

Abstract
Erythropoietin (EPO) was shown to have protective effects after myocardial infarction (MI) by neovascularization and antiapoptotic mechanisms. Beside direct receptor-dependent mechanisms, mobilization and homing of bone marrow-derived cells (BMCs) may play a pivotal role in this regard. In this study, we intended to track different subpopulations of BMCs and to assess serially myocardial perfusion changes in EPO-treated mice after MI. To allow tracking of BMCs, we used a chimeric mouse model. Therefore, mice (C57BL/6J) were sublethally irradiated, and bone marrow (BM) from green fluorescent protein transgenic mice was transplanted. Ten weeks later coronary artery ligation was performed to induce MI. EPO was injected for 3 days with a total dose of 5000 IU/kg. Subpopulations (CD31, c-kit, CXCR-4 and Sca-1) of EGFP+ cells were studied in peripheral blood, bone marrow and hearts by flow cytometry. Myocardial perfusion was serially investigated in vivo by pinhole single-photon emission computed tomography (SPECT) at days 6 and 30 after MI. EPO-treated animals revealed an enhanced mobilization of BMCs into peripheral blood. The numbers of these cells in BM remained unchanged. Homing of all BMCs subpopulations to the ischaemic myocardium was significantly increased in EPO-treated mice. Among the investigated subpopulations, EPO predominantly affected migration of CXCR-4+ (4.3-fold increase). Repetitively SPECT analyses revealed a reduction of perfusion defects after EPO treatment over time. Our study shows that EPO treatment after MI enhances the migration capacity of BMCs into ischaemic tissue, which may attribute to an improved perfusion and reduced size of infarction, respectively.
Keywords: erythropoietin, myocardial infarction, bone marrow-derived cells, SPECT
Introduction
Erythropoietin (EPO) is a glycoprotein hormone that is mainly known as the principal regulator of erythropoiesis promoting survival, proliferation and differentiation of erythroid progenitor cells. The expression of EPO is induced by hypoxia-inducible transcription factors stimulated by decreased oxygen delivery as a result of anaemia or hypoxaemia [1, 2]. Beyond this function, EPO reveals protective properties on other tissues because its receptor is expressed on a variety of other cells [3].
Different pre-clinical models of ischaemic disease showed beneficial effects after EPO treatment through activation of EPO receptor-related pathways resulting in antiapoptotic effects and therefore strengthened cell survival [4–6]. In addition, EPO induces neovascularization in experimental models of ischaemic diseases [7–9]. This is related to increased mobilization of bone marrow-derived cells (BMCs), and subsequently migration into the ischaemic tissue [9, 10].
Reduced apoptosis and enhanced neovascularization are the main effects of EPO treatment after myocardial infarction (MI). These effects result in improved cardiac function associated with reduced size of infarction and increased vessel density [11].
Because the influence of EPO on BMCs may play a crucial role for these effects after MI, in this study we focused on mobilization and migration of BMCs. Therefore, we used a chimeric mouse model with BM expressing EGFP considered gold standard for tracking BMCs.
To analyse the effects on myocardial perfusion, we additionally performed repetitively analyses of infarcted mice by pinhole single-photon emission computed tomography (SPECT). For experimental preclinical studies, this is an innovative method allowing non-invasive serial analysis of myocardial perfusion. This indirectly reflects vascularization, perfusion defects and infarct sizes, respectively [12].
Materials and methods
Chimeric mouse model
To allow tracking of BMCs, chimeric mice with BM replaced by transgenic cells expressing green fluorescent protein (GFP) were used. Therefore, C57BL/6 mice (Charles River Laboratories, Sulzbach, Germany) were irradiated and underwent BM transplantation thereafter. Donor mice (C57BL/6) express a transgene coding for GFP under control of the human ubiquitin C promoter (UBI-GFP/BL6) [13]. BM was harvested from the femurs and tibias of donor mice. Cell mixtures of erythrocyte-lysed 5 × 106 BM cells were supplemented with 1 × 106 splenic cells from donors, resuspended in Leibovitz L-15 medium (Life Technologies, Grand Island, NY, USA) and transplanted into recipients via tail vein infusion (0.25 ml total volume). Before transplantation, host mice received 14 Gy of total body irradiation (accelerator, 150 cGy/min) delivered in two fractions, separated by 3 hrs, to reduce gastrointestinal toxicity. Mice were subsequently housed in sterilized microisolator cages and received normal chow and autoclaved hyperchlorinated water for the first 2 weeks after BMT and filtered water thereafter.
