

Abstract
Progranulin haploinsufficiency is associated with frontotemporal dementia in humans. Deficiency of progranulin led to exaggerated inflammation and premature aging in mice. The role of progranulin in adaptations to nerve injury and neuropathic pain are still unknown. Here we found that progranulin is up-regulated after injury of the sciatic nerve in the mouse ipsilateral dorsal root ganglia and spinal cord, most prominently in the microglia surrounding injured motor neurons. Progranulin knockdown by continuous intrathecal spinal delivery of small interfering RNA after sciatic nerve injury intensified neuropathic pain-like behaviour and delayed the recovery of motor functions. Compared to wild-type mice, progranulin-deficient mice developed more intense nociceptive hypersensitivity after nerve injury. The differences escalated with aging. Knockdown of progranulin reduced the survival of dissociated primary neurons and neurite outgrowth, whereas addition of recombinant progranulin rescued primary dorsal root ganglia neurons from cell death induced by nerve growth factor withdrawal. Thus, up-regulation of progranulin after neuronal injury may reduce neuropathic pain and help motor function recovery, at least in part, by promoting survival of injured neurons and supporting regrowth. A deficiency in this mechanism may increase the risk for injury-associated chronic pain.
Keywords: nerve injury; growth factor; microglia; neuroinflammation; pain, dorsal root ganglia; spinal cord
Introduction
Progranulin (PGRN) is a cysteine-rich pleiotropic protein affecting processes as diverse as embryonic development, wound healing, inflammation, and tumourigenesis [1-4]. PGRN has attracted a significant attention in the neuroscience research field since 2006, when loss-of-function mutations of PGRN were discovered as the cause of familial ubiquitin-positive frontotemporal dementia (FTD) [5, 6]. A reduction of PGRN by traumatic brain injury may also increase the risk for FTD [7]. Besides FTD, the plasma level of PGRN has been suggested to serve as a marker for other neurodegenerative diseases including Alzheimer’s disease [8, 9] or motor neuron degeneration [10]. Little is known about the role of PGRN in modulating persistent chronic pain after peripheral or central nerve injury or degeneration.
PGRN-deficient mice showed behavioural deficits and progressive neuropathology [11] and signs of premature aging [12]. These mice displayed exaggerated inflammation in the brain, with greater activation of microglia and astrocytes than their wild-type counterparts [13]. Because microglia plays a crucial role in the initiation of neuropathic pain [14-18], we hypothesized that PGRN may contribute to adaptive processes after nerve injury and modify the development or maintenance of neuropathic pain. Here we showed that PGRN expression was up-regulated after neuronal injury in the spared nerve injury (SNI) model of neuropathic pain. We also found that PGRN deficiency increases nociceptive hypersensitivity after nerve injury and reduces survival and outgrowth of primary neurons. PGRN may therefore contribute to favourable endogenous adaptations that help to prevent chronic pain after nerve injury.
Material and methods
Mice
Male C57BL/6 mice were purchased from the Charles River (Sulzfeld, Germany). PGRN-deficient (Grn−/−) and matched wild-type mice (Grn+/+) were generated as described [13]. Animals had free access to food and water and were maintained in climate controlled rooms at a 12-hr light–dark cycle. Behavioural experiments were performed between 10 a.m. and 1 p.m. The experiments were approved by the local Ethics Committee for animal research (Darmstadt, Germany), adhered to the guidelines for pain research in conscious animals of the International Association for the Study of PAIN (IASP) and are in line with the European and German regulations for animal research.
Culture of primary dorsal root ganglia (DRG) neurons
Primary adult dissociated DRG neuron-enriched cultures were prepared by dissecting mouse DRGs into HBSS (Ham’s balanced salt solution; Dulbecco, GibcoBRL, Karlsruche, Germany) and 10 mM HEPES, followed by digestion with 5 mg/ml collagenase A and 1 mg/ml dispase II (Roche Diagnostics, Mannheim, Germany) prior to treatment with 0.25% trypsin (GibcoBRL, Karlsruhe, Germany). Triturated cells were centrifuged through a 10% bovine serum albumin solution prior to plating on poly-L-lysine and laminin-coated cover slips in Neurobasal medium (GibcoBRL) containing 2% (v/v) B27 supplement (GibcoBRL), 50 μg/ml Pen-Strep, 10 μM Ara-C, 100 ng/ml nerve growth factor (NGF) and 200 mM l-glutamine. After incubation for 2 hrs, 2 ml complete Neurobasal medium was added and neurons were incubated for 24 hrs. Cells were kept at 37°C, 5% CO2, 95% humidity. Lentivirus particles were added at 1 moi to cultured primary neurons and incubated for 2–7 days (depending on the experiment) with half exchange of the medium at 3 days. Lentivirus transduction efficiency was analysed by enhanced green fluorescent protein (EGFP) immunofluorescence.
Nerve injury
Surgery and injections were done under 1.5–2% isoflurane anaesthesia. For the SNI model of neuropathic pain, two of the three peripheral branches of the sciatic nerve, the common peroneal and the tibial nerves, were ligated with silk (6–0) and distally cut, leaving the sural nerve intact [19]. For the chronic constriction injury of the sciatic nerve (CCI), the sciatic nerve was constricted with three silk ligatures providing about 50% constriction of the nerve diameter [20]. For the spinal nerve ligation (SNL) model, the L5 spinal nerve was sectioned. For the crush model the sciatic nerve was crushed for 25 sec. with a blunt forceps providing constant pressure without damaging the myelin sheath. In the axotomy model, the sciatic nerve was ligated and sectioned, proximal to the separation into its branches. Mechanical and cold pain sensitivity and motor functions were determined before and after nerve injury up to three months after SNI.
Treatments of interfering RNA (siRNA)
The PGRN small interfering RNA (siRNA) was administered by continuous intrathecal delivery through a spinal catheter (1.5 pmol, 0.25 μl/hr) with a subcutaneously implanted osmotic pump (Alzet model 2004; Charles River) for 4 weeks. The control group received corresponding scramble siRNA at identical infusion rates. Nine mice were used per group. To enable intrathecal delivery at the level of lumbar spinal segments, a polytetrafluoroethylene catheter (PTFE Sub-Lite Wall Tubing 0.05 mm I.D. × 0.15 mm O.D.; Braintree Scientific Inc., MA, USA) was stereotactically inserted after hemilaminectomy at S1–S2 under isoflurane anaesthesia. The tip of the catheter was positioned few millimetres proximal of L4/5. The intrathecal catheter was attached to a silicone tube, which was connected to the outlet of the Alzet mini-pump. The Alzet pump was inserted into the subcutaneous space at the left flank. Correct positioning of the catheter tip was confirmed at the end of the treatment period by microscopic inspection. Green BLOCK-iT™ Fluorescent Oligo (Invitrogen, Darmstadt, Germany) was added into siRNA and controls to visualize the correct delivery.
| Name | Start | Sense RNA sequence 5′-3′ | Region | GC% |
|---|---|---|---|---|
| NM_008175_stealth_349 | 349 | CACUGUAGUGCAGAUGGGAAAUCCU | ORF | 48 |
| NM_008175_stealth_control_349 | CACGAUCGUAGAGGUAAGUAUGCCU | 48 | ||
| NM_008175_stealth_733 | 733 | CCAAUGCCCAAUGCCAUCUGCUGUU | ORF | 52 |
| NM_008175_stealth_control_733 | CCACCCGGUAAUACCCGUCUUAGUU | 52 |
Nociception and motor functions
All tests were performed by an investigator blinded to the treatments or mouse genotypes. After habituation, we determined the latency for paw withdrawal using a Dynamic Plantar Aesthesiometer (Ugo Basile, Comerio, Italy) to assess the sensitivity to mechanical stimulation. The steel rod was pushed towards the paw with ascending force (0–5 g over a 10 sec. period, 0.2 g/sec.) and then maintained at 5 g until the paw was withdrawn. The paw withdrawal latency was the mean of three consecutive trials with at least 30 sec. intervals.
