

Abstract
A heart attack kills off many cells in the heart. Parts of the heart become thin and fail to contract properly following the replacement of lost cells by scar tissue. However, the notion that the same adult cardiomyocytes beat throughout the lifespan of the organ and organism, without the need for a minimum turnover, gives way to a fascinating investigations. Since the late 1800s, scientists and cardiologists wanted to demonstrate that the cardiomyocytes cannot be generated after the perinatal period in human beings. This curiosity has been passed down in subsequent years and has motivated more and more accurate studies in an attempt to exclude the presence of renewed cardiomyocytes in the tissue bordering the ischaemic area, and then to confirm the dogma of the heart as terminally differentiated organ. Conversely, peri-lesional mitosis of cardiomyocytes were discovered initially by light microscopy and subsequently confirmed by more sophisticated technologies. Controversial evidence of mechanisms underlying myocardial regeneration has shown that adult cardiomyocytes are renewed through a slow turnover, even in the absence of damage. This turnover is ensured by the activation of rare clusters of progenitor cells interspersed among the cardiac cells functionally mature. Cardiac progenitor cells continuously interact with each other, with the cells circulating in the vessels of the coronary microcirculation and myocardial cells in auto-/paracrine manner. Much remains to be understood; however, the limited functional recovery in human beings after myocardial injury clearly demonstrates weak regenerative potential of cardiomyocytes and encourages the development of new approaches to stimulate this process.
Keywords: cardiac regeneration, cardiomyocyte turnover, tissue plasticity, heart
Introduction
The adult human heart has been viewed as a static terminally differentiated organ, formed of oriented mature cardiomyocytes that allow for the conduction of electrical signals and the synchronous contractile function aimed at maintaining the physiological systemic perfusion. Adult human myocardium has been considered a tissue capable of increasing its mass by hypertrophy of existing myocytes in response to increased after- and/or pre-load. Indeed, historical evidence highlighted that, in physiological or pathological conditions, a post-natal mammalian cardiomyocyte could not re-enter the cell cycle and replace itself. However, no clear evidence in favour of the above possibility was provided prompting the international scientific community to reveal the hidden truth in definitive way. Since the late 1800s, the question of regeneration of cardiac muscle fibres has often been discussed. Goldenberg [1] reviewed the previous reports and concluded that longitudinal splitting of cardiomyocytes could occur, yet the increase of cardiac tissue was largely by hypertrophy of muscle fibres. Conversely, other investigators demonstrated that the increase of the heart mass was mainly due to hypertrophy rather than hyperplasia of cardiomyocytes, on the basis of the lack of evidence of myocardial mitotic figures [2–5]. In the early 1920s, counts and measurements of muscle fibres made by Collier [6] and Karsner et al. [7] confirmed that myocardial hypertrophy and hyperplasia are occurred. Thereafter, Macmahon [8] indicated that the principal difficulty was failure to demonstrate mitotic figures in adult cardiomyocyte nuclei, but not in myocardium of infants and children. Thus opinions on cardiomyocyte turnover in adult heart was divided for many years, but in the late 1960s and early 1970s the evaluation of DNA synthesis in cardiomyocyte nuclei was shown to be negligible, strengthening the notion that cardiomyocytes can increase in volume but not in number [9, 10].
The steps forward of the molecular cardiology and the development of advanced microscopy techniques have modified the scale of analysis and allowed conceivable that the early cardiomyocyte response to homeostatic and pathophysiological environments is a dynamic consequence of alterations in the effector pathways, which regulate myocyte growth and proliferation [11–14]. Intercellular signals are generated from resident cells functionally mature or immature, and tend to maintain a homeostatic code in order to preserve the myocardial architecture. The dissection of the mechanisms is still in progress and this is changing dramatically our understanding of cardiac biology by witnessing the principles dictating initial cardiac genesis during development so as to exclude any artefact.
Adult cardiomyocyte turnover
At birth the number of cardiomyocytes in the heart of mammals is largely determined. The hyperplastic growth occurring during embryonic life is quickly replaced by a hypertrophic process soon after birth. Studies based on the method of incorporation of thymidine showed that DNA synthesis accompanied by cytokinesis in mice decreases from 70% at E8 to 45% at E11 and almost disappears 1–2 days after birth [15]. In the second phase, starting just after the birth, karyokinesis occurs in the absence of cytokinesis thus determines an increasing percentage of binucleated cardiomyocytes from 2.5% at day 2, to 80% at day 14 [16]. These two distinct phases in the heart development are reflected in the expression levels of regulators of cell cycle such as cyclin-dependent kinases (Cdks) and their inhibitors. The expression patterns of cell cycle regulatory proteins are well reviewed by Pasumarthi K.B. & Field L.J. [11]. In general, Cdks exert their positive effects in terms of promoting mitosis of cardiac cells during the embryonic life while they are down-regulated in the adult heart. Therefore, the replicative capacity of the heart suffers a sharp slowdown after birth and starting from this certainty, several scientists have wondered if the adult heart was still able to retain a regenerative capacity in adult life. Recently, two independent studies have addressed the issue following a similar approach but in two different species [11, 17]. Although it was known that many invertebrates such as the zebrafish can regenerate the heart after amputation up to 20%[18]. Jopling et al. first established that the contribution of existing cardiomyocytes to regenerate tissue is of paramount importance. Using a CRE/lox approach they demonstrated that after partial amputation of the ventricle, survived cardiomyocytes adjacent to the damaged region express regulators of cell-cycle progression, such as plk1 and mps1, de-differentiate detaching from one another. They lose their sarcomeric structure and re-enter the cell cycle [19]. A more recent paper by Porrello et al. showed that similar events occur in 1-day-old mouse when the ventricular apex is resected. The region of resection is progressively interested by a robust inflammatory response followed by the formation of blood clot that is subsequently replaced by new functional heart, which derives mainly from the pre-existing cardiomyocytes. At 21 days the majority of the new formed cardiomyocytes were positive for the mitotic index, phosphohistone H3 and the aurora B kinase. The mouse heart loses its regenerative potential within the first week of postnatal life [17]. While demonstrating the absolute importance of the existing cardiomyocytes in regenerating new cardiac tissue, none of the above-cited papers can exclude the contribution of resident progenitor/stem cells.