Induction of MI
Ten weeks after transplantation, MI was induced as described previously [14]. In brief, MI was induced by surgical occlusion of the left anterior descending artery (LAD) through a left anterolateral approach. Mice were anaesthetized by i.p. injection of a mixture of 100 mg/kg ketamine (Sigma-Aldrich Corp., St. Louis, MO, USA) and 5 mg/kg xylazine/Sigma-Aldrich Corp.), intubated, and artificially ventilated by a mouse ventilator (Hugo Sachs Elektronik, March-Hugstetten, Germany) with 200 strokes/min and 200 μl/stroke. Animal care and all experimental procedures were performed in strict accordance to the German and National Institutes of Health animal legislation guidelines and were approved by the local animal care and use committees.
Experimental protocol
Mice were divided into the following groups: (1) subcutaneous (s.c.) administration of saline daily for 3 days; (2) administration of EPO (Epoetin alpha; Janssen-Cilag, Neuss, Germany). EPO treatment was initiated immediately after the surgical procedure (day 1) with a starting dose of 3000 IU/kg s.c. Treatment was continued with a dose of 1000 IU/kg at days 2 and 3, respectively. Flow cytometry (FACS) was performed in both groups at day 6 and perfusion defects were analysed in all animals at day 6 and at day 30.
Flow cytometry
Different organs (BM, peripheral blood and heart) of chimeric mice were analysed by FACS. At day 6, 1 ml of peripheral blood was harvested from each mouse by aspirating the carotid artery [15, 16]. BM cells were obtained by flushing the tibias and femurs from the euthanized mice. Mononuclear cells were separated by density-gradient centrifugation using Histopaque solution (1.077 g/ml; Sigma-Aldrich Chemicals, St. Louis, MO, USA), purified and resuspended in phosphate-buffered saline containing 1% bovine serum albumin. For cardiac FACS analyses, infarcted hearts of chimeric mice were explanted at day 6 and retrogradely perfused with saline (0.9% NaCl) to wash out circulating blood cells. Thereafter, a ‘myocyte-depleted’ cardiac cell population was prepared, incubating minced myocardium in 0.1% collagenase IV (Gibco BrL, Grand Island, NY, USA) 30 min at 37°C, lethal to most adult mouse cardiomyocytes. Cells were then filtered through a 70-μm mesh. To exclude spurious effects of enzymatic digestion, BM cells with or without collagenase treatment were stained revealing no significant changed staining of labelled cell antigens. Cells from BM, PB and heart were incubated for 40 minutes in the dark at 4°C with the phycoerythrin (PE) conjugated monoclonal antibodies: CD31-PE, c-kit-PE, CXCR-4-PE, Sca-1-PE (all from BD Pharmingen, Heidelberg, Germany). Matching isotype antibodies (BD Pharmingen) served as controls. Cells were analysed by 3-colour flow cytometry using a Coulter® Epics® XL-MCLTM flow cytometer (Beckman Coulter, Krefeld, Germany). Each analysis included 50,000 events.
Cytokine serum levels
Serum levels of VEGF in saline treated and in EPO-treated mice were determined by ELISA following the protocol of the company (RayBiotech, Norcross, GA, USA).
