

Abstract
More than 1000 microRNAs (miRNAs) are expressed in human cells, some tissue or cell type specific, others considered as house-keeping molecules. Functions and direct mRNA targets for some miRNAs have been relatively well studied over the last years. Every miRNA potentially regulates the expression of numerous protein-coding genes (tens to hundreds), but it has become increasingly clear that not all miRNAs are equally important; diverse high-throughput screenings of various systems have identified a limited number of key functional miRNAs over and over again. Particular miRNAs emerge as principal regulators that control major cell functions in various physiological and pathophysiological settings. Since its identification 3 years ago as the miRNA most commonly and strongly up-regulated in human brain tumour glioblastoma [1], miR-21 has attracted the attention of researchers in various fields, such as development, oncology, stem cell biology and aging, becoming one of the most studied miRNAs, along with let-7, miR-17–92 cluster (‘oncomir-1’), miR-155 and a few others. However, an miR-21 knockout mouse has not yet been generated, and the data about miR-21 functions in normal cells are still very limited. In this review, we summarise the current knowledge of miR-21 functions in human disease, with an emphasis on its regulation, oncogenic role, targets in human cancers, potential as a disease biomarker and novel therapeutic target in oncology.
Keywords: human disease, cancer, glioma, oncogene, non-coding RNA, post-transcriptional regulation
miR-21 expression in cancer and other diseases
MicroRNA-21 (miR-21) has been identified as the best hit in a number of medium-scale and high-scale profiling experiments designed for the detection of miRNAs dysregulated in cancer. The first indication of miR-21's aberrant expression came from the miRNA profiling of human glioblastoma (GBM), the most malignant brain tumour of glial origin [1]. miR-21 was strongly elevated in all high-grade glioma samples tested, including tumour tissues from patients and early passage GBM cultures established from additional patients. Similar increases in miR-21 expression were found in six commonly used model cell lines derived from GBM, an important finding since GBM tumours and cell lines are genetically extremely diverse, with a number of tumour suppressor genes and proto-oncogenes often mutated, lost or amplified, and no common genetic marker was identified prior to the discovery of miR-21. In most cases, the concomitant up-regulation of the 72-nt miRNA precursor (pre-miR-21) was also apparent on Northern blots. The miR-21 up-regulation in glioma was in comparison to a variety of controls including non-neoplastic adult human and mouse brain tissues (cortexes and white matters), foetal human and mouse brain tissue at multiple stages of development, rat primary neurons and astrocytes, mouse embryonic stem cells, embryoid bodies, neural precursors and their neuronal and glial derivatives, P19 neuronal cells, as well as mouse astrocytic and oligodendrocytic cells differentiated from adult hippocampal progenitor cell [1]. All of these controls showed either trace or no expression of miR-21 compared with glioma samples. Elevated expression of miR-21 in GBM was further confirmed by an independent study [2].
In a large-scale profiling of miRNA expression in 540 human samples derived from 363 specimens representing six types of solid tumours and 177 respective normal control tissues [3], miR-21 was the only miRNA up-regulated in all types of the analysed tumours, including the breast, colon, lung, pancreas, prostate, and stomach. Additional studies demonstrated elevated miR-21 expression in hepatocellular carcinomas [4], gastric cancer [5], ovarian cancer [6, 7], cervical carcinoma [8], multiple head and neck cancer cell lines [9], papillary thyroid carcinoma [10] and some other solid tumours. More recent studies indicate that miR-21 is also up-regulated in leukaemic cancers. Its expression is dramatically higher (up to 10-fold) in patients with chronic lymphocytic leukaemia (CLL) than in normal CD19+ lymphocytes [11]. It is also overexpressed in aggressive diffuse large B-cell lymphoma (DLBCL), in both de novo and transformed cases, and follicular center lymphoma cases compared with normal B cells [12]. It is consistently up-regulated in different subgroups of the disease as heterogeneous as acute myeloid leukaemia (AML), regardless of their cytogenetic status and the presence of specific mutations [13]. miR-21 is also overexpressed in both Hodgkin lymphoma lymph nodes and the human Hodgkin lymphoma cell lines [14]. Generally, miR-21 expression levels are also very high in most cancer cell lines of various origins, and in some lines, it accounts for up to 15–25% of the cellular miRNA content [15]. Therefore, abundant miR-21 may be a general, albeit not universal, feature of tumour cells (Table 1). miR-21 is also strongly up-regulated in Epstein–Barr virus-infected human B lymphocytes [16] and hepadnavirus-associated hepatocellular carcinoma [17], suggesting the possibility that it is also involved in viral infections and virus-linked proliferative disorders.
1.
miR-21 regulation and function in human cancer
| Cancer | miR-21 expression in human tissues/cells | miR-21 involvement in biological process | miR-21 targets | References |
|---|---|---|---|---|
| Glioma | Up-regulation in GBM tumours, primary cells and glioma cell lines | Invasion and cell growth | PDCD4, RECK, TIMP3?*, NFIB, APAF1?, STAT3? | [1, 2, 49] |
| Breast cancer | Up-regulation | Cell growth, apoptosis, angiogenesis and invasion | PDCD4, TPM1, maspin | [3, 53, 58, 60] |
| Ovarian cancer | Up-regulation | [6, 7] | ||
| Colorectal cancer | Up-regulation | Cellular outgrowth, migration, invasion and metastasis | PDCD4, NFIB, SPRY2 | [3, 25, 52, 86] |
| Stomach/gastric cancer | Up-regulation | RECK | [3, 5] | |
| Hepatocellular carcinoma | Up-regulation | Cell migration and invasion and proliferation | PTEN? | [4, 51] |
| Prostate cancer | Up-regulation | [3] | ||
| Pancreas cancer | Up-regulation | [3] | ||
| Lung cancer | Up-regulation | [3] | ||
| Head and neck cancer | Up-regulation in cell lines | [9] | ||
| Thyroid carcinoma | Up-regulation | [10] | ||
| Cervical cancer | Up-regulation | [8] | ||
| Cholangiocarcinoma | PTEN? | [87] | ||
| Leukaemia | Up-regulation in CLL and AML patients | [11, 13] | ||
| B-cell and Hodgkin lymphoma | Up-regulation in patients and cell lines | [12, 14] |
Question mark depicts direct targeting to be further validated.
Interestingly, high levels of miR-21 may not only characterise cancer cells but also represent a common feature of pathological cell growth or cell stress. For example, miR-21 is up-regulated in several models of mouse hypertrophic heart including thoracic aortic banding, which induces hypertrophy by increased afterload on the heart, and in transgenic mice expressing constitutively active calcineurin A in the heart muscle, which results in a severe, well-characterised form of hypertrophy [18–20]. It is also elevated in vascular walls after balloon injury, a model of vascular neointimal lesion formation [21]. One group also demonstrated a five-fold up-regulation in hypertrophic left cardioventricular tissue from human patients with end-stage systolic heart failure [22]. In vitro, miR-21 was the most up-regulated miRNA in cultured rat neonatal cardiac myocytes stimulated with the hypertrophic agents angiotensin II and phenylephrine [18]. While there is a good agreement among independent studies of miR-21 expression in hypertrophic heart and vasculature, the data regarding its functional effects obtained by several groups appear more controversial. For example, inhibition of endogenous miR-21 by antisense 2′O-Me molecules slows down the hypertrophic growth in a model of induced hypertrophy in cultured cardiomyocytes [18]. Down-regulation of aberrantly expressed miR-21 also reduces neointima formation in rat carotid artery after angioplasty by affecting both proliferation and apoptosis of vascular smooth muscle cells (VSMCs) [21]. Conversely, miR-21 overexpression by its synthetic mimics transfected in cultured cardiomyocytes in combination with miR-212 and miR-129 mimics modified a set of foetal cardiac genes and led to the development of cellular hypertrophy [22]. In contrast, a study by Tatsuguchi et al. [19] suggests that miR-21 has a subtle yet reproducible inhibitory effect on cardiac hypertrophy, whereas LNA-based miR-21 inhibitors may induce hypertrophy. Regardless of its role, miR-21 is clearly up-regulated in cardiac hypertrophy and in a variety of other human proliferative disorders, implying a function in regulating cell growth.
This idea is further supported by evidence of miR-21 induction associated with cellular dedifferentiation. An interesting example is the restricted thyroid cell line FRTL-5 that depends on the presence of thyroid-stimulating hormone (TSH). Oncogenic Ras induction leads to dedifferentiation and TSH-independent proliferation of the thyroid cells and up-regulation of miR-21 after 7 days from 0.3% to 11% total miRNA content [15]. Ras-induced expression of miR-21 can be mediated through signal transducer and activator of transcription (STAT3) and/or SMADs signalling (see the next section). The understanding of this subject, however, is further complicated by unexpected patterns of miR-21 expression during differentiation: in cell lines expressing low or undetectable levels of miR-21 (such as mouse embryonic stem cells, neuroblastoma human SHSY5Y, NTera2 or mouse NG, N1E, N2A and myeloid line HL-60), its expression is induced by differentiation signals such as phorbol 12-myristate 13-acetate (PMA) and retinoic acid [15, 23–25]. It is also induced in adherent mammosphere cultures differentiating on collagen [26].
One interesting observation was made by Fujita [25] using PMA-induced terminally differentiating HL-60 cells, whose differentiation into monocytes/macrophages is concomitant with miR-21 induction. Within 24 hrs of PMA treatment, wild-type HL-60 cells attach to the substrate and dramatically perturb their cell division rate in association with macrophage differentiation. However, the HL-60-overexpressing exogenous miR-21 neither attach to the substrate nor exhibit cell division arrest, even at 72 hrs after lentivirus vector transduction. Instead, they demonstrate cell division stimulation as a result of forced expression of miR-21. miR-21 overexpression in these cells therefore does not allow a characteristic exit from the cell cycle, but enhances proliferation, suggesting that accurate control of miR-21 levels and function is critical for balancing cellular proliferation and differentiation. These findings led to the speculation that relatively low levels of miR-21 may be temporary and spatially required for differentiation and development, whereas high levels may have an oncogenic potential. Therefore, the central question posed by numerous studies described above is how miR-21 expression is regulated and what are the mechanisms leading to its deregulation in human disease.
