

Abstract
Asthma is a prevalent disease of chronic inflammation in which endogenous counter-regulatory signaling pathways are dysregulated. Recent evidence suggests that innate lymphoid cells (ILCs), including natural killer (NK) cells and type 2 innate lymphoid cells (ILC2), can participate in the regulation of allergic airways responses, in particular airway mucosal inflammation. Here, we have identified both NK cells and ILC2 in human lung and peripheral blood in healthy and asthmatic subjects. NK cells were highly activated in severe asthma, linked to eosinophilia and interacted with autologous eosinophils to promote their apoptosis. ILC2 generated antigen-independent IL-13 in response to the mast cell product prostaglandin D2 (PGD2) alone and in a synergistic manner with the airway epithelial cytokines IL-25 and IL-33. Both NK cells and ILC2 expressed the pro-resolving ALX/FPR2 receptors. Lipoxin A4, a natural pro-resolving ligand for ALX/FPR2 receptors, significantly increased NK cell mediated eosinophil apoptosis and decreased IL-13 release by ILC2. Together, these findings indicate that ILCs are targets for lipoxin A4 to decrease airway inflammation and mediate the catabasis of eosinophilic inflammation. Because lipoxin A4 generation is decreased in severe asthma, these findings also implicate unrestrained ILC activation in asthma pathobiology.
Introduction
Asthma is characterized by chronic airway inflammation with mucosal infiltration of eosinophils, T lymphocytes, mast cells and release of pro-inflammatory cytokines and lipid mediators (1). In health, the resolution of inflammation is now appreciated to involve active biochemical programs that enable inflamed tissues to return to homeostasis (2). Counter-regulatory lipid mediators are rapidly generated from essential fatty acids during inflammation to promote resolution. Lipoxins are the lead members of this new class of pro-resolving mediators (3) with cell type specific actions that include inhibition of neutrophil activation and promotion of macrophage engulfment of apoptotic neutrophils for the resolution of acute tissue inflammation. Lipoxins are generated in asthma (4), and defects in the production of pro-resolving mediators have been associated with chronic inflammatory diseases, including severe asthma (4, 5).
Cellular targets for lipoxins to regulate asthmatic airway responses remain to be determined. Innate lymphoid cells (ILCs) serve protective roles in immune responses (6). Natural Killer (NK) cells are members of the ILC family that serve essential roles in host defense (7), including cytokine secretion, contact-dependent cell-cell signaling and direct killing of other immune cells. NK cells display functional diversity and both disease-controlling and disease-promoting roles have been implicated for NK cells in chronic inflammatory disease (reviewed in (8)). Potential roles for NK cells in asthma and allergic diseases are undefined; however, recent evidence in model systems suggests that NK cells can participate in the down-regulation of allergic airways responses, in particular airway mucosal inflammation (9).
In addition to NK cells, the ILC family also includes type 2 innate lymphoid cells (ILC2), which have been implicated in allergic responses (6). In an antigen-independent manner, ILC2 can generate the cytokines IL-5 and IL-13 that were previously linked to Th2 lymphocytes. ILC2 were recently identified in humans as a population of Lin−CD127+CD161+ ILCs, which also express the chemoattractant receptor-homologous molecule expressed on Th2 lymphocytes (CRTH2) (10). Several studies in murine models of lung disease have demonstrated a role for ILC2s in the development of airway inflammation (11, 12).
Here, we have identified both NK cells and ILC2 in human lung and peripheral blood from healthy and asthmatic subjects. NK cells were highly activated in severe asthma, linked to eosinophilia and interacted with autologous eosinophils to promote their apoptosis. ILC2 generated IL-13 in response to the mast cell product prostaglandin D2 (PGD2) alone and in a synergistic manner with the airway epithelial cytokines IL-25 and IL-33. In addition, both NK cells and ILC2 expressed pro-resolving receptors. A natural pro-resolving mediator lipoxin A4 increased NK cell mediated eosinophil clearance and decreased IL-13 release by ILC2. Together, these findings establish two new cellular targets for pro-resolving mediators and assign critical roles to innate immune lymphoid cells in asthma pathobiology.
Results
Severe asthmatic subjects have lower lung function and more symptoms despite increased use of corticosteroids
Subject characteristics are described in Table 1. The Asthma Control Questionnaire (ACQ) score was higher and lung function (i.e., FEV1) was lower in the subjects with severe asthma compared with mild asthma. None of the subjects with severe asthma were taking oral corticosteroids. Most of the asthmatics were on daily inhaled corticosteroids, and the total daily dose was higher in severe asthma (Table 1). All the patients with severe asthma were treated with long acting β2-agonists, 18% with leukotriene antagonists and 18% with omalizumab.
Table 1.
Subject characteristics.
| Mild Asthma | Severe Asthma | |
|---|---|---|
| No. of subjects | 11 | 11 |
| Clinical data | ||
| Age (years) | 30.6±10.2 (21–54) | 41.5±11.8 (26–60)* |
| Gender (M:F) | 3:8 | 2:9 |
| Race (% white) | 45 | 55 |
| Ethnicity (% Hispanic) | 9 | 18 |
| Inhaled corticosteroids (Fluticasone equivalent, μg per day) | 152.73±143.74 (0–360) | 1085.46±303.06 (640–1760)* |
| Oral corticosteroids (%) | 0 | 0 |
| Long Acting Beta-Agonist (%) | 0 | 100* |
| Leukotriene Antagonists (%) | 0 | 18* |
| Omalizumab (%) | 0 | 18* |
| ACQ Score | 1.18±0.58 (0.43–2.43) | 2.52±0.64 (1.57–3.57)* |
| FEV1 (L) | 3.25±0.39 (2.74–3.93) | 2.14±0.57 (1.27–3.35)* |
| FEV1 (% predicted) | 96.8±8.5 (82–106) | 71.9±17.3 (45–101)* |
Definition of abbrevations: ACQ = Asthma Control Questionnaire.