Cardiac tissue generation: contribution of resident interstitial cells
The adult myocardium is a well-organized tissue composed by different resident cell types forming a morpho-functional unit [20]: cardiomyocytes, endothelial cells, smooth muscle cells and a variety of interstitial cells including cardiac resident stem/progenitor cells (Fig. 1). Among the interstitial cells, cardiac fibroblasts, which account for two-thirds of total myocardial cells, play important physiological role in ensuring a spatial orientation, a structural organization and mutual interplay of resident cardiac cells, which work interdependently in order to promote an efficient global and regional function of the heart [21]. The intrinsic characteristics of tension-generating fibroblasts also have a continuous mechanical interplay with their extrinsic environment [22]. Moreover, the cardiac fibroblasts are the main myocardial source of collagen, which composes the post-ischaemic infarct scar, and release several humoral mediators involved in myocardial remodelling: proinflammatory cytokines, including tumour necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6) [23, 24], growth factors involved in myocardial remodelling, such as platelet-derived growth factor [25], high molecular weight fibroblast growth factor-2 (FGF-2) [26], insulin-like growth factor-1 [27] and vascular endothelial growth factor (VEGF) [28]. Cardiac fibroblasts are able to interact with other interstitial cells resident into the myocardium, such as mast cells (MCs) [29] and telocytes (TCs) [30].
Fig 1.
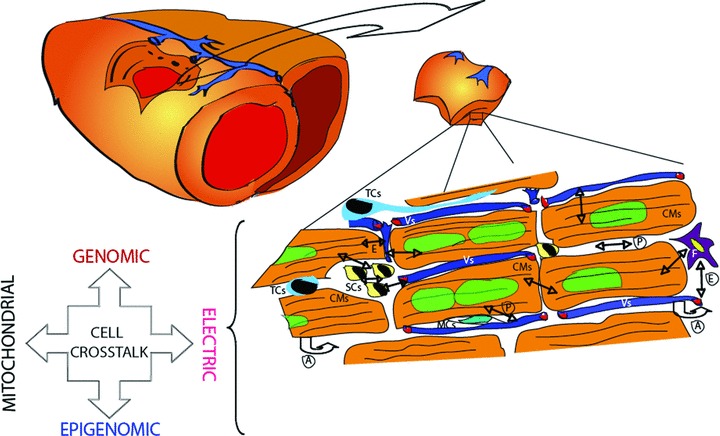
Cross-talk of resident cardiac cells. Schematic representation of resident cardiac cells potentially involved in myocardial regeneration and modalities of intercellular cross-talk. Vs: vessels; CMs: cardiomyocytes; SCs: stem cells; F: fibroblast; TCs: telocytes; MCs: mast cells; P: paracrine action; A: autocrine action; E: endocrine action.
Mast cells are multifunctional cells releasing different humoral mediators, such as cytokines, and histamine, and they have been identified in injured myocardium [31]. MCs have been suggested also to play a role in adverse remodelling in human failing heart [32]. Chymase, which is an enzyme stored in the cardiac MCs, is insensitive to ACE inhibitors and promotes interstitial fibrosis by affecting collagen metabolism via transforming growth factor-β. Chronic chymase inhibition decreases the cardiac angiotensin II levels, myocardial fibrosis and contractile failure [33]. Conversely, Kwon et al. [34] recently showed that the intramyocardial injection of MC granules prolonged survival of cardiomyocytes and promoted angiogenesis in rodent infarcted hearts. However, the role of cardiac MCs during remodelling, which are also CD117(+), remains controversial and some investigators even suggested them as cardiac progenitor cells (CPCs) [35].
Telocytes have been recently described as a novel different types of interstitial cell type in adult mammalian heart [36, 37]. They are characterized to shed exceptionally long cellular processes – telopodes – which form an interstitial network, connecting different types of cell from myocardium, epicardium and endocardium. TCs seem to be involved in a long distance intercellular signalling, due to both membrane-to-membrane junction and release of vesicles [30, 36]. TCs presumably contribute to neo-angiogenesis via paracrine secretion of VEGF and microRNAs (e.g. let-7e, 10a, 21, 27b, 100, 126–3p, 130a, 143, 155) [38]. Particularly important is the role of TCs in supporting the renewal and survival of CPCs [30].
Cardiac progenitor cells reside in the heart as well as in other solid organs, within niches that are composed of extracellular matrix, stem/supporting cells and blood vessels [39, 40]. In the last decade, several independent groups have tried to isolate progenitor cells from adult heart tissue with a view to reuse them as cell source for autologous cell transplantation in injured heart [41–46]. With the heterogeneity of markers that have been chosen to define cardiac progenitor populations, few scientists have bothered to study the role of these progenitors in the normal cardiac physiology. So far it is not clear if these populations are related and which is their origin. The in vitro condition that better recapitulates the features of CPC niche is the three-dimensional culture of CPCs as cardiospheres, in which the cell–cell and cell–matrix interactions are conserved and the cell survival and functional benefits are ensured [47, 48].
An indirect evidence that precursors may contribute to generate new cardiomyocytes in the area of the injury derives from paper by Hsieh et al. [49]. They used an elegant ‘fate-mapping’ approach establishing a double transgenic mouse in which all the cells positive for the cardiac specific promoter Mhy6 were constitutively expressing the β-galactosidase reporter gene (β-gal+). The latest is replaced by the GFP when mice are pulsed with 4-OH tamoxifen. Theoretically, all the cardiomyocytes that were β-gal+ become GFP+ after pulse. The findings consist in the fact that following the induction of the infarct in the transgenic mice, the number of GFP+ myocytes were significantly decreased with respect to the β-gal+ cells indicating the presence of new cardiomyocytes deriving from the differentiation of progenitor cells. Although authors cannot exclude that some progenitors were labelled by the 4-OH-tamoxifen, the percentage of new cardiac cells in myocardial infarction border areas that were not derived from the mitosis of pre-existing cardiomyocytes, reached 34%. Thus cardiomyocytes can be generated by activation of resident [50] or in part by migration of progenitor cells from distant organs [51]. Similar genetic mapping approach to track cell fate in injured heart has been followed by Loffredo et al. [52]. In this article, authors establish that the dilution in GFP+ and complimentary increase in β-gal + cardiomyocytes in infarcted transgenic mice is associated with an increase in the number of nkx2.5- and GATA4-positive progenitor cells. Moreover, in the same set of experiments they were able to asses that the delivery of exogenous bone marrow-derived c-kit+ cells exert effects by releasing a paracrine factors able to stimulate regenerative capacity of progenitors rather than proliferation of pre-existing cardiomyocytes [52]. However, recent findings showed that the function and potential plasticity of stem cells, potentially activated or recruited after injury into the myocardium, is impaired by aging [53] and severity of cardiac disease [54].
The consistency of cell turnover due to CPC differentiation was analysed in mouse by the in vivo incorporation of the BrdU [40]. BrdU retaining myocyte was 19%, 15% and 10% in the apex, atria and base–midregion, respectively [40].
In human beings who live much longer than rodents, it is reasonable to expect a slow cell turnover that implies cardiomyocytes replacement. Clear evidence that cell division occurs in human heart is shown by Bergmann et al. [55]. During the Cold War the concentration of 14C in the atmosphere had increased sharply following an exponential fluctuation with a peak around 1963. By relating this information with the levels of 14C in the DNA of cardiomyocytes of individuals born before and after the nuclear bomb tests, authors reached a conclusion that the amount of 14C present in heart cells of individuals correspond to atmospheric levels several years after the persons’ birth. It follows that the DNA of cardiomyocytes has been synthesized many years after the birth thus implicating cell renewal. Using elaborate mathematical models and excluding contamination of other cell types by cell-sorting, authors came to the following conclusions: at the age of 50, 55% of the cardiomyocytes remain from the time birth and 45% have been generated later; cardiomyocytes are renewed at a rate of ∼1% per year at the age of 25, 0.45% at the age of 75; the age of cardiomyocytes is on average 6 years younger than the individual.
This article was recently criticized in numbers and methodology by Kajstura J. et al.[56]. They use a different approach based on the detection of the iododeoxyuridine, analogue of thymidine, into the cardiac cells of patients undergoing infusion of this radiosensitizer. The results were ‘dramatically’ different with an average of myocytes renewal of about 22% per year suggesting that the heart is capable to renew itself several times during the course of life [56]. Moreover, the myocyte turnover increases further with age reaching the 40% in a heart of female of 100 years of age.
The above experimental evidence, obtained by the use of more accurate technologies than optical microscopy, seemed to have answered the question that tormented too long cardiovascular pathologists. Over the next two decades, the significant lack of further technological progress had mitigated the interest in the field of regenerative cardiology maturing awareness that a deep understanding of the mechanisms should be guaranteed only by a change of point of view of the biological phenomena.
Signalling of miocardiogenesis: a developmental perspective
Unravelling how different signalling pathways dictate cardiac precursor specification and subsequent behaviour is critical for predicting how damaged myocardium will self-renew (Fig. 1). To date, studies on cardiogenesis offered new hypothesis to highlight the mechanisms underlying the regeneration of adult myocardium. Several signalling pathways integrate and cross-talk during cardiogenesis and it is conceivable that multiple molecular inputs regulate the balance in the earliest steps of cardiogenesis through mesoderm specification and allocation.
TNF-related weak inducer of apoptosis (TWEAK), a member of the TNF-α family, regulates proliferation in multiple cell types and its effects are mediated through the activation of fibroblast growth factor-inducible molecule 14 (FN14) receptor [57]. TWEAK caused a dose-dependent increase in DNA synthesis of cardiomyocytes and down-regulates the cell cycle inhibitor p27KIP1. TWEAK/FN14-mediated patterning involves the activation of ERK, and PI3K, as well as inhibition of GSK-3β which in turn led to stabilization and accumulation of total β-catenin and accumulation of dephosphorylated β-catenin in the nucleus. However, TWEAK does not affect proliferation of adult rat cardiomyocytes due to progressive down-regulation of FN14 gene and protein expression after birth. Therefore, a guided post-natal overexpression of FN14 receptor is able to induce an efficient cell cycle reentry in adult cardiomyocytes on the basis of the presence of endogenous TWEAK protein in these cells [14]. Terminal differentiation of organ involves the retinoblastoma family of tumour-suppressor pocket proteins and Cdks that modulate their function [58]. However, the role of these proteins has been substantiated in cardiac cell cycle control [59] and needs the activity of telomerase reverse transcriptase (TERT). Continuous cell division erodes the telomeric repeat because of incomplete replication of the distal 39 strand during active replicative growth [60]. In adult human beings, telomerase activity is found predominantly in germ cells, tumour cells and stem cells but not somatic cells that ultimately senesce. Failure to express TERT is the principal mechanism for low levels of telomerase activity seen with replicative senescence. Telomerase activity and TERT expression are lacking or are markedly decreased in the adult heart, but not during cardiogenesis [61]. On the basis of this notion, Oh et al. [62] showed that the replacement of ancestral telomerase activity in adult cardiomyocytes delays cell cycle exit, induces hypertrophy and promotes cardiac myocyte survival.
Other investigations showed that Nodal/Activin/TGF-b, bone morphogenetic proteins (BMPs) and FGFs drive cardiogenesis [63] and suggested how their re-activation might improve the self-regenerative capacity of the heart.
BMPs promote mesoderm formation from embryonic stem cells through a Wnt/Activin/Nodal-dependent mechanism [64]. Thereafter, Notch regulates CPC proliferation and fate through an interaction with Wnt/b-catenin signalling [64]. Previous study showed that cardiomyogenesis in the adult heart depends on the activation and differentiation of cardiac stem cells [65]. In accordance with the traditional model of development of self-renewing organs, stem cells give rise to progenitor precursor cells, which then proliferate and eventually reach terminal differentiation and growth arrest. Newly generated cardiomyocytes might proliferate and concurrently differentiate, mimicking cellular hyperplasia and hypertrophy [66]. Activation of the Notch1 cascade is crucial for cell-to-cell interaction during cardiovascular development and may influence differentiation, proliferation and apoptotic events of CPC [67, 68]. Alteration of Notch1 pathway hampers coronary vessel wall maturation during myocardium growth and development [69] and even predisposes to the onset of lethal cardiomyopathy [70]. Notch1 signalling is limited by the abundance of Notch1, which is transcriptionally controlled by Nrf2 (nuclear factor erythroid-derived 2-related factor 2), which is a prosurvival transcription factor that plays a pivotal role in maintaining cellular homeostasis following oxidative stress [71–73] and pro-inflammatory state [74] in adult heart. Nrf2-mediated regulation of Notch1 expression may be most critical in postnatal stages, rather than during development, because Nrf2 disruption does not produce altered phenotypes during early development stages. Nrf2 is abundant in tissues containing stem cells, such as liver, kidney and gastrointestinal-tract. Wakabayashi et al. [75] showed original evidence that Nrf2 facilitates tissue regeneration in the liver by regulating Notch1 expression. Moreover, recent experimental evidence showed that an increased expression of Nrf2 improves the effectiveness of cell therapy delivered in infarcted rodent heart [76].
Wnt signalling has been shown to influence the development of the heart. Recent data suggested canonical Wnts promote the emergence and expansion of cardiac progenitors in the pregastrula embryo. Martin et al. showed that canonical Wnt signalling up-regulates expression of the cardiac stem cell marker c-kit and pluripotency genes Oct25 and Oct60 without a corresponding inhibition of cardiogenic differentiation [77].
During cardiomyogenesis, Notch and TGF-β signalling pathways are involved in endothelial-to-mesenchymal transition of endothelial cells. Notch activation modulates TGF-β signalling pathways in a receptor-activated Smad-specific manner [78]. In particular, Notch increases SMAD3 mRNA expression and cooperatively induces histone H4 acetylation in cooperation with TGF-β. Recently, Maioli et al. [79] showed that the transcription of the cardiogenic gene Nkx-2.5 in embryonic stem cells is enhanced through Smad4 binding to its own consensus Smad site. The above signalling pathway driving cardiogenesis is modulated by novel molecules, which maintains acetylation of histone H4 in stem cells and adult cardiomyocytes, such as hyaluronan mixed esters of butyric and retinoic acids [80].
Imaging of self-renewed myocardium
The phenomenon of myocardial self-regeneration needs more detectable and quantifiable evidence in response to historical controversies [81]. In the past, it was very difficult to determine the sequence of the process of myocardial remodelling and/or repair after an injury as well as the plausible interference of a therapy during the healing’s phase without the use of histological analysis and, therefore, the sacrifice of a large number of animals. In addition, the tools available were not advanced enough to promote accurate morphological and functional study at regional and molecular level. So, it was common opinion that the no detection of a global cardiac effect, such as following a potential healing treatment, reflects the absence of an adaptive response post-injury (repair/regeneration), which was rather active regionally to a different scale. To date, the technological progress in non-invasive diagnosis allowed the development of more accurate tools for the ongoing detection of new forming coronary vessels or myocardium as well as for the real-time assessment at higher magnification of contractile function and structure in vivo[82–84]. The process of myocardial remodelling is initiated with a number of dynamic adverse effects in injured heart. Firstly, the non-contracting tissue expands and becomes thinner. The extreme thinning of the infarcted cardiac wall is easily detectable in both animal models and human beings. Secondly, the reactive growth of the cardiac tissue causes an increased volume load, after which compensatory mechanisms also lead to an increased pressure load on the heart. The mechanical overload results in LV hypertrophy and then dilation with increasing of LV cavity volume.
Finally, the occurrence of myoarchitectural disarray in the ventricular wall, i.e. the presence of abnormally organized cardiomyocytes, characterizes the remodelled myocardium in cardiovascular disease, such as hypertrophic cardiomyopathy, hypertension and myocardial infarction [85]. The cardiac muscle architecture lies at the basis of the mechanical and electrical properties of the heart, and dynamic alterations in fibre structure are known to be of prime importance in healing and remodelling after myocardial infarction. The amount of disarray present in the hearts after myocardial infarction, in both the border areas and the scar tissue formed by the infarction, is quantifiable by magnetic resonance imaging (MRI). In fact, diffusion tensor imaging offers a non-destructive tool for the study of myocardial fibre orientations [86, 87] as well as for the evaluation of structural tissue changes caused by ischaemia [88].
Myocardial wall anatomy and function of normal/injured cardiac regions are also accurately assessed by MRI using velocity encoding, tagging or DENSE (displacement encoding with stimulated echo) imaging [89–92]. The delayed enhancement-MRI of the infarcted myocardial wall after injection of a gadolinium-based contrast agent has been demonstrated to correlate with infarct size [93].
Regional myocardial wall perfusion in infarcted heart is quantified by positron emission tomography (PET) after intravenous injection of specific radiotracers, such as 13NH3 (ammonia) [91, 94], 11C-acetate [95] or (15)O-labelled water [96]. Recent investigations also demonstrated that it is feasible to quantify regional myocardial perfusion in ischaemic heart using gated single photon emission computed tomography (SPECT) [97] or hybrid SPECT/coronary computed tomography angiography technology [98]. The nuclear medicine has made possible an accurate assessment of the myocardial viability through a monitoring of its metabolism. Radionuclide modalities currently used in the assessment of viability include (201)Tl SPECT, (99m)Tc-based SPECT imaging and (18)F-fluorodexoyglucose ((18)F-FDG)-PET imaging. The initial myocardial uptake early after systemic delivery of thallium reflects the magnitude of the regional blood flow. However, redistribution of 201Tl is related to the rate of 201Tl ‘washout’ from the myocardium, dependent on the thallium concentration gradient between myocytes, blood pool and the integrity of the membrane-based Na+/K+ ATPase pump [99].
Despite 201Tl, 99mTc-based radiopharmaceuticals have a worse relationship to flow and minimal redistributing properties, and the mitochondrial uptake of these compounds requires an intact mitochondrial membrane and oxidative metabolism, thus the basis for sensitive viability detection [100]. Recent study compared the detection of viable myocardium by 18F-FDG, MRI and 99mTc-sestamibi (MIBI) and demonstrated that a significantly higher number of segments were identified as scar by MIBI than by either 18F-FDG or CMR [101]. However, the greatest discrepancy between MIBI and 18F-FDG and MRI was in detection of scar of the inferior and lateral wall. Cardiac PET utilizing 18F-FDG is considered the most sensitive modality for detecting scar and viable myocardium. To date, 18F-FDG PET is considered the most sensitive means of assessing viable myocardium and hence predicting LV functional recovery post-coronary revascularization [102].
However, no single imaging modality can provide all the information required to monitor the repair or regeneration of injured myocardium; hence, there is a necessity for combining complementary imaging methodologies. Indeed, the combination of MRI and PET modalities allows the acquisition of anatomical, physiological and metabolic information from the same subject. Recent study showed that multimodal imaging approach allowed an accurate and regional assessment of the new myocardium regenerated after cardiac transplantation of stem cells [103–106] or administration of new drugs, which elicit self-repair of the myocardium, such as hyaluronan mixed esters of butyric and retinoic acid [80]. These studies showed that the myocardial structure, function and viability of infarcted heart following regenerative treatment is equal to uninjured myocardium and the infarct scar size is significantly reduced.
Conclusions
Since ancient times, the heart’s ability to regenerate itself has always intrigued the human curiosity. Hyginus has narrated the myth of Prometheus, the Titan punished by the wrath of Zeus for stealing the fire from the gods. The Prometheus’s heart grows back during the night after being eaten by eagle on the day. The mythology seems to have foreseen that the human heart could not beat for the whole life with the same cardiomyocytes. The heart is an organ exposed to constant and variable mechanical forces and so it is potentially exposed to wear. The heart, similar to all adult tissues, has a plasticity that provides a partial renewal, yet it is weak, slow and limited. The plasticity of the heart depends both on rare cardiac progenitors resident into orderly architecture of the myocardium as well as on continuous cross-talk with other cells that shape the adult myocardium (cardiomyocytes, endothelial cells, fibroblasts), and it is also due to own ability of the myocardium to recruit and activate circulating inflammatory and progenitor cells. Some pathways of intercellular communication have been revealed ensuring the development of new pharmacological modulators. The future perspective will be to identify the complete code by which heart cells communicate with each other to promote a slow and continuous regeneration of the cells worn by mechanical forces and vascular insults. This new frontier of research in the cardiovascular field will lead to the affirmation of the pro-regenerative drugs for the treatment of cardiac dysfunction.
Acknowledgments
This work was supported by grant from the Ministry of Health of Italy (GR-2007-683407).
Conflict of interest
Authors certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.
References
- 1.Goldenberg B. Ueber Atrophie und Hypertrophie der Muskelfasern des Herzens. Virchows Arch Pathol Anat Physiol Klin, Med. 1886;103:88–130. [Google Scholar]
- 2.Kaufmann E. Lehrbuch der spez. path. Anat (Berl) 1928;1:56. [Google Scholar]
- 3.Nicholls JA. The Principles of Pathology. Vol. 2. Philadelphia: Lea & Febiger; 1909. p. 158. [Google Scholar]
- 4.Tangl F. Ueber die Herzhypertrophie u. das Physiologische Wachstum des Herzens. Virchows Arch Pathol Anat. 1889;116:432. [Google Scholar]
- 5.Wideroe S. Histologische Studien über die Muskulatur des Herzens. Virchows Arch Pathol Anat. 1911;204:190–6. [Google Scholar]
- 6.Collier WD. The adaptive changes of heart muscle. J Med Res. 1922;43:207–51. [PMC free article] [PubMed] [Google Scholar]
- 7.Karsner HT, Saphir O, Todd TW. The state of the cardiac muscle in hypertrophy and atrophy. Am J Pathol. 1925;1:351–72. [PMC free article] [PubMed] [Google Scholar]
- 8.Macmahon HE. Hyperplasia and regeneration of the myocardium in infants and in children. Am J Pathol. 1937;13:845–54. [PMC free article] [PubMed] [Google Scholar]
- 9.Zak R. Development and proliferative capacity of cardiac muscle cells. Circ Res. 1974;35:17–26. [PubMed] [Google Scholar]
- 10.Grove D, Zak R, Nair KG, et al. Biochemical correlates of cardiac hypertrophy. IV. Observations on the cellular organization of growth during myocardial hypertrophy in the rat. Circ Res. 1969;25:473–85. doi: 10.1161/01.res.25.4.473. [DOI] [PubMed] [Google Scholar]
- 11.Pasumarthi KB, Field LJ. Cardiomyocyte cell cycle regulation. Circ Res. 2002;90:1044–54. doi: 10.1161/01.res.0000020201.44772.67. [DOI] [PubMed] [Google Scholar]
- 12.Lionetti V, Bianchi G, Recchia FA, et al. Control of autocrine and paracrine myocardial signals: an emerging therapeutic strategy in heart failure. Heart Fail Rev. 2010;15:531–42. doi: 10.1007/s10741-010-9165-7. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Li TS, Lee ST, et al. Dedifferentiation and proliferation of mammalian cardiomyocytes. PloS One. 2010;5:e12559. doi: 10.1371/journal.pone.0012559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Novoyatleva T, Diehl F, van Amerongen MJ, et al. TWEAK is a positive regulator of cardiomyocyte proliferation. Cardiovasc Res. 2010;85:681–90. doi: 10.1093/cvr/cvp360. [DOI] [PubMed] [Google Scholar]
- 15.Dowell JD, Field LJ, Pasumarthi KB. Cell cycle regulation to repair the infarcted myocardium. Heart Fail Rev. 2003;8:293–303. doi: 10.1023/a:1024738104722. [DOI] [PubMed] [Google Scholar]
- 16.Kang MJ, Kim JS, Chae SW, et al. Cyclins and cyclin dependent kinases during cardiac development. Mol Cells. 1997;7:360–6. [PubMed] [Google Scholar]
- 17.Porrello ER, Mahmoud AI, Simpson E, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–80. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298:2188–90. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- 19.Jopling C, Sleep E, Raya M, et al. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. 2010;464:606–9. doi: 10.1038/nature08899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ausoni S, Sartore S. The cardiovascular unit as a dynamic player in disease and regeneration. Trends Mol Med. 2009;15:543–52. doi: 10.1016/j.molmed.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 21.Jacquemet V, Henriquez CS. Loading effect of fibroblast-myocyte coupling on resting potential, impulse propagation, and repolarization: insights from a microstructure model. Am J Physiol Heart Circ Physiol. 2008;294:H2040–52. doi: 10.1152/ajpheart.01298.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curtis MW, Russell B. Micromechanical regulation in cardiac myocytes and fibroblasts: implications for tissue remodeling. Pflugers Arch. 2011;462:105–17. doi: 10.1007/s00424-011-0931-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–78. doi: 10.1016/j.pharmthera.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 24.Turner NA, Warburton P, O’Regan DJ, et al. Modulatory effect of interleukin-1alpha on expression of structural matrix proteins, MMPs and TIMPs in human cardiac myofibroblasts: role of p38 MAP kinase. Matrix Biol. 2010;29:613–20. doi: 10.1016/j.matbio.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao W, Zhao T, Huang V, et al. Platelet-derived growth factor involvement in myocardial remodeling following infarction. J Mol Cell Cardiol. 2011;51:830–8. doi: 10.1016/j.yjmcc.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Santiago JJ, Ma X, McNaughton LJ, et al. Preferential accumulation and export of high molecular weight FGF-2 by rat cardiac non-myocytes. Cardiovasc Res. 2011;89:139–47. doi: 10.1093/cvr/cvq261. [DOI] [PubMed] [Google Scholar]
- 27.Santini MP, Lexow J, Borsellino G, et al. IGF-1Ea induces vessel formation after injury and mediates bone marrow and heart cross-talk through the expression of specific cytokines. Biochem Biophys Res Commun. 2011;410:201–7. doi: 10.1016/j.bbrc.2011.05.081. [DOI] [PubMed] [Google Scholar]
- 28.Zhao L, Eghbali-Webb M. Release of pro- and anti-angiogenic factors by human cardiac fibroblasts: effects on DNA synthesis and protection under hypoxia in human endothelial cells. Biochim Biophys Acta. 2001;1538:273–82. doi: 10.1016/s0167-4889(01)00078-7. [DOI] [PubMed] [Google Scholar]
- 29.Dixon IM, Cunnington RH. Mast cells and cardiac fibroblasts: accomplices in elevation of collagen synthesis in modulation of fibroblast phenotype. Hypertension. 2011;58:142–4. doi: 10.1161/HYPERTENSIONAHA.111.174748. [DOI] [PubMed] [Google Scholar]
- 30.Gherghiceanu M, Popescu LM. Cardiomyocyte precursors and telocytes in epicardial stem cell niche: electron microscope images. J Cell Mol Med. 2010;14:871–7. doi: 10.1111/j.1582-4934.2010.01060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sperr WR, Bankl HC, Mundigler G, et al. The human cardiac mast cell: localization, isolation, phenotype, and functional characterization. Blood. 1994;84:3876–84. [PubMed] [Google Scholar]
- 32.Patella V, Marino I, Arbustini E, et al. Stem cell factor in mast cells and increased mast cell density in idiopathic and ischemic cardiomyopathy. Circulation. 1998;97:971–8. doi: 10.1161/01.cir.97.10.971. [DOI] [PubMed] [Google Scholar]
- 33.Matsumoto T, Wada A, Tsutamoto T, et al. Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure. Circulation. 2003;107:2555–8. doi: 10.1161/01.CIR.0000074041.81728.79. [DOI] [PubMed] [Google Scholar]
- 34.Kwon JS, Kim YS, Cho AS, et al. The novel role of mast cells in the microenvironment of acute myocardial infarction. J Mol Cell Cardiol. 2011;50:814–25. doi: 10.1016/j.yjmcc.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 35.Zhou Y, Pan P, Yao L, et al. CD117-positive cells of the heart: progenitor cells or mast cells. J Histochem Cytochem. 2010;58:309–16. doi: 10.1369/jhc.2009.955146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gherghiceanu M, Popescu LM. Heterocellular communication in the heart: electron tomography of telocyte-myocyte junctions. J Cell Mol Med. 2011;15:1005–11. doi: 10.1111/j.1582-4934.2011.01299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Popescu LM, Faussone-Pellegrini MS. TELOCYTES – a case of serendipity: the winding way from Interstitial Cells of Cajal (ICC), via Interstitial Cajal-Like Cells (ICLC) to TELOCYTES. J Cell Mol Med. 2010;14:729–40. doi: 10.1111/j.1582-4934.2010.01059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manole CG, Cismasiu V, Gherghiceanu M, et al. Experimental acute myocardial infarction: telocytes involvement in neo-angiogenesis. J Cell Mol Med. 2011 doi: 10.1111/j.1582-4934.2011.01449.x. doi: 10.1111/j.1582-4934.2011.01449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Popescu LM, Gherghiceanu M, Manole CG, et al. Cardiac renewing: interstitial Cajal-like cells nurse cardiomyocyte progenitors in epicardial stem cell niches. J Cell Mol Med. 2009;13:866–86. doi: 10.1111/j.1582-4934.2009.00758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Urbanek K, Cesselli D, Rota M, et al. Stem cell niches in the adult mouse heart. Proc Natl Acad Sci USA. 2006;103:9226–31. doi: 10.1073/pnas.0600635103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beltrami AP, Barlucchi L, Torella D, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–76. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 42.Hierlihy AM, Seale P, Lobe CG, et al. The post-natal heart contains a myocardial stem cell population. FEBS Lett. 2002;530:239–43. doi: 10.1016/s0014-5793(02)03477-4. [DOI] [PubMed] [Google Scholar]
- 43.Laugwitz KL, Moretti A, Lam J, et al. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature. 2005;433:647–53. doi: 10.1038/nature03215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oh H, Bradfute SB, Gallardo TD, et al. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci USA. 2003;100:12313–8. doi: 10.1073/pnas.2132126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ott HC, Matthiesen TS, Brechtken J, et al. The adult human heart as a source for stem cells: repair strategies with embryonic-like progenitor cells. Nat Clin Pract Cardiovasc Med. 2007;1:S27–39. doi: 10.1038/ncpcardio0771. [DOI] [PubMed] [Google Scholar]
- 46.Smith RR, Barile L, Cho HC, et al. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation. 2007;115:896–908. doi: 10.1161/CIRCULATIONAHA.106.655209. [DOI] [PubMed] [Google Scholar]
- 47.Li TS, Cheng K, Lee ST, et al. Cardiospheres recapitulate a niche-like microenvironment rich in stemness and cell-matrix interactions, rationalizing their enhanced functional potency for myocardial repair. Stem Cells. 2010;28:2088–98. doi: 10.1002/stem.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Altomare C, Barile L, Marangoni S, et al. Caffeine-induced Ca(2+) signaling as an index of cardiac progenitor cells differentiation. Basic Res Cardiol. 2010;105:737–49. doi: 10.1007/s00395-010-0111-6. [DOI] [PubMed] [Google Scholar]
- 49.Hsieh PC, Segers VF, Davis ME, et al. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med. 2007;13:970–4. doi: 10.1038/nm1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barile L, Messina E, Giacomello A, et al. Endogenous cardiac stem cells. Progr Cardiovasc Dis. 2007;50:31–48. doi: 10.1016/j.pcad.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 51.Barile L, Cerisoli F, Frati G, et al. Bone marrow-derived cells can acquire cardiac stem cells properties in damaged heart. J Cell Mol Med. 2011;15:63–71. doi: 10.1111/j.1582-4934.2009.00968.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Loffredo FS, Steinhauser ML, Gannon J, et al. Bone marrow-derived cell therapy stimulates endogenous cardiomyocyte progenitors and promotes cardiac repair. Cell Stem Cell. 2011;8:389–98. doi: 10.1016/j.stem.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cesselli D, Beltrami AP, D’Aurizio F, et al. Effects of age and heart failure on human cardiac stem cell function. Am J Pathol. 2011;179:349–66. doi: 10.1016/j.ajpath.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fortini C, Toffoletto B, Fucili A, et al. Circulating stem cell vary with NYHA stage in heart failure patients. J Cell Mol Med. 2011;15:1726–36. doi: 10.1111/j.1582-4934.2010.01195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bergmann O, Bhardwaj RD, Bernard S, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kajstura J, Urbanek K, Perl S, et al. Cardiomyogenesis in the adult human heart. Circ Res. 2010;107:305–15. doi: 10.1161/CIRCRESAHA.110.223024. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Winkles JA. The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat Rev. 2008;7:411–25. doi: 10.1038/nrd2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lipinski MM, Jacks T. The retinoblastoma gene family in differentiation and development. Oncogene. 1999;18:7873–82. doi: 10.1038/sj.onc.1203244. [DOI] [PubMed] [Google Scholar]
- 59.Mac Lellan WR, Schneider MD. Genetic dissection of cardiac growth control pathways. Annu Rev Physiol. 2000;62:289–319. doi: 10.1146/annurev.physiol.62.1.289. [DOI] [PubMed] [Google Scholar]
- 60.Shay JW, Wright WE. Telomeres and telomerase: implications for cancer and aging. Radiat Res. 2001;155:188–93. doi: 10.1667/0033-7587(2001)155[0188:tatifc]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 61.Borges A, Liew CC. Telomerase activity during cardiac development. J Mol Cell Cardiol. 1997;29:2717–24. doi: 10.1006/jmcc.1997.0503. [DOI] [PubMed] [Google Scholar]
- 62.Oh H, Taffet GE, Youker KA, et al. Telomerase reverse transcriptase promotes cardiac muscle cell proliferation, hypertrophy, and survival. Proc Natl Acad Sci USA. 2001;98:10308–13. doi: 10.1073/pnas.191169098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Harvey RP, Lai D, Elliott D, et al. Homeodomain factor Nkx2-5 in heart development and disease. Cold Spring Harb Symp Quant Biol. 2002;67:107–14. doi: 10.1101/sqb.2002.67.107. [DOI] [PubMed] [Google Scholar]
- 64.Kwon C, Qian L, Cheng P, et al. A regulatory pathway involving Notch1/beta-catenin/Isl1 determines cardiac progenitor cell fate. Nat Cell Biol. 2009;11:951–7. doi: 10.1038/ncb1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hosoda T, D’Amario D, Cabral-Da-Silva MC, et al. Clonality of mouse and human cardiomyogenesis in vivo. Proc Natl Acad Sci USA. 2009;106:17169–74. doi: 10.1073/pnas.0903089106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boni A, Urbanek K, Nascimbene A, et al. Notch1 regulates the fate of cardiac progenitor cells. Proc Natl Acad Sci USA. 2008;105:15529–34. doi: 10.1073/pnas.0808357105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 67.Nemir M, Pedrazzini T. Functional role of Notch signaling in the developing and postnatal heart. J Mol Cell Cardiol. 2008;45:495–504. doi: 10.1016/j.yjmcc.2008.02.273. [DOI] [PubMed] [Google Scholar]
- 68.Collesi C, Zentilin L, Sinagra G, et al. Notch1 signaling stimulates proliferation of immature cardiomyocytes. J Cell Biol. 2008;183:117–28. doi: 10.1083/jcb.200806091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.del Monte G, Casanova JC, Guadix JA, et al. Differential Notch signaling in the epicardium is required for cardiac inflow development and coronary vessel morphogenesis. Circ Res. 2011;108:824–36. doi: 10.1161/CIRCRESAHA.110.229062. [DOI] [PubMed] [Google Scholar]
- 70.Urbanek K, Cabral-da-Silva MC, Ide-Iwata N, et al. Inhibition of notch1-dependent cardiomyogenesis leads to a dilated myopathy in the neonatal heart. Circ Res. 2010;107:429–41. doi: 10.1161/CIRCRESAHA.110.218487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li J, Ichikawa T, Villacorta L, et al. Nrf2 protects against maladaptive cardiac responses to hemodynamic stress. Arterioscler Thromb Vasc Biol. 2009;29:1843–50. doi: 10.1161/ATVBAHA.109.189480. [DOI] [PubMed] [Google Scholar]
- 72.Dreger H, Westphal K, Weller A, et al. Nrf2-dependent upregulation of antioxidative enzymes: a novel pathway for proteasome inhibitor-mediated cardioprotection. Cardiov Res. 2009;83:354–61. doi: 10.1093/cvr/cvp107. [DOI] [PubMed] [Google Scholar]
- 73.He X, Kan H, Cai L, et al. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. J Mol Cell Cardiol. 2009;46:47–58. doi: 10.1016/j.yjmcc.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 74.Zakkar M, Van der Heiden K, Luong le A, et al. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler Thromb Vasc Biol. 2009;29:1851–7. doi: 10.1161/ATVBAHA.109.193375. [DOI] [PubMed] [Google Scholar]
- 75.Wakabayashi N, Shin S, Slocum SL, et al. Regulation of notch1 signaling by nrf2: implications for tissue regeneration. Sci Signal. 2010;3:ra52. doi: 10.1126/scisignal.2000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gorbunov N, Petrovski G, Gurusamy N, et al. Regeneration of infarcted myocardium with resveratrol-modified cardiac stem cells. J Cell Mol Med. 2011 doi: 10.1111/j.1582-4934.2011.01281.x. doi: 10.1111/j.1582-4934.2011.01281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martin LK, Mezentseva NV, Bratoeva M, et al. Canonical WNT signaling enhances stem cell expression in the developing heart without a corresponding inhibition of cardiogenic differentiation. Stem Cells Dev. 2011 doi: 10.1089/scd.2010.0490. doi: 10.1089/scd.2010.0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fu Y, Chang A, Chang L, et al. Differential regulation of transforming growth factor beta signaling pathways by Notch in human endothelial cells. J Biol Chem. 2009;284:19452–62. doi: 10.1074/jbc.M109.011833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maioli M, Santaniello S, Montella A, et al. Hyaluronan esters drive Smad gene expression and signaling enhancing cardiogenesis in mouse embryonic and human mesenchymal stem cells. PloS One. 2010;5:e15151. doi: 10.1371/journal.pone.0015151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lionetti V, Cantoni S, Cavallini C, et al. Hyaluronan mixed esters of butyric and retinoic acid affording myocardial survival and repair without stem cell transplantation. J Biol Chem. 2010;285:9949–61. doi: 10.1074/jbc.M109.087254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alexander JM, Bruneau BG. Lessons for cardiac regeneration and repair through development. Trends Mol Med. 2010;16:426–34. doi: 10.1016/j.molmed.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dawson DK, Maceira AM, Raj VJ, et al. Regional thicknesses and thickening of compacted and trabeculated myocardial layers of the normal left ventricle studied by cardiovascular magnetic resonance. Circulation. 2011;4:139–46. doi: 10.1161/CIRCIMAGING.110.960229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sosnovik DE, Nahrendorf M, Panizzi P, et al. Molecular MRI detects low levels of cardiomyocyte apoptosis in a transgenic model of chronic heart failure. Circulation. 2009;2:468–75. doi: 10.1161/CIRCIMAGING.109.863779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Agostini S, Recchia FA, Lionetti V. Molecular advances in reporter genes: the need to witness the function of stem cells in failing heart in vivo. Stem Cell Rev. 2011 doi: 10.1007/s12015-011-9296-9. doi: 10.1007/s12015-011-9296-9. [DOI] [PubMed] [Google Scholar]
- 85.Chen J, Song SK, Liu W, et al. Remodeling of cardiac fiber structure after infarction in rats quantified with diffusion tensor MRI. Am J Physiol Heart Circ Physiol. 2003;285:H946–54. doi: 10.1152/ajpheart.00889.2002. [DOI] [PubMed] [Google Scholar]
- 86.Toussaint N, Sermesant M, Stoeck CT, et al. In vivo human 3D cardiac fibre architecture: reconstruction using curvilinear interpolation of diffusion tensor images. Med Image Comput Comput Assist Interv. 2010;13:418–25. doi: 10.1007/978-3-642-15705-9_51. [DOI] [PubMed] [Google Scholar]
- 87.Munoz-Moreno E, Cardenes R, Frangi AF. Analysis of the helix and transverse angles of the muscle fibers in the myocardium based on diffusion tensor imaging. Conf Proc IEEE Eng Med Biol Soc. 2010;2010:5720–3. doi: 10.1109/IEMBS.2010.5627867. [DOI] [PubMed] [Google Scholar]
- 88.Strijkers GJ, Bouts A, Blankesteijn WM, et al. Diffusion tensor imaging of left ventricular remodeling in response to myocardial infarction in the mouse. NMR Biomed. 2009;22:182–90. doi: 10.1002/nbm.1299. [DOI] [PubMed] [Google Scholar]
- 89.Henson RE, Song SK, Pastorek JS, et al. Left ventricular torsion is equal in mice and humans. Am J Physiol Heart Circ Physiol. 2000;278:H1117–23. doi: 10.1152/ajpheart.2000.278.4.H1117. [DOI] [PubMed] [Google Scholar]
- 90.Liu W, Ashford MW, Chen J, et al. MR tagging demonstrates quantitative differences in regional ventricular wall motion in mice, rats, and men. Am J Physiol Heart Circ Physiol. 2006;291:H2515–21. doi: 10.1152/ajpheart.01016.2005. [DOI] [PubMed] [Google Scholar]
- 91.Lionetti V, Guiducci L, Simioniuc A, et al. Mismatch between uniform increase in cardiac glucose uptake and regional contractile dysfunction in pacing-induced heart failure. Am J Physiol Heart Circ Physiol. 2007;293:H2747–56. doi: 10.1152/ajpheart.00592.2007. [DOI] [PubMed] [Google Scholar]
- 92.Lionetti V, Aquaro GD, Simioniuc A, et al. Severe mechanical dyssynchrony causes regional hibernation-like changes in pigs with nonischemic heart failure. J Card Fail. 2009;15:920–8. doi: 10.1016/j.cardfail.2009.06.436. [DOI] [PubMed] [Google Scholar]
- 93.Wagner A, Mahrholdt H, Thomson L, et al. Effects of time, dose, and inversion time for acute myocardial infarct size measurements based on magnetic resonance imaging-delayed contrast enhancement. J Am Coll Cardiol. 2006;47:2027–33. doi: 10.1016/j.jacc.2006.01.059. [DOI] [PubMed] [Google Scholar]
- 94.Bellina CR, Parodi O, Camici P, et al. Simultaneous in vitro and in vivo validation of nitrogen-13-ammonia for the assessment of regional myocardial blood flow. J Nucl Med. 1990;31:1335–43. [PubMed] [Google Scholar]
- 95.Hata T, Nohara R, Fujita M, et al. Noninvasive assessment of myocardial viability by positron emission tomography with 11C acetate in patients with old myocardial infarction. Usefulness of low-dose dobutamine infusion. Circulation. 1996;94:1834–41. doi: 10.1161/01.cir.94.8.1834. [DOI] [PubMed] [Google Scholar]
- 96.Vermeltfoort IA, Raijmakers PG, Lubberink M, et al. Feasibility of subendocardial and subepicardial myocardial perfusion measurements in healthy normals with (15)O-labeled water and positron emission tomography. J Nucl Cardiol. 2011;18:650–6. doi: 10.1007/s12350-011-9375-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Arruda-Olson AM, Roger VL, Jaffe AS, et al. Troponin T levels and infarct size by SPECT myocardial perfusion imaging. JACC Cardiovasc Imaging. 2011;4:523–33. doi: 10.1016/j.jcmg.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rispler S, Aronson D, Abadi S, et al. Integrated SPECT/CT for assessment of haemodynamically significant coronary artery lesions in patients with acute coronary syndrome. Eur J Nucl Med Mol Imaging. 2011;38:1917–25. doi: 10.1007/s00259-011-1856-3. [DOI] [PubMed] [Google Scholar]
- 99.Strauss HW, Harrison K, Langan JK, et al. Thallium-201 for myocardial imaging. Relation of thallium-201 to regional myocardial perfusion. Circulation. 1975;51:641–5. doi: 10.1161/01.cir.51.4.641. [DOI] [PubMed] [Google Scholar]
- 100.Beanlands RS, Dawood F, Wen WH, et al. Are the kinetics of technetium-99m methoxyisobutyl isonitrile affected by cell metabolism and viability. Circulation. 1990;82:1802–14. doi: 10.1161/01.cir.82.5.1802. [DOI] [PubMed] [Google Scholar]
- 101.Crean A, Khan SN, Davies LC, et al. Assessment of myocardial scar; comparison between F-FDG PET, CMR and Tc-sestamibi. Clin Med Cardiol. 2009;3:69–76. doi: 10.4137/cmc.s730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Inaba Y, Chen JA, Bergmann SR. Quantity of viable myocardium required to improve survival with revascularization in patients with ischemic cardiomyopathy: a meta-analysis. J Nucl Cardiol. 2010;17:646–54. doi: 10.1007/s12350-010-9226-2. [DOI] [PubMed] [Google Scholar]
- 103.Simioniuc A, Campan M, Lionetti V, et al. Placental stem cells pre-treated with a hyaluronan mixed ester of butyric and retinoic acid to cure infarcted pig hearts: a multimodal study. Cardiovasc Res. 2011;90:546–56. doi: 10.1093/cvr/cvr018. [DOI] [PubMed] [Google Scholar]
- 104.Lee AS, Xu D, Nguyen PK, et al. Preclinical derivation and imaging of autologously transplanted canine induced pluripotent stem cells. J Biol Chem. 2011;286:32697–704. doi: 10.1074/jbc.M111.235739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gyongyosi M, Blanco J, Marian T, et al. Serial noninvasive in vivo positron emission tomographic tracking of percutaneously intramyocardially injected autologous porcine mesenchymal stem cells modified for transgene reporter gene expression. Circulation. 2008;1:94–103. doi: 10.1161/CIRCIMAGING.108.797449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.de Silva R, Raval AN, Hadi M, et al. Intracoronary infusion of autologous mononuclear cells from bone marrow or granulocyte colony-stimulating factor-mobilized apheresis product may not improve remodelling, contractile function, perfusion, or infarct size in a swine model of large myocardial infarction. Eur Heart J. 2008;29:1772–82. doi: 10.1093/eurheartj/ehn216. [DOI] [PMC free article] [PubMed] [Google Scholar]