Perfusion measurement by pinhole SPECT
Animals with EPO treatment as well as infarcted control animals (each n = 6) were scanned. Imaging was performed 6 and 30 days after LAD-ligation as described previously [12]. Briefly, after induction of anaesthesia by intraperitoneal injection of a mixture containing medetomidine (0.714 mg/kg), midazolam (7.14 mg/kg) and fentanyl (0.07 mg/kg), each mouse was injected with approximately 370 MBq [99mTc] sestamibi (Cardiolite; Bristol-Myers Squibb Medical Imaging, Inc., North Billerica, MA, USA) in the tail vein. Forty-five minutes after injection, the mouse was positioned in the scanner. Left ventricular perfusion was measured using a clinically used triple-headed single-photon emission computed tomography system (Prism 3000XP; Philips Medical Systems, Hamburg, Germany), each detector head equipped with a 0.5-mm-diameter custom-made tungsten knife-edge pinhole collimator (Nuclear Fields, Des Plaines, IL, USA). The radius of rotation was set to 4 cm (magnification 4), and data were acquired over 20 projections angles (120° for each head), 90 sec per projection, giving a total acquisition time of 30 min. Zoom factor was set as 2.0. Centre of rotation error was corrected by scanning a multiple point phantom and iteratively adjusting the centre-of-rotation offsets. The same source was used to measure the spatial resolution of the system for 99mTc. Images consisted of a matrix of 128 × 128 × 128 with an isotropic voxel size of 0.445 mm. All the images were reconstructed using six iterations. Dedicated software was used to generate transverse slices. A volumetric sampling tool was applied to create polar maps of the relative distribution of activity throughout the left ventricle [17]. Each polar map was adjusted for its own maximal value. The size of the defect was calculated with the use of a threshold of 60%, which was derived from the histological infarct sizes as described previously [12]. Defect size was indicated as percent of left ventricular myocardium. To investigate prospectively, the effectiveness of the treatment protocols the primary endpoint was chosen as change of defect size from 6 to 30 days after MI. The analysis of SPECT images was performed in a blinded manner by one independent person in the Department of Nuclear Medicine.
Statistical analyses
Results were expressed as mean ± S.E.M. Multiple group comparisons were performed by one-way anova followed by the Bonferroni procedure for comparison of means. Comparisons between two groups were performed using the unpaired Student's t-test. Data were considered statistically significant at a value of P ≤ 0.05.
Results
FACS analysis of EGFP+ cell populations in peripheral blood
To investigate different subpopulations of BMCs in peripheral blood, flow cytometry of circulating mononuclear cells was performed. FACS analysis of BM and peripheral blood revealed that >99% of mononuclear cells were positive for EGFP demonstrating that BM was completely replaced by transgenic cells expressing GFP (Fig. 1A).
Fig 1.
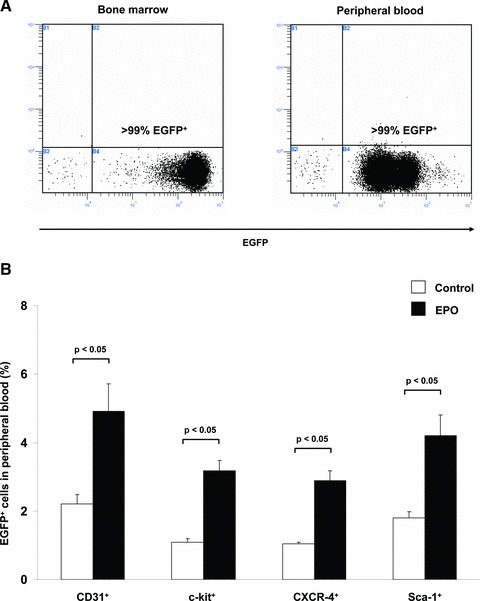
EPO administration after myocardial infarction increased mobilization of EGFP+ BMCs from BM into peripheral blood in GFP-transgenic mice. (A) Representative FACS analyses of EGFP+ cells in bone marrow (left) and peripheral blood (right) of wild-type mice 10 weeks after BM-transplantation showing successful BM replacement. (B) Bar graph representing the percentage of EGFP+ cell populations (subclassified by CD31, c-kit, CXCR-4, Sca-1) in peripheral blood of infarcted control mice (white bar) or EPO-treated mice (black bar). All data represent mean ± S.E.M. (n = 8).
In peripheral blood, we found a significant increase of all investigated subtypes of EGFP+ cells after EPO treatment compared to the control: EGFP+CD31+: 2.6-fold (4.91% versus 2.21%; P < 0.05); EGFP+c-kit+: 2.6-fold (3.18% versus 1.09%; P < 0.01); EGFP+CXCR-4+: 3.1-fold (2.89% versus 1.04%; P < 0.01); EGFP+Sca-1+: 3.4-fold (4.21% versus 1.80%; P < 0.01) (Fig. 1B).