Results are expressed as mean ± SD (range)
P < 0.05 when compared with subjects with mild asthma.
Natural Killer cells are activated in asthma
Natural Killer cells (NK cells) were identified as a lymphoid morphology cell population that expressed NKp46 but not CD3 (Fig. 1A). Relative to healthy subjects, total NK cell numbers in peripheral blood were decreased in mild and severe asthma (Fig. 1A). In contrast, the number of peripheral T lymphocytes (CD3+), and their CD4+ and CD8+ subsets were similar in healthy subjects and mild and severe asthma (Fig. S1). NK cells were further subdivided based on their surface expression of CD56 and CD16 (Fig. 1B). The numbers of CD56dim NK cells were decreased in both mild and severe asthma (Fig. 1B), while the numbers of CD56brightCD16neg and CD56brightCD16pos NK cells were similar in all cohorts (Fig. S2). To determine the proportion of CD56dim and CD56bright NK cells at sites of inflammation, NK cell subsets were enumerated in BALF samples from subjects with severe asthma. When compared to peripheral blood samples from the same subjects, the CD56bright subset was highly enriched in BALF samples (Fig. 1C). The percentage of CD3+CD4+ and CD3+CD8+ cells was similar in PBMC and BALF samples (Fig. S3).
Fig. 1. NK cells are activated in asthma.
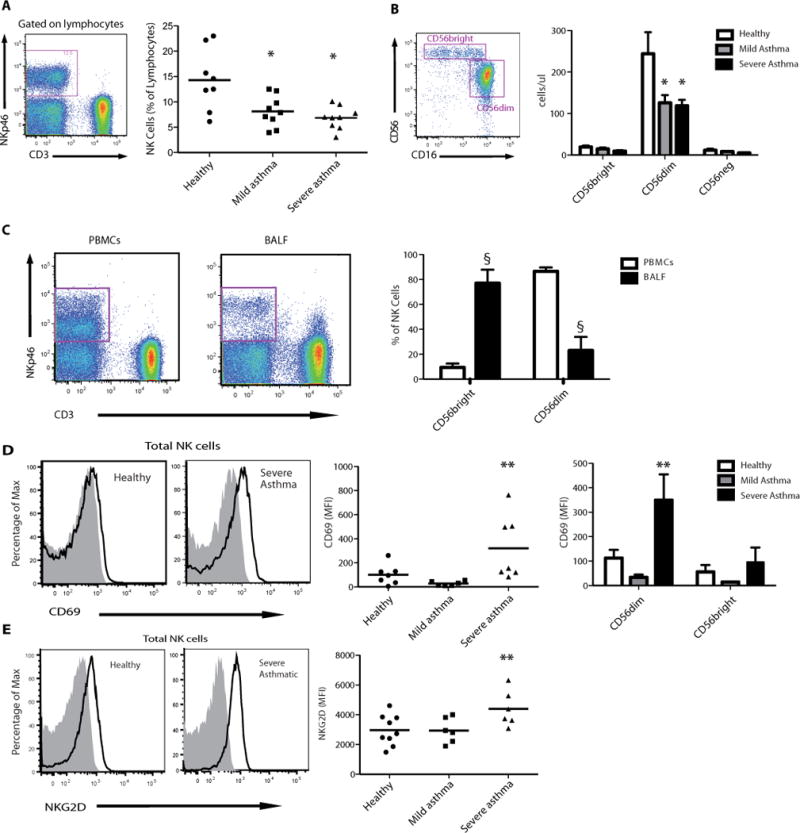
Peripheral venous blood was obtained from healthy subjects and individuals with mild and severe asthma. (A) NK cells were identified as a NKp46+CD3− cell population in a lymphocyte morphology gate based on SSC and FSC characteristics (violet box, left panel) and NK cells as a percentage of total lymphocytes were enumerated (right panel), bar indicates mean value. (B) NK cells were further phenotyped by flow cytometry analysis of CD56 expression for CD56bright and CD56dim subsets (violet boxes, left panel) in samples from healthy and asthmatic subjects (right panel). (C) Paired samples of BALF and blood were obtained on a subset of severe asthmatics. Representative FACS plots show an enriched NKp46bright cell population in BALF compared to PBMCs (left panel). BALFs were also enriched for CD56bright subset of NK cells (n=5) (right panel). (D) Representative FACS histograms showing CD69 expression in peripheral blood NK cells (left panel) that was upregulated in severe asthma (middle panel), in particular in the CD56dim subset (right panel). (E) Representative FACS histograms showing NKG2D expression in peripheral NK cells (left panel) that was upregulated in severe asthma (right panel). Values are expressed as mean ± s.e.m.; *, P < 0.05, compared with healthy subjects (one-way ANOVA); §, P < 0.0001, compared with PBMCs (one-way ANOVA); **, P < 0.05, compared with healthy and mild asthmatic subjects (one-way ANOVA).
Peripheral blood NK cells were highly activated in severe asthma relative to cells from mild asthmatics or healthy subjects (Fig. 1D and 1E). In severe asthma, the NK cell activation marker CD69 was markedly increased (Fig. 1D), in particular in the CD56dim subset (Fig. 1D). The NK cell activating receptor NKG2D, which specifically interacts with several stress-induced self-ligands (13), was also selectively increased in severe asthma (Fig. 1E). There was a similar increase in the expression of this receptor in severe asthma in both CD56dim and CD56bright NK subsets (Fig. S4).