To assess the sensitivity to cold, we recorded the latency of paw licking or withdrawal on a Cold Plate at 10°C (AHP-1200CPHC; Teca, Chicago, IL, USA). We also analysed cold allodynia employing the acetone test. A drop of acetone was applied to the plantar surface of the hindpaw with help of an angled feeding tube. The mouse was sitting on a mesh floor and was observed with bottom and side mirrors. The time the mouse spent licking, flinching or shaking the paw was measured with a stopwatch during a period of 90 sec. starting immediately after acetone application.
Heat hyperalgesia was analysed in the Hargreaves test employing a radiant heat source placed underneath the paw with help of a mirror system (IITC Plantar Analgesia Meter). The heat source shuts off automatically upon withdrawal of the paw or at a pre-set cut-off time if the animal had not responded. The paw withdrawal latency was the mean of three consecutive trials with at least 30 sec. intervals.
We performed Rota Rod tests at constant speed (60 rpm) to assess sensorimotor functions and motor coordination. The time the animal kept running was determined with a stopwatch, with 1.5 min. running time as upper limit.
Quantitative RT-PCR (QRT-PCR)
Total RNA was extracted from homogenized tissue according to the protocol provided in the RNAeasy tissue Mini Kit (Qiagen, Hilden, Germany), and reverse transcribed using poly-dT as a primer to obtain cDNA fragments. QRT-PCR was performed with an ABI prism 7700 TaqMan thermal cycler (Applied Biosystems, Germany) using the Sybrgreen detection system with primer sets detailed later. Specific PCR product amplification was confirmed with gel electrophoresis. Transcript regulation was determined using the relative standard curve method according to the manufacturer’s instructions (Applied Biosystems). Amplification was achieved at 64°C for 35 cycles using two specific primer pairs for mouse PGRN:
pair-1 (spanning nucleotides 169–285; the A of ATG is referred to as nucleotide no. 1)
forward: 5′-CTAGATGGCTCCTGCCAGAC-3′; reverse: 5′-GCCATCACCACAAGACACAC-3′
pair-2: spanning nucleotides 1817–1955
forward: 5′-CCGAGGGTACCCACTACTCA-3′; reverse: 5′-GCCACAGCCTTCTTTCCATA-3′
Western blot analysis
Whole cell protein extracts were prepared in RIPA lysis buffer (Sigma, Steinheim, Germany), containing a protease inhibitor cocktail and 1 mM PMSF. Spinal cord and DRG tissue samples were homogenized in PhosphoSafe buffer (Sigma), and protease inhibitor mixture (Complete™, Roche, Germany). Proteins were separated by 12% SDS-PAGE (30 μg/lane) and transferred to nitrocellulose membranes (Amersham Pharmacia Biotech, Freiburg, Germany) by Western blotting. After blocking in Odyssey blocking buffer (LI-COR Biosciences, Bad Homburg, Germany), proteins were detected using the following antibodies directed against: PGRN N19 (N-terminal peptide N19; Santa Cruz Biotechnology, Heidelber, Germany) detects full-length PGRN and granulins 1, 2 and 7), Hsp90 (Becton Dickinson, Germany) and β-actin (Sigma) and secondary antibodies conjugated with IRDye 680 or 800 (1:10,000; LI-COR Biosciences). Primary antibodies were used at 1:500 dilutions in blocking buffer and overnight incubation at 4°C, secondary antibodies 1:1000 for 2 hrs at room temperature. To confirm the specificity of the N19 goat PGRN antibody, antigen-competition was carried out by pre-adsorbing the antibody at 1:500 with the blocking peptide (Santa Cruz Biotechnology) and with recombinant protein. In further control experiments, the antibody was pre-absorbed with protein extracts of PGRN knockout mice before use. Blots were visualized and analysed on the Odyssey Imaging System (LI-COR Biosciences). The ratio of the respective protein band to the control band was used for semi-quantitative analysis.
In situ hybridization
Freshly frozen DRGs and spinal cord were cut at 14 μm, fixed for 20 min. in 4% paraformaldehyde (PFA) in 0.1M phosphate buffered saline (PBS) and acetylated. Sense and anti-sense riboprobes for mouse PGRN (nucleotides 55–692, length 637) were obtained by cloning PCR products into the pCR4 TOPO sequencing vector (Invitrogen), and subsequently subjected to in vitro transcription and labelling with digoxigenin (Dig-labeling kit, Roche). Sections were pre-hybridized for 2 hrs at room temperature and hybridized at 70°C for 16 hrs with 200 ng/ml of sense and anti-sense probes in the pre-hybridization mix (50% formamide, 5× SSC, 5× Denhardt’s solution, 500 μg/ml herring sperm DNA, 250 μg/ml yeast tRNA) [21], washed in 0.2% SSC at 60°C and incubated with anti–Dig-AP or anti–Dig-FITC (1:1000, Roche) in 0.12M maleic acid buffer with 0.15M NaCl, pH 7.5 and 1% Blocking Reagent (Roche), washed in TBS, equilibrated in alkaline buffer (0.1M Tris-HCl, 0.1M NaCl, 0.05M MgCl2, pH 9.5, 2 mM levamisole), and developed with BM Purple AP substrate (Roche Diagnostics) or NBT/BCIP (Sigma). Slides were embedded in glycerol/gelatine or processed for post in situ immunohistochemistry and analysed on a fluorescence microscope (AxioImager, Zeiss, Germany).
Immunofluorescence
We perfused terminally anaesthetized mice transcardially with 0.9% saline followed by 4% PFA in 0.1M PBS (pH 7.4). The L4 and L5 spinal cord segments and DRGs were dissected and post-fixed for 2 hrs and then transferred into 20% sucrose in PBS for overnight cryoprotection at 4°C. The tissue was embedded in Tissue-Tek® O.C.T. Compound (Science Services, Munich, Germany) and cut in transverse sections (10 μm for DRGs, 14 μm spinal cord) on a cryotome. Sections were permeabilized for 5 min. in PBST (0.1% Triton X-100 in 0.1M PBS), blocked for 1 hr with 1% blocking reagent containing casein (Roche Diagnostics) in PBST, and incubated overnight at 4°C with primary antibodies dissolved in 1% blocking reagent in PBST. Antibodies directed against PGRN (N19, 1F5 from Abnova, Heidelberg, Germany), Iba-1 (Dianova, Hamburg, Germany), peripherin (Chemicon, Hofheim, Germany), GFAP (Sigma), ATF-3 (Santa Cruz Biotechnology) and F4/80-Cy5 (Becton Dickinson) were used. After washes in PBS, we incubated the sections for 2 hrs at room temperature with species-specific secondary antibodies conjugated with Alexa dyes (Invitrogen) or Cy3 (Sigma). To reduce lipofuscin-like autofluorescence slides were briefly immersed in 0.1% Sudan black B (in 70% ethanol) [22], rinsed in PBS and cover slipped in antifade medium. Sections were analysed on a fluorescent microscope (AxioImager).
Generation of PGRN small hairpin RNA (shRNA) expressing lentiviral constructs
The siRNA sequence was designed as described previously [23] employing an mRNA sequence of 19–23 nucleotides complimentary to the target PGRN cDNA [sequence AAG (N18–22) TT]. The first guanine base is required to recreate the +1 site of the U6 promoter in the lentiviral vector, thereby making it functional for the transcription of shRNAs. We used the lentiviral vector pLentiLox3.7 (pLL-3.7) [23]. To generate the pLL-shGrn construct, two complimentary oligonucleotides consisting of the Grn-siRNA sequence were annealed and inserted into the HpaI and XhoI sites of pLL-3.7. The oligomer annealing reaction was performed at an established thermal cycle protocol consisting of 95°C for 2 min., 65°C for 10 min., 37°C for 10 min., 20°C for 20 min. and final incubation at 4°C for 10 min. Each step was run once. We tested three different siRNAs for progranulin (sequence later). Because progranulin knockdown was most efficient with siRNA-Grn C (Fig. S1) further experiments were carried out with this one. Scramble-shGrn-pLL-3.7 and ‘empty’ pLL-3.7 were used in control experiments. After confirmation that scramble-shGrn did not affect progranulin expression (Fig. S1) we used ‘empty’ pLL-3.7 as control.
| Accession NM_008175 | Sequence of small interfering RNAs | Start | Region |
|---|---|---|---|
| siRNA-Grn a | GGGTGTGTCTTGTGGTGAT | 303 | ORF |
| siRNA-Grn b | GGCCGTGTGTTGTGAGGATCACA | 966 | ORF |
| siRNA-Grn c | GGTTGGGAATGTGGAGTGTG | 1572 | ORF |
Cloning of lentiviral vectors and generation of stable cell lines expressing PGRN
To generate lentiviral constructs expressing PGRN, full-length mouse and human PGRN cDNAs were cloned into a modified lentiviral pLL-3.7 vector (Fig. S1). The ubiquitin promoter-MCS (multiple cloning site) cassette was inserted in frame 5’ upstream of the U6 promoter of pLL-3.7 via SpeI and XhoI sites. The modified pLL-3.7 was kindly provided by Dr. Kwon (Ajou University, Suwon, South Korea) and referred to as FUMU6W-Lox3.7 (short: pUW-lox3.7). Hence, the backbone of the pLL-3.7 was unaltered so that the empty pLL-3.7 could be used as control for viruses silencing and overexpressing PGRN. The pLL-3.7 contains an open reading frame for the EGFP under a CMV promoter, making identification of transduced cells straightforward. PGRN cDNA sequences were confirmed by sequencing using the pCR4 TOPO sequencing vector (Invitrogen) before final subcloning into the lentiviral vector. F11 hybridoma cells and primary sensory neurons were transduced in foetal calf serum free medium with lentivirus particles. Transduced cells were cultured for 5 days, harvested in lysis buffer or RNA extraction buffer and monitored for PGRN expression by Western blot analysis and RT-PCR.
Virus amplification and production of high-titre lentivirus stocks
Lentiviral particles were produced by transient cotransfection of HEK 293T cells with pLL-shGrn or pUGrnW-lox3.7 together with three helper plasmids, pMDL/RRE, RSV-Rev, pMD2G-VSVG, using the calcium–phosphate method according to standard protocols [23]. For transduction, 1.8 × 106 293T cells were seeded in 6-cm tissue culture dishes the day before transduction. Fresh medium was added 2–4 hrs prior to precipitation. The transfection mix consisted of 10 μg purified endogen-free DNAs of shGrn-LL3.7 or pUGrnW-lox3.7, 5 μg of pMDLg/RRE, 5 μg RSV-Rev and 3 μg of pMD2G-VSVG per culture plate. The supernatants containing lentivirus particles were harvested at 24 and 48 hrs after transduction, passed through a 0.45-μm filter, concentrated by ultracentrifugation at 25,000 rpm for 90 min. at 48°C and resuspended in 20 μl 0.1M PBS. The titre was determined by transducing F11 cells followed by flow cytometric analysis of EGFP expression, because the parent vector pLL-3.7 expressed EGFP (Fig. S1A).
Culture of F11 hybridoma cells
F11 cells are derived by fusion of mouse DRG neurons and mouse neuroblastoma cells and they have maintained several characteristics of primary DRG neurons [24]. F11 hybridoma cells were cultured in DMEM, 10% foetal calf serum and 2 mM glutamine. All cells were kept in an incubator at 37°C, 95% humidity and 5% CO2 atmosphere.
Neurite outgrowth and survival
Neuron cultures were fixed in 4% PFA in 0.1M PBS and detected by immunostaining for NF200 and subsequent Cy3-labeled secondary antibody. Successful lentiviral transduction was assessed by EGFP immunofluorescence. Neurite outgrowth was determined by measuring the area covered by neurites, the length of the longest neurite, and total length of all neurites, the number of central neurites and the area of the cell soma. The analysis was done using the AutMess modul of AxioVision (Zeiss, Jena, Germany) and adapted to automatically detect neurites and neuronal bodies in collaboration with S.CO LifeScience GmbH (Hohenkirchen, Germany). Examples of the quality of the identification of neurites and soma are shown in Figure S2.
Cell survival was assayed by counting the number of surviving neurons and reported as percent of baseline, taken immediately before transduction. We captured each four representative images of primary neuronal cultures before transduction and then every 24 hrs up to 5 days until control cultures showed a substantial decline of the number of surviving neurons. An inverted AxioImager.Z1 fluorescence microscope was used and cell counts obtained with AxioVision 4.2 analysis software (AutMess modul).
For starvation experiments in adult primary neurons we replaced the medium at 2 hrs after plating cells with NGF-free medium with or without recombinant PGRN (100 ng/ml), NGF (200 ng/ml) or both. Images were taken daily up to 120 hrs and surviving neurons were counted as described earlier.
Statistics
We used SPSS 18.0 for statistical evaluation. Data are presented as means ± S.E.M. Time courses of behavioural data were analysed using ANOVA for repeated measurements. In case of a significant difference of the time course between genotypes, differences at individual time points were subsequently assessed with Student’s t-tests employing a Bonferroni correction of the α-level for repeated comparisons. Counts of neurons, area of neurites, QRT-PCR and Western blot results were analysed with Student's t-tests (for two groups) or one-way ANOVA and subsequent Bonferroni t-tests. P < 0.05 was considered to be statistically significant.
Results
Up-regulation of PGRN in the spinal cord and DRGs after sciatic nerve injury
A strong up-regulation of PGRN mRNA was previously observed in rats in a microarray screen to assess transcriptional changes in the DRGs and spinal cord after peripheral sciatic nerve injury [25]. We now performed QRT-PCR in mice and confirmed that there was a statistically significant increase of PGRN mRNA in the L4/5 DRGs and spinal cord ipsilateral to the sciatic nerve lesion after SNI (Fig. 1A). A protein at 47–48 kD position, detected in the Western blot using an antibody recognizing PGRN, granulin 1, 2 and 7 and intermediary fragments, was also found increased in both spinal cord and DRGs after neuron injury (Fig. 1B). The specificity of this antibody was established by pre-adsorption experiments (Fig. 1C). It is possible that this is a PGRN partial degradation product. The same PGRN product was also found up-regulated after nerve injury in other different sciatic nerve injury models 7 days after the injury as compared with sham-surgery controls (Fig. 1D). These include chronic constriction injury model, spinal nerve ligation model, crush model and axotomy model. Thus, PGRN up-regulation in response to nerve injuries seem to be a general phenomenon.
Fig 1.
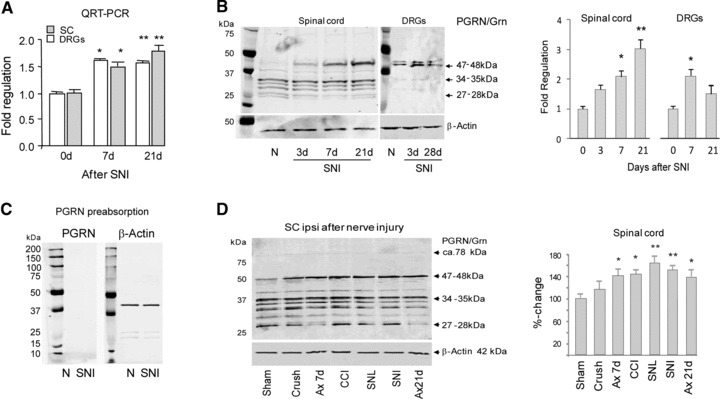
Up-regulation of PGRN expression in spinal cord (SC) and dorsal root ganglia (DRG) after nerve injury. (A) Time course of mRNA levels of PGRN in the L4/L5 DRGs and the L4/5 dorsal horn of the lumbar spinal cord ipsilateral to a sciatic nerve lesion in the SNI model by QRT-PCR (n = 3 per group). (B) Western blot analysis of PGRN in mouse SC and DRGs (n = 6 per time point) using anti-PGRN (N19); densitometric analysis is shown on the right panel. (C) Western blot analysis of PGRN in the spinal cord in naïve and SNI treated mice with N19 pre-absorbed with recombinant PGRN. (D) Western blot analysis of PGRN in the spinal cord seven days after sciatic nerve injury in five different injury models (n = 4 per model). Ax: sciatic nerve transection (axotomy). SNL: spinal nerve ligation; CCI: chronic constriction injury. Densitometric analysis is shown on the right. Results are mean ± S.E.M. *P < 0.05; **P < 0.01; ANOVA and subsequent Bonferroni t-tests.
PGRN up-regulation in microglia and neurons
We next used in situ hybridization and immunostaining to identify the location and the cell types related to the up-regulation of PGRN. In naïve mice, there was a weak expression of PGRN in neurons of the spinal cord (Fig. 2A) and the DRG (Fig. 3A), and very little PGRN was seen in microglia. After nerve injury using the SNI model, PGRN mRNA was mostly detected in the ipsilateral dorsal and ventral horn neurons and in activated microglia on the side ipsilateral to the nerve lesion (Figs 2B-D and 4A). Confocal staining using an antibody against microglia activation marker Iba-1 identified microglia as important sources of PGRN up-regulation (Fig. 2G). Interestingly, the highest levels of PGRN were found in microglia surrounding injured motor neurons that were identified by ATF-3 and peripherin staining (Fig. 2E and F). Injured DRGs also expressed an increased level of PGRN (Fig. 3B), where small neurons with unmyelinated C-fibres were identified by staining with isolectin B4-FITC (IB4) and injured neurons by anti–ATF-3. Roughly 50–60% of all DRGs neurons were PGRN positive, consistent with the number of injured neurons in the SNI model [21]. In addition, we observed a strong PGRN up-regulation in satellite glial cells (SGC) in the DRGs (Fig. 3C and D), SGCs were identified by their morphology, F4/80 and Iba-1 immunofluorescence (not shown). These findings suggest that microglia surrounding injured neurons are the major source of up-regulated PGRN.
Fig 2.
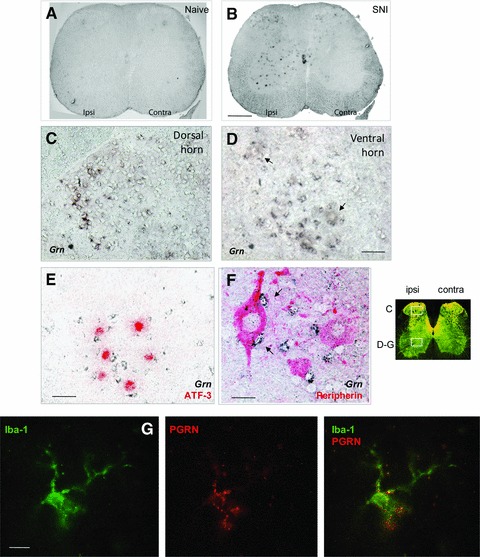
Up-regulation of PGRN expression in mouse spinal cord 7 days after SNI. (A–F) In situ hybridization of PGRN mRNA in L5 spinal cord (A, B), ipsilateral dorsal (C) and ventral horn (D–F) from naïve (A) or injured (B–F) mice. Arrows indicate motor neurons (D) and surrounding microglia (F). Injured motoneurons were identified by immunoreactivity with ATF-3 (red in E) or peripherin (red in F). (G) Co-staining of PGRN (red) and microglia activation marker Iba-1 (green) in the ventral horn of injured mice. Representative images of n = 5 mice. Scale bars: 500 μm (A, B), 50 μm (C–E), 20 μm (F) and 10 μm (G).
Fig 3.
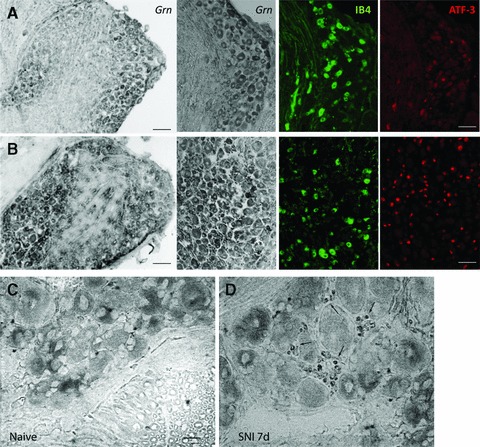
Up-regulation of PGRN expression in dorsal root ganglia seven days after SNI. In situ hybridization of PGRN mRNA in naïve (A, C) and injured (B, D) mice. Unmyelinated neurons were detected with isolectin B4-FITC (IB4, green) and injured neurons were identified by immunostaining with anti-ATF-3 (red) (A, B). Arrows in (D) point to satellite glial cells with up-regulated PGRN after SNI. Representative images of n = 5 mice. Scale bars: 100 μm (A and B, left panel), 50 μm (A and B, right three panels), 20 μm (C, D).
PGRN gene silencing intensified nociception and impaired motor function recovery
Given the strong up-regulation of PGRN in DRGs and spinal cord after injury, we hypothesized that PGRN up-regulation may be an important mechanism of endogenous pain defence. To assess the effect of PGRN down-regulation on nociceptive sensitivity after injury, we subjected mice to SNI. Then the injured mice were divided into two groups. One received continuous intrathecal delivery of PGRN siRNA for 4 weeks through a spinal catheter using a subcutaneously implanted osmotic pump. The control group received scramble oligos in the same fashion. PGRN siRNA effectively blocked the up-regulation of PGRN in the spinal cord, as determined by QRT-PCR and in situ hybridization (Fig. 4A). Nociceptive sensitivity was compared between PGRN siRNA treated and scramble-siRNA treated mice. The mice receiving PGRN siRNA showed heightened hyperalgesia, that is they were more sensitive to mechanical stimulation (Fig. 4B), spent more time licking, flinching or shaking their paws in response to acetone (Fig. 4C), and spent less time on a cold plate before withdrawing their paws (Fig. 4D). The pro-nociceptive effects of PGRN-knockdown were evident throughout the second to fourth week of the testing period and escalated towards the end. The differences in the nociceptive behaviour between siRNA treated and scramble-siRNA control groups were significant for all tests (NM_008175_stealth_349 versus control: mechanical P < 0.001; acetone P = 0.001; Cold Plate P = 0.026), although endogenous PGRN did not affect the onset (day 3 after SNI) of injury-induced hyper-nociception. Similar results were obtained with NM_008175_stealth_733 versus control (not shown).
Fig 4.
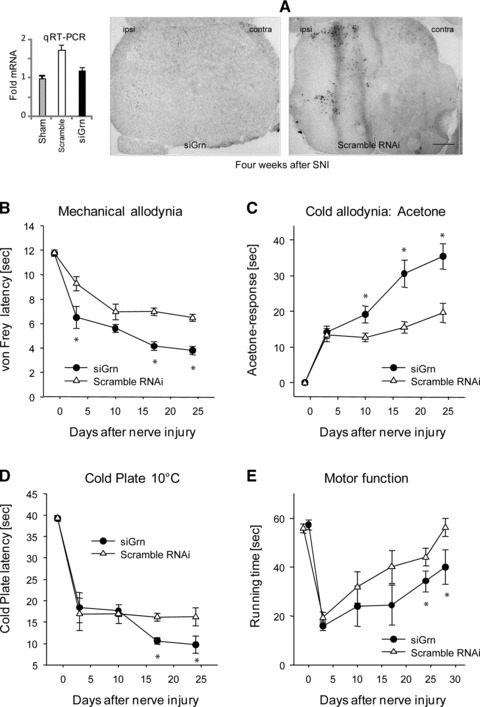
Silencing of PGRN enhanced pro-nociceptive behaviour after nerve injury. Adult male C57BL/6 mice were subjected to SNI as described in Material and methods. Mice then received continuous intrathecal delivery of PGRN siRNA or scramble oligos for four weeks (n = 9 per group) through a spinal catheter using a subcutaneously implanted osmotic pump. (A) Silencing of PGRN by siRNA in vivo. PGRN levels were measured by QRT-PCR (n = 6) (left) or in situ hybridization (right) of the L5 spinal cord in 15-weeks old male mice treated with scramble RNAi or PGRN siRNA for four weeks (NM_008175_ stealth_349). Mice were tested for their nociceptive behaviour to mechanical stimulation (B), for cold allodynia (C), or cold hyperalgesia (D) and motor functions in the RotaRod test (E) as described in Material and methods. Comparison of the time courses (ANOVA for repeated measurements) revealed statistically significant differences between groups for all tests. The asterisks indicate significant time points (P < 0.05).
We also assessed effects of PGRN silencing on motor function recovery and compensation after sciatic nerve injury in the SNI model (Fig. 4E). In the Rota Rod test, the initial drop of the running time after nerve injury did not differ between PGRN siRNA and scramble-siRNA treated mice. However, control mice rapidly regained running performance with almost complete recovery 30 days after SNI. Mice treated with PGRN siRNA only reached partial recovery within this period. The motor function recovery differed significantly between the two groups (P = 0.003). These findings suggest that PGRN has a beneficial role in motor neuron survival and function after axonal injury.
Late onset of hypernociception and impaired motor recovery in PGRN-deficient mice
siRNAs sometimes can have off-target effects [26]. To confirm that observed intensified nociception and impaired motor function recovery after injury directly resulted from a PGRN-deficiency, we then tested mice with homozygous Grn gene deletion. Adult PGRN knockout mice and their age- and sex-matched wild-type controls were subjected to SNI, and compared for their responses to mechanical or cold allodynia (Fig. 5A and B), heat hyperalgesia (Fig. 5C) and performance in the RotaRod test (Fig. 5D). The baseline nociceptive sensitivity and RotaRod running performance did not differ between PGRN-deficient and wild-type mice. Their nociceptive behaviour in the first 2 weeks after SNI was also similar. However PGRN-deficient mice developed stronger sensitivity to mechanical (Fig. 5A), cold (Fig. 5B) and heat stimulation (Fig. 5C) than wild-type mice starting about 3 weeks after the nerve injury. The differences persisted and escalated towards the end the observation period, when these mice were 6–7 months old. Statistically, nociceptive sensitivity after SNI was enhanced in PGRN-deficient mice for mechanical (F11.8, df1, P = 0.0009), heat (F4.07, df1, P = 0.0464) and cold (F13.6, df1, P = 0.0004) stimulation (Fig. 5) as compared with wild-type controls. In addition, motor function recovery after SNI was significantly impaired in PGRN-deficient mice (Fig. 5D). Wild-type mice recovered their motor function one week after the injury, whereas the recovery of the PGRN-deficient mice only reached 50% and became worse seven weeks after injury. ANOVA for repeated measurements revealed significant differences between the two genotypes (F12.71, df1, P = 0.0006).
Fig 5.
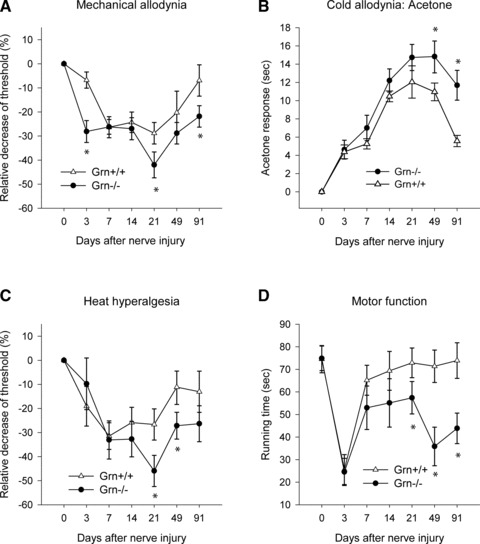
Progranulin-deficient (Grn−/−) mice showed stronger nociceptive sensitivity after nerve injury than wild-type mice. PGRN knockout (Grn−/−) mice and their wild-type control mice (n = 8 per group, four male, four female, 10–12 weeks at the time of surgery) were tested for their mechanical allodynia (A), cold allodynia (B), heat hyperalgesia (C) and motor functions (D) as in Figure 4. Data are means ± S.E.M. Comparison of the results with ANOVA revealed statistically significant differences between Grn−/− and wild-type mice for the percentage change of mechanical, cold and heat nociception and for the RotaRod running time as indicated with asterisks (P < 0.05).
PGRN down-regulation by RNA interference reduced survival of primary sensory neurons
To probe the mechanisms of PGRN-mediated anti-nociceptive effects, we altered PGRN levels in primary DRG neurons and assessed their survival and growth. Down-regulation and up-regulation of PGRN was achieved by transduction of primary neurons with lentiviral particles expressing PGRN shRNA (shGrn-LL3.7 a, b and c) or PGRN cDNA (pUGrnW-lox3.7; Fig. S1A). We tested three different PGRN shRNA constructs (Fig. S1). The efficacy of PGRN silencing or up-regulation with these lentiviral particles was confirmed in cultured F11 hybridoma cells at both mRNA and protein levels (Fig. S1B–D). Down-regulation of PGRN was most effective with shGrn-LL3.7c. We therefore used this construct for further experiments in primary neurons. Although knockdown of PGRN with shGrn-LL3.7c was very effective, increase in PGRN expression with pUGrnW-lox3.7 was only moderate. Because the parental viral vector pLL-3.7 expressed EGFP, transduction efficiency was determined by fluorescence staining. In primary mouse DRG neuron culture, 80–90% of NF200 positive cells expressed EGFP (Fig. 6A). shGrn-transduced cultures presented with a network of thin neurites extending from neurons with small somata (Fig. 6A). Representative life images of the DRG cultures 48 hrs after tranduction are shown (Fig. 6B). Primary neuron numbers declined in the culture (Fig. 6C). Transduction with viruses facilitated this decline, which was not rescued by forced expression of PGRN (Fig. 6B and C). The inactivity of transduction with pUGrnW-lox3.7 viruses may be, in part, due to their limited PGRN overexpression (Fig. 1B and D). Down-regulation of PGRN, however, significantly reduced the number of surviving neurons as compared with neurons treated with control viral particles (Fig. 6A–C). We next asked whether recombinant PGRN could rescue adult primary DRG neurons after NGF withdrawal. In the culture, the numbers of surviving neurons were reduced by 75–80% compared to the baseline 96 hrs after NGF withdrawal (Fig. 6D). Recombinant human PGRN completely restored the survival of these neurons in the absence of NGF, whereas the combination of NGF plus recombinant PGRN provided no further benefit.
Fig 6.
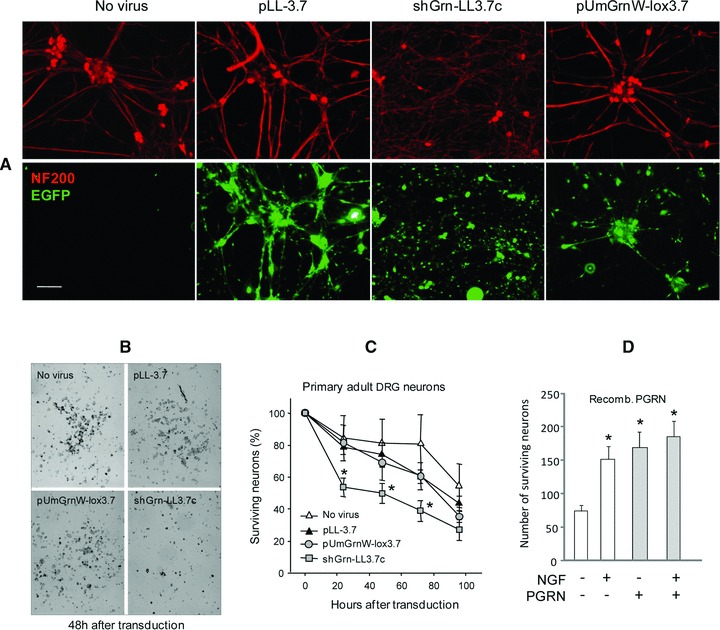
PGRN promotes survival of primary DRG neurons in the culture. (A) Immunofluorescence staining of primary DRG neurons transduced with indicated lentivirus particles and counterstained for neurofilament of 200 kD (red, NF200) and EGFP (green). (B) Representative life images of primary DRG cultures 48 hrs after transduction with indicated lentiviral particles. (C) Time courses of percentage survival of adult primary DRG neuron cultures (n = 4 cultures per treatment) after transduction with indicated viruses. Asterisks indicate significant differences between shGrn versus control virus (pLL-3.7), P < 0.05. (D) Numbers of surviving primary DRG neurons 96 hrs after NGF withdrawal from the culture medium, with or without recombinant human PGRN, as measured by counting the number of surviving neurons and expressed as percentage of baseline, taken immediately before transduction. At baseline images captured 298 ± 17 neurons per microscopic field. Data are means ± S.E.M. of triplicates. Asterisks indicate significant differences versus NGF-free/PGRN-free cultures, P < 0.05. ANOVA with subsequent Bonferroni t-tests.
Suppression of PGRN expression impaired neurite outgrowth
Axonal regeneration after injury is an indispensable component of a successful healing process. To test whether PGRN has a role in the axonal regeneration, we examined the neurite outgrowth in adult primary DRG neurons after injury under PGRN silencing or overexpression. Dissociated primary adult DRG neurons were cultured for 24 hrs to allow for initiation of neurite outgrowth. Cultured neurons were then transduced with lentiviral particles to silence or express PGRN. Figure 7A shows representative images of transduced neurons from each group. The length, width and area of the neurites and soma were scored with help of the imaging analysis software (S.CO LifeScience) and comparison was made between neurons transduced with different viral particles. Figure S2 shows examples of the quality of the automatic identification of neurites and soma from which quantitative data of length, width and area of the neurites and soma were computed. We analysed exclusively growing neurons 2 days after transduction. Quantification revealed that silencing of PGRN led to a statistically significant reduction of the overall number and length of neurites and the area of the neuronal somata (Fig. 7A and B). In contrast, overexpression of PGRN resulted in enhanced neurite outgrowth (Fig. 7A and B). Transduction with empty vector pLL-3.7 or scramble-shRNA lentivirus (not shown) had no significant effect on neuron survival or neurite outgrowth as compared to untransduced DRG cultures.
Fig 7.

PGRN supports neurite outgrowth from primary adult DRG neurons in vitro. (A) Representative images demonstrate the morphology of neurons transduced with lentivirus particles expressing pUmGrn-lox3.7 (PGRN overexpression), shGrn-pLL3.7c (PGRN silencing) or empty vector pLL-3.7. Neurons were immunostained with anti-NF200. (B) Quantitative analysis of the length and number of neurites and the area of the neurites. Only EGFP positive neurons extending neurites of >two fold length of the neuronal soma diameter were analysed. The results are shown as box plots where the line indicates the median, the box the interquartile range, whiskers 5–95th percentile and open dots individual outliers (n = 15–19 neurons per group). *P < 0.05, ANOVA with subsequent Bonferroni t-tests.
Discussion
To our knowledge, this study is the first to show that progranulin has a role in neuropathic pain defense. Peripheral or central nerve injury or degeneration is a frequent cause of persistent chronic, pathological pain. Particularly, the elderly have a high risk of post-zoster, ischaemic or metabolic neuralgias with persistent and substantial impairment of quality of life. A loss of axonal transport of growth factors from the peripheral target region to the soma of a sensory neuron contributes to the death of subsets of primary and secondary neurons [27-30]. Surviving neurons develop a state of hyperexcitability and enhancement of the synaptic transmission of nociceptive input [31], giving rise to the development of neuroinflammation [17] and neuropathic pain [14,18]. Endogenous mechanisms that attenuate or prevent secondary neuronal damage are essential to regain synaptic stability and to prevent the development of irreversible nociceptive hypersensitivity. It is still incompletely understood why neuropathic pain persists in some people and why this risk increases with age.
We showed in this study that injury to the peripheral nerve increased PGRN expression in injured neurons and glial cells at both the mRNA and protein levels. Our findings are consistent with the report by Matzilevich et al. that PGRN was up-regulated in a high-density microarray analysis of hippocampal gene expression following traumatic brain injury [32]. We found that microglial cells surrounding the injured neurons were the major source of up-regulated PGRN in the SNI model, which is reminiscent of the effect seen after axotomy of motor neurons [33], or in the mid-thoracic contusion spinal cord injury model [34]. These authors found elevated levels of PGRN throughout the injury epicentre, and co-localization of PGRN staining with myeloid cell marker CD11b and CD68. In addition, we observed a strong PGRN up-regulation in SGC in the DRGs. These cells have been suggested to be activated after nerve injury [35-37]. Although considered as originated from the neuroectoderm, SGC resemble microglia and dendritic cells in the expression of myeloid marker proteins such as Iba-1 and CD11b [38] and might act as PGRN-secreting cells.
Up-regulation of PGRN after injury appears to be physiologically important at least for endogenous pain defense. When this up-regulation was abolished by RNA interference or Grn gene deletion, we found injury-induced late-phase nociception becoming much intense and the recovery of motor functions delayed. Thus, an enhanced production of PGRN likely constitutes an endogenous adaptation that helps to fend off chronic pain and help motor function recovery. PGRN may be particularly important for neuronal survival under challenging conditions such as those evoked by axonal injury. Sufficient supply with PGRN in vivo may rely on a concomitant PGRN up-regulation in injured neurons, surrounding microglia and SGCs. Despite this up-regulation however, animals still developed nociceptive hypersensitivity; and PGRN knockout animals had normal nociceptive responses in the early phase after nerve injury. These findings suggest that PGRN may not directly modulate the excitability of nociceptive neurons, but rather play an important role in facilitating the recovery of injured neurons, hence preventing persistence of chronic neuropathic pain associated with injuries.
The mechanisms by which PGRN modulates pain responses remain unclear. As a secreted protein PGRN can act as an autocrine and paracrine growth factor by inducing signals via a putative membrane-bound receptor. The β-hairpin structure of granulin peptides has a substantial similarity to epidermal growth factor [39]. In fact, PGRN was originally named as the PC-cell derived growth factor because of its mitogenic activity towards a teratoma-derived adipogenic cell line [40]. A recent study indicated recombinant PGRN could function as a neurotrophic factor in promoting survival and neurite outgrowth of motor neurons in vitro [41]. Consistent with this observation, we showed here that suppression of PGRN with siRNA specifically reduced the survival of primary DRG neurons and impaired their neurite outgrowth. Another route for PGRN to enter neurons is by transmembrane transport and sorting to intracellular sites via sortilin [42]. It was suggested that PGRN might act as a cargo for proteins undergoing lysosomal or autophagosomal degradation and contribute to the intracellular clearance of misfolded or dysfunctional proteins such as phosphorylated TDP-43 [43]. Whether PGRN/sortilin interaction has a role in modulating pain defense after nerve injury remains to be determined.
The brain pathology of PGRN knockout mice revealed an age-dependent exaggerated activation of microglia and astrocytes, together with an increase in various pro-inflammatory factors including interleukin-6 and macrophage chemoattractant factor (MCP-1/CCL2) [13]. These factors are involved in the transformation of protective microglia into phagocyte-like microglia in the spinal cord after sciatic nerve injury [44] and promote the development of neuropathic pain [45-47]. Enhanced levels of PGRN in response to injury may thus be particularly important in maintaining a balanced, well-controlled inflammatory homeostasis in the CNS. Insufficient PGRN supply, particularly at the injured lesion may lead to exaggerated inflammation and subsequent enhancement of nociceptive responses.
In summary, we showed that PGRN expression was induced in the spinal cord and DRGs after nerve injury in a mouse SNI model. Blockage of this induction by either siRNA or gene deletion led to an enhancement of nociceptive responses to mechanical, cold or heat stimulation. PGRN up-regulation may thus represent one of the endogenous adaptive mechanisms that help to prevent chronic pain through promoting survival and regrowth of injured neurons.
Acknowledgments
We thank Dr. Kwon (Ajou University, Suwon, South Korea) for the lentiviral construct, FUMU6W-Lox3.7. We acknowledge the financial support of the Deutsche Forschungsgemeinschaft (SFB 815 A12 and CRC 971, IT), the LOEWE Lipid Signaling Forschungszentrum Frankfurt (LiFF), the Heinrich and Fritz Riese foundation and the Interdisciplinary Center of Neuroscience Frankfurt (ICNF).
Conflict of interest
The authors confirm that there are no conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Schematic diagram of the modified lentiviral vector, pLL-3.7 used for overexpression of progranulin. The ubiquitin promoter-MCS (multiple cloning site) cassette was inserted in frame 5′ upstream of the U6 promoter of pLL-3.7 via SpeI and XhoI sites.
Fig S2 Representative images showing the automated identification of neurites (yellow) and neuronal body (red) from which neurite area, total length, number of central neurites, neurite thickness, and area, diameter and circumference of the soma were calculated.
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Bateman A, Bennett HP. The granulin gene family: from cancer to dementia. Bioessays. 2009;31:1245–54. doi: 10.1002/bies.200900086. [DOI] [PubMed] [Google Scholar]
- 2.Zhu J, Nathan C, Jin W, et al. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell. 2002;111:867–78. doi: 10.1016/s0092-8674(02)01141-8. [DOI] [PubMed] [Google Scholar]
- 3.He Z, Ong CH, Halper J, et al. Progranulin is a mediator of the wound response. Nat Med. 2003;9:225–9. doi: 10.1038/nm816. [DOI] [PubMed] [Google Scholar]
- 4.Daniel R, Daniels E, He Z, et al. Progranulin (acrogranin/PC cell-derived growth factor/granulin-epithelin precursor) is expressed in the placenta, epidermis, microvasculature, and brain during murine development. Dev Dyn. 2003;227:593–9. doi: 10.1002/dvdy.10341. [DOI] [PubMed] [Google Scholar]
- 5.Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–4. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 6.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–9. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 7.Jawaid A, Rademakers R, Kass JS, et al. Traumatic brain injury may increase the risk for frontotemporal dementia through reduced progranulin. Neurodegener Dis. 2009;6:219–20. doi: 10.1159/000258704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sleegers K, Brouwers N, Van Broeckhoven C. Role of progranulin as a biomarker for Alzheimer’s disease. Biomark Med. 2010;4:37–50. doi: 10.2217/bmm.09.82. [DOI] [PubMed] [Google Scholar]
- 9.Sleegers K, Brouwers N, Van Damme P, et al. Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol. 2009;65:603–9. doi: 10.1002/ana.21621. [DOI] [PubMed] [Google Scholar]
- 10.Philips T, De Muynck L, Thu HN, et al. Microglial upregulation of progranulin as a marker of motor neuron degeneration. J Neuropathol Exp Neurol. 2010;69:1191–200. doi: 10.1097/NEN.0b013e3181fc9aea. [DOI] [PubMed] [Google Scholar]
- 11.Yin F, Dumont M, Banerjee R, et al. Behavioral deficits and progressive neuropathology in progranulin-deficient mice: a mouse model of frontotemporal dementia. Faseb J. 2010;24:4639–47. doi: 10.1096/fj.10-161471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmed Z, Sheng H, Xu YF, et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggests a rold for progranulin in successful aging. Am J Pathol. 2010;177:311–24. doi: 10.2353/ajpath.2010.090915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yin F, Banerjee R, Thomas B, et al. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010;207:117–28. doi: 10.1084/jem.20091568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–8. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- 15.Coull JA, Beggs S, Boudreau D, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–21. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 16.Tsuda M, Shigemoto-Mogami Y, Koizumi S, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–83. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- 17.Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci. 2005;28:101–7. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 18.Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–58. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- 20.Bennett GJ, Chung JM, Honore M, et al. Models of neuropathic pain in the rat. Curr Protoc Neurosci. 2003. Chapter 9: Unit 9.14. [DOI] [PubMed]
- 21.Tegeder I, Costigan M, Griffin RS, et al. GTP cyclohydrolase and tetrahydrobiopterin regulate pain sensitivity and persistence. Nat Med. 2006;12:1269–77. doi: 10.1038/nm1490. [DOI] [PubMed] [Google Scholar]
- 22.Schnell SA, Staines WA, Wessendorf MW. Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J Histochem Cytochem. 1999;47:719–30. doi: 10.1177/002215549904700601. [DOI] [PubMed] [Google Scholar]
- 23.Rubinson DA, Dillon CP, Kwiatkowski AV, et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–6. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- 24.Goswami C, Hucho T. TRPV1 expression-dependent initiation and regulation of filopodia. J Neurochem. 2007;103:1319–33. doi: 10.1111/j.1471-4159.2007.04846.x. [DOI] [PubMed] [Google Scholar]
- 25.Griffin RS, Costigan M, Brenner GJ, et al. Complement induction in spinal cord microglia results in anaphylatoxin C5a-mediated pain hypersensitivity. J Neurosci. 2007;27:8699–708. doi: 10.1523/JNEUROSCI.2018-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- 27.Mannion RJ, Costigan M, Decosterd I, et al. Neurotrophins: peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc Natl Acad Sci U S A. 1999;96:9385–90. doi: 10.1073/pnas.96.16.9385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson-Gerwing TD, Dmyterko MV, Zochodne DW, et al. Neurotrophin-3 suppresses thermal hyperalgesia associated with neuropathic pain and attenuates transient receptor potential vanilloid receptor-1 expression in adult sensory neurons. J Neurosci. 2005;25:758–67. doi: 10.1523/JNEUROSCI.3909-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chuang HH, Prescott ED, Kong H, et al. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957–62. doi: 10.1038/35082088. [DOI] [PubMed] [Google Scholar]
- 30.Hoheisel U, Unger T, Mense S. Excitatory and modulatory effects of inflammatory cytokines and neurotrophins on mechanosensitive group IV muscle afferents in the rat. Pain. 2005;114:168–76. doi: 10.1016/j.pain.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 31.Scholz J, Broom DC, Youn DH, et al. Blocking caspase activity prevents transsynaptic neuronal apoptosis and the loss of inhibition in lamina II of the dorsal horn after peripheral nerve injury. J Neurosci. 2005;25:7317–23. doi: 10.1523/JNEUROSCI.1526-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matzilevich DA, Rall JM, Moore AN, et al. High-density microarray analysis of hippocampal gene expression following experimental brain injury. J Neurosci Res. 2002;67:646–63. doi: 10.1002/jnr.10157. [DOI] [PubMed] [Google Scholar]
- 33.Moisse K, Volkening K, Leystra-Lantz C, et al. Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: implications for TDP-43 in the physiological response to neuronal injury. Brain Res. 2009;1249:202–11. doi: 10.1016/j.brainres.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 34.Naphade SB, Kigerl KA, Jakeman LB, et al. Progranulin expression is upregulated after spinal contusion in mice. Acta Neuropathol. 2010;119:123–33. doi: 10.1007/s00401-009-0616-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanani M. Satellite glial cells in sensory ganglia: from form to function. Brain Res Brain Res Rev. 2005;48:457–76. doi: 10.1016/j.brainresrev.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 36.Capuano A, De Corato A, Lisi L, et al. Proinflammatory-activated trigeminal satellite cells promote neuronal sensitization: relevance for migraine pathology. Mol Pain. 2009;5:43. doi: 10.1186/1744-8069-5-43. Doi: 10.1186/1744-8069-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cherkas PS, Huang TY, Pannicke T, et al. The effects of axotomy on neurons and satellite glial cells in mouse trigeminal ganglion. Pain. 2004;110:290–8. doi: 10.1016/j.pain.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 38.van Velzen M, Laman JD, Kleinjan A, et al. Neuron-interacting satellite glial cells in human trigeminal ganglia have an APC phenotype. J Immunol. 2009;183:2456–61. doi: 10.4049/jimmunol.0900890. [DOI] [PubMed] [Google Scholar]
- 39.Hrabal R, Chen Z, James S, et al. The hairpin stack fold, a novel protein architecture for a new family of protein growth factors. Nat Struct Biol. 1996;3:747–52. doi: 10.1038/nsb0996-747. [DOI] [PubMed] [Google Scholar]
- 40.Zhou J, Gao G, Crabb JW, et al. Purification of an autocrine growth factor homologous with mouse epithelin precursor from a highly tumorigenic cell line. J Biol Chem. 1993;268:10863–9. [PubMed] [Google Scholar]
- 41.Van Damme P, Van Hoecke A, Lambrechts D, et al. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol. 2008;181:37–41. doi: 10.1083/jcb.200712039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu F, Padukkavidana T, Vaegter CB, et al. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68:654–67. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang YJ, Xu YF, Dickey CA, et al. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci. 2007;27:10530–4. doi: 10.1523/JNEUROSCI.3421-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michelucci A, Heurtaux T, Grandbarbe L, et al. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol. 2009;210:3–12. doi: 10.1016/j.jneuroim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 45.Zhang J, Shi XQ, Echeverry S, et al. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27:12396–406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thacker MA, Clark AK, Bishop T, et al. CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur J Pain. 2009;13:263–72. doi: 10.1016/j.ejpain.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 47.Ledeboer A, Sloane EM, Milligan ED, et al. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Schematic diagram of the modified lentiviral vector, pLL-3.7 used for overexpression of progranulin. The ubiquitin promoter-MCS (multiple cloning site) cassette was inserted in frame 5′ upstream of the U6 promoter of pLL-3.7 via SpeI and XhoI sites.
Fig S2 Representative images showing the automated identification of neurites (yellow) and neuronal body (red) from which neurite area, total length, number of central neurites, neurite thickness, and area, diameter and circumference of the soma were calculated.