

Abstract
Chrysin is a natural and biologically active flavonoid with anticancer effects. However, little is known about the adaptive response of cancer cells to chrysin. Chrysin reportedly has proteasome inhibitor activity. Previous studies demonstrated that proteasome inhibitors might induce endoplasmic reticulum (ER) stress response. In this study, we aimed to determine the effects of chrysin on hepatoma cells and roles of the ER-resident protein GRP78 (glucose-regulated protein 78) in its action. Also, we investigated the effects of green tea polyphenol (-)-epigallocatechin-3-gallate (EGCG), a natural GRP78 inhibitor, on the sensitivity of hepatoma cells to chrysin. Here, we report that chrysin inhibits hepatoma cells growth and induces apoptosis in a dose-dependent manner. Chrysin induces GRP78 overexpression, X-box binding protein-1 splicing and eukaryotic initiation factor 2α phosphorylation, hallmarks of the unfolded protein response. GRP78 knockdown potentiates chrysin-induced caspase-7 cleavage in hepatoma cells and enhances chrysin-induced apoptosis. EGCG overcomes chrysin-induced GRP78 expression. Combination of EGCG potentiates chrysin-induced caspase-7 and poly (ADP-ribose) polymerase (PARP) cleavage. Finally, EGCG sensitizes hepatoma cells to chrysin through caspase-mediated apoptosis. These data suggest that chrysin triggers the unfolded protein response. Abrogation of GRP78 induction may improve the anticancer effects of chrysin. Combination of EGCG and chrysin represents a new regimen for cancer chemoprevention and therapeutics.
Keywords: chrysin, endoplasmic reticulum stress, EGCG, glucose-regulated protein 78, hepatoma
The stress-inducible glucose-regulated protein 78 (GRP78) is a multifunctional protein overexpressed in a variety of cancer cells [1]. In progressively growing tumours, GRP78 is highly induced by endoplasmic reticulum (ER) stress stimuli such as hypoxia, glucose starvation and low pH. Overexpression of GRP78 can stimulate tumour cells proliferation, survival, invasion and metastasis [2]. As an ER-resident chaperone, GRP78 interacts with transmembrane ER stress sensors such as inositol restriction enzyme 1, pancreatic ER kinase (PERK) and activation transcription factor 6 and control their activation; maintain Ca2+ homeostasis and target misfolded proteins for proteasomal degradation [3]. Moreover, GRP78 may help to maintain ER integrity and inhibit ER-stress-induced apoptosis by preventing the activation of several pro-apoptosis molecules such as caspase-4, caspase-7 and Bik [4–6]. ER stress-induced autophagy is also dependent on GRP78 [7]. In addition to responding to the pathophysiological insults that disturb ER homeostasis, GRP78 confers resistance to chemotherapeutic drugs such as adriamycin, etoposide, temozolomide and fluorouracil [5, 8]. GRP78 expression can be induced by anticancer agents such as microtubules-targeting agents [9], Hsp70 inhibitor [10], histone deacetylase inhibitor [11] and anti-angiogenesis inhibitors [12, 13]. Blockade of GRP78 may sensitize cancer cells to these agents.
GRP78 is one of the targets for (-)-Epigallocatechin gallate (EGCG), a green tea polyphenol with cancer chemopreventive potential [14]. Mounting studies showed that EGCG inhibited the survival rate of malignant cells and induced apoptosis of malignant cells via the mitochondrial signal transduction pathway [15–17]. As a natural compound, EGCG is a promising agent for cancer chemoprevention. Also, EGCG may be of utility in cancer chemotherapeutics. Preclinical studies demonstrated that EGCG could sensitize tumour cells to temozolomide [8], quercetin [10], TNF-related apoptosis-inducing ligand (TRAIL) [18], paclitaxel and vinblastine [9, 19]. EGCG is a natural inhibitor of GRP78 ATPase activity [14]. As a GRP78 inhibitor, EGCG reportedly overcame resistance to ER stress-induced cell death in vitro[14]. In addition to GRP78, EGCG can target other molecules such as IGF-IR [20], Hsp90 [21] and BCL-2 [22], which play important roles in cell growth and survival.
Chrysin (5,7-dihydroxyflavone) is a natural and biologically active flavonoid in many plants, honey and propolis. Previous studies have demonstrated that chrysin possesses potent anti-inflammatory and anti-oxidant properties [23, 24]. As an anti-oxidant, chrysin possesses vitamin-like activity in the body. Inhibition of the Cox-2 pathway is a mechanism underlying chrysin anti-inflammatory activity [24]. In light of these beneficial activities, chrysin has been described as disease-preventing dietary supplement. More importantly, chrysin may promote cancer cell death or perturb cell cycle progression [25, 26]. Although chrysin have activity as cancer-preventive agents, the molecular mechanisms underlying chrysin anticancer effects remain to be understood. Meanwhile, the elucidation of cellular adaptive response to chrysin may provide a novel strategy to sensitize cancer cells to this bioflavonoid. The combination of chrysin and other natural products or chemotherapeutic agents may be more effective in treating selective types of cancer.
Here, we provide evidence that chrysin inhibits hepatoma cell growth. The unfolded protein response is induced by chrysin in hepatoma cells. Chrysin induces the expression of GRP78. GRP78 knockdown sensitizes hepatoma cells to chrysin by potentiating chrysin-induced apoptosis. EGCG overcomes chrysin-induced GRP78 expression and enhances chrysin-induced cell death.
Materials and methods
Reagents
Chrysin and EGCG were purchased from MUST Biotech. (Chengdu, China). The caspase inhibitor z-VAD-fmk was from Beyotime Institute of Biotechnology (Jiangsu, China). Anti-GRP78, anti-β-actin and anti-XBP-1 (X-box binding protein-1) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-phosphorylated eukaryotic initiation factor 2α (eIF2α), anti-caspase-7 and anti-PARP antibodies were provided by Cell Signaling Technology, Inc. (Beverly, MA, USA).
Cell culture
Hepatoma cells HepG2 and SMMC-7721 were purchased from Cell Lines Bank, Chinese Academy of Science (Shanghai, China). Cells were grown in tissue culture flasks at 37°C in a humidified atmosphere of 5% CO2 and were maintained as monolayer cultures in DMEM supplemented with 5% foetal bovine serum and 100 U/ml penicillin and 100 μg/ml streptomycin.
Transfection of siRNA
The target sequence used for knockdown of GRP78 was 5′-GGAGCGCAUUGAUACUAGA-3′. The negative control siRNA was purchased from Ribobio Co., Ltd. (Guangzhou, China). The double-stranded siRNA duplex was dissolved in DEPC-treated water. For transfection, 5000 cells were plated into 24-well plates and incubated for two days. LipofecTAMINE 2000 reagent (Invitrogen, Carlsbad, CA, USA) was diluted in 50 μl of Opti-MEM I reduced serum medium and incubated at room temperature for 5 min. In addition, siRNA duplex was diluted in 50 μl of Opti-MEM I reduced serum medium and mixed with the pre-diluted LipofecTAMINE 2000. The mixture was incubated at room temperature for 20 min., 50 nmol/l of siRNA was added into each well and incubated at 37°C.
Western blotting
Cells were washed twice with phosphate buffered saline and harvested with cold lysis buffer containing protease inhibitors. Cell lysates were collected from culture plates using a rubber policeman, and protein collected by centrifugation. Protein concentrations were determined by bicinchoninic acid (BCA) protein assay (Pierce Biotechnology, Rockford, IL, USA). Forty micrograms of total protein were boiled in 2× loading buffer (0.1 M Tris-Cl, pH 6.8, 4% SDS, 0.2% bromophenyl blue, 20% glycerol) for 10 min., then loaded into Tris-HCl-Polyacrylamide gels and transferred electrophoretically to Immobilon-P membrane (Millipore Corporation, Billerica, MA, USA). Membranes were incubated with primary antibodies and appropriate horseradish peroxidase (HRP)-linked secondary antibodies. Membranes were additionally probed with an antibody against actin to normalize loading of protein among samples. The secondary antibodies were detected by chemiluminescent agents (Pierce Biotechnology).
Cell viability assay
Hepatoma cells were plated in 96-well plates at 5000 cells per well. The next day, cells were treated with or without chrysin and EGCG in four replicates. After 48 hrs, cell viability was assessed by incubating cells with CCK-8 (Cell Counting Kit-8) reagents (Dojindo Laboratory Co., Ltd., Kumamoto, Japan) for 2–4 hrs and measuring the absorbance at 490 nm, and at 630 nm as reference, with a microplate reader (Bio-Rad, Hercules, CA, USA).
Cell apoptosis assay
Cell apoptosis was assessed by the Hoechst 33342 (Sigma-Aldrich, Inc., St. Louis, MO, USA) staining. Briefly, replicate cultures of hepatoma cells were plated in tissue culture plates. The cells were treated with or without chrysin and EGCG. After a change of fresh medium 24 or 48 hrs later, the cells were incubated with Hoechst 33342 solution at 37°C for 10 min., followed by examination under a fluorescence microscope. Strong fluorescence and condensed or fragmented nuclei can be observed in the nuclei of apoptotic cells, while weak fluorescence was observed in live cells. Quantification of apoptotic cells was performed by taking the images in random fields and counting at least 200 cells in four random fields in each well.
Statistical analysis
One-way anova with least significant difference post hoc test was used to test for the differences in cell viability and apoptosis rate. All statistical tests were two-tailed, and difference to be considered statistically significant when P < 0.05.
Results
Chrysin inhibits hepatoma cell growth
To determine the effects of chrysin on hepatoma cell growth, HepG2 cells were treated with increasing doses of chrysin ranging from 2.5 to 40 μM for 48 hrs, followed by CCK-8 assay. Obvious change in cell shape was also detected after chrysin treatment. Whereas the untreated HepG2 cells displayed the cubic cell shape, many spindle cells were observed in chrysin-treated cells (Fig. 1A). Chrysin inhibited HepG2 cell growth in a dose-dependent manner (Fig. 1B). In addition, the inhibitory effects of chrysin on SMMC-7721 cells were observed, while SMMC-7721 cells were less sensitive to chrysin than HepG2 cells (Fig. 1C).
Fig 1.
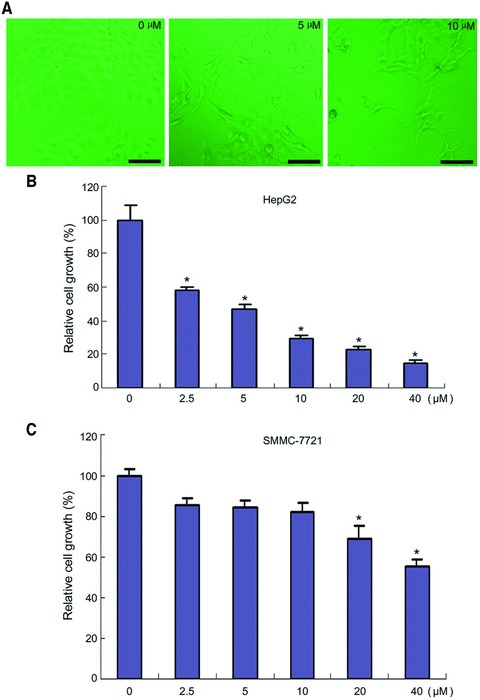
Inhibition of hepatoma cells growth by chrysin. (A) HepG2 cells were treated with chrysin at the indicated dosages for 48 hrs. The cells were observed under a phase-contrast microscopy. Bar: 100 μM. (B) HepG2 cells were seeded in a 96-well plate at 5000 cells per well. The next day, the cells were treated with chrysin at the indicated dosages for 48 hrs. Cell viability was assessed by CCK-8 assay. The relative cell growth was plotted. Cell viability in vehicle-treated group was set as 100%. Points: mean of four replicates; bars: S.E. *P < 0.05 versus control. (C) SMMC-7721 cells were seeded in a 96-well plate at 5000 cells per well. The next day, the cells were treated with chrysin at the indicated dosages for 48 hrs. Cell viability was assessed by CCK-8 assay. The relative cell growth was plotted. Cell viability in vehicle-treated group was set as 100%. Points: mean of four replicates; bars: S.E. *P < 0.05 versus untreated control. A representative of three experiments was shown.
Next, we investigated whether chrysin induced hepatoma cell apoptosis. HepG2 cells were treated with increasing concentration of chrysin ranging from 2.5 to 40 μM for 48 hrs, followed by Hoechst 33342 staining. Although HepG2 cells growth was significantly inhibited by 2.5 μM chrysin (Fig. 1B), the apoptosis was not induced until the dosage of chrysin reached 10 μM. Increasing apoptosis rate was detected when the cells were treated with chrysin at higher dosages (Fig. 2). These data suggested that chrysin inhibited cellular proliferation at relatively lower concentration, and induced apoptosis at relatively higher concentration.
Fig 2.
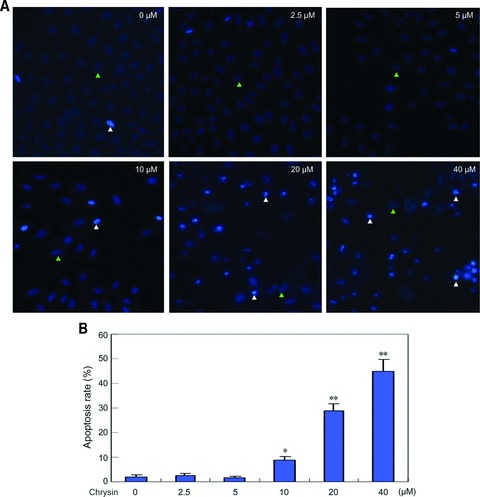
Induction of hepatoma cells apoptosis by chrysin. (A) HepG2 cells were treated with chrysin at the indicated dosages for 48 hrs, and apoptosis was assessed by Hoechst 33342 staining. The apoptotic cells with strong fluorescence, fragmented or condensed nuclei were observed under fluorescent microscopy. White triangle: representative of normal nuclei; green triangle: representative of apoptotic nuclei. (B) Quantification of apoptotic cells was performed by taking the images in random fields and counting apoptotic nuclei. The apoptosis rate was plotted. Columns, mean percentage of apoptotic cells; bars: S.E. *P < 0.05 versus untreated control. **P < 0.001 versus untreated control. A representative of three experiments was shown.
Chrysin induces the unfolded protein response
Previous study indicated that chrysin possessed proteasome inhibitor activity [27]. Because proteasome inhibitor may induce ER stress, we investigated whether chrysin would induce ER stress or the unfolded protein response in cancer cells. HepG2 cells were treated with different doses of chrysin for 24 hrs, and then subjected to Western blot analysis. The results revealed that GRP78 expression was stimulated by chrysin in a dose-dependent manner (Fig. 3A). Similar effects were observed in another hepatoma cell line SMMC-7721 (Fig. 3A).
Fig 3.
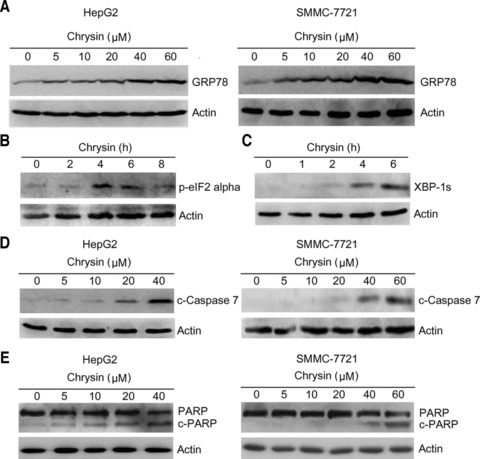
Induction of the unfolded protein response by chrysin. (A) HepG2 and SMMC-7721 cells were treated with chrysin at the indicated dosages for 24 hrs. Cell lysates were harvested and subjected to Western blot analysis with anti-GRP78 antibody. β-actin was probed as a loading control. (B) HepG2 cells were treated with 20 μM chrysin for indicated periods. Total proteins were harvested and subjected to Western blot analysis of the phosphorylated eIF2α. (C) HepG2 cells were treated with 20 μM chrysin for indicated periods. Total proteins were harvested and subjected to Western blot analysis of the spliced XBP-1 (XBP-1s). (D) HepG2 and SMMC-7721 cells were treated with indicated doses of chrysin for 24 hrs. Total proteins were harvested and subjected to Western blot analysis of cleaved caspase-7. (E) HepG2 and SMMC-7721 cells were treated with indicated doses of chrysin for 24 hrs. Total proteins were harvested and subjected to Western blot analysis of PARP.
Next, we investigated the effects of chrysin on other ER stress responsive proteins including phosphorylated eIF2α and spliced XBP-1. During the unfolded protein response, a transient translation arrest is induced upon phosphorylation of eIF2α by PERK. The phosphorylation of eIF2α was observed after treatment of HepG2 cells with chrysin for 4 hrs (Fig. 3B). In addition, treatment of HepG2 cells with chrysin induced XBP-1 splicing, a hallmark of the unfolded protein response (Fig. 3C). Thus, chrysin is identified as an inducer of the unfolded protein response.
Caspase-7 is one of executioner caspases that mediate ER stress-induced apoptosis. To determine whether chrysin induce caspase-7 cleavage, HepG2 and SMMC-7721 cells were treated with different doses of chrysin for 24 hrs, followed by Western blot analysis of caspase-7 cleavage. Chrysin induced caspase-7 cleavage in a dose-dependent manner (Fig. 3D). In addition, treatment with chrysin induced PARP cleavage (Fig. 3E), a hallmark of cell apoptosis.
GRP78 knockdown potentiates chrysin-induced apoptosis
Because GRP78 represents a pro-survival arm in the UPR, down-regulation of GRP78 may break the balance of pro-survival signals and pro-apoptosis molecules. GRP78 can interact with caspase-7 and prevent its activation. To investigate whether GRP78 down-regulation potentiates the activation of caspase-7 by chrysin, the effects of GRP78 knockdown by small interfering RNA on the cleavage of caspase-7 were examined. Treatment of HepG2 and SMMC-7721 cells with chrysin up-regulated GRP78 expression, but weakly induced caspase-7 cleavage. In contrast, GRP78 knockdown resulted in dramatic increase in chrysin-induced caspase-7 cleavage (Fig. 4). These data indicate that GRP78 blockade can potentiate the activation of caspase-7 by chrysin.
Fig 4.
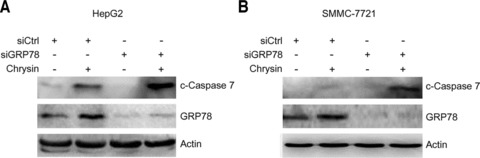
Potentiation of chrysin-induced caspase-7 cleavage by GRP78 knockdown. (A) HepG2 cells were transfected with negative control siRNA (siCtrl) or siRNA to GRP78 (siGRP78). Forty-eight hours later, the cells were treated with 10 μM chrysin for further 24 hrs. Total proteins were harvested and subjected to Western blot analysis of cleaved caspase-7, GRP78 and β-actin. (B) SMMC-7721 cells were transfected with negative control siRNA (siCtrl) or siRNA to GRP78 (siGRP78). Forty-eight hours later, the cells were treated with 20 μM chrysin for further 24 hrs. Total proteins were harvested and subjected to Western blot analysis of cleaved caspase-7, GRP78 and β-actin.
Next, we investigated the effects of GRP78 knockdown on chrysin-induced apoptosis. siRNA-mediated knockdown of GRP78 significantly enhanced chrysin-induced apoptosis in both HepG2 and SMMC-7721 cells. Treatment with caspase inhibitor abrogated the effects of chrysin and GRP78 knockdown on apoptosis (Fig. 5). These data suggested that GRP78 knockdown could sensitize hepatoma cells to chrysin by promoting caspase activation.
Fig 5.
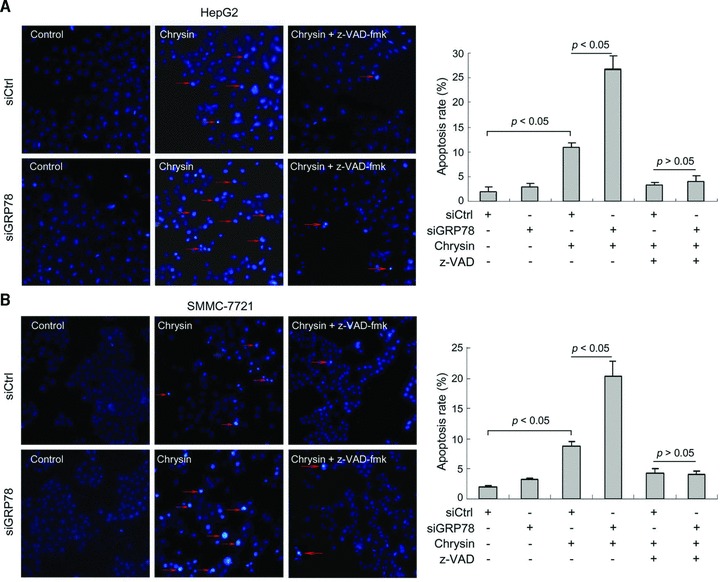
Potentiation of chrysin-induced apoptosis by GRP78 knockdown. (A) HepG2 cells were seeded in a 24-well plate at 5000 cells per well. Two days later, cells were transfected with siRNA to GRP78 (siGRP78) or negative control siRNA (siCtrl). Forty-eight hours after transfection, the cells were treated with 10 μM chrysin for 48 hrs. Apoptosis was assessed by Hoechst 33342 staining. Red arrow: representatives of apoptotic cells. Quantification of apoptotic cells was performed by taking the images in random fields and counting cells with strong fluorescence, condensed or fragmented nuclei. The apoptosis rate was plotted. Columns, mean percentage of apoptotic cells; bars: S.E. (B) SMMC-7721 cells were seeded in a six-well plate at 5000 cells per well. The next day, cells were transfected with siRNA to GRP78 (siGRP78) or negative control siRNA (siCtrl). Forty-eight hours after transfection, the cells were treated with 20 μM chrysin for 48 hrs. Apoptosis was assessed by Hoechst 33342 staining. Red arrow, representatives of apoptotic cells. The apoptosis rate was plotted. Columns: mean percentage of apoptotic cells; bars: S.E. A representative of two independent experiments was shown.
EGCG overcomes chrysin-induced GRP78 expression and potentiates the activation of caspase-7 by chrysin
Previous studies have identified EGCG, the major component of green tea, as a natural compound that overcomes paclitaxel-induced GRP78 expression and directly interacts with the ATP-binding domain of GRP78, blocks the interaction between GRP78 and procaspase-7, and suppresses the protective function of GRP78 [14, 19]. To investigate whether EGCG can overcome chrysin-induced GRP78 expression and enhance chrysin-induced caspase-7 activation, HepG2 cells were treated with EGCG, chrysin or both. Treatment with EGCG alone had no effects on GRP78 expression and caspase-7 cleavage. However, combination of EGCG suppressed chrysin-induced GRP78 expression, and resulted in an increase in caspase-7 cleavage compared to that in cells treated with chrysin alone (Fig. 6A). Similar effects were observed in SMMC-7721 cells (Fig. 6A). EGCG also potentiated chrysin-induced PARP cleavage (Fig. 6B). These results demonstrated that EGCG could overcome chrysin-induced GRP78 expression and potentiate the pro-apoptotic signals induced by chrysin.
Fig 6.
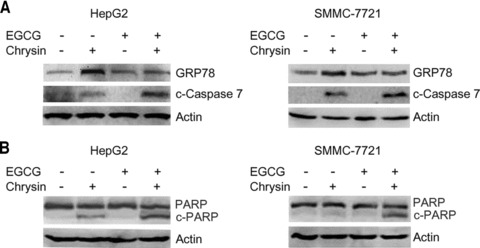
Inhibition of chrysin-induced GRP78 expression and stimulation of chrysin-induced caspase-7 and PARP cleavage by EGCG. (A) HepG2 and SMMC-7721 cells were plated in 25 ml flasks. HepG2 cells were treated with 20 μM EGCG and 20 μM chrysin, and SMMC-7721 cells were treated with 20 μM EGCG and 40 μM chrysin for 24 hrs. Total proteins were harvested and subjected to Western blot analysis of GRP78, cleaved caspase-7, and β-actin. (B) In addition, total proteins were subjected to Western blot analysis of PARP and β-actin.
EGCG sensitizes hepatoma cells to chrysin
To determine whether combination of EGCG and chrysin could synergistically inhibit hepatoma cell growth, HepG2 and SMMC-7721 cells were treated with EGCG, chrysin or both for 48 hrs, followed by CCK-8 assay. Treatment of HepG2 and SMMC-7721 cells with 20 μM EGCG had little effects on cell growth. Treatment with 5 μM chrysin inhibited HepG2 cell growth. Combination of EGCG and chrysin yielded significantly more profound inhibition of cell growth than single agents (Fig. 7A). Whereas treatment of SMMC-7721 cells with 10 μM chrysin poorly inhibited cell growth, combination of EGCG and chrysin resulted in significant inhibition of SMMC-7721 cell growth (Fig. 7B).
Fig 7.

Stimulation of chrysin-induced cell growth arrest by EGCG. (A) HepG2 cells were seeded in 96-well plates at 5000 cells per well. The next day, the cells were treated with 20 μM EGCG, 5 μM chrysin for 48 hrs. Cell viability was assessed by CCK-8 assay. The relative cell growth was plotted. Cell viability in vehicle-treated group was set as 100%. Points: mean of four replicates; bars: S.E. *P < 0.01 versus untreated control. ΔP < 0.01 versus chrysin alone. (B) SMMC-7721 cells were seeded in 96-well plates at 5000 cells per well. The next day, the cells were treated with 20 μM EGCG, 10 μM chrysin for 48 hrs. Cell viability was assessed by CCK-8 assay. The relative cell growth was plotted. Cell viability in vehicle-treated group was set as 100%. Points: mean of four replicates; bars: S.E. *P < 0.01 versus chrysin alone. A representative of two independent experiments was shown.
To determine whether EGCG potentiated chrysin-induced apoptosis, HepG2 cells were treated with EGCG, chrysin or both for 48 hrs, followed by Hoechst 33342 staining. Whereas EGCG alone had little effects on apoptosis, combination of EGCG and chrysin resulted in significantly higher apoptotic rate than chrysin alone. The stimulatory effects of EGCG on chrysin-induced apoptosis could be abrogated by caspase inhibitor (Fig. 8A). EGCG also potentiated chrysin-induced apoptosis in SMMC-7721 cells (Fig. 8B). These data indicated that EGCG could potentiate chrysin-induced apoptosis through caspase activation.
Fig 8.
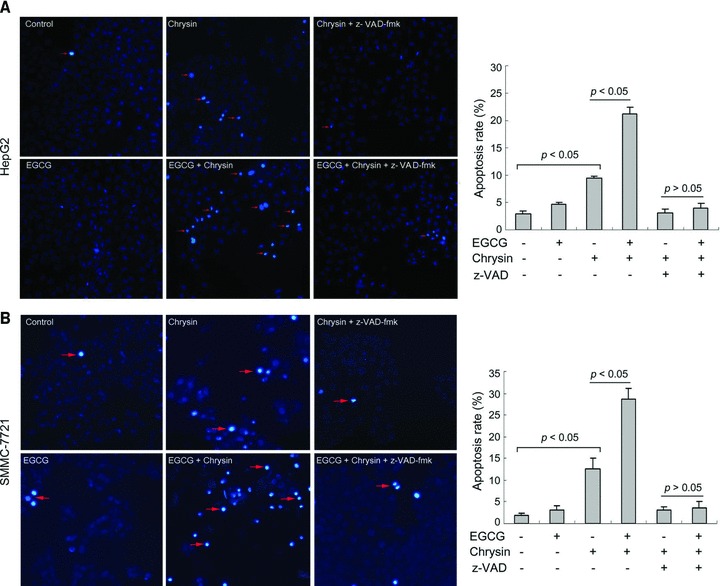
EGCG potentiates chrysin-induced apoptosis. (A) HepG2 cells were treated with or without 20 μM EGCG, 10 μM chrysin and 20 μM z-VAD-fmk for 48 hrs. Apoptosis was assessed by Hoechst 33342 staining. Red arrow: representatives of apoptotic cells. Quantification of apoptotic cells was performed by taking the images in random fields and counting cells with strong fluorescence, condensed or fragmented nuclei. The apoptosis rate was plotted. Columns: mean percentage of apoptotic cells; bars: S.E. (B) SMMC-7721 cells were treated with or without 20 μM EGCG, 20 μM chrysin and 20 μM z-VAD-fmk for 48 hrs. Apoptosis was assessed by Hoechst 33342 staining. Red arrow: representatives of apoptotic cells. The apoptosis rate was plotted. Columns: mean percentage of apoptotic cells; bars: S.E. A representative of three experiments was shown.
Discussion
Previous studies have suggested that both flavonoids and green tea polyphenol can reduce the incidence of many types of cancer. Chrysin, a common flavonoid in plants, has been proposed as an antitumour agent. EGCG, the main constituent of green tea, also shows chemopreventive effects in animal models, as well as in epidemiological studies [27–29]. In this study, we have shown that chrysin inhibits hepatoma cell growth. Chrysin can induce ER stress response in hepatoma cells, including up-regulation of GRP78 expression, induction of eIF-2α phosphorylation and XBP-1 splicing. GRP78 knockdown potentiates chrysin-induced apoptosis. EGCG overcomes chrysin-induced GRP78 expression and synergistically promoted chrysin-induced apoptosis. These evidence may help to understand the mechanisms underlying the anticancer effects of chrysin.
Induction of ER stress is a mechanism underlying the anticancer effects of chemotherapeutic agents such as proteasome inhibitors and non-steroidal anti-inflammatory drugs [31, 32]. Upon ER stress, multiple signalling cascades may be activated to respond to perturbations in ER homeostasis. On one hand, ER stress response can promote cell survival through attenuation of protein synthesis and up-regulation of chaperones and folding enzymes [33]; on the other hand, prolonged or severe ER stress may ultimately overcome or bypass the cellular protective mechanisms, triggering cell death. We found that chrysin induced GRP78 expression, eIF-2α phosphorylation and XBP-1 splicing, hallmarks of the unfolded protein response. Chrysin is a known proteasome inhibitor [27]. Given that proteasome inhibition induces the unfolded protein response [31], it is reasonable to speculate that inhibition of proteasome may contribute, at least in part, to the induction of the unfolded protein response by chrysin. Tumour cells are generally more resistant to ER stress-induced apoptosis compared with un-transformed cells [34]. Overexpression of molecular chaperones in cancer cells may confer resistance to ER stress [35]. For example, GRP78 is a multifunctional chaperone that can protect cells from ER stress-induced apoptosis [3]. Mechanistically, GRP78 interacts with caspase-4, caspase-7 and BIK, and inhibits their activation [4–6]. Chrysin can induce the activation of pro-apoptotic signals, such as caspase-7 and PARP cleavage. In agreement with the inhibitory effect of GRP78 on caspase-7 activation [5], GRP78 knockdown enhanced chrysin-induced activation of caspase-7. Furthermore, GRP78 knockdown potentiated chrysin-induced apoptosis. Therefore, it can be concluded that GRP78 may protect hepatoma cells from chrysin-induced apoptosis.
Up-regulation of GRP78 by chrysin may represent an adaptive response. However, in higher concentrations like 40 to 60 μM of chrysin, casapse-7 cleavage and apoptosis was still observed in the presence of abundant GRP78 induction, suggesting that the protective effects of GRP78 may be overcome or compromised by deterious effects in the higher dosages. This is in agreement with the scenario that the protective branches in ER stress responses may attenuate but not completely abolish cell death upon severe or prolonged ER stress. Severe ER stress may ultimately overcome or bypass the cellular protective mechanisms, triggering cell death. Nonetheless, blockade of the protective arms of ER stress response may maximize the anticancer effects of chrysin. This is in consistent with previous studies that show GRP78 blockade can sensitize tumour cells to etoposide, temozolomide, quercetin and paclitaxel [8–10, 14].
The combination of natural products in cancer chemoprevention and therapy may prove to be more effective than single agents. Given that cancer cells is capable of eliciting robust response to resist exogenous insults including chemotherapeutic agents, down-regulation of the protective stress proteins may improve the therapeutic efficacy. The combination of a compound that may induce stress proteins and another compound that antagonizes these stress proteins may have synergistic effects on cancer therapy. One example of this scenario is Hsp90 inhibitor 17-Allylamino-demethoxy geldanamycin (17-AAG). 17-AAG inhibits the association of hsp90 with the heat shock factor-1 (HSF-1) and thereby rescues the inhibition of HSF-1 by Hsp90, which leads to Hsp70 overexpression [36, 37]. Hsp70 may inhibit several steps of the apoptotic cascades including release of cytochrome C and apoptosis-inducing factor from the mitochondria, nuclear import of AIF, activation of procaspases-9 and -3 and even downstream of active caspase-3 [38–41]. Abrogation of Hsp70 induction significantly enhances the anti-leukaemia activity of 17-AAG [42]. The green tea polyphenol EGCG is known as an inhibitor of stress protein GRP78 [14]. Combination of EGCG with chemotherapeutic agents that induce the unfolded protein response can enhance tumour cells sensitivity in these agents [8, 9, 14]. EGCG also synergistically promotes quercetin-induced cell death [10]. In this study, we found that EGCG overcame chrysin-induced GRP78 expression. These results suggest that EGCG may have dual effects on GRP78, including inhibition of the ATPase activity of GRP78 and inhibition of stress-induced GRP78 expression. Hence, combination of EGCG and chrysin may be more effective than single agents. Indeed, EGCG sensitizes hepatoma cells to chrysin-induced apoptosis, which is associated with increased caspase and PARP cleavage. Thus, we speculate that combination of chrysin and EGCG may be a novel regimen for cancer prevention or therapeutics.
Acknowledgments
We thank the National Natural Science Foundation of China (grant 81001011) for supporting this study.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Li J, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med. 2006;6:45–54. doi: 10.2174/156652406775574523. [DOI] [PubMed] [Google Scholar]
- 2.Dong D, Ni M, Li J, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008;68:498–505. doi: 10.1158/0008-5472.CAN-07-2950. [DOI] [PubMed] [Google Scholar]
- 3.Lee AS. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res. 2007;67:3496–9. doi: 10.1158/0008-5472.CAN-07-0325. [DOI] [PubMed] [Google Scholar]
- 4.Jiang CC, Chen LH, Gillespie S, et al. Inhibition of MEK sensitizes human melanoma cells to endoplasmic reticulum stress-induced apoptosis. Cancer Res. 2007;67:9750–61. doi: 10.1158/0008-5472.CAN-07-2047. [DOI] [PubMed] [Google Scholar]
- 5.Reddy RK, Mao C, Baumeister P, et al. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem. 2003;278:20915–24. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- 6.Fu Y, Li J, Lee AS. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007;67:3734–40. doi: 10.1158/0008-5472.CAN-06-4594. [DOI] [PubMed] [Google Scholar]
- 7.Li J, Ni M, Lee B, et al. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 2008;15:1460–71. doi: 10.1038/cdd.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pyrko P, Schonthal AH, Hofman FM, et al. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007;67:9809–16. doi: 10.1158/0008-5472.CAN-07-0625. [DOI] [PubMed] [Google Scholar]
- 9.Wang J, Yin Y, Hua H, et al. Blockade of GRP78 sensitizes breast cancer cells to microtubules-interfering agents that induce the unfolded protein response. J Cell Mol Med. 2009;13:3888–97. doi: 10.1111/j.1582-4934.2009.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li M, Wang J, Jing J, et al. Synergistic promotion of breast cancer cells death by targeting molecular chaperone GRP78 and heat shock protein 70. J Cell Mol Med. 2009;13:4540–50. doi: 10.1111/j.1582-4934.2008.00575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baumeister P, Dong D, Fu Y, et al. Transcriptional induction of GRP78/BiP by histone deacetylase inhibitors and resistance to histone deacetylase inhibitor-induced apoptosis. Mol Cancer Ther. 2009;8:1086–94. doi: 10.1158/1535-7163.MCT-08-1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong D, Ko B, Baumeister P, et al. Vascular targeting and antiangiogenesis agents induce drug resistance effector GRP78 within the tumor microenvironment. Cancer Res. 2005;65:5785–91. doi: 10.1158/0008-5472.CAN-05-0754. [DOI] [PubMed] [Google Scholar]
- 13.Virrey JJ, Dong D, Stiles C, et al. Stress chaperone GRP78/BiP confers chemoresistance to tumor-associated endothelial cells. Mol Cancer Res. 2008;6:1268–75. doi: 10.1158/1541-7786.MCR-08-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ermakova SP, Kang BS, Choi BY, et al. (-)-Epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Res. 2006;66:9260–9. doi: 10.1158/0008-5472.CAN-06-1586. [DOI] [PubMed] [Google Scholar]
- 15.Qanungo S, Das M, Haldar S, et al. Epigallocatechin-3-gallate induces mitochondrial membrane depolarization and caspase-dependent apoptosis in pancreatic cancer cells. Carcinogenesis. 2005;26:958–67. doi: 10.1093/carcin/bgi040. [DOI] [PubMed] [Google Scholar]
- 16.Chen C, Shen G, Hebbar V, et al. Epigallocatechin-3-gallate-induced stress signals in HT-29 human colon adenocarcinoma cells. Carcinogenesis. 2003;24:1369–78. doi: 10.1093/carcin/bgg091. [DOI] [PubMed] [Google Scholar]
- 17.Roy AM, Baliga MS, Katiyar SK. Epigallocatechin-3-gallate induces apoptosis in estrogen receptor-negative human breast carcinoma cells via modulation in protein expression of p53 and Bax and caspase-3 activation. Mol Cancer Ther. 2005;4:81–90. [PubMed] [Google Scholar]
- 18.Siddiqui IA, Malik A, Adhami VM, et al. Green tea polyphenol EGCG sensitizes human prostate carcinoma LNCaP cells to TRAIL-mediated apoptosis and synergistically inhibits biomarkers associated with angiogenesis and metastasis. Oncogene. 2008;27:2055–63. doi: 10.1038/sj.onc.1210840. [DOI] [PubMed] [Google Scholar]
- 19.Luo T, Wang J, Yin Y, et al. (-)-Epigallocatechin gallate sensitizes breast cancer cells to paclitaxel in a murine model of breast carcinoma. Breast Cancer Res. 2010;12:R8. doi: 10.1186/bcr2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li M, He Z, Ermakova S, et al. Direct inhibition of insulin-like growth factor-I receptor kinase activity by (-)-epigallocatechin-3-gallate regulates cell transformation. Cancer Epidemiol Biomarkers Prev. 2007;16:598–605. doi: 10.1158/1055-9965.EPI-06-0892. [DOI] [PubMed] [Google Scholar]
- 21.Yin Z, Henry EC, Gasiewicz TA. (-)-Epigallocatechin-3-gallate is a novel Hsp90 inhibitor. Biochemistry. 2009;48:336–45. doi: 10.1021/bi801637q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leone M, Zhai D, Sareth S, et al. Cancer prevention by tea polyphenols is linked to their direct inhibition of antiapoptotic Bcl-2-family proteins. Cancer Res. 2003;63:8118–21. [PubMed] [Google Scholar]
- 23.Lapidot T, Walker MD, Kanner J. Antioxidant and prooxidant effects of phenolics on pancreatic beta-cells in vitro. J Agric Food Chem. 2002;50:7220–5. doi: 10.1021/jf020615a. [DOI] [PubMed] [Google Scholar]
- 24.Cho H, Yun CW, Park WK, et al. Modulation of the activity of pro-inflammatory enzymes, COX-2 and iNOS, by chrysin derivatives. Pharmacol Res. 2004;49:37–43. doi: 10.1016/s1043-6618(03)00248-2. [DOI] [PubMed] [Google Scholar]
- 25.Woo KJ, Jeong YJ, Park JW, et al. Chrysin-induced apoptosis is mediated through caspase activation and Akt inactivation in U937 leukemia cells. Biochem Biophys Res Commun. 2004;325:1215–22. doi: 10.1016/j.bbrc.2004.09.225. [DOI] [PubMed] [Google Scholar]
- 26.Weng MS, Ho YS, Lin JK. Chrysin induces G1 phase cell cycle arrest in C6 glioma cells through inducing p21Waf1/Cip1 expression: involvement of p38 mitogen-activated protein kinase. Biochem Pharmacol. 2005;69:1815–27. doi: 10.1016/j.bcp.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 27.Lu G, Liao J, Yang G, et al. Inhibition of adenoma progression to adenocarcinoma in a 4-(Methylnitrosamino)-1-(3-Pyridyl)-1-Butanone–induced lung tumorigenesis model in A/J mice by tea polyphenols and caffeine. Cancer Res. 2006;66:11494–501. doi: 10.1158/0008-5472.CAN-06-1497. [DOI] [PubMed] [Google Scholar]
- 28.Xiao H, Hao X, Simi B, et al. Green tea polyphenols inhibit colorectal aberrant crypt foci (ACF) formation and prevent oncogenic changes in dysplastic ACF in azoxymethane-treated F344 rats. Carcinogenesis. 2008;29:113–9. doi: 10.1093/carcin/bgm204. [DOI] [PubMed] [Google Scholar]
- 29.Katiyar SK, Mukhtar H. Tea in chemoprevention of cancer: epidemiological and experimental studies. Int J Oncol. 1996;8:221–38. doi: 10.3892/ijo.8.2.221. [DOI] [PubMed] [Google Scholar]
- 30.Bonfili L, Cecarini V, Amici M, et al. Natural polyphenols as proteasome modulators and their role as anti-cancer compounds. FEBS J. 2008;275:5512–26. doi: 10.1111/j.1742-4658.2008.06696.x. [DOI] [PubMed] [Google Scholar]
- 31.Fribley A, Zeng Q, Wang CY. Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Mol Cell Biol. 2004;24:9695–704. doi: 10.1128/MCB.24.22.9695-9704.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pyrko P, Kardosh A, Liu YT, et al. Calcium-activated endoplasmic reticulum stress as a major component of tumor cell death induced by 2,5-dimethyl-celecoxib, a non-coxib analogue of celecoxib. Mol Cancer Ther. 2007;6:1262–75. doi: 10.1158/1535-7163.MCT-06-0629. [DOI] [PubMed] [Google Scholar]
- 33.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–98. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe. Nat Rev Cancer. 2004;4:966–77. doi: 10.1038/nrc1505. [DOI] [PubMed] [Google Scholar]
- 35.Zou J, Guo Y, Guettouche T, et al. Repression of heat shock transcription factor HSF1 activation by Hsp90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998;94:471–80. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
- 36.Bagatell R, Paine-Murrieta GD, Taylor CW, et al. Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin Cancer Res. 2000;6:3312–8. [PubMed] [Google Scholar]
- 37.Mosser DD, Caron AW, Bourget L, et al. Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol Cell Biol. 1997;17:5317–27. doi: 10.1128/mcb.17.9.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ravagnan L, Gurbuxani S, Susin SA, et al. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol. 2001;3:839–43. doi: 10.1038/ncb0901-839. [DOI] [PubMed] [Google Scholar]
- 39.Gurbuxani S, Schmitt E, Cande C, et al. Heat shock protein 70 binding inhibits the nuclear import of apoptosis-inducing factor. Oncogene. 2003;22:6669–78. doi: 10.1038/sj.onc.1206794. [DOI] [PubMed] [Google Scholar]
- 40.Beere HM, Wolf BB, Cain K, et al. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–75. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- 41.Jaattela M, Wissing D, Kokholm K, et al. Hsp70 exerts its anti-apoptotic function downstream of caspase-3-like proteases. EMBO J. 1998;17:6124–34. doi: 10.1093/emboj/17.21.6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo F, Rocha K, Bali P, et al. Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. Cancer Res. 2005;65:10536–44. doi: 10.1158/0008-5472.CAN-05-1799. [DOI] [PubMed] [Google Scholar]