Activated NK cells interact with eosinophils
Severe asthmatics had a higher peripheral blood total white blood cell count than mild asthmatics and healthy subjects (P < 0.05, one-way ANOVA) (Fig. S5A) with increased numbers of circulating eosinophils (P < 0.05, one-way ANOVA) (Fig. S5B). In severe asthmatics, there was a positive relationship between the total number of peripheral blood NK cells and eosinophils (Fig. 2A). In addition, NK cell surface expression of CD69 and NKG2D were also positively correlated to peripheral blood eosinophil count (Fig. 2B and 2C).
Fig. 2. NK cells interact with eosinophils.
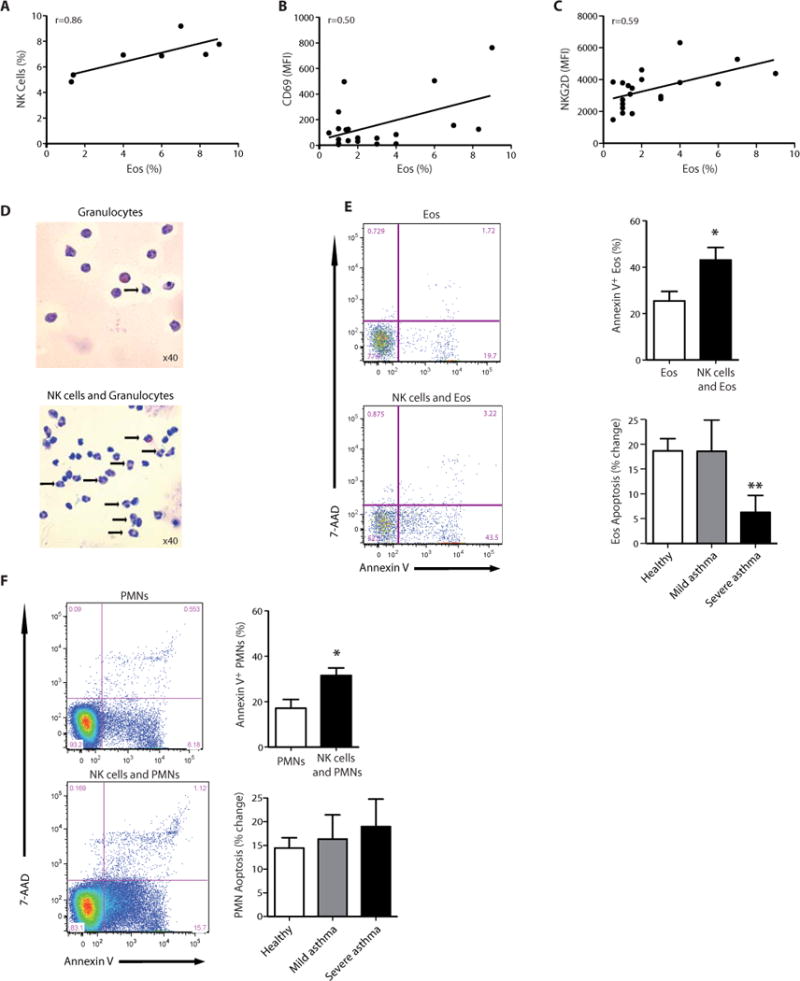
(A–C) The percentage of peripheral blood eosinophils in severe asthma was correlated to (A) the percentage of peripheral blood NK cells; Spearman r=0.86, P < 0.05. (B) NK cell CD69 expression; Pearson r=0.50, P < 0.05, and (C) NK cell NKG2D expression, Spearman r=0.59, P < 0.01. (D–F) Incubation (4h, 37°C) of autologous peripheral blood granulocytes with NK cells (1:5 ratio) triggered increased granulocyte apoptosis compared to granulocytes incubated alone by (D) morphological appearance on cytospin (arrows, apoptotic granulocytes) and (E,F) by FACS criteria (Annexin V+7-AAD−) for (E) eosinophils (Eos) and (F) neutrophils (PMN) in samples of peripheral blood from healthy subjects and individuals with mild or severe asthma. Results are expressed as mean ± SEM.; n=5 individual healthy donors, n=5 mild asthmatic donors, n=5 severe asthmatic donors; *, P < 0.05, compared with granulocytes alone and **, P < 0.05 compared with healthy donors (Student’s t-test).
To determine the impact of NK cells on granulocyte function, freshly isolated NK cells were incubated (4h, 37°C) with autologous granulocytes. Cytospins of these incubations revealed clear evidence of granulocyte cell death (cell shrinkage and nuclear condensation) that was more prominent in the presence of NK cells (Fig. 2D). Quantitative analyses by flow cytometry indicated that NK cells triggered apoptosis of both eosinophils (Fig. 2E) and neutrophils (Fig. 2F). During co-incubations, NK cells from healthy and mild asthmatic subjects led to similar increases in both eosinophil and neutrophil apoptosis. In sharp contrast, NK cells from severe asthmatic subjects had a significantly reduced capacity to augment eosinophil apoptosis (Fig. 2E) despite the increase in NK cell expression of CD69 and NKG2D (Fig. 1). This impairment in severe asthma NK cell responses was selective for eosinophils, as NK cell mediated neutrophil apoptosis was similar for all three cohorts (Fig. 2F). Of interest, NK cells that were incubated with eosinophils and neutrophils did not express surface CD107, which was distinct from similar incubations with the K562 myeloid leukemia cells (Fig. S6).