FACS analysis of EGFP+ cell populations in bone marrow
To investigate the effects of EPO on BM, FACS analysis of the subpopulations named earlier was performed. The data revealed no significant difference of these BMC subpopulations between control mice and EPO-treated mice: EGFP+CD31+: 25.7% versus 23.7% (P = 0.55); EGFP+c-kit+: 13.2% versus 10.1% (P = 0.07); EGFP+CXCR-4+: 2.7% versus 2.2% (P = 0.17); EGFP+Sca-1+: 8.1% versus 9.5% (P = 0.55).
FACS analysis of a myocyte depleted fraction of cardiac cells
To compare the impact of EPO on migration of EGFP+ subpopulations into the infracted hearts, we isolated a myocyte depleted fraction of cardiac cells and flow cytometry was performed of these cells. In EPO-treated animals, the number of migrated BMCs was significantly increased in comparison to the control group: EGFP+CD31+: 6.31% versus 3.47% (P < 0.05); EGFP+c-kit+: 2.52% versus 1.38% (P < 0.05); EGFP+CXCR-4+: 3.24% versus 0.76 (P < 0.05); EGFP+Sca-1+: 7.11% versus 4.80% (P < 0.05) (Fig. 2A).
Fig 2.
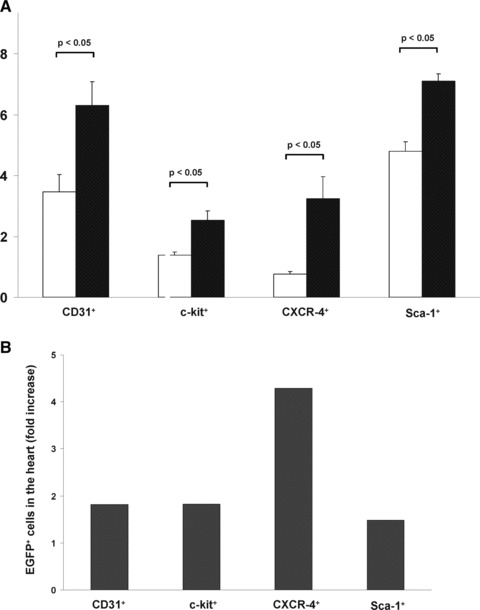
EPO administration after myocardial infarction increased homing of EGFP+ BMCs into ischaemic myocardium in GFP-transgenic mice. (A) Bar graph representing the percentage of myocardial EGFP+ cell populations (subclassified by CD31, c-kit, CXCR-4, Sca-1) of infarcted control mice (white bar) or EPO-treated mice (black bar). All data represent mean ± S.E.M. (n = 8). (B) Bar graph representing the fold-increase of EGFP+ subpopulations in the hearts of infarcted control mice (white bar) or EPO-treated mice (black bar). The represented value is the ratio of the mean of control mice and the mean of EPO-treated mice.
Interestingly, the extent of migration of EGFP+ cells was different between the subpopulations. The EGFP+CXCR-4+ subpopulation showed the strongest increase (4.3-fold), whereas the other analysed subpopulations showed also a significant increase, but with less extent (EGFP+CD31+: 1.8-fold; EGFP+c-kit+: 1.8-fold; EGFP+Sca-1+: 1.5-fold) (Fig. 2B).
Effect on serum VEGF levels
To investigate the impact of EPO treatment on VEGF as a potential mediator of neovascularization, we performed an ELISA of saline treated and EPO-treated mice. Serum VEGF levels were significantly increased in EPO-treated mice compared to control (141.9 pg/ml versus 87.2 pg/ml; P < 0.05) (Fig. 3).
Fig 3.
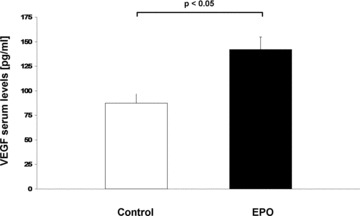
VEGF serum levels. Bar graph representing the serum levels of VEGF of infracted control mice (white bar) and EPO-treated mice (black bar). Data represent mean ± S.E.M. (n = 6).
