine treatment significantly than those in the mutant BiP brains by densitometry (B, lower panel, arbitrary unit, +/+ C; n= 11, 1/1 M5; n= 11, B/1 C; n= 10, B/+ M5, n= 13). **P < 0.01, ***P < 0.001, one-way ANOVA with the Bonferroni post hoc test.
These observations suggest that chronic MOR stimulation by repetitive morphine injection may activate GSK-3β and that the activation of GSK-3β may be related to the development of morphine tolerance. Mice with the mutant BiP may be defective in the activation of GSK-3β.
The mutant BiP may attenuate GSK-3β in mice
In order to evaluate the effect of the deletion of the carboxyl-terminal KDEL sequence from BiP on GSK-3β activation in vivo, we examined embryonic brains of homozygous mutant BiP mice which did not express the wild-type BiP. Western blotting revealed prominent phosphorylation at Ser9 of GSK-3β and less phosphorylation at Tyr216 in the homozygous mutant BiP brain compared to those in the wild-type brain (Fig. 6). Akt inactivates GSK-3β by phosphorylation at Ser9 [23]. A similar extent of Akt phosphorylation at Ser473 was seen in embryonic brains (Fig. 6). These results suggest that the mutant BiP lacking the KDEL sequence may attenuate the activation of GSK-3βin vivo.
Fig 6.
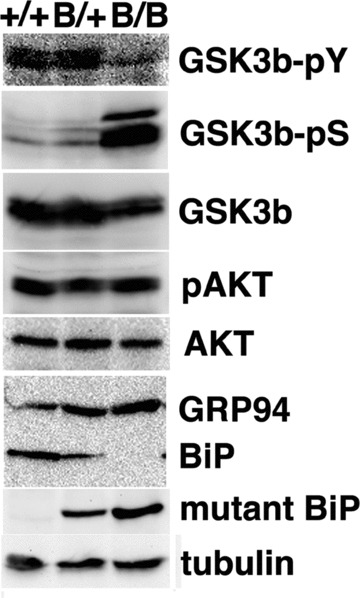
GSK-3b is attenuated in homozygous mutant BiP embryos. The expressions of phospho-tyrosine216 GSK3β (GSK3β-pY), phospho-serine9 GSK3β (GSK3β-pS), GSK3β, phospho-serine473 Akt (pAKT), Akt, GRP94, BiP, mutant BiP and g-tubulin in embryonic brains (day 18.5) from wild-type (1/1), heterozygous (B/1) and homozygous (B/B) mutant BiP mice were evaluated by Western blotting.
GSK-3β is attenuated in the mutant BiP MEF
ER stress has been reported to induce the dephosphorylation at phospho-Ser9 of GSK-3β[21]. We examined MEFs from wild-type and homozygous mutant embryos in order to evaluate the effect of ER stress on GSK-3β activation. Thapsigargin inhibits calcium transport to the ER and perturbs protein maturation in the ER, resulting in the induction of the UPR. Although the dephosphorylation at Ser9 of GSK-3β was observed in both types of MEFs upon stimulation with thapsigargin, the phosphorylation at Ser9 of GSK-3β in the homozygous mutant BiP MEFs was more prominent, suggesting that the deletion of the carboxyl-terminal KDEL sequence from BiP may attenuate GSK-3β activation (Fig. 7).
Fig 7.
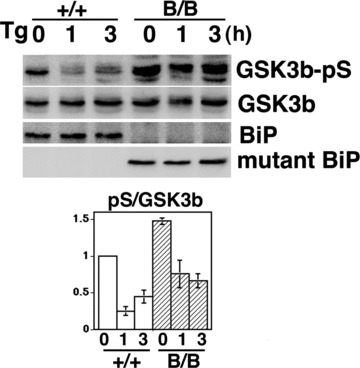
GSK3β is attenuated in homozygous mutant BiP MEFs. Thapsigargin-treated (Tg, 1 μM for 0, 1 or 3 hrs) MEFs from homozygous (B/B) and wild-type (+/+) embryos were collected. Expressions of phospho-serine9 GSK3β (GSK3β-pS), GSK3β, BiP and mutant-BiP were determined by Western blotting. The inactivation of GSK3β was assessed by comparing the relative density (arbitrary units) of the band corresponding to phospho-serine 9 GSK3β to the GSK3β band (lower panel, pS/GSK3β). Mean ± S.E., N 5 3. The pS/GSK3β values of homozygous mutant MEF were significantly larger than those of wild-type MEF. P < 0.05, two-way ANOVA.
Chemical chaperone attenuates the development of morphine tolerance
In order to confirm that an ER chaperone mediates the development of morphine tolerance, we examined the effect of a chemical chaperone on morphine tolerance. Tauroursodeoxycholic acid (TUDCA) is a derivative of endogenous bile acids that is thought to increase ER folding capacity and suppresses the expression of BiP [29, 30]. We administered TUDCA together with 20 mg/kg morphine twice a day for 5 days in wild-type mice, and hot plate tests were performed at the first and the tenth treatments. The response latencies of the mice receiving both TUDCA and morphine were significantly longer than those of control mice with morphine alone after the tenth treatment (Fig. 8A). The mean percentage MPE of mice treated with TUDCA and morphine was significantly greater than that of the mice treated with morphine alone (Fig. 8B). Thus, TUDCA prevented the development of morphine tolerance, suggesting a mechanistic relationship between an ER chaperone and morphine tolerance.
Fig 8.
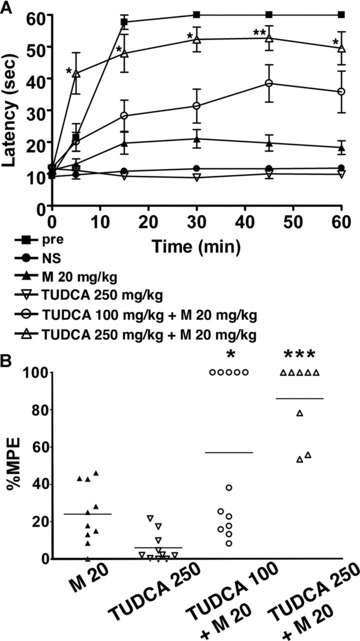
A chemical chaperone attenuates the development of morphine tolerance. Wild-type mice were injected intraperitoneally with TUDCA together with 20 mg/kg morphine twice a day for 5 days, and the hot plate test was performed to evaluate analgesia at the tenth injection on day 5. (A) The graph represents the response latencies (0–60 sec.) of the mice with saline control (NS, N= 10), morphine alone (M, N= 10), 250 mg/kg TUDCA alone (N= 10), 100 mg/kg TUDCA and morphine (N= 12), 250 mg/kg TUDCA and morphine (N= 8). ‘Pre’ represents control mice evaluated by the hot plate test after the first morphine injection on day 1 (N= 10). The response latencies of the mice receiving both TUDCA (250 mg/kg) and morphine were significantly longer than those of control mice with morphine alone after the tenth treatment. *P < 0.05, **P < 0.01, two-way ANOVA with the Bonferroni post hoc test. (B) The graph represents the distribution of percentage MPE after the treatment of wild-type mice with morphine alone (N= 10), 250 mg/kg TUDCA alone (N= 10), 100 mg/kg TUDCA and morphine (N= 12), 250 mg/kg TUDCA and morphine (N= 8). The mean percentage MPE of mice treated with TUDCA and morphine was significantly larger than that of the mice treated with morphine alone. *P < 0.05, ***P < 0.001, one-way ANOVA with the Bonferroni post hoc test.
Discussion
In this study, we showed that chronic morphine administration caused the development of morphine antinociceptive tolerance in wild-type mice, and that the inhibition of GSK-3β suppressed the development. We also found that heterozygous mutant BiP mice showed less morphine tolerance, which seemed to be related with the attenuation of GSK-3β. Furthermore, TUDCA, a chemical chaperone, suppressed the development of morphine tolerance.
BiP, one of the most abundant ER chaperones, plays a central role in ER function, assisting in protein translocation, folding and degradation, and the regulation of UPR [24]. ER chaperones are localized to the ER by two mechanisms: retention and retrieval [36]. BiP is retained in the ER through interaction with other ER proteins and the ER matrix. When misfolded proteins accumulate in the ER, BiP dissociates from some ER membrane proteins, such as inositol-requiring kinase-1, PKR-like ER-associated kinase and activating transcription factor 6. BiP dissociation activates these kinases and transcription factors, and initiates UPR [37]. When BiP is secreted from the ER along with misfolded proteins [38, 39], the carboxyl-terminal KDEL sequence of BiP is recognized by the KDEL receptor, thereby facilitating the retrieval of BiP from post-ER compartments to the ER [26, 27].
The mutant BiP protein lacks the retrieval KDEL sequence. Deletion of the retrieval sequence from BiP and the consequent lack of mutant BiP recycling could have two possible effects. First, the folding environment in the ER may be impaired. However, mutant BiP is functional as long as it remains in the ER. Therefore, constitutive activation of UPR could compensate for the altered folding environment by producing mutant BiP in quantities sufficient for cell function. Indeed, we found cell surface expression of MOR in the homozygous mutant BiP MEF. Furthermore, naive heterozygous mutant BiP mice are as sensitive as wild-type mice to thermal injury and morphine analgesia. Therefore, the impairment of protein folding in the ER may have a minimal effect on morphine analgesia in mutant BiP mice. Second, signal transduction during UPR could be affected. In addition to retrieval, the recognition of the KDEL sequence of BiP by the KDEL receptor leads to signal transduction. The activation of the KDEL receptor may trigger subsequent activation of signalling molecules such as GTPase-activating protein for ADP-ribosylation factor 1 [39], src [40], protein kinase A [41] and mitogen-activated protein kinases [31].
The persistent accumulation of misfolded proteins beyond the capacity of ER quality control mechanisms causes ER stress, necessitating UPR to accommodate the protein overload [18]. A further overload of misfolded proteins initiates apoptosis, leading to diverse human disorders [42, 43], such as neurodegenerative diseases [44, 45]. Another distinct mechanism for human disorders caused by ER stress is the alteration of signal transduction pathways during UPR. Obesity causes ER stress that induces UPR, which may cause insulin resistance in type II diabetes [19]. On the other hand, chronic morphine administration may cause altered signal transduction through persistent MOR activation. A mechanism similar to that occurring in type II diabetes would be possible in the crosstalk between MOR analgesic signal transduction and UPR, where BiP may play an important role. In this sense, TUDCA may increase ER folding capacity, and suppress the expression of BiP and other ER chaperones.
GSK-3β plays important roles in a variety of human disorders such as inflammation, Alzheimer’s disease, mood disorders, diabetes and cancer [15]. ER stress also induces the activation of GSK-3β[21, 22]. The results from homozygous mutant mice in Figs 6 and 7 suggest that the mutant BiP lacking the KDEL sequence may attenuate the function of GSK-3β. In an embryonic brain, neurons may be stimulated by various growth factors, which activates growth factor receptors, leading to the activation of the PI3K/Akt signalling pathway. Akt inactivates GSK-3β by phosphorylation at Ser9 [23]. Because a similar extent of Akt phosphorylation at Ser473 was seen in embryonic brains (Fig. 6), the effect of the BiP mutation on GSK-3β phosphorylation might be separate from the activation of the PI3K/Akt signalling pathway by growth factors. BiP is a luminal chaperone that resides inside the ER lumen and transport vesicles such as coat protein complex (COP)I and COPII coated vesicles. On the other hand, GSK-3β is a cytosolic protein that resides in the cytosol and on the cytosolic side of the membranes. Therefore, it seems difficult that BiP and GSK-3β interact directly with each other. Although the way in which BiP modulates the analgesic signal transduction through MOR needs further investigation, the regulatory mechanism between BiP and GSK-3β may possibly contribute to the development of morphine tolerance.
The present study suggests a novel function of BiP on the development of morphine tolerance in vivo. The modulation of morphine analgesia by TUDCA reveals a potential clinical application of chemical chaperones that can modulate ER functions for the prevention of morphine tolerance.
Acknowledgments
This work was supported by Grants-in-Aid for Science Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan to T.D, T.N. and T.A.
References
- 1.Somogyi AA, Barratt DT, Coller JK. Pharmacogenetics of opioids. Clin Pharmacol Ther. 2007;81:429–44. doi: 10.1038/sj.clpt.6100095. [DOI] [PubMed] [Google Scholar]
- 2.Dickenson AH, Kieffer B. Opiates: basic mechanisms. In: McMahon S, Koltzenburg M, editors. Wall and Melzack’s text book of pain. 5. Amsterdam: Elsevier; 2006. pp. 427–42. [Google Scholar]
- 3.Zhang J, Ferguson SS, Barak LS, et al. Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc Natl Acad Sci USA. 1998;95:7157–62. doi: 10.1073/pnas.95.12.7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson EE, Christie MJ, Connor M. The role of opioid receptor phosphorylation and trafficking in adaptations to persistent opioid treatment. Neurosignals. 2005;14:290–302. doi: 10.1159/000093044. [DOI] [PubMed] [Google Scholar]
- 5.Bohn LM, Lefkowitz RJ, Gainetdinov RR, et al. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286:2495–8. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- 6.Gintzler AR, Chakrabarti S. Post-opioid receptor adaptations to chronic morphine; altered functionality and associations of signaling molecules. Life Sci. 2006;79:717–22. doi: 10.1016/j.lfs.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 7.Martini L, Whistler JL. The role of mu opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr Opin Neurobiol. 2007;17:556–64. doi: 10.1016/j.conb.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Zollner C, Mousa SA, Fischer O, et al. Chronic morphine use does not induce peripheral tolerance in a rat model of inflammatory pain. J Clin Invest. 2008;118:1065–73. doi: 10.1172/JCI25911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finn AK, Whistler JL. Endocytosis of the mu opioid receptor reduces tolerance and a cellular hallmark of opiate withdrawal. Neuron. 2001;32:829–39. doi: 10.1016/s0896-6273(01)00517-7. [DOI] [PubMed] [Google Scholar]
- 10.Chakrabarti S, Regec A, Gintzler AR. Biochemical demonstration of mu-opioid receptor association with Gsalpha: enhancement following morphine exposure. Brain Res Mol Brain Res. 2005;135:217–24. doi: 10.1016/j.molbrainres.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 11.Granados-Soto V, Kalcheva I, Hua X, et al. Spinal PKC activity and expression: role in tolerance produced by continuous spinal morphine infusion. Pain. 2000;85:395–404. doi: 10.1016/S0304-3959(99)00281-X. [DOI] [PubMed] [Google Scholar]
- 12.Trujillo KA, Akil H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science. 1991;251:85–7. doi: 10.1126/science.1824728. [DOI] [PubMed] [Google Scholar]
- 13.Parkitna JR, Obara I, Wawrzczak-Bargiela A, et al. Effects of glycogen synthase kinase 3beta and cyclin-dependent kinase 5 inhibitors on morphine-induced analgesia and tolerance in rats. J Pharmacol Exp Ther. 2006;319:832–9. doi: 10.1124/jpet.106.107581. [DOI] [PubMed] [Google Scholar]
- 14.Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 15.Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res. 2007;32:577–95. doi: 10.1007/s11064-006-9128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–91. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 17.Schroder M, Kaufman RJ. The Mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 18.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 19.Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–61. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 20.Bridges JP, Xu Y, Na CL, et al. Adaptation and increased susceptibility to infection associated with constitutive expression of misfolded SP-C. J Cell Biol. 2006;172:395–407. doi: 10.1083/jcb.200508016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song L, De Sarno P, Jope RS. Central role of glycogen synthase kinase-3beta in endoplasmic reticulum stress-induced caspase-3 activation. J Biol Chem. 2002;277:44701–8. doi: 10.1074/jbc.M206047200. [DOI] [PubMed] [Google Scholar]
- 22.Qu L, Huang S, Baltzis D, et al. Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3beta. Genes Dev. 2004;18:261–77. doi: 10.1101/gad.1165804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muller DL, Unterwald EM. In vivo regulation of extracellular signal-regulated protein kinase (ERK) and protein kinase B (Akt) phosphorylation by acute and chronic morphine. J Pharmacol Exp Ther. 2004;310:774–82. doi: 10.1124/jpet.104.066548. [DOI] [PubMed] [Google Scholar]
- 24.Hendershot LM. The ER function BiP is a master regulator of ER function. Mt Sinai J Med. 2004;71:289–97. [PubMed] [Google Scholar]
- 25.Mimura N, Hamada H, Kashio M, et al. Aberrant quality control in the endoplasmic reticulum impairs the biosynthesis of pulmonary surfactant in mice expressing mutant BiP. Cell Death Differ. 2007;14:1475–85. doi: 10.1038/sj.cdd.4402151. [DOI] [PubMed] [Google Scholar]
- 26.Munro S, Pelham HR. A C-terminal signal prevents secretion of luminal ER proteins. Cell. 1987;48:899–907. doi: 10.1016/0092-8674(87)90086-9. [DOI] [PubMed] [Google Scholar]
- 27.Lewis MJ, Pelham HR. A human homologue of the yeast HDEL receptor. Nature. 1990;348:162–3. doi: 10.1038/348162a0. [DOI] [PubMed] [Google Scholar]
- 28.Mimura N, Yuasa S, Soma M, et al. Altered quality control in the endoplasmic reticulum causes cortical dysplasia in knock-in mice expressing a mutant BiP. Mol Cell Biol. 2008;28:293–301. doi: 10.1128/MCB.00473-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xie Q, Khaoustov VI, Chung CC, et al. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatology. 2002;36:592–601. doi: 10.1053/jhep.2002.35441. [DOI] [PubMed] [Google Scholar]
- 30.Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–40. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamamoto K, Hamada H, Shinkai H, et al. The KDEL receptor modulates the endoplasmic reticulum stress response through mitogen-activated protein kinase signaling cascades. J Biol Chem. 2003;278:34525–32. doi: 10.1074/jbc.M304188200. [DOI] [PubMed] [Google Scholar]
- 32.Hamada H, Suzuki M, Yuasa S, et al. Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol Cell Biol. 2004;24:8007–17. doi: 10.1128/MCB.24.18.8007-8017.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yaksh TL, Yeung JC, Rudy TA. Systematic examination in the rat of brain sites sensitive to the direct application of morphine: observation of differential effects within the periaqueductal gray. Brain Res. 1976;114:83–103. doi: 10.1016/0006-8993(76)91009-x. [DOI] [PubMed] [Google Scholar]
- 34.Bagley EE, Chieng BC, Christie MJ, et al. Opioid tolerance in periaqueductal gray neurons isolated from mice chronically treated with morphine. Br J Pharmacol. 2005;146:68–76. doi: 10.1038/sj.bjp.0706315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morgan MM, Fossum EN, Levine CS, et al. Antinociceptive tolerance revealed by cumulative intracranial microinjections of morphine into the periaqueductal gray in the rat. Pharmacol Biochem Behav. 2006;85:214–9. doi: 10.1016/j.pbb.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 36.Sonnichsen B, Fullekrug J, Nguyen Van P, et al. Retention and retrieval: both mechanisms cooperate to maintain calreticulin in the endoplasmic reticulum. J Cell Sci. 1994;107:2705–17. doi: 10.1242/jcs.107.10.2705. [DOI] [PubMed] [Google Scholar]
- 37.Bertolotti A, Zhang Y, Hendershot LM, et al. Dynamic interaction of BiP and ER stress transducers in the unfolded- protein response. Nat Cell Biol. 2000;2:326–32. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 38.Hammond C, Helenius A. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J Cell Biol. 1994;126:41–52. doi: 10.1083/jcb.126.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto K, Fujii R, Toyofuku Y, et al. The KDEL receptor mediates a retrieval mechanism that contributes to quality control at the endoplasmic reticulum. EMBO J. 2001;20:3082–91. doi: 10.1093/emboj/20.12.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bard F, Mazelin L, Pechoux-Longin C, et al. Src regulates Golgi structure and KDEL receptor-dependent retrograde transport to the endoplasmic reticulum. J Biol Chem. 2003;278:46601–6. doi: 10.1074/jbc.M302221200. [DOI] [PubMed] [Google Scholar]
- 41.Cabrera M, Muniz M, Hidalgo J, et al. The retrieval function of the KDEL receptor requires PKA phosphorylation of its C-terminus. Mol Biol Cell. 2003;14:4114–25. doi: 10.1091/mbc.E03-04-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–98. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Curr Opin Cell Biol. 2006;18:444–52. doi: 10.1016/j.ceb.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 44.Katayama T, Imaizumi K, Sato N, et al. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat Cell Biol. 1999;1:479–85. doi: 10.1038/70265. [DOI] [PubMed] [Google Scholar]
- 45.Imai Y, Soda M, Inoue H, et al. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]