NK cells express pro-resolving receptors and respond to lipoxin A4
With this evidence that NK cells display the pro-resolving capacity to inactivate granulocytes by inducing apoptosis (Fig. 2), their expression of specific receptors for pro-resolving mediators was next determined. Circulating NK cells expressed ALX/FPR2 (Fig. 3A), a receptor for Lipoxin A4 (14). Both CD56dim and CD56bright NK cell subsets expressed ALX/FPR2 at similar levels (Fig. S7). Surface expression of this receptor was increased in severe asthma (Fig. 3A). Peripheral blood NK cells also expressed CMKLR1, a receptor for Resolvin E1 (15). Distinct from ALX/FPR2, the cell surface expression of CMKLR1 receptors was similar in all subjects (Fig. 3B).
Fig. 3. NK cells express pro-resolving receptors and respond to LXA4.
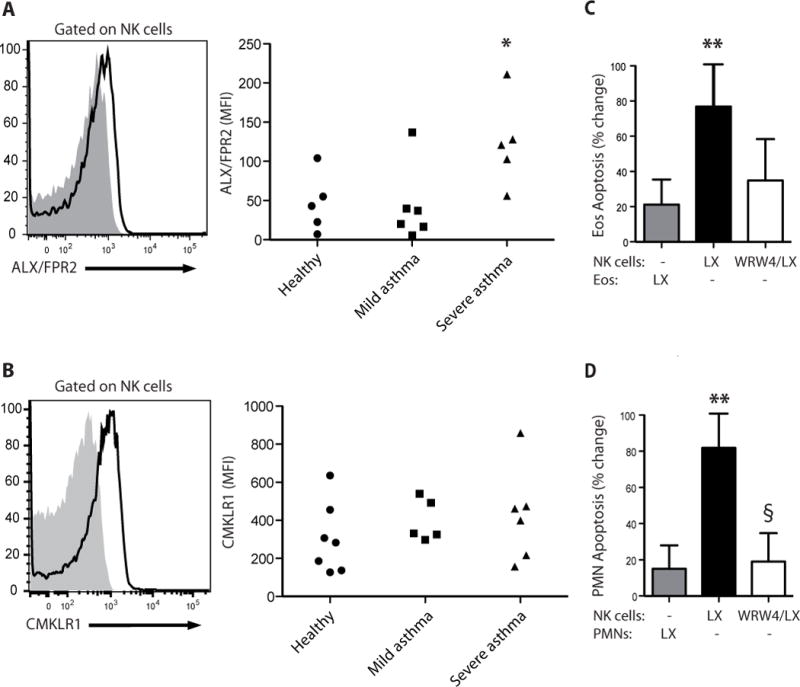
(A) Flow cytometry histograms for anti-ALX (FPRL1) Ab (white) and isotype control (gray) for peripheral blood NK cells (left panel) and MFI for ALX expression on peripheral NK cells in healthy subjects and mild and severe asthmatic subjects (right panel). (B) Flow cytometry histograms for anti-CMKLR1 Ab (white) and isotype control (gray) for peripheral blood NK cells (left panel) and MFI for CMKLR1 expression on peripheral NK cells in healthy subjects and mild and severe asthmatic subjects (right panel). *, P < 0.05, compared with healthy and mild asthmatic subjects (one-way ANOVA). (C, D) Autologous granulocytes and NK cells from healthy subjects were selectively exposed to lipoxin A4 (LX, 100 nM, 15 min, 37°C) in the absence or presence of the ALX/FPR2 antagonist (WRW4, 230nM) prior to co-incubation and the percent change in apoptosis was determined for (C) eosinophils (Eos) and (D) neutrophils (PMN) (see Methods). Results are expressed as mean ± SEM; n≥4; **, P < 0.05, compared with control (one-way ANOVA); §, P < 0.05, compared with NK(LX).
To determine if lipoxin A4 could influence NK cell mediated granulocyte apoptosis, NK cells or granulocytes were selectively exposed to lipoxin A4 (100 nM) prior to cell-cell interactions during their co-incubations. When NK cells were exposed (15 min) to lipoxin A4, NK cell induced apoptosis of both eosinophils and neutrophils was significantly increased (Fig. 3C and 3D). This effect was not observed if the granulocytes were exposed to lipoxin A4 prior to their co-incubations with NK cells (Fig. 3C and 3D). Lipoxin A4’s actions were inhibited in part by concomitant exposure to the ALX/FPR2 receptor antagonist WRW4 (Fig. 3C and 3D).
Type 2 Innate Lymphoid Cells are present in peripheral blood of healthy and asthmatic subjects and express pro-resolving receptors
In addition to NK cells, other ILC family members, including type 2 innate lymphoid cells (ILC2) are present in human peripheral blood (10). To determine potential roles for these cells in asthma pathobiology, samples of human peripheral blood from healthy and asthmatic subjects were analyzed by flow cytometry for the presence of helper ILCs (Fig. 4). Peripheral blood mononuclear cells were first depleted of most T cells (with anti-CD3), B cells (with anti-CD19) and monocytes (with anti-CD14). After gating on lineage negative (Lin−) cells, a population of CD127+ cells distinct from CD56-expressing NK cells was identified (Fig. 4A). In the Lin−CD127+ population, we were able to further distinguish subsets of CD117+ and CD117− cells (Fig. 4A) that expressed the NK cell receptors CD161, NKG2D (Fig. 4B) and NKp46 (Fig. 4C). A subpopulation of Lin−CD127+ cells were identified as ILC2 by the expression of CRTH2 receptors (Fig. 4D and 4E), which ranged between 1.78 – 27.9% in healthy subjects, 1.08 – 24.2% in mild asthmatics and 1.08 – 17.8% in severe asthmatics (Fig. 4E). These Type 2 ILCs (Lin−CD127+CRTH2+ cells) also expressed both ALX/FPR2 and CMKLR1 receptors (Fig 4F).