Perfusion defects in EPO-treated mice
After showing enhanced homing of BMCs after EPO treatment, we evaluated the impact of EPO on cardiac perfusion after MI. We therefore used the SPECT technique which has recently been validated for non-invasive, repetitive and quantitative infarct size quantification in mice [12]. At day 6 after MI perfusion defects were similar in both groups. Evaluating change of defect size from day 6 (baseline) to day 30 after MI we found a relative reduction of perfusion defect sizes after EPO treatment (−3.44%, Fig. 4), whereas an increase in perfusion defects (+2.43%; Fig. 4E–G) was detected in control animals (P < 0.05).
Fig 4.
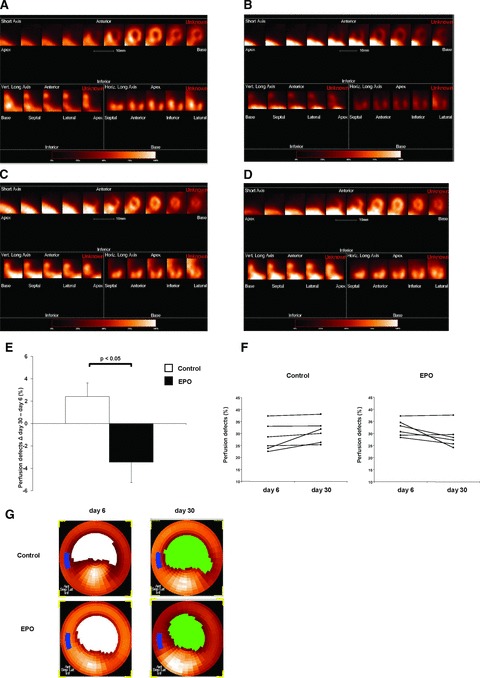
Quantification of myocardial perfusion defects by pinhole SPECT. (A) Representative short-axis, vertical long-axis and horizontal long-axis slices from a myocardial perfusion study of a mouse with myocardial infarction and EPO treatment at day 6 and (B) day 30. (C) Representative short-axis, vertical long-axis and horizontal long-axis slices from a myocardial perfusion study of an infarcted control mouse at day 6 and (D) day 30. A large antero-lateral perfusion defect is evident extending from the apex to the mid-ventricular region. (E) Bar graph representing absolute change of perfusion defect size between baseline (day 6) and day 30 of control and EPO-treated mice (each n = 6), all values are mean ± S.E.M. (F) Perfusion defect size at day 6 and day 30 of each control and EPO-treated mouse. (G) Representative repetitive pinhole SPECT bull's eye polar maps of control (upper row) and EPO-treated mice (bottom) at day 6 and day 30 after MI. White or green area represents LV perfusion defects with signal intensities below the threshold.
Discussion
In this study, we focused on the effects of EPO treatment after MI on mobilization and homing of BMCs as well as the impact on myocardial perfusion. In this regard, we used a chimeric mouse model with BM expressing EGFP allowing a precise tracking of BMCs. Our main findings are the following: EPO treatment after MI results in (1) significant mobilization of BMCs into peripheral blood; (2) no depletion of bone marrow; (3) improved homing of BMCs into the infarcted myocardium; (4) improved BMC recruitment predominantly of CXCR-4+ cells; (5) increased serum VEGF levels and (6) and reduced perfusion defects.
Our data on circulating EGFP+ BMCs showed an increase of all investigated BMC subpopulations after EPO treatment in peripheral blood. This finding confirms previous studies demonstrating increased numbers of circulating CD45+CD34+ cells or endothelial progenitor cells (EPCs) after EPO administration in infarcted mice [9, 11]. Our model using chimeric mice with BM cells expressing EGFP proves that the origin of circulating and migrating cells is the BM. The same populations that were analysed in peripheral blood were also investigated in BM. The number of these cells remained unchanged after EPO administration despite its highly significant mobilizing property. This finding suggests a proliferating effect within bone marrow. This may be an advantage compared to the other mobilizing agents such as G-CSF, which is known to result in depletion of BM after release of BMCs [18, 19].
To investigate the fate of the mobilized BMCs after MI, we performed flow cytometry of a myocyte depleted cell fraction of the infarcted hearts. Our data show that EPO treatment may promote the migration of EGFP+ BMCs. All investigated subpopulations of BMCs were significantly up-regulated after EPO treatment in the ischaemic hearts. By far, the most distinctive increase was noticed in the population of EGFP+CXCR-4+ cells. Previous studies could demonstrate, that the expression of SDF-1, which is the interacting ligand of CXCR-4, is increased after EPO treatment. Systemic administration of EPO results in an up-regulation of SDF-1 in ischaemic myocardium [11]. Also local intracardiac injections of EPO after MI lead to increased expression of SDF-1 [20]. Our present finding supports the hypothesis that EPO may enhance migration of BMCs via the SDF-1/CXCR-4 axis. Therefore, the cardioprotective effects may be attributed to the strengthening of this axis, which was demonstrated in several studies to play a major role in homing, chemotaxis, engraftment and retention of BMCs in the ischaemic myocardium [21, 22].
To achieve a translational level towards a clinical use of EPO, we analysed myocardial perfusion by pinhole SPECT, an appropriate imaging method for therapy monitoring and follow-up examinations. SPECT indirectly reflects vascularization, perfusion defects and infarct sizes, respectively [12]. Therefore, this imaging method reflects the main effects of EPO treatment after MI. On the one hand, reduced size of infarction is because of reduced apoptosis. Stimulation of the EPO receptor results in an activation of antiapoptotic pathways, which were shown to be involved in cardioprotective effects of EPO treatment after MI [4–6]. On the other hand, improved perfusion over time is because of enhanced neovascularization, which is related to increased mobilization and recruitment of BMCs and augmented expression of angiogenic factors, predominatly VEGF in the ischaemic myocardium [9, 11, 23, 24]. Previous histological data showed an increase of vessel density in the infarct border zone [11]. This neovascularization was demonstrated to be related to migrated BMCs [9, 10]. Two factors may play a pivotal role for BMC recruitment and therefore neovascularization. As discussed earlier, the SDF-1/CXCR-4 axis is strengthened by EPO treatment resulting in increased numbers of migrated CXCR-4+ BMCS. The second important factor is VEGF, that was shown to affect BMC migration to the ischaemic hearts and whose expression in the myocardium correlates with the extent of neovascularization [9, 23].
Our data generated by the clinical SPECT device reflect these mechanisms, which are involved in perfusion changes after EPO treatment in infarcted mice. Six days after MI, perfusion defects were similar in EPO- and placebo-treated animals. However, the change of perfusion defect size from day 6 to 30 after MI showed a significant difference between both groups. Perfusion defects after EPO treatment were reduced over time, which suggest a long sustained improvement of cardiac function attenuating the development of an ischaemic cardiomyopathy after MI.
First clinical trials on EPO treatment after MI reveal very heterogeneous results. As some trials cannot show any effect after EPO treatment, others demonstrate an increase of ejection fraction or a decrease of major adverse cardiac events [25–28]. This heterogeneity may be because of the usage of different EPO derivatives or different treatment regimes. To optimize EPO treatment for ischaemic diseases, further clinical trials will have to address this aspect.
Our preclinical study shows that short-term EPO treatment after MI enhances mobilization and homing of BMCs into the ischaemic myocardium. The effect is most prominent on CXCR-4+ cells indicating a strengthening of the SDF-1/CXCR-4− axis. Increased homing of CXCR-4+ cells was associated with a reduction of perfusion defects determined by pinhole SPECT. Further preclinical studies using transgenic mice will have to unravel the question to which extent direct receptor-dependent pathways are involved in these EPO induced cardioprotective effects.
Acknowledgments
The authors thank Judith Arcifa and Barbara Markieton for excellent technical assistance. The position of Barbara Markieton was funded by the Fritz-Bender-Stiftung. Additional financial support was provided by the Förderprogramm für Forschung und Lehre (FöFoLe) and the Else Kröner-Fresenius-Stiftung.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Jelkmann W, Wagner K. Beneficial and ominous aspects of the pleiotropic action of erythropoietin. Ann Hematol. 2004;83:673–86. doi: 10.1007/s00277-004-0911-6. [DOI] [PubMed] [Google Scholar]
- 2.Zwezdaryk KJ, Coffelt SB, Figueroa YG, et al. Erythropoietin, a hypoxia-regulated factor, elicits a pro-angiogenic program in human mesenchymal stem cells. Exp Hematol. 2007;35:640–52. doi: 10.1016/j.exphem.2007.01.044. [DOI] [PubMed] [Google Scholar]
- 3.Maiese K, Li F, Chong ZZ. New avenues of exploration for erythropoietin. JAMA. 2005;293:90–5. doi: 10.1001/jama.293.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cai Z, Semenza GL. Phosphatidylinositol-3-kinase signaling is required for erythropoietin-mediated acute protection against myocardial ischemia/reperfusion injury. Circulation. 2004;109:2050–3. doi: 10.1161/01.CIR.0000127954.98131.23. [DOI] [PubMed] [Google Scholar]
- 5.Hanlon PR, Fu P, Wright GL, et al. Mechanisms of erythropoietin-mediated cardioprotection during ischemia-reperfusion injury: role of protein kinase C and phosphatidylinositol 3-kinase signaling. FASEB J. 2005;19:1323–5. doi: 10.1096/fj.04-3545fje. [DOI] [PubMed] [Google Scholar]
- 6.Wright GL, Hanlon P, Amin K, et al. Erythropoietin receptor expression in adult rat cardiomyocytes is associated with an acute cardioprotective effect for recombinant erythropoietin during ischemia-reperfusion injury. FASEB J. 2004;18:1031–3. doi: 10.1096/fj.03-1289fje. [DOI] [PubMed] [Google Scholar]
- 7.Heeschen C, Aicher A, Lehmann R, et al. Erythropoietin is a potent physiologic stimulus for endothelial progenitor cell mobilization. Blood. 2003;102:1340–6. doi: 10.1182/blood-2003-01-0223. [DOI] [PubMed] [Google Scholar]
- 8.Iwai M, Cao G, Yin W, et al. Erythropoietin promotes neuronal replacement through revascularization and neurogenesis after neonatal hypoxia/ischemia in rats. Stroke. 2007;38:2795–803. doi: 10.1161/STROKEAHA.107.483008. [DOI] [PubMed] [Google Scholar]
- 9.Westenbrink BD, Lipsic E, van der Meer P, et al. Erythropoietin improves cardiac function through endothelial progenitor cell and vascular endothelial growth factor mediated neovascularization. Eur Heart J. 2007;28:2018–27. doi: 10.1093/eurheartj/ehm177. [DOI] [PubMed] [Google Scholar]
- 10.Kocher AA, Schuster MD, Szabolcs MJ, et al. Neovascularization of ischemic myocardium by human bone-marrow-derived angioblasts prevents cardiomyocyte apoptosis, reduces remodeling and improves cardiac function. Nat Med. 2001;7:430–6. doi: 10.1038/86498. [DOI] [PubMed] [Google Scholar]
- 11.Brunner S, Winogradow J, Huber BC, et al. Erythropoietin administration after myocardial infarction in mice attenuates ischemic cardiomyopathy associated with enhanced homing of bone marrow-derived progenitor cells via the CXCR-4/SDF-1 axis. FASEB J. 2009;23:351–61. doi: 10.1096/fj.08-109462. [DOI] [PubMed] [Google Scholar]
- 12.Wollenweber T, Zach C, Rischpler C, et al. Myocardial perfusion imaging is feasible for infarct size quantification in mice using a clinical single-photon emission computed tomography system equipped with pinhole collimators. Mol Imaging Biol. 2010;12:427–34. doi: 10.1007/s11307-009-0281-5. [DOI] [PubMed] [Google Scholar]
- 13.Schaefer BC, Schaefer ML, Kappler JW, et al. Observation of antigen-dependent CD81 T-cell/dendritic cell interactions in vivo. Cell Immunol. 2001;214:110–22. doi: 10.1006/cimm.2001.1895. [DOI] [PubMed] [Google Scholar]
- 14.Zaruba MM, Huber BC, Brunner S, et al. Parathyroid hormone treatment after myocardial infarction promotes cardiac repair by enhanced neovascularization and cell survival. Cardiovasc Res. 2008;77:722–31. doi: 10.1093/cvr/cvm080. [DOI] [PubMed] [Google Scholar]
- 15.Deindl E, Zaruba MM, Brunner S, et al. G-CSF administration after myocardial infarction in mice attenuates late ischemic cardiomyopathy by enhanced arteriogenesis. FASEB J. 2006;20:956–8. doi: 10.1096/fj.05-4763fje. [DOI] [PubMed] [Google Scholar]
- 16.Zaruba MM, Theiss HD, Vallaster M, et al. Synergy between CD26/DPP-IV inhibition and G-CSF improves cardiac function after acute myocardial infarction. Cell Stem Cell. 2009;4:313–23. doi: 10.1016/j.stem.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Nekolla SG, Miethaner C, Nguyen N, et al. Reproducibility of polar map generation and assessment of defect severity and extent assessment in myocardial perfusion imaging using positron emission tomography. Eur J Nucl Med. 1998;25:1313–21. doi: 10.1007/s002590050301. [DOI] [PubMed] [Google Scholar]
- 18.Freedman A, Neuberg D, Mauch P, et al. Cyclophosphamide, doxorubicin, vincristine, prednisone dose intensification with granulocyte colony-stimulating factor markedly depletes stem cell reserve for autologous bone marrow transplantation. Blood. 1997;90:4996–5001. [PubMed] [Google Scholar]
- 19.van Os R, Robinson S, Sheridan T, et al. Granulocyte colony-stimulating factor enhances bone marrow stem cell damage caused by repeated administration of cytotoxic agents. Blood. 1998;92:1950–6. [PubMed] [Google Scholar]
- 20.Klopsch C, Furlani D, Gabel R, et al. Intracardiac injection of erythropoietin induces stem cell recruitment and improves cardiac functions in a rat myocardial infarction model. J Cell Mol Med. 2009;13:664–79. doi: 10.1111/j.1582-4934.2008.00546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abbott JD, Huang Y, Liu D, et al. Stromal cell-derived factor-1alpha plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation. 2004;110:3300–5. doi: 10.1161/01.CIR.0000147780.30124.CF. [DOI] [PubMed] [Google Scholar]
- 22.Askari AT, Unzek S, Popovic ZB, et al. Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet. 2003;362:697–703. doi: 10.1016/S0140-6736(03)14232-8. [DOI] [PubMed] [Google Scholar]
- 23.Westenbrink BD, Oeseburg H, Kleijn L, et al. Erythropoietin stimulates normal endothelial progenitor cell-mediated endothelial turnover, but attributes to neovascularization only in the presence of local ischemia. Cardiovasc Drugs Ther. 2008;22:265–74. doi: 10.1007/s10557-008-6094-y. [DOI] [PubMed] [Google Scholar]
- 24.Westenbrink BD, Ruifrok WP, Voors AA, et al. Vascular endothelial growth factor is crucial for erythropoietin-induced improvement of cardiac function in heart failure. Cardiovasc Res. 2010;87:30–9. doi: 10.1093/cvr/cvq041. [DOI] [PubMed] [Google Scholar]
- 25.Ott I, Schulz S, Mehilli J, et al. Erythropoietin in patients with acute ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention: a randomized, double-blind trial. Circ Cardiovasc Interv. 2010;3:408–13. doi: 10.1161/CIRCINTERVENTIONS.109.904425. [DOI] [PubMed] [Google Scholar]
- 26.Ozawa T, Toba K, Suzuki H, et al. Single-dose intravenous administration of recombinant human erythropoietin is a promising treatment for patients with acute myocardial infarction-randomized controlled pilot trial of EPO/AMI-1 study. Circ J. 2010;74:1415–23. doi: 10.1253/circj.cj-10-0109. [DOI] [PubMed] [Google Scholar]
- 27.Voors AA, Belonje AM, Zijlstra F, et al. A single dose of erythropoietin in ST-elevation myocardial infarction. Eur Heart J. 2010;31:2593–600. doi: 10.1093/eurheartj/ehq304. [DOI] [PubMed] [Google Scholar]
- 28.Yoshimura N, Toba K, Ozawa T, et al. A novel program to accurately quantify infarction volume by (99m)Tc MIBI SPECT, and its application for re-analyzing the effect of erythropoietin administration in patients with acute myocardial infarction. Circ J. 2010;74:2741–3. doi: 10.1253/circj.cj-10-0912. [DOI] [PubMed] [Google Scholar]