Mechanisms of miR-21 elevation in cancer: multi-level regulatory control
The mature miR-21 is perfectly conserved in mammals, as many other miRNAs are, and is encoded by a single gene. The human miR-21 gene is relatively well characterised and mapped to chromosome 17q23.2, where it overlaps with the protein-coding gene VMP1 (or TMEM49), a human homologue of rat vacuole membrane protein [25, 27]. Common up-regulation of miR-21 expression in cancer led us to examine the possible amplification of this genomic locus in cancer.
The amplification of the 17q chromosomal region is associated with a number of cancers, including breast [28] and prostate cancer [29], and occurs in ∼50% of medulloblastoma cases [30]. The 17q region has also been associated with frequent gains in Hodgkin lymphoma [31]. However, the genomic locus encoding miR-21 is not amplified in most cancers including those expressing very high levels of miR-21, such as GBM and CLL [11, 32]. In particular, an analysis of genomic DNA from CLL patients and healthy donors showed that an increase in miR-21 expression was not paralleled by the corresponding locus amplification. Similarly, there is no current evidence of association of the 17q chromosomal region with GBM, although amplification of 17q23 is frequent and predictive in neuroblastomas [33], one of the few ‘atypical’ types of cancers with low miR-21 levels. Altogether, there is no clear correlation between the amplification of miR-21 genomic locus and its elevated expression in cancer, suggesting that deregulation in the expression of this miRNA occurs at either the transcriptional or the post-tran-scriptional level or both.
Interestingly, the miR-21 gene is located in the fragile site FRA17B within the 17q23.2 chromosomal region, which is one of the HPV16 integration loci [34]. It is known that HPV integration into the host cell genome can cause genetic and epigenetic alterations, suggesting that the mapping of miR-21 gene at or near HPV integration sites may contribute to its elevation in cancer. Infection with HPV16 or 18 is a major risk factor for developing cervical cancer, and common fragile sites are preferential targets for HPV16 integrations in cervical tumours [35]. miR-21 up-regulation in cervical carcinoma therefore may be associated with HPV16 integration. Nevertheless, the question remains how miR-21 is regulated in various (other) tumours and if there is a common mechanism. To start addressing these questions, we will review the information about miR-21 transcription and processing.
Transcriptional control
Since mature miR-21 is abundant in most cancerous cell lines, it was among the first miRNAs used as a model for studying miRNA expression and maturation [27]. Several primary (pri-miR-21) transcripts have been identified in a number of cell types by applying RACE and primer extension analyses. In 293T cells, transcribed by RNA polymerase II, capped and polyadenylated unspliced ∼3.5-kb pri-miR-21 was detected [27], and in PMA-induced HL-60 cells, a different promoter was identified whose PMA-dependent utilisation led to the transcription of the longer ∼4.3-kb pri-miR-21 [25]. This pri-miR-21 is transcribed independently from the overlapping protein-coding VMP1 gene, since the last does not respond to PMA treatment and is polyadenylated before reaching the miR-21 hairpin region [25].
An analysis of the consensus sequences within the miR-21 promoter region identified several conserved enhancer elements (Fig. 1), including the binding sites for activation protein 1 (AP-1; composed of Fos and Jun family nuclear oncogenes), Ets/PU.1, C/EBP-α (key factors governing haematopoietic lineage differentiation), nuclear factor I (NFI), SRF, p53 and signal transducer and activator of transcription 3 (STAT3) [25]. The human miR-21 promoter retains all of these elements, and their high conservation among vertebrates suggests that highly conserved transcriptional regulatory mechanisms operate on the promoter. Experiments with heterologous luciferase reporters bearing different enhancer elements, either wild-type or mutated, in their promoter region revealed that AP-1, induced by PMA, triggers the transcription of pri-miR-21, and that c-Fos and c-Jun are the principal contributors among the AP-1 components induced [25]. In addition, two Ets/PU.1 elements in the miR-21 promoter enhance its transcriptional activation by AP-1. Oncogenic transformation is frequently associated with the enhancement of endogenous AP-1 activity through various signal transduction pathways, and AP-1 activation strongly contributes to the oncogenic potential. Therefore, up-regulated miR-21 expression in multiple types of cancers may reflect the elevated AP-1 activity in these carcinomas. In addition, miR-21 transcription is induced by STAT3, another factor whose activation is essential for the transforming potential of many oncogenes. STAT3-dependent miR-21 transcription was demonstrated in IL-6-stimulated XG-1 and INA-6 myeloma and HepG2 hepatocellular carcinoma cells [36].
1.
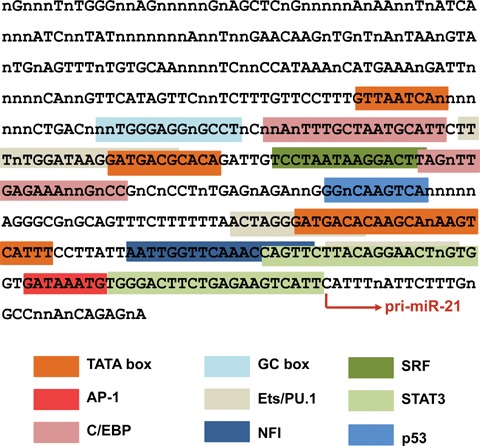
The consensus sequence of putative promoter region of miR-21. Conserved bases across vertebrates are shown in capitals and non-conserved bases or deletions are denoted by ‘n’. The arrow indicates the transcription start site of pri-miR-21. Conserved regions of various transcription factors are indicated by different colours. Two additional RE-1-binding elements responding to transcription factor REST are located at 7214 and 7100 bp upstream of the miR-21 transcription start site [38]. This figure is reproduced from reference 25.
On the other hand, NFIB and C/EBP-α binding to the miR-21 promoter contribute to the repression of the basal-level transcription of miR-21 [25]. Dissociation of these factors from the promoter occurs quickly (within 4 hrs) after PMA stimulation of HL-60 cells and leads to enhanced promoter activity. Important and distinct roles of AP-1/PU.1 for monocyte and C/EBP-α for granulocyte differentiation may be partly mediated by miR-21. Moreover, interactions among stimulatory (e.g. AP-1 and STAT3) and inhibitory (NFIB and C/EBP-α) transcription factors may determine the activity of the miR-21 promoter not only in myeloid but in other cellular settings as well. For example, low levels of miR-21 in the normal brain [15] may be explained by its repressed transcription caused by NFIB, a factor abundantly expressed in the brain and involved in brain development [37].
An additional regulator of miR-21 transcription is RE-1-silencing transcription (REST) factor, which is believed to be a major transcriptional repressor of neurogenesis. It is associated with and blocks transcription from the miR-21 promoter in mouse ES cells [38]. However, REST activity did not influence miR-21 expression in Hdh7/7 mouse cell line derived from embryonic striatum [39], suggesting additional cell-specific factors that may affect REST interaction with the miR-21 promoter.
An additional mechanism by which miR-21 expression may be increased in specific cells is the epigenetic modification of its transcriptional regulatory sequences. One study demonstrated that miR-21 was among several miRNAs strongly induced in ovarian cell line OVCAR3 by treatment with a demethylating agent 5-AZA, and therefore suggested that the hypomethylation could be the mechanism responsible for its overexpression in vivo[6]. Further research is clearly required to investigate the epigenetic mechanisms of miR-21 induction in disease.
Transcriptional control of miR-21 expression, especially in cancer, seems to be rather an exceptional phenomenon. For the majority of miRNAs dysregulated in cancer, the changes in the expression levels of mature miRNAs do not correlate with the levels of their primary precursors, mostly unchanged, indicating that most of the regulation takes place after transcription [40]. However, for miR-21, at least during development, there is a good correlation between pri-miR-21 and miR-21 levels, suggesting that (i) transcription is indeed an important regulatory step for miR-21 expression and function, (ii) miR-21 transcription and processing must be tightly coupled and, consequently, (iii) miR-21 processing is highly efficient. An analysis of multiple cancers revealed that the expression of numerous miRNAs is repressed in human cancers [41], a phenomenon referred as ‘global repression of miRNAs in cancers’. The fact that this repression does not coincide with reductions in the primary miRNA transcripts suggests that altered regulation of the miRNA-processing machinery might occur in human cancers. If true, specific pri-miRNA/pre-miRNA sequences, such as the miR-21 precursor, may serve as preferential substrates for Drosha and/or Dicer in such conditions of reduced or impaired activity of the miRNA-processing machinery. The unusually efficient processing of the miR-21 precursor would explain why the mature molecule is strongly up-regulated, whereas the expression of many other miRNAs is reduced.
Post-trancriptional regulation
A recent study of TGF-β- and BMP-induced miR-21 expression in VSMCs has revealed additional mechanisms that control miR-21 processing, which again makes this molecule outstanding [42]. In the BMP4-treated human primary pulmonary artery smooth muscle cells, mature miR-21 was up-regulated at the expense of many other tested miRNAs. This elevation was after transcription, likely at the level of processing of the primary transcript by the Drosha microprocessor complex. After ligand stimulation, receptor-specific SMAD signal transducers (SMAD1/5 and SMAD2/3) were recruited to pri-miR-21 in a complex with the RNA helicase p68, a component of the Drosha microprocessor complex. This led to a fast (within 30 min) SMAD4-independent processing of pri-miR-21 to pre-miR-21, followed by its subsequent maturation, resulting in an active miR-21 molecule [42].
Since TGF-β expression is often increased in cancer cells, where it promotes epithelial-to-mesenchymal transition (EMT) and metastatic behaviour, it is conceivable that a similar mechanism may operate in cancer cells as well. Indeed, TGF-p/BMP4-induced pri-miR-21 processing and up-regulation of the mature miR-21 was also observed in MDA-MB-468 breast carcinoma cells [42]. Similarly, miR-21 was also induced in TGF-β- stimulated human HaCaT keratinocytes, a model of EMT, recapitulating epithelial injury and progression of epithelial tumours [43]. It seems that miR-21 is one of the only few miRNAs whose processing is regulated by TGF-β. An open question remains regarding the determinants of SMAD specificity in their selection of pri-miR-21, that is, how unique is the pri-miR-21 in this regard? The MH1 domain of R-SMADs binds DNA by specifically recognizing a sequence element. It was also observed that the MH1 domain of SMAD1 associates with pri-miR-21 despite its inability to interact with p68 [42]. One could therefore speculate that the SMAD MH1 domain may recognise an RNA sequence or a structural element and thus provide specificity in the selection of BMP and TGF-β target miRNA.
Interestingly, miR-21 is one of the miRNAs consistently induced in response to hypoxia, as demonstrated in breast and colon cancer cells [44]. The hypoxia-induced factor 1 (HIF-1)-binding site is present in the pri-miR-21 promoter [44], but the possibility that miR-21 is directly regulated by this transcription factor remains to be tested. Alternatively, hypoxia may regulate miR-21 expression in an HIF-1-independent manner, for example, through AP-1 transcription (the pathway observed in [45]) or by stimulating TGF-β signalling and miR-21 maturation. These mechanisms may cooperate in miR-21 induction since TGF-β can enhance both AP-1 and HIF-1 DNA-binding activities [46]. Given that hypoxia is an essential factor of the neoplastic microenvironment, and of cardiovascular pathology, these data provide an additional link between cell physiology and the stress associated with pathological cell growth and control of miR-21 gene expression.
miR-21 functions in cancer
How does miR-21 work? What genes does it regulate? Do the proteins whose expression is regulated by miR-21 function coordinate in one or multiple signalling pathways? Recent studies have begun to shed light on the molecular mechanisms by which miR-21 regulates cellular processes. To study miRNA function, both gain- and loss-of-function approaches are commonly utilised. miRNAs are typically overexpressed by transfecting the cells either with the so-called miRNA mimics (synthetic dsRNA duplexes similar to pre-miRNA hairpin) or with pri-miRNA-like vectors that are processed to produce the mature miRNA. For miRNA knockdown, various synthetic chemically modified antisense oligonucleotides (ASOs) are applied. Although both miR-21 overexpression and inhibition have been used by several groups to investigate its functions and targets, it is difficult to compare or merge the results of these studies due to a number of common issues.
First, both miRNA overexpression and inhibition with synthetic oligonucleotides must be validated. Reporter assays designed to validate modulated functional activity of the miRNA are optimally suited for this purpose. Such assays are usually based on luciferase- or GFP-encoding vectors with an miRNA-binding site (miRNA antisense sequence) inserted into the 3′UTR of the reporter. However, it is still quite common to solely test miRNA levels after transfection with miRNA mimics or ASOs by qRT-PCR or Northern blotting and report the results as a confirmation of successful gain- or loss-of-function. These techniques can easily be misleading since the oligonucleotides used for the overexpression and inhibition may interfere with the detection and create false-positive results. In many cases, the results of such expression analysis overestimate the degree of functional miRNA over-expression or inhibition.
Second, overexpression of double-stranded miRNA mimics may lead to RISC incorporation and functional activity of the second (unintended) strain. For some miRNAs, the design of a duplex with the functionally active ‘right’ strand is straightforward; for others, however, the undesired passenger strand may become more stable and preferentially active. The rules of strand selection for some pre-miRNA-like duplexes may be more complex than currently appreciated, and therefore validation of the functional overexpression of the ‘right’ strand should be applied. In particular, our data suggest that overexpression of miR-21 using synthetic duplexes often leads to a preferential passenger strand activity. In this case, phenotypic effects observed as a result of such artificial activity should not be interpreted as a function of miR-21 signalling.
Last, ASOs used for miRNA inhibition vary greatly in their potency as well as their specificity. Based on published data [47–49] and our own unpublished observations, commonly used LNA ASOs are more potent as miRNA inhibitors than 2′O-Me ASOs. However, because of their high affinity to a target, they may produce a wide range of non-miRNA-mediated off-target effects if designed suboptimally. A careful analysis is required to discriminate such effects from genuine miRNA functions (see below). We currently use 2′-O-MOE ASOs that seem optimal in terms of both their potency and their specificity [47, 49]. In the rapidly developing miRNA field, however, the phenotypic effects and changes in protein-coding gene expression caused by miRNA ASOs are often immediately interpreted as results of the miRNA knockdown. Although a number of studies have been performed on miR-21 using these approaches, not all of them validated bona fide modulation of miR-21 activity. Therefore, the results should be translated with a certain degree of caution.
With these notes in mind, we believe that the following studies provide supportive evidence for an oncogenic role of miR-21. Overexpression of miR-21 from the expression vector pSIF carrying the miR-21 gene driven by the H1 RNA polymerase III promoter led to an approximately two-fold increase in anchorage-independent colony formation of human MCF7 breast carcinoma and murine JB6 epidermal cells, both serving well-characterised models of neoplastic transformation [50]. Elevation of miR-21 using an expression vector significantly promoted survival and reduced cytokine dependency of myeloma cells [36]. Enhanced miR-21 expression by transfection with precursor miR-21 increased tumour cell proliferation, migration and invasion in cultured human hepatocellular cancer cells [51] and invasion of colon cancer cells [52]. Conversely, inhibition of miR-21 expression by various ASOs reduced anchorage-independent colony formation, proliferation and invasion while inducing apoptosis of hepatocellular carcinoma cells [17, 51]. In addition, reduced proliferation and tumour growth of MCF7 breast cancer cells [53, 54], motility and invasiveness of glioma [49] and invasion, intravasation and metastatic capacity of colon cancer cells [52] were reported. This overwhelming spectrum of data clearly implies that miR-21 is a key molecule on the roadmap of carcinogenesis. It is also noticeable that miR-21, as one of the miRNAs induced by hypoxia and up-regulated in cancer, possesses pro-survival and anti-apoptotic properties; its expression may therefore represent an adaptation to a hypoxic environment that favours cancer cell survival.
Identification of direct miR-21 targets
Since there is just partial complementarity between miRNAs and their targets in animal cells, the identification of specific target genes for a given miRNA still represents a major challenge in our understanding of miRNA function. Several computational algorithms predict hundreds of mRNAs as possible targets for miR-21 [55–57]; however, relatively few have been experimentally validated. In different cellular contexts, one miRNA perhaps can regulate diverse pathways and cause various phenotypes depending on the availability of a certain population of mRNA targets. Both gene prediction-based and systematic screening approaches have been used to identify miR-21 targets. Since miRNAs can regulate both mRNA stability and translation into protein, direct targets can be identified among either mRNAs or proteins whose expression is affected by miR-21. For this reason, mRNA array expression [49, 53] and proteomics [58], each with certain advantages and flaws, have been used after cell treatments with anti-miR-21.
While mRNA array analysis following miRNA inhibition or over-expression is a relatively simple and robust method for target identification, this approach cannot, as per definition, identify mRNAs subjected exclusively to translational repression. This apparent limitation may not be as strong as initially thought, since recent data suggest that the majority of miRNA regulation can be detected at mRNA levels (see also the examples below) [59]. Proteomics, on the other hand, can potentially identify targets regulated at the translational level as well. However, the sensitivity and resolution of currently available proteomic tools typically allow identification of ∼1000 proteins by a two-dimensional differentiation in-gel electrophoresis or, maximally, 2000–5000 proteins by a recently developed quantitative mass spectrometry-based approach using stable isotope labelling with amino acids in cell culture (SILAC) compared with ∼30,000 protein-coding mRNAs identified by a traditional Affimetrix or Agilent gene expression arrays. Generally, whole-genome profiling approaches like mRNA expression arrays have an additional advantage. They allow for the determination of enrichment of miRNA seed-containing mRNAs (putative targets) among negatively regulated genes, and thus validate the specificity of miRNA manipulation (e.g. inhibition). Both mRNA and protein analyses have been utilised for identification of miR-21 targets, resulting in tropomyosin 1 (TPM-1) identified by proteomics and programmed cell death protein 4 (PDCD4), and reversion-inducing cysteine-rich protein with kazal (RECK) motifs by mRNA arrays. Below, we briefly describe validated miR-21 direct targets that meet at least the following criteria: (i) their expression correlates inversely with miR-21 levels and activity (i.e. increased in anti-miR-21-treated cells and reduced in miR-21-overexpressing cells); (ii) they have miR-21 binding site(s) with a complementary seed and are capable of directly binding to miR-21, as detected in a luciferase reporter assay. The luciferase constructs with a target 3′UTR are specifically responsive to miR-21 overexpression or anti-miR-21 treatment (or both). Finally, deletion or mutation of the miR-21 binding site from the 3′UTR abolishes the miR-21 regulation (the targets are summarized in Table 1).
PDCD4 is one of the principal miR-21 targets validated independently by several groups. It has a single highly conserved miR-21 target site within its 3′UTR, and its regulation by miR-21 has been reported in a number of human cancer cells including breast cancer [53, 60], colorectal cancer [52] and glioma [49], as well as in a murine JB6 epidermal model of neoplastic transformation [50]. Reduced PDCD4 expression has been reported in at least six human tumour types or cancer cell lines (lung, brain, renal, breast, colon and pancreas) [61–64] in which miR-21 is overexpressed [1, 3, 65–67], indicating that miR-21-PDCD4 is likely to be a clinically significant oncogene/tumour suppressor pair in the induction and progression of human carcinomas. PDCD4 is also a functional target of miR-21 involved in the BMP-mediated induction of smooth muscle cell markers in the differentiation of vascular smooth muscle cells [42].
Initially discovered as a gene that is up-regulated in apoptosis in response to a number of inducers [68, 69], PDCD4 was further characterised as a potent tumour suppressor. PDCD4 inhibits PMA-induced neoplastic transformation [70] and tumour promotion and progression [71] and inhibits invasion and intravasation [72]. It is down-regulated in a number of cancers, and its suppression in lung and colorectal cancers is associated with poor patient prognosis [61, 73]. PDCD4 interacts with translation initiation factors eIF4A and eIF4G and inhibits translation initiation by displacing eIF4G and RNA from eIF4A [74–76]. Specific molecules regulated by PDCD4 include p21 [77], Cdk4, ornithine decarboxylase [71], carbonic anhydrase II [78] and JNK/c-Jun/AP-1 [79, 80].
RECK is a membrane-anchored inhibitor of metalloproteinases (MMPs) whose reduced expression or inactivation seems to be critical for the invasiveness and metastasis of various cancers, including glioma [81, 82]. Its expression level is also an important prognostic factor for multiple cancer types [83]. miR-21 regulation of RECK expression was detected in glioma and osteoblas-toma cancer cells [49] and gastric cancers [5]. In glioma, RECK appears to be a principal target that mediates miR-21 invasiveness and possibly angiogenesis by inhibiting activities of MMP-2, MMP-9 and other MMPs. Interestingly, RECK also appears as the major miR-21 target and MMP regulator in mouse uterus during embryo implantation and in endometrial adenocarcinoma Ishikawa cells [84]. Therefore, miR-21 may be a key regulator of normal cell motility and invasiveness during developmental processes (e.g. blastocyst implantation), as well as of cancer cell invasiveness.
Another recently identified miR-21 target, perhaps more confined to several cell types, is mammary serine protease inhibitor or maspin [60], a non-inhibitory serpin with tumour-suppressive properties. The molecular mechanisms underlying maspin's pro-apoptotic, anti-angiogenic and anti-metastatic functions are diverse [85]: transcriptional control by regulation of chromatin remodelling activities and direct interactions with transcription factors, regulation of GSH redox system and thus maintaining cellular homeostasis and responding to cellular stress and regulation of integrin profile and invasiveness of the cell. So far, miR-21 regulation of maspin expression has been demonstrated exclusively in metastatic breast MDA-MB-231 cells.
NFIB, a phylogenetically conserved miR-21 target, is a member of the NFI gene family, often functioning as a versatile transcriptional repressor of many promoters either through competition with other transcriptional factors for binding or through changes in the nucleosome structure. This protein is essential for lung maturation and brain development, but its function in cancer is not well studied yet. miR-21 regulation over NFIB mRNA has been demonstrated in HCT-116 colon carcinoma, HL-60 myeloid cells [25] and glioma [49].
Tropomyosin 1 (TPM1), a protein with potential of suppressing cell growth and invasiveness of breast carcinoma, is the only miR-21 target identified in the analysis of MCF-7 tumours by a proteomics approach [58]. Perhaps this fact can be explained by the high expression of this actin-binding microfilament-stabilising protein. Interestingly, although miR-21 inhibits TPM1 protein translation in MCF7 cells, steady-state TPM1 mRNA levels are not affected by miR-21. However, in human glioma cells, TPM1 mRNA levels were up-regulated by anti-miR-21 [49]. It should be noted though that the miR-21 binding site within TPM1 mRNA is not conserved in rodent cells and therefore TPM1 may represent a human-specific target.
Sprouty2 (SPRY2), a protein that affects cellular outgrowths, branching and migration and is down-regulated in a number of cancers expressing high miR-21 levels, has been described as a direct miR-21 target in cardiocytes and colon cancer SW480 cells [86]. It can represent a physiologically relevant miR-21 target in cardiac hypertrophy and perhaps some developmental processes and specific forms of cancer.
miR-21 also regulates expression of the well-known tumour suppressor phosphatase and tensin (PTEN) homologue and downstream PI3-kinase signalling in human cholangiocarcinoma and hepatocellular carcinoma cells [51, 87]. Both miR-21 and anti-miR-21 modulate PTEN levels and a luciferase construct containing PTEN 3′UTR. Furthermore, down-regulation of PTEN by siRNA attenuates the effects of anti-miR-21 on hepatocellular carcinoma cell growth and invasion. Given the importance of the PTEN pathway and the frequency of PTEN mutations or silencing in a variety of cancers, the effects of miR-21 on PTEN expression have been tested in additional cell lines. Modulation of PTEN protein expression by miR-21 was detected in a colon cancer cell line [52] and VSMCs [21], but not in the MCF-7 breast cancer [53], A549 non-small cell lung cells [88] or glioma cells [49]. It is still unclear whether miR-21 may regulate PTEN directly or not since the miR-21 binding site in the PTEN mRNA has not been characterised yet. In addition, so far, no mutation analysis abolishing a direct binding and targeting by miR-21 has been performed. Whether direct or not, miR-21 regulation over PTEN appears to be cell specific rather than being common to a number of cancers.
miR-21 in gliomas: targeting cell cycle, apoptosis and invasion
Our recent work on a glioma cell model, in which miR-21 was either inhibited by the most potent and specific 2′O-MOE ASO or overexpressed with a synthetic duplex RNA, allowed us to survey miR-21 signalling in a more detailed way [49]. One interesting question emerging from our studies referred to miR-21's role in targeting the expression of functionally related proteins; in other words, whether it regulates a specific signalling pathway in a certain cellular environment.
Transcriptional profiling of cells after miR-21 knockdown revealed changes in the expression of ∼570 genes (P< 0.05) associated with various biological functions [49]. DNA damage response genes, regulators of cell cycle arrest and positive regulators of apoptosis were enriched among the genes that were up-regulated within 24 hrs. Among down-regulated genes, those involved in stress response, apoptosis, regulation of signal transduction (particularly, JNK cascade, MAPKKK cascade and stress-activated protein kinase pathway) and, most significantly, genes associated with blood vessel morphogenesis and development were strongly enriched (e < 10−4). This molecular profiling suggested that miR-21 regulates multiple genes involved in several cellular programmes in glioma cells. From a technical perspective, accurate analysis of mRNA expression profiling after miR-21 knockdown and overexpression in combination with bioinformatics analysis enabled discrimination between direct targets and indirect downstream effects. Notably, the previously identified miR-21 targets TPM1 and NFIB, whose mRNA levels previously seemed unchanged by miR-21 [25, 58], were detected by our arrays, suggesting that destabilisation of mRNA targets is a general (though a weak) mechanism of miRNA regulation.
In addition to several validated miR-21 targets described above (PDCD4, RECK, TPM1 and NFIB), many seed-containing computationally predicted (by commonly used algorithms TargetScan, PicTar Miranda and RNA22) targets indeed respond to both increased and decreased levels of miR-21 [49]. Among them are the following: STAT3, SOX2, PELI1, Yod1, PPARA, GPR64, RASGRP1, FAM63B, TIMP3, CDC25A, GLCCI1, TRIM59, CCDC14, PLEKHA1, CPEB3, MSH2, TNFRSF11B, ANKRD46, Sesn1, FAM3c and APAF1. Several of these genes play important roles in glioma biology and in carcinogenesis, and may likely represent direct miR-21 targets yet to be validated. For example, APAF1, the apoptotic protease activating fac-tor-1, is the molecular core of the apoptosome. It is typically required for activation of those caspases that initiate apoptosis [89, 90]. APAF1 3′UTR contains a strong miR-21 binding site (9-mer binding at miR-21 5′ end), and therefore it is likely one of the direct miR-21 targets. In gliomas, APAF1 is often inactivated or down-regulated [91], and our data suggest that these effects can be at least partly due to miR-21 regulation, in addition to the reported chromosome 12q22–23 LOH and hypermethylation [91]. Overexpression of APAF1 by viral transduction could induce apoptosis in glioma cells and may be beneficial in glioma treatment [92]. STAT3, the other gene that may have a tumour suppressor function in GBM [93], is also negatively regulated by miR-21, according to the microarray data, and is a predicted miR-21 target [49, 57, 94]. TIMP3, a tissue inhibitor of MMPs that inhibits angiogenesis and tumour cell infiltration and induces apoptosis [95, 96], is also extensively regulated by miR-21 in glioma and MCF7 breast cancer and U2OS osteobastoma cells [49]. Though TIMP3 3′UTR has two putative miR-21 binding sites, we were unable to validate its direct binding using a luciferase reporter system. Nevertheless, TIMP3 down-regulation in a number of cancers, including GBM, hepatocellular carcinoma and adenocarcinoma, is associated with tumour cell invasiveness and increased angiogenesis and is clearly caused, at least partly, by miR-21.
The role of other genes associated with cancer and (possibly directly) regulated by miR-21, such as TGFB2, CDC25a, PPARA, SKP2, MEIS1, LIFR and CPEB3, and their contribution to miR-21 pleiotropic function have to be further investigated [49]. It is also worth noting that miR-21 inhibition leads to the reduced expression of several critical oncogenes, including MYC, Jun, RELB and LIF, and MYC reduction was also detected by a similar analysis performed on breast cancer MCF7 cells [53]. Therefore, it appears that multiple critical proteins associated with the glioma cell cycle, apoptosis and invasion, rather than a single signalling pathway, are regulated by miR-21.
miR-21 networking and feedback regulation
Interestingly, miR-21 seems to be involved in a number of positive and negative feedback loops, and therefore is a part of the complex regulatory network operating in both normal and diseased cells (Fig. 2). These complex regulations may explain why miR-21 is probably one of the most dynamic miRNAs responsive to various stimuli.
2.
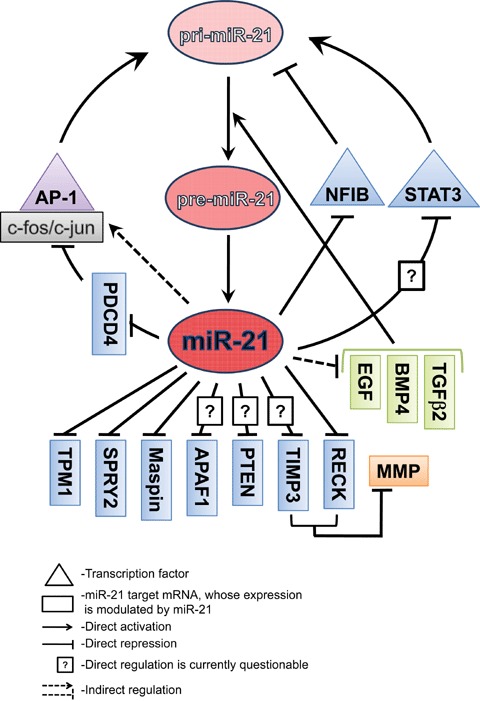
Model of miR-21 network and feedback regulation. Maturation of miR-21 from pri-miR-21 is shown in the center of the model. miR-21 direct target genes are depicted on blue background. Genes shown on green background are regulated (probably indirectly) by miR-21 and are involved in miR-21 processing from pri-miR-21 to pre-miR-21.
One evolutionary conserved regulatory module consists of miR-21 and its direct target NFIB [25]. NFIB is a transcriptional repressor that suppresses basal expression of the miR-21 gene. In stimulated or cancer cells, it can be displaced from the miR-21 promoter (e.g. by AP-1 in PMA-induced cells), which may lead to elevation of miR-21 levels, miR-21 binding to NFIB mRNA, down-regulation of NFIB production and further up-regulation of miR-21 expression.
Another related mechanism of sustained miR-21 expression might involve its transcriptional inducer AP-1. As previously discussed, AP-1 mediates transcriptional activation of the miR-21 promoter [25]. We hypothesize that miR-21, in turn, is capable of inducing AP-1 activity and AP-1-dependent transcription by two, likely independent, mechanisms. First, miR-21 represses expression of PDCD4, a protein that blocks the transactivation of AP-1 by interfering with c-Jun phosphorylation and activation [79]. In addition, miR-21 knockdown in glioma cells leads to down-regulation of c-Jun mRNA [49], suggesting that miR-21 indirectly activates expression of c-Jun and thus may also induce AP-1-dependent transcription. Since AP-1 itself acts as an miR-21 inducer in cancer cells, it can in fact initiate the self-perpetuating circle of AP-1-dependent transcription of cancer genes.*
Both feedback mechanisms, the first double-negative and the second double-positive, may contribute to high levels of miR-21 expression in cancer and suggest a self-sustained machinery of miR-21 expression. It was noted, though, that overexpression of exogenous miR-21 caused only a moderate increase in the production of endogenous miR-21 [25]. While this topic requires further investigation, additional negative regulatory mechanisms that help stabilise miR-21 levels in the normal cellular environment may exist, one of them being STAT3-mediated IL-6–miR-21 autocrine feedback. STAT3-dependent miR-21 transcription has been demonstrated in several cell types [36], and it may be one of the factors inducing miR-21 expression in some cancers. miR-21, in turn, may down-regulate STAT3, since it has two conserved miR-21 binding sites, and STAT3 mRNA levels were regulated in glioma cells by both miR-21 inhibition and overexpression [49]. Such a regulatory loop between miR-21 and IL-6/STAT3 may provide a feedback mechanism for stabilizing miR-21 expression and balancing STAT3 signalling.
Further work is also required to explore the potential relationship between miR-21 and TGF-β signalling. As discussed earlier miR-21 maturation is induced by TGF-β and BMP4 ligands [42]. Analysis of our arrays indicates that miR-21 may regulate, either directly or indirectly, TGF-p2, BMP4 and EGF factors, as well as receptors TGF-pR1 and TGF-pR2 (that are predicted as direct miR-21 targets) [49]. Recent data by Papagiannakopoulos et al. also suggest the regulation of the TGF-β pathway by miR-21 [97]. If validated, the involvement of miR-21 in the TGF-β pathway will be important for understanding complex molecular networks associated with oncogenic and tumour-suppressive properties of these molecules. Particularly, it would be very interesting to investigate whether miR-21 accumulation in cancer progression leads to reduced expression of TGF-β receptors, which may result in resistance to growth inhibition by TGF-β, explaining the characteristic but poorly understood switch of TGF-β from tumour suppressor to tumour promoter.
miR-21 as a diagnostic and prognostic marker
Strongly elevated expression of miR-21 in a variety of human neoplastic disorders and its demonstrated regulatory potential in targeting a number of important tumour suppressor genes suggest that miR-21 can be used as a diagnostic biomarker. Furthermore, if miR-21 expression is causal to the progression of cancer, its elevated levels may be associated with more advanced stages of the disease and may be prognostic. Since this miRNA is one of the most abundant in a variety of cancer cells, and thus easily detectable, data from multiple studies suggest that it could be uniquely suited as a biomarker.
Generally, more advanced/malignant tumours indeed express higher levels of miR-21. For example, miR-21 expression is significantly up-regulated in glioma progression from low grades to GBM (most malignant grade IV glioma) [1, 49]. In breast cancer, miR-21 overexpression correlates significantly with advanced clinical stage, lymph node metastasis and patient's poor prognosis [98]. miR-21 expression is significantly higher in colon adenocarcinomas than in their precursor stage adenomas and correlates with the adenoma staging [99] and the development of metastasis [100]. Moreover, in a large study performed by Schetter et al. [99] on two independent cohorts totaling ∼200 colon adenocarcinoma patients, miR-21 was the only miRNA associated confidently with poor survival and poor therapeutic outcome. In pancreatic endocrine tumours, high miR-21 levels correlate with more aggressive tumours, as signified by an increased Ki67 proliferation index and the presence of liver metastases [66]. A group of pancreatic ductal adenocarcinoma patients with lower miR-21 expression demonstrated a 50% longer survival than the remainder of the patients tested, though it was not statistically significant due to the small number of tumours analysed [101]. In a study performed on 48 pairs of non-small cell lung cancer (NSCLC) specimens, miR-21 overexpression correlated inversely with overall survival of the patients, suggesting that a high level of miR-21 is an independent negative prognostic factor for survival in NSCLC patients [102]. However, in gastric carcinomas in which miR-21 can serve as a diagnostic marker, its levels did not seem to have prognostic value [103]. Strikingly, a recent report about miR-21 expression in patients with DLBCL suggests that high levels of tumoural miR-21 were associated with a better prognostic outcome [12]. Moreover, high expression levels of miR-21 in DLBCL patient sera were found to be associated with improved relapse-free survival time, though not with overall survival [104]. Why DLBCL patients with lower miR-21 levels have a poorer clinical prognosis remains to be determined. Overall, these combined data clearly indicate that the miR-21 molecule could match the rigorous criteria of an ideal biomarker in our search for non-invasive tools for the diagnosis and management of cancer.
Recent advances in the characterisation of tumour-derived exosomes (also called microvesicles) further extend miR-21's utility as a biomarker. Exosomes are the ‘bioactive vesicles’ released by many tumours (as well as some normal cells of various origins) that are taken up by surrounding host cells, and therefore function to promote intercellular communication [105]. Tumours also release exosomes into peripheral circulation, and exosomes can be readily isolated from patients’ blood by differential centrifugation or using tumour markers such as epithelial cell adhesion molecule (EpCAM). Exosomes contain specific sets of proteins and RNA and seem to be particularly enriched in miRNAs. Recent studies performed on circulating tumour exosomes from ovarian and lung cancer patients indicate a high degree of correlation between the miRNA profiles of the tumour and its corresponding exosomes [106]. Similarly, miRNAs elevated in biopsies of GBM patients were also detected in corresponding serum-derived exosomes [107]. In both studies, miR-21 was one of the most abundant miRNAs detected in patients’ circulating exosomes. These data suggest that miR-21 levels in exosomes can be used as a surrogate marker for diagnostic or prognostic biopsy profiling. While validation studies will be necessary prior to bypassing the use of tumour mass biopsies, it is possible that, for a number of human neoplasias, miR-21 levels in peripheral circulation may serve as a measure of cancer stage or for the monitoring of therapeutic response or disease recurrence.
Potential therapeutic target
Ideal therapeutic targets should be causally associated with disease and suitable for designing therapeutic interventions. In this review, we have described a function of miR-21 associated with tumour cell invasiveness and resistance to apoptosis and its direct regulation of multiple tumour suppressor genes, pro-apoptotic and anti-invasive. The accumulated data support a very attractive idea that sequence-specific inhibition of a single oncomir, miR-21, can provide a novel therapeutic approach for ‘physiological’ modulation of multiple proteins whose expression is de-regulated in cancer. The findings of in vivo efficacy of miR-21 inhibitors against breast carcinoma suggest therapeutic potential for such modulation. Indeed, treatment with anti-miR-21 oligonucleotides reduced breast cancer MCF-7 xenograft growth by approximately 50% for up to 2 weeks [54]. miR-21 inhibition also induces apoptosis and blocks anchorage-independent growth of hepatocellular carcinoma [17]. While the data suggesting pro-apoptotic effect of miR-21 inhibitors on glioma cells in vitro and in vivo[1, 108] were not validated by application of the potent and specific 2′O-MOE inhibitor, this inhibitor, nevertheless, had significant effects on glioma cell migration and invasion and reduced MMP activities in a mouse model of human glioma xenografts [49]. Effects of miR-21 inhibitors on proliferation and apoptosis of VSMCs also suggested miR-21 as a new therapeutic target for proliferative vascular diseases such as atherosclerosis, post-angioplasty restenosis, transplantation arteriopathy and stroke [21].
miR-21 modulation may also sensitise cells and play a role in modulating drug response. Several reports suggest that miR-21 is one of the key miRNAs playing a broad role in sensitivity to chemotherapeutic agents. It has been demonstrated that suppression of miR-21 in a cholangiocarcinoma cell line increased sensitivity to gemcitabine [87]. Similarly, growth inhibition of breast cancer MCF7 cells by topotecan, a clinical camptothecin analogue, was increased by 40% by transfection with miR-21 antisense oligonucleotides [54]. In a study of miRNA effects on drug response performed on three cancer cell lines (non-small lung A549, glioma SNB19 and ovarian OVCAR3), the effects were most prominent for miR-21, whose levels significantly shifted the growth-inhibitory activity of 6 out of 10 compounds tested [88]. The effects were generally consistent among the three different cell lines tested, that is, if decreased levels of miR-21 increased the sensitivity of A549 to a compound, they also increased the sensitivity of the other cell lines. The shifts in drug potency detected did not exceed four-fold in terms of the differences between cells treated with miRNA precursor and with inhibitor, perhaps due to the relatively weak miR-21 inhibitor used in this study. Nevertheless, even small changes in activity could make a difference between the success and failure of cancer chemotherapy. Further research is obviously required to address the therapeutic potential of modulating miR-21 alone and/or in combination with other targets. As for any targeted miRNA, in vivo tests should carefully consider benefits of the miR-21 modulation while taking into account a variety of molecular effects produced at the whole-RNome level (including possible effects not caused by miR-21 but rather associated with expression changes in miR-21-unrelated genes or off-targets).
If the miR-21 modulation approach is found effective, more prolonged effects of its inhibition on tumour growth and invasiveness will be tested and more delivery systems will be developed in the future. Currently, synthetic chemically modified antisense oligonucleotides that can be delivered either systemically or locally [109–111], and particularly cholesterol-conjugated antagomirs, represent the most powerful tool for silencing a specific miRNA in vivo. At first glance, the task of miRNA inhibitor delivery would seem feasible in the era of RNAi and targeted gene silencing. On closer examination, however, this approach will pose at least one serious problem: miR-21 inhibitor drugs may have undesirable side effects including those associated with inhibition of miR-21 in normal non-cancerous cells. Though miR-21 levels are usually low in normal adult cells, functions of miR-21 in normal cells of different origins and effects of miR-21 inhibition in these cells have to be further explored in the future. Cell-specific viral delivery of miRNA ‘sponges’[112] for targeted inhibition of miR-21 in diseased (e.g. tumour) cells may represent a valuable alternative approach for miRNA inhibition. In any case, strong association of miR-21 with multiple human diseases and its function in controlling a number of key cancer genes make this small molecule an excellent target for future research and likely for gene therapy.
* Comment: when this review was submitted for publication, a paper describing RAS/AP-1/miR-21/PDCD4 relationship and confirming AP-1/miR-21 regulatory feedback loop has been published (Talotta F, Cimmino A, Matarazzo MR, Casalino L, DE Vita G, D'Esposito M, Di Lauro R, Verde P. An autoregulatory loop mediated by miR-21 and PDCD4 controls the AP-1 activity in RAS transformation. Oncogene. 2008 Oct 13, epub ahead of print).
Acknowledgments
We thank Krichevsky laboratory members for insightful discussions and Dr. Kai-Christian Sonntag for his careful reading of the manuscript. The work in the laboratory is supported by grants from the National Cancer Institute and the Brain Tumor Society.
References
- 1.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–33. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 2.Ciafrè SA, Galardi S, Mangiola A, Ferracin M, Liu CG, Sabatino G, Negrini M, Maira G, Croce CM, Farace MG. Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem Biophys Res Commun. 2005;334:1351–8. doi: 10.1016/j.bbrc.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 3.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM. A micro-RNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006;103:2257–61. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kutay H, Bai S, Datta J, Motiwala T, Pogribny I, Frankel W, Jacob ST, Ghoshal K. Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J Cell Biochem. 2006;99:671–8. doi: 10.1002/jcb.20982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Z, Li Z, Gao C, Chen P, Chen J, Liu W, Xiao S, Lu H. miR-21 plays a pivotal role in gastric cancer pathogenesis and progression. Lab Invest. 2008 doi: 10.1038/labinvest.2008.94. [DOI] [PubMed] [Google Scholar]
- 6.Iorio MV, Visone R, Di Leva G, Donati V, Petrocca F, Casalini P, Taccioli C, Volinia S, Liu CG, Alder H, Calin GA, Ménard S, Croce CM. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007;67:8699–707. doi: 10.1158/0008-5472.CAN-07-1936. [DOI] [PubMed] [Google Scholar]
- 7.Nam EJ, Yoon H, Kim SW, Kim H, Kim YT, Kim JH, Kim JW, Kim S. MicroRNA expression profiles in serous ovarian carcinoma. Clin Cancer Res. 2008;14:2690–5. doi: 10.1158/1078-0432.CCR-07-1731. [DOI] [PubMed] [Google Scholar]
- 8.Lui WO, Pourmand N, Patterson BK, Fire A. Patterns of known and novel small RNAs in human cervical cancer. Cancer Res. 2007;67:6031–43. doi: 10.1158/0008-5472.CAN-06-0561. [DOI] [PubMed] [Google Scholar]
- 9.Tran N, McLean T, Zhang X, Zhao CJ, Thomson JM, O'Brien C, Rose B. MicroRNA expression profiles in head and neck cancer cell lines. Biochem Biophys Res Commun. 2007;358:12–7. doi: 10.1016/j.bbrc.2007.03.201. [DOI] [PubMed] [Google Scholar]
- 10.Tetzlaff MT, Liu A, Xu X, Master SR, Baldwin DA, Tobias JW, Livolsi VA, Baloch ZW. Differential expression of miRNAs in papillary thyroid carcinoma compared to multinodular goiter using formalin fixed paraffin embedded tissues. Endocr Pathol. 2007;18:163–73. doi: 10.1007/s12022-007-0023-7. [DOI] [PubMed] [Google Scholar]
- 11.Fulci V, Chiaretti S, Goldoni M, Azzalin G, Carucci N, Tavolaro S, Castellano L, Magrelli A, Citarella F, Messina M, Maggio R, Peragine N, Santangelo S, Mauro FR, Landgraf P, Tuschl T, Weir DB, Chien M, Russo JJ, Ju J, Sheridan R, Sander C, Zavolan M, Guarini A, Foà R, Macino G. Quantitative technologies establish a novel microRNA profile of chronic lymphocytic leukemia. Blood. 2007;109:4944–51. doi: 10.1182/blood-2006-12-062398. [DOI] [PubMed] [Google Scholar]
- 12.Lawrie CH, Soneji S, Marafioti T, Cooper CD, Palazzo S, Paterson JC, Cattan H, Enver T, Mager R, Boultwood J, Wainscoat JS, Hatton CS. MicroRNA expression distinguishes between germinal center B cell-like and activated B cell-like subtypes of diffuse large B cell lymphoma. Int J Cancer. 2007;121:1156–61. doi: 10.1002/ijc.22800. [DOI] [PubMed] [Google Scholar]
- 13.Jongen-Lavrencic M, Sun SM, Dijkstra MK, Valk PJ, Lowenberg B. MicroRNA expression profiling in relation to the genetic heterogeneity of acute myeloid leukemia. Blood. 2008;111:5078–85. doi: 10.1182/blood-2008-01-133355. [DOI] [PubMed] [Google Scholar]
- 14.Navarro A, Gaya A, Martinez A, Urbano-Ispizua A, Pons A, Balagué O, Gel B, Abrisqueta P, Lopez-Guillermo A, Artells R, Montserrat E, Monzo M. MicroRNA expression profiling in classic Hodgkin lymphoma. Blood. 2008;111:2825–32. doi: 10.1182/blood-2007-06-096784. [DOI] [PubMed] [Google Scholar]
- 15.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, Lin C, Socci ND, Hermida L, Fulci V, Chiaretti S, Foà R, Schliwka J, Fuchs U, Novosel A, Müller RU, Schermer B, Bissels U, Inman J, Phan Q, Chien M, Weir DB, Choksi R, De Vita G, Frezzetti D, Trompeter HI, Hornung V, Teng G, Hartmann G, Palkovits M, Di Lauro R, Wernet P, Macino G, Rogler CE, Nagle JW, Ju J, Papavasiliou FN, Benzing T, Lichter P, Tam W, Brownstein MJ, Bosio A, Borkhardt A, Russo JJ, Sander C, Zavolan M, Tuschl T. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–14. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mrazek J, Kreutmayer SB, Grasser FA, Polacek N, Huttenhofer A. Subtractive hybridization identifies novel differentially expressed ncRNA species in EBV-infected human B cells. Nucleic Acids Res. 2007;35:e73. doi: 10.1093/nar/gkm244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Connolly E, Melegari M, Landgraf P, Tchaikovskaya T, Tennant BC, Slagle BL, Rogler LE, Zavolan M, Tuschl T, Rogler CE. Elevated expression of the miR-17–92 polycistron and miR-21 in hepadnavirus-associated hepatocellular carcinoma contributes to the malignant phenotype. Am J Pathol. 2008;173:856–64. doi: 10.2353/ajpath.2008.080096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am J Pathol. 2007;170:1831–40. doi: 10.2353/ajpath.2007.061170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, Rojas M, Hammond SM, Wang DZ. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2007;42:1137–41. doi: 10.1016/j.yjmcc.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA. 2006;103:18255–60. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of microRNA in vascular neointimal lesion formation. Circ Res. 2007;100:1579–88. doi: 10.1161/CIRCRESAHA.106.141986. [DOI] [PubMed] [Google Scholar]
- 22.Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, Van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, Bauersachs J. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–67. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- 23.Houbaviy HB, Murray MF, Sharp PA. Embryonic stem cell-specific microRNAs. Dev Cell. 2003;5:351–8. doi: 10.1016/s1534-5807(03)00227-2. [DOI] [PubMed] [Google Scholar]
- 24.Kasashima K, Nakamura Y, Kozu T. Altered expression profiles of microRNAs during TPA-induced differentiation of HL-60 cells. Biochem Biophys Res Commun. 2004;322:403–10. doi: 10.1016/j.bbrc.2004.07.130. [DOI] [PubMed] [Google Scholar]
- 25.Fujita S, Ito T, Mizutani T, Minoguchi S, Yamamichi N, Sakurai K, Iba H. miR-21 gene expression triggered by AP-1 is sustained through a double-negative feedback mechanism. J Mol Biol. 2008;378:492–504. doi: 10.1016/j.jmb.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 26.Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J, Song E. Let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–23. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 27.Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA. 2004;10:1957–66. doi: 10.1261/rna.7135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu GJ, Sinclair CS, Paape J, Ingle JN, Roche PC, James CD, Couch FJ. 17q23 amplifications in breast cancer involve the PAT1, RAD51C, PS6K, and SIGma1B genes. Cancer Res. 2000;60:5371–5. [PubMed] [Google Scholar]
- 29.Kasahara K, Taguchi T, Yamasaki I, Kamada M, Yuri K, Shuin T. Detection of genetic alterations in advanced prostate cancer by comparative genomic hybridization. Cancer Genet Cytogenet. 2002;137:59–63. doi: 10.1016/s0165-4608(02)00552-6. [DOI] [PubMed] [Google Scholar]
- 30.Griffin CA, Hawkins AL, Packer RJ, Rorke LB, Emanuel BS. Chromosome abnormalities in pediatric brain tumors. Cancer Res. 1988;48:175–80. [PubMed] [Google Scholar]
- 31.Chui DT, Hammond D, Baird M, Shield L, Jackson R, Jarrett RF. Classical Hodgkin lymphoma is associated with frequent gains of 17q. Genes Chromosomes Cancer. 2003;38:126–36. doi: 10.1002/gcc.10266. [DOI] [PubMed] [Google Scholar]
- 32.Roversi G, Pfundt R, Moroni RF, Magnani I, Van Reijmersdal S, Pollo B, Straatman H, Larizza L, Schoenmakers EF. Identification of novel genomic markers related to progression to glioblastoma through genomic profiling of 25 primary glioma cell lines. Oncogene. 2006;25:1571–83. doi: 10.1038/sj.onc.1209177. [DOI] [PubMed] [Google Scholar]
- 33.Saito-Ohara F, Imoto I, Inoue J, Hosoi H, Nakagawara A, Sugimoto T, Inazawa J. PPM1D is a potential target for 17q gain in neuroblastoma. Cancer Res. 2003;63:1876–83. [PubMed] [Google Scholar]
- 34.Thorland EC, Myers SL, Gostout BS, Smith DI. Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene. 2003;22:1225–37. doi: 10.1038/sj.onc.1206170. [DOI] [PubMed] [Google Scholar]
- 35.Zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–50. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 36.Löffler D, Brocke-Heidrich K, Pfeifer G, Stocsits C, Hackermüller J, Kretzschmar AK, Burger R, Gramatzki M, Blumert C, Bauer K, Cvijic H, Ullmann AK, Stadler PF, Horn F. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood. 2007;110:1330–3. doi: 10.1182/blood-2007-03-081133. [DOI] [PubMed] [Google Scholar]
- 37.Piper M, Dawson AL, Lindwall C, Barry G, Plachez C, Richards LJ. Emx and Nfi genes regulate cortical development and axon guidance in the telencephalon. Novartis Found Symp. 2007;288:230–42. discussion 42–5, 76–81. [PubMed] [Google Scholar]
- 38.Singh SK, Kagalwala MN, Parker-Thornburg J, Adams H, Majumder S. REST maintains self-renewal and pluripotency of embryonic stem cells. Nature. 2008;453:223–7. doi: 10.1038/nature06863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson R, Zuccato C, Belyaev ND, Guest DJ, Cattaneo E, Buckley NJ. A microRNA-based gene dysregulation pathway in Huntington's disease. Neurobiol Dis. 2008;29:438–45. doi: 10.1016/j.nbd.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 40.Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 2006;20:2202–7. doi: 10.1101/gad.1444406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 42.Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zavadil J, Narasimhan M, Blumenberg M, Schneider RJ. Transforming growth factor-beta and microRNA: mRNA regulatory networks in epithelial plasticity. Cells Tissues Organs. 2007;185:157–61. doi: 10.1159/000101316. [DOI] [PubMed] [Google Scholar]
- 44.Kulshreshtha R, Ferracin M, Wojcik SE, Garzon R, Alder H, Agosto-Perez FJ, Davuluri R, Liu CG, Croce CM, Negrini M, Calin GA, Ivan M. A microRNA signature of hypoxia. Mol Cell Biol. 2007;27:1859–67. doi: 10.1128/MCB.01395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salnikow K, Kluz T, Costa M, Piquemal D, Demidenko ZN, Xie K, Blagosklonny MV. The regulation of hypoxic genes by calcium involves c-Jun/AP-1, which cooperates with hypoxia-inducible factor 1 in response to hypoxia. Mol Cell Biol. 2002;22:1734–41. doi: 10.1128/MCB.22.6.1734-1741.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shih SC, Claffey KP. Role of AP-1 and HIF-1 transcription factors in TGF-beta activation of VEGF expression. Growth Factors. 2001;19:19–34. doi: 10.3109/08977190109001073. [DOI] [PubMed] [Google Scholar]
- 47.Davis S, Lollo B, Freier S, Esau C. Improved targeting of miRNA with antisense oligonucleotides. Nucleic Acids Res. 2006;34:2294–304. doi: 10.1093/nar/gkl183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orom UA, Kauppinen S, Lund AH. LNA-modified oligonucleotides mediate specific inhibition of microRNA function. Gene. 2006;372:137–41. doi: 10.1016/j.gene.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 49.Gabriely G, Wurdinger T, Kesari S, Esau CC, Burchard J, Linsley PS, Krichevsky AM. MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol Cell Biol. 2008;28:5369–80. doi: 10.1128/MCB.00479-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu Z, Liu M, Stribinskis V, Klinge CM, Ramos KS, Colburn NH, Li Y. MicroRNA-21 promotes cell transformation by targeting the programmed cell death 4 gene. Oncogene. 2008;27:4373–9. doi: 10.1038/onc.2008.72. [DOI] [PubMed] [Google Scholar]
- 51.Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133:647–58. doi: 10.1053/j.gastro.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S, Allgayer H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27:2128–36. doi: 10.1038/sj.onc.1210856. [DOI] [PubMed] [Google Scholar]
- 53.Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem. 2008;283:1026–33. doi: 10.1074/jbc.M707224200. [DOI] [PubMed] [Google Scholar]
- 54.Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY. miR-21-mediated tumor growth. Oncogene. 2007;26:2799–803. doi: 10.1038/sj.onc.1210083. [DOI] [PubMed] [Google Scholar]
- 55.Griffiths-Jones S, Grocock RJ, Van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–4. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, Da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 58.Zhu S, Si ML, Wu H, Mo YY. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1) J Biol Chem. 2007;282:14328–36. doi: 10.1074/jbc.M611393200. [DOI] [PubMed] [Google Scholar]
- 59.Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu S, Wu H, Wu F, Nie D, Sheng S, Mo YY. MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell Res. 2008;18:350–9. doi: 10.1038/cr.2008.24. [DOI] [PubMed] [Google Scholar]
- 61.Chen Y, Knösel T, Kristiansen G, Pietas A, Garber ME, Matsuhashi S, Ozaki I, Petersen I. Loss of PDCD4 expression in human lung cancer correlates with tumour progression and prognosis. J Pathol. 2003;200:640–6. doi: 10.1002/path.1378. [DOI] [PubMed] [Google Scholar]
- 62.Lee S, Bang S, Song K, Lee I. Differential expression in normal-adenoma-carcinoma sequence suggests complex molecular carcinogenesis in colon. Oncol Rep. 2006;16:747–54. [PubMed] [Google Scholar]
- 63.Ma G, Guo KJ, Zhang H, Ozaki I, Matsuhashi S, Zheng XY, Dong M. Expression of programmed cell death 4 and its clinicopathological significance in human pancreatic cancer. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2005;27:597–600. [PubMed] [Google Scholar]
- 64.Jansen AP, Camalier CE, Stark C, Colburn NH. Characterization of programmed cell death 4 in multiple human cancers reveals a novel enhancer of drug sensitivity. Mol Cancer Ther. 2004;3:103–10. [PubMed] [Google Scholar]
- 65.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Ménard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–70. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 66.Roldo C, Missiaglia E, Hagan JP, Falconi M, Capelli P, Bersani S, Calin GA, Volinia S, Liu CG, Scarpa A, Croce CM. MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J Clin Oncol. 2006;24:4677–84. doi: 10.1200/JCO.2005.05.5194. [DOI] [PubMed] [Google Scholar]
- 67.Diederichs S, Haber DA. Sequence variations of microRNAs in human cancer: alterations in predicted secondary structure do not affect processing. Cancer Res. 2006;66:6097–104. doi: 10.1158/0008-5472.CAN-06-0537. [DOI] [PubMed] [Google Scholar]
- 68.Shibahara K, Asano M, Ishida Y, Aoki T, Koike T, Honjo T. Isolation of a novel mouse gene MA-3 that is induced upon programmed cell death. Gene. 1995;166:297–301. doi: 10.1016/0378-1119(95)00607-9. [DOI] [PubMed] [Google Scholar]
- 69.Zhang Z, DuBois RN. Detection of differentially expressed genes in human colon carcinoma cells treated with a selective COX-2 inhibitor. Oncogene. 2001;20:4450–6. doi: 10.1038/sj.onc.1204588. [DOI] [PubMed] [Google Scholar]
- 70.Cmarik JL, Min H, Hegamyer G, Zhan S, Kulesz-Martin M, Yoshinaga H, Matsuhashi S, Colburn NH. Differentially expressed protein Pdcd4 inhibits tumor promoter-induced neoplastic transformation. Proc Natl Acad Sci USA. 1999;96:14037–42. doi: 10.1073/pnas.96.24.14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jansen AP, Camalier CE, Colburn NH. Epidermal expression of the translation inhibitor programmed cell death 4 suppresses tumorigenesis. Cancer Res. 2005;65:6034–41. doi: 10.1158/0008-5472.CAN-04-2119. [DOI] [PubMed] [Google Scholar]
- 72.Leupold JH, Yang HS, Colburn NH, Asangani I, Post S, Allgayer H. Tumor suppressor Pdcd4 inhibits invasion/intravasation and regulates urokinase receptor (u-PAR) gene expression via Sp-transcription factors. Oncogene. 2007;26:4550–62. doi: 10.1038/sj.onc.1210234. [DOI] [PubMed] [Google Scholar]
- 73.Mudduluru G, Medved F, Grobholz R, Jost C, Gruber A, Leupold JH, Post S, Jansen A, Colburn NH, Allgayer H. Loss of programmed cell death 4 expression marks adenoma-carcinoma transition, correlates inversely with phosphorylated protein kinase B, and is an independent prognostic factor in resected colorectal cancer. Cancer. 2007;110:1697–707. doi: 10.1002/cncr.22983. [DOI] [PubMed] [Google Scholar]
- 74.Yang HS, Cho MH, Zakowicz H, Hegamyer G, Sonenberg N, Colburn NH. A novel function of the MA-3 domains in transformation and translation suppressor Pdcd4 is essential for its binding to eukaryotic translation initiation factor 4A. Mol Cell Biol. 2004;24:3894–906. doi: 10.1128/MCB.24.9.3894-3906.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zakowicz H, Yang HS, Stark C, Wlodawer A, Laronde-Leblanc N, Colburn NH. Mutational analysis of the DEAD-box RNA helicase eIF4AII characterizes its interaction with transformation suppressor Pdcd4 and eIF4GI. RNA. 2005;11:261–74. doi: 10.1261/rna.7191905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Suzuki C, Garces RG, Edmonds KA, Hiller S, Hyberts SG, Marintchev A, Wagner G. PDCD4 inhibits translation initiation by binding to eIF4A using both its MA3 domains. Proc Natl Acad Sci USA. 2008;105:3274–9. doi: 10.1073/pnas.0712235105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Goke R, Barth P, Schmidt A, Samans B, Lankat-Buttgereit B. Programmed cell death protein 4 suppresses CDK1/cdc2 via induction of p21(Waf1/Cip1) Am J Physiol Cell Physiol. 2004;287:C1541–6. doi: 10.1152/ajpcell.00025.2004. [DOI] [PubMed] [Google Scholar]
- 78.Lankat-Buttgereit B, Gregel C, Knolle A, Hasilik A, Arnold R, Goke R. Pdcd4 inhibits growth of tumor cells by suppression of carbonic anhydrase type II. Mol Cell Endocrinol. 2004;214:149–53. doi: 10.1016/j.mce.2003.10.058. [DOI] [PubMed] [Google Scholar]
- 79.Bitomsky N, Bohm M, Klempnauer KH. Transformation suppressor protein Pdcd4 interferes with JNK-mediated phosphorylation of c-Jun and recruitment of the coactivator p300 by c-Jun. Oncogene. 2004;23:7484–93. doi: 10.1038/sj.onc.1208064. [DOI] [PubMed] [Google Scholar]
- 80.Yang HS, Matthews CP, Clair T, Wang Q, Baker AR, Li CC, Tan TH, Colburn NH. Tumorigenesis suppressor Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase kinase 1 expression to suppress colon carcinoma cell invasion. Mol Cell Biol. 2006;26:1297–306. doi: 10.1128/MCB.26.4.1297-1306.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Echizenya M, Kondo S, Takahashi R, Oh J, Kawashima S, Kitayama H, Takahashi C, Noda M. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogen-esis. Cell. 2001;107:789–800. doi: 10.1016/s0092-8674(01)00597-9. [DOI] [PubMed] [Google Scholar]
- 82.Correa TC, Brohem CA, Winnischofer SM, Da Silva Cardeal LB, Sasahara RM, Taboga SR, Sogayar MC, Maria-Engler SS. Downregulation of the RECK-tumor and metastasis suppressor gene in glioma invasiveness. J Cell Biochem. 2006;99:156–67. doi: 10.1002/jcb.20917. [DOI] [PubMed] [Google Scholar]
- 83.Clark JC, Thomas DM, Choong PF, Dass CR. RECK—a newly discovered inhibitor of metastasis with prognostic significance in multiple forms of cancer. Cancer Metastasis Rev. 2007;26:675–83. doi: 10.1007/s10555-007-9093-8. [DOI] [PubMed] [Google Scholar]
- 84.Hu SJ, Ren G, Liu JL, Zhao ZA, Yu YS, Su RW, Ma XH, Ni H, Lei W, Yang ZM. MicroRNA expression and regulation in mouse uterus during embryo implantation. J Biol Chem. 2008;283:23473–84. doi: 10.1074/jbc.M800406200. [DOI] [PubMed] [Google Scholar]
- 85.Bailey CM, Khalkhali-Ellis Z, Seftor EA, Hendrix MJ. Biological functions of maspin. J Cell Physiol. 2006;209:617–24. doi: 10.1002/jcp.20782. [DOI] [PubMed] [Google Scholar]
- 86.Sayed D, Rane S, Lypowy J, He M, Chen IY, Vashistha H, Yan L, Malhotra A, Vatner D, Abdellatif M. MicroRNA-21 targets Sprouty2 and promotes cellular outgrowths. Mol Biol Cell. 2008;19:3272–82. doi: 10.1091/mbc.E08-02-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Meng F, Henson R, Lang M, Wehbe H, Maheshwari S, Mendell JT, Jiang J, Schmittgen TD, Patel T. Involvement of human micro-RNA in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology. 2006;130:2113–29. doi: 10.1053/j.gastro.2006.02.057. [DOI] [PubMed] [Google Scholar]
- 88.Blower PE, Chung JH, Verducci JS, Lin S, Park JK, Dai Z, Liu CG, Schmittgen TD, Reinhold WC, Croce CM, Weinstein JN, Sadee W. MicroRNAs modulate the chemosensitivity of tumor cells. Mol Cancer Ther. 2008;7:1–9. doi: 10.1158/1535-7163.MCT-07-0573. [DOI] [PubMed] [Google Scholar]
- 89.Johnson CE, Huang YY, Parrish AB, Smith MI, Vaughn AE, Zhang Q, Wright KM, Van Dyke T, Wechsler-Reya RJ, Kornbluth S, Deshmukh M. Differential Apaf-1 levels allow cytochrome c to induce apoptosis in brain tumors but not in normal neural tissues. Proc Natl Acad Sci USA. 2007;104:20820–5. doi: 10.1073/pnas.0709101105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bratton SB, Walker G, Srinivasula SM, Sun XM, Butterworth M, Alnemri ES, Cohen GM. Recruitment, activation and retention of caspases-9 and -3 by Apaf-1 apoptosome and associated XIAP complexes. EMBO J. 2001;20:998–1009. doi: 10.1093/emboj/20.5.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Watanabe T, Hirota Y, Arakawa Y, Fujisawa H, Tachibana O, Hasegawa M, Yamashita J, Hayashi Y. Frequent LOH at chromosome 12q22–23 and Apaf-1 inactivation in glioblastoma. Brain Pathol. 2003;13:431–9. doi: 10.1111/j.1750-3639.2003.tb00474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shinoura N, Sakurai S, Asai A, Kirino T, Hamada H. Transduction of Apaf-1 or cas-pase-9 induces apoptosis in A-172 cells that are resistant to p53-mediated apoptosis. Biochem Biophys Res Commun. 2000;272:667–73. doi: 10.1006/bbrc.2000.2839. [DOI] [PubMed] [Google Scholar]
- 93.De La Iglesia N, Konopka G, Puram SV, Chan JA, Bachoo RM, You MJ, Levy DE, Depinho RA, Bonni A. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008;22:449–62. doi: 10.1101/gad.1606508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 95.Baker AH, George SJ, Zaltsman AB, Murphy G, Newby AC. Inhibition of invasion and induction of apoptotic cell death of cancer cell lines by overexpression of TIMP-3. Br J Cancer. 1999;79:1347–55. doi: 10.1038/sj.bjc.6690217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Qi JH, Ebrahem Q, Moore N, Murphy G, Claesson-Welsh L, Bond M, Baker A, Anand-Apte B. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat Med. 2003;9:407–15. doi: 10.1038/nm846. [DOI] [PubMed] [Google Scholar]
- 97.Papagiannakopoulos T, Shapiro A, Kosik KS. MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res. 2008;68:8164–72. doi: 10.1158/0008-5472.CAN-08-1305. [DOI] [PubMed] [Google Scholar]
- 98.Yan LX, Huang XF, Shao Q, Huang MY, Deng L, Wu QL, Zeng YX, Shao JY. MicroRNA miR-21 overexpression in human breast cancer is associated with advanced clinical stage, lymph node metastasis and patient poor prognosis. RNA. 2008 doi: 10.1261/rna.1034808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, Bowman ED, Yanaihara N, Yuen ST, Chan TL, Kwong DL, Au GK, Liu CG, Calin GA, Croce CM, Harris CC. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon ade-nocarcinoma. JAMA. 2008;299:425–36. doi: 10.1001/jama.299.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Slaby O, Svoboda M, Fabian P, Smerdova T, Knoflickova D, Bednarikova M, Nenutil R, Vyzula R. Altered expression of miR-21, miR-31, miR-143 and miR-145 is related to clinicopathologic features of colorectal cancer. Oncology. 2007;72:397–402. doi: 10.1159/000113489. [DOI] [PubMed] [Google Scholar]
- 101.Bloomston M, Frankel WL, Petrocca F, Volinia S, Alder H, Hagan JP, Liu CG, Bhatt D, Taccioli C, Croce CM. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA. 2007;297:1901–8. doi: 10.1001/jama.297.17.1901. [DOI] [PubMed] [Google Scholar]
- 102.Markou A, Tsaroucha EG, Kaklamanis L, Fotinou M, Georgoulias V, Lianidou ES. Prognostic value of mature microRNA-21 and microRNA-205 overexpression in non-small cell lung cancer by quantitative realtime RT-PCR. Clin Chem. 2008;54:1696–704. doi: 10.1373/clinchem.2007.101741. [DOI] [PubMed] [Google Scholar]
- 103.Chan SH, Wu CW, Li AF, Chi CW, Lin WC. miR-21 microRNA expression in human gastric carcinomas and its clinical association. Anticancer Res. 2008;28(2A):907–11. [PubMed] [Google Scholar]
- 104.Lawrie CH, Gal S, Dunlop HM, Pushkaran B, Liggins AP, Pulford K, Banham AH, Pezzella F, Boultwood J, Wainscoat JS, Hatton CS, Harris AL. Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. Br J Haematol. 2008;141:672–5. doi: 10.1111/j.1365-2141.2008.07077.x. [DOI] [PubMed] [Google Scholar]
- 105.Keller S, Sanderson MP, Stoeck A, Altevogt P. Exosomes: from biogenesis and secretion to biological function. Immunol Lett. 2006;107:102–8. doi: 10.1016/j.imlet.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 106.Taylor DD, Gercel-Taylor C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol Oncol. 2008;110:13–21. doi: 10.1016/j.ygyno.2008.04.033. [DOI] [PubMed] [Google Scholar]
- 107.Skog J, Würdinger T, Van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT, Jr, Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and protein that promote tumor growth and provide diagnostic biomarkers. Nat Cell Biol. 2008 doi: 10.1038/ncb1800. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Corsten MF, Miranda R, Kasmieh R, Krichevsky AM, Weissleder R, Shah K. MicroRNA-21 knockdown disrupts glioma growth in vivo and displays synergistic cytotoxicity with neural precursor cell delivered S-TRAIL in human gliomas. Cancer Res. 2007;67:8994–9000. doi: 10.1158/0008-5472.CAN-07-1045. [DOI] [PubMed] [Google Scholar]
- 109.Elmén J, Lindow M, Schütz S, Lawrence M, Petri A, Obad S, Lindholm M, Hedtjärn M, Hansen HF, Berger U, Gullans S, Kearney P, Sarnow P, Straarup EM, Kauppinen S. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452:896–9. doi: 10.1038/nature06783. [DOI] [PubMed] [Google Scholar]
- 110.Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–9. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 111.Esau CC. Inhibition of microRNA with antisense oligonucleotides. Methods. 2008;44:55–60. doi: 10.1016/j.ymeth.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 112.Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–6. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]