Fig. 4. ILC2 are present in peripheral blood of healthy and asthmatic subjects and express pro-resolving receptors.
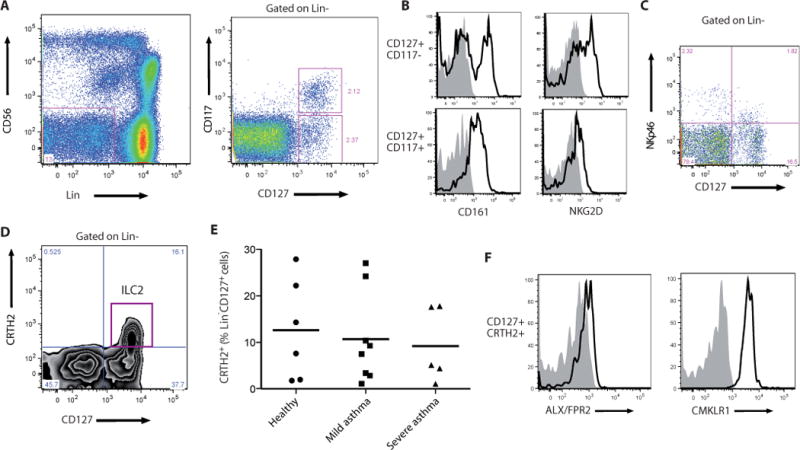
(A–D) Flow cytometry gating strategy for the identification of helper ILC and the ILC2 subset. PBMCs were depleted of most T and B lymphocytes and monocytes, and a lymphoid morphology cell population was selected based on their forward and side scatter characteristics. (A) Flow cytometry analysis showing the presence of a Lin− cell population (violet box, CD3−TCRαβ−TCRγδ−CD19−CD14−CD16−CD34−CD123−CD11c−) different from NK cells (Lin−CD56+), left panel; identification of a Lin−CD127+CD117+/− population, right panel. (B) Lin−CD127+CD117+/− expressed NK cell receptors, including CD161, left panel; NKG2D, right panel, and (C) NKp46. (D) ILC2 were identified as a subpopulation of Lin−CD127+ cells that also expressed CRTH2. (E) The number of Lin−CD127+CRTH2+ ILC2 cells was determined in healthy and mild and severe asthmatic subjects. (F) ILC2 also expressed the pro-resolving receptors ALX (left panel) and CMKLR1 (right panel) on their surface (see Methods).
IL-13 generation by ILC2 is induced by PGD2 and inhibited by Lipoxin A4
Distinct from NK cells, Lin−CD127+ circulating cells stimulated with PMA plus a divalent cation ionophore (A23187) produced bioactive quantities of IL-13 that were in the pg range (8.68 +/− 3.58 (SEM) pg IL-13/1000 cells) (Fig. 5A). IL-13 generation by helper ILCs is linked to ILC2 that express CRTH2 receptors (10). To determine the impact of PGD2, a ligand for CRTH2 (16), Lin−CD127+ circulating cells were incubated in the presence of PGD2 (100 nM) alone or in combination with select cytokines that can induce IL-13 release by ILC2 (10). Relative to control incubations with only vehicle, PGD2 significantly induced IL-13 production by 72.6% (Mean), which was quantitatively similar to the increase in IL-13 levels with exposure to IL-25 and IL-33 (54.6%, Mean). Of note, when ILCs were exposed to PGD2 in combination with IL-25 and IL-33, there was a synergistic increase in IL-13 generation by 500.7% (Mean) (3.9 +/− 2.4 (SEM) pg IL-13/1000 cells) (Fig. 5B).
Fig. 5. IL-13 production by peripheral blood ILC2 is enhanced by PGD2and inhibited by lipoxin A4.
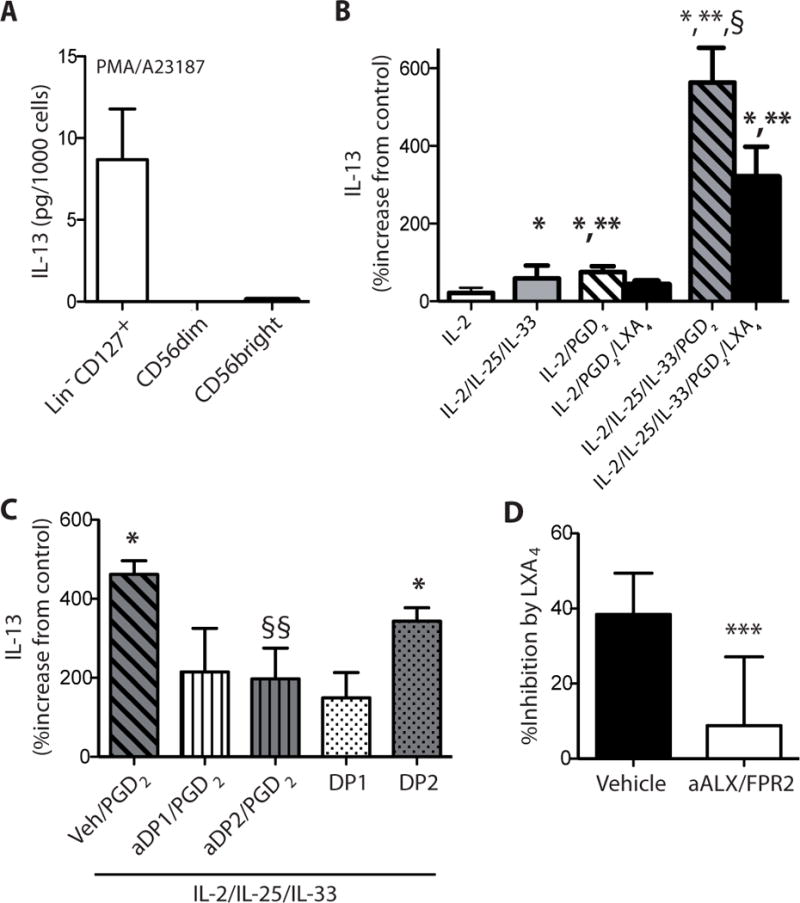
IL-13 was quantitated by ELISA in the cell-free supernatants from incubations with (A) Lin−CD127+ cells, CD56bright NK cells and CD56dim NK cells that were sorted by flow cytometry and cultured for 24h alone (Control) or with PMA (50 ng/ml) and A23187 (500 ng/ml). Results are expressed per 1,000 cells. (B) In separate incubations, IL-13 release was determined for Lin−CD127+ cells that were incubated (15 min, 37°C) with lipoxin A4 (100 nM) or vehicle (0.1% ethanol) prior to (24h, 37°C) media alone, IL-2 (2 ng/ml) (IL-2), IL-2 (2 ng/ml), IL-25 (50 ng/ml) and IL-33 (50 ng/ml) (IL-2/IL-25/IL-33), IL-2 (2 ng/ml) and PGD2 (100 nM) (IL2/PGD2), or IL-2 (2 ng/ml), IL-25 (50 ng/ml), IL-33 (50 ng/ml) and PGD2 (100 nM) (IL-2/IL-25/IL-33/PGD2). (C) To assess PGD2 receptor signaling, ILCs were incubated with IL-2/IL-25/IL-33/PGD2 in the presence of a DP1 receptor antagonist (BWA868C, aDP1) or DP2 receptor antagonist (BAYu3405, aDP2). Some cells were exposed to IL-2/IL-25/IL-33 in conjunction with a selective DP1 receptor agonist (BW245C, DP1) or DP2 receptor agonist (15(R)-methyl-PGD2, DP2) without PGD2. (D) To determine the role of ALX/FPR2 signaling, cells were exposed (15 min, 37°C) to the receptor antagonist WRW4 (aALX/FPR2) prior to LXA4 and IL-2/IL-25/IL-33/PGD2. Results are expressed as the percent increase from control (mean ± s.e.m.); n≥3 individual healthy donors; *, P < 0.01, compared with control; **, P < 0.05, compared with IL-2; §, P < 0.05, compared with IL-2/IL-25/IL-33, §§, P < 0.05, compared with IL-2/IL-25/IL-33/PGD2; ***, P < 0.05, compared with vehicle.
In addition to CRTH2, ILC2 expressed ALX/FPR2 receptors (Fig. 4F), so the impact of lipoxin A4 on IL-13 generation was next determined. The PGD2 and cytokine induced IL-13 production was significantly inhibited by equimolar amounts of lipoxin A4 (P< 0.05) (Fig. 5B). IL-13 generation triggered by PGD2 alone was decreased 42.5 +/− 3.9% by lipoxin A4 (Mean +/− SEM) and IL-13 levels after PGD2, IL-25 and IL-33 were decreased 42.7 +/− 4.4% by lipoxin A4 (Mean +/− SEM), suggesting a common mechanism for ILC2 regulation.
PGD2 can activate cells via either DP1 or DP2 (CRTH2) receptors, so Lin−CD127+ cells were exposed to PGD2 in the presence of either a DP1 receptor antagonist (BWA868C) or DP2 receptor antagonist (BAYu3405). Both antagonists partially inhibited IL-13 release by the ILCs (Fig. 5C); however, only inhibition with the DP2 antagonist reached statistical significance. To further explore PGD2 receptor-mediated ILC generation of IL-13, ILCs were exposed to DP1 and DP2 selective agonists. Relative to control incubations with only IL-2, IL-25 and IL-33, the DP2 agonist (15R-methyl-PGD2) but not the DP1 agonist (BW245C) led to significant increases in IL-13 release (Fig. 5C). Together, these findings suggest a dominant, but not exclusive role for the DP2 receptor in PGD2-mediated ILC2 activation. Of note, regulation of PGD2’s actions by LXA4 was significantly inhibited (30%) by an ALX/FPR2 receptor antagonist WRW4 (Fig. 5D). Together, the marked increase in IL-13 release with PGD2 and its regulation by lipoxin A4 indicate that ILC2 functional responses are selectively regulated by lipid mediators.
Innate Lymphoid Cells are present in human lung
To identify ILCs and related cells in lung tissue, samples of perfused human lung from a previously healthy individual were fixed and stained for immunofluorescence microscopy. Because unique surface markers for each of these cell types were not available, a combination surface markers was used to identify and distinguish helper ILCs (cKit+, CD161+, tryptase−), NK cells (cKit−CD161+tryptase−) and mast cells (cKit+, CD161−, tryptase+) (see Methods). NK cells were also positively identified by perforin. Sensitive and specific antibody for CRTH2 was not available for immunohistochemistry, so the ILC2 subset was not targeted in these experiments. Cells with the distinct immunofluorescence staining characteristics of helper ILCs and NK cells were present near medium and small airways (Fig. 6, Fig. S8). In addition, ILCs were in proximity of tissue mast cells in these anatomic locations (Fig. S8).
Fig. 6. Innate lymphoid cells are present in human lung.
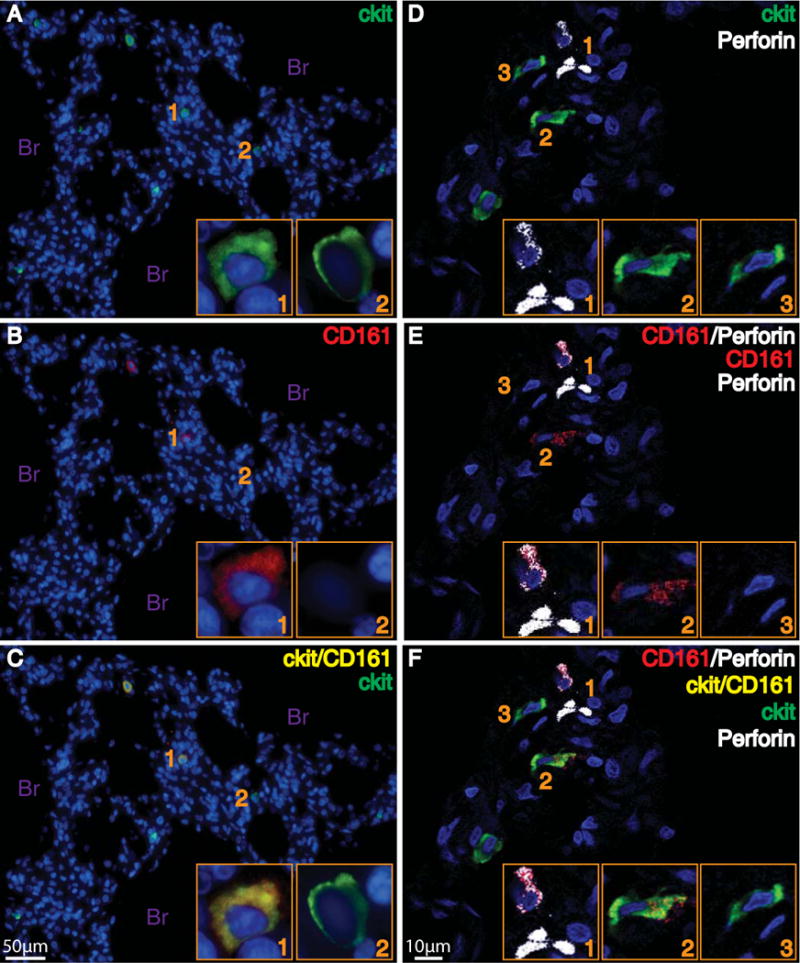
Immune cells were assessed in the distal lung using immunofluorescent staining with antibodies for cKit, CD161, and perforin to identify cells with the staining characteristics of ILC (cKit+, CD161+) and NK cells (cKit−, CD161+, perforin+). Cell nuclei were stained with DAPI (blue). Panels on the left show cells that stain positively for ckit (green, Panel A) and CD161 (red, Panel B). When images A and B are merged, cells stain for ckit and CD161 (yellow, Panel C) or ckit alone (green, Panel C). Light microscopy images taken with 20X objective, and 100X objective for inset. Panels on the right show cells that that stain positively for ckit (green, Panel D) or Perforin (white, Panel D). Panel E depicts cells that are positive for Perforin (white), CD161 (red), or both Perforin and CD161 (white and red). When images D and E are merged, cells are present in Panel F that stains for Perforin and CD161 (white and red), Perforin alone (white), ckit alone (green), or ckit and CD161 (yellow). Confocal images taken with 60X objective. Inset images provide higher power views of the cells, and correspond numerically with the lower power images. Br, bronchioles
Discussion
While evidence from murine model systems has uncovered critical roles for ILCs (NK cells and ILC2) in regulating allergic airway inflammation (9, 12, 17–19), only limited translational findings are available in human asthma. Here, total circulating NK cells were decreased in asthma. Relative to healthy subjects, the decrements were most significant in the CD56dim subset, which displays potent cytotoxic actions (7). In severe asthma, peripheral blood NK cells had significantly increased expression of CD69 and NKG2D, especially in CD56dim NK cells, indicating that the cells were activated. BALF NK cell subsets were in sharp contrast to the distribution of circulating NK cells and displayed a further decrease in the CD56dim subset. There is no prior information on NK cells in severe asthma, but asthmatics in general have increased in vitro NK cell activity (20, 21) that can be reduced by fluticasone propionate (22); however, severe asthma is defined, in part, as a corticosteroid refractory condition (23). Despite the use of high dose inhaled corticosteroids, samples from our subjects with severe asthma had circulating NK cells that were significantly activated. While peripheral blood NK cells were decreased here in asthma relative to healthy subjects, these findings do not preclude a substantial increase in NK cells during asthma exacerbations, which are often caused by viral infection. NK cells play important roles in anti-viral host defense (7) and NK cells transiently increase in PBMCs from asthmatic children during acute exacerbations (24). Together, these findings indicate that asthmatics, in particular severe asthmatics, have a relative decrease in the numbers of peripheral blood and lung cytotoxic NK cells (ie, CD56dim) and severe asthma is distinguished from milder asthma and health by increased NK cell activation (i.e., CD69 and NKG2D expression). This suggests a tissue specific and dynamic regulation of NK cell phenotype that is unrestrained in severe asthma.
While the peripheral blood numbers of CD56dim NK cells were significantly decreased in asthma relative to healthy subjects, their numbers were still significantly greater than CD56bright NK cells. In contrast to the relative abundance of CD56dim NK cells in peripheral blood, the majority of NK cells found in peripheral lymphoid tissue are CD56bright (25). Here, paired analyses of severe asthma NK cell subsets in peripheral blood and BALF also uncovered a marked switch in CD56 subsets with increased CD56bright and decreased CD56dim NK cells in the airway relative to the circulation. There are biological implications to this distribution, because, in general, the functional responses of these NK cell subsets are distinct - the CD56dim subset exhibits potent cytotoxicity and the CD56bright subset secretes large amounts of cytokines and chemokines but displays limited cytotoxic capacity (7). Affected joints from patients with rheumatoid arthritis also contain an enriched population of CD56bright NK cells (26, 27) with an activated phenotype (CD69+) (28), suggesting a common mechanism for NK cell tissue recruitment in chronic inflammation. Of interest, NK cells from healthy subjects obtained from BAL or lung tissue are profoundly impaired in their cytotoxic capacity (29, 30), likely reflecting an enriched CD56bright population of NK cells. Moreover, patients with sarcoidosis have increased CD56bright NK cells in their BALF without differences in peripheral blood (31). Accumulation of CD56bright NK cells to the lung or at sites of inflammation may result from selective recruitment, as CD56bright NK cells and CD56dim NK cells differ in their pattern of expression of chemokine receptors and adhesion molecules (32, 33). Evidence from rodent models of allergic asthma support an important role for selective NK cell homing to the lung and associated lymph nodes (9, 34). NK cell cytotoxicity is critical for the timely clearance of antigen-specific T cells in the lung (9), so decreased CD56dim NK cells would predispose to chronic airway inflammation.
Most human asthma phenotypes are characterized by increased eosinophils in peripheral blood, lung tissue and bone marrow and peripheral blood eosinophilia generally correlates with disease severity (35). Preclinical evidence links NK cells to eosinophilia. NK cell depletion in mice leads to persistent allergic airway inflammation with significantly increased numbers of antigen-specific T cells and eosinophils in BALF after allergen challenge (9) and cytokine-primed murine NK cells can suppress airway hyper-responsiveness and eosinophilia (36). When activated, eosinophils secrete an array of cytotoxic cationic proteins that can cause significant lung tissue damage, so induction of eosinophil apoptosis is crucial to avoid inadvertent tissue injury. Because granulocyte apoptosis can accelerate resolution of tissue inflammation (37), induction of eosinophil apoptosis would likely participate in the timely resolution of airway inflammation in controlled asthmatic responses. NK cells appear to be linked to eosinophil clearance, as peripheral eosinophilia in asthmatic subjects correlated with NK cell numbers and activation (ie, CD69, NKG2D). Similar to neutrophils (38), when NK cells from healthy subjects or mild asthmatic subjects were incubated ex vivo with autologous eosinophils, there was a significant increase in eosinophil apoptosis. Of interest, eosinophil apoptosis induced by severe asthma NK cells was significantly decreased despite their increased expression of CD69 and NKG2D. The lysosomal-associated membrane protein-1 (LAMP-1 or CD107a), a degranulation marker, was not upregulated on NK cells during co-incubations with granulocytes, suggesting that the NK cells triggered granulocyte apoptosis in a manner distinct from cytotoxicity. For neutrophils, NK cells utilize a caspase-dependant mechanism that is mediated by the activating NK cell receptor NKp46 and Fas pathway (38), and a similar mechanism is likely for eosinophils. Of note, a positive association between NK cells and eosinophils has also been observed in BALF from patients with eosinophilic pneumonias (39).
In addition to NK cells, ILC2s are innate lymphocytes that have been linked in murine studies to asthma pathobiology. Amongst the larger family of ILCs, ILC2 share a lymphoid morphology, lack specific antigen receptors and depend on the transcriptional repressor Id2 and the common γ–chain of the IL-2-receptor (6). In addition to their roles in protective immunity to helminths (6), ILC2s constitute a significant and early source of type 2 cytokines in several murine models of experimental asthma and are instrumental in mediating inflammatory aspects of airway hyperresponsiveness (12, 17–19). ILC2s can serve as an innate source of IL-13 that directly induces airway hyperresponsiveness in the absence of IL-13–producing CD4+ T cells (17). This ILC2 subset has been recently identified in humans in the gut, lung and blood of healthy individuals and was strikingly enriched in nasal polyps from patients with chronic rhinosinusitis, a typical type 2 mediated disease (10). Here, healthy and asthmatic subjects had CRTH2+ ILC2 in their peripheral blood, and cells with the staining characteristics of ILCs were also identified in human lung near medium and small airways in close proximity to cells with the staining pattern for NK cells and mast cells, suggesting that these cells interact in vivo. When activated by PMA and A23187, peripheral blood ILC2, but not NK cells, generated measureable levels of the type 2 cytokine IL-13. Located in the lung near mast cells and airway epithelial cells, the ILC2 released significant amounts of IL-13 in response to receptor-mediated, soluble stimuli from these cell types, namely PGD2 (mast cells) and IL-25 and IL-33 (airway epithelia). ILC2 express CRTH2 (DP2) and PGD2’s activation of IL-13 release by ILC2 was predominantly mediated via this receptor. As antigen independent sources of IL-13 in response to environmental cues from activated cells in the human airway, ILC2 are well poised to participate in the genesis of asthmatic responses, including eosinophilic inflammation and airway hyperresponsiveness.
Allergic airway responses, including eosinophilic inflammation, airway hyperresponsiveness and mucus metaplasia, are regulated by select members of a new genus of anti-inflammatory and pro-resolving mediators, including lipoxin A4 and resolvin E1 (14, 40). Lipoxin A4 is enzymatically derived from arachidonic acid and serves as an agonist at ALX/FPR2 receptors to mediate cell type-specific actions in acute inflammatory responses, including inhibition of neutrophil activation, promotion of macrophage engulfment of apoptotic neutrophils and regulation of epithelial cell cytokine release (41–44). Resolvin E1 is enzymatically derived from the omega-3 fatty acid eicosapentaenoic acid and serves as a pro-resolving agonist at CMKLR1 receptors (15), including for the resolution of airway inflammation in a murine model of allergic asthma (40). CMKLR1 also serves as a receptor for chemerin, a chemotactic protein found in inflammatory exudates (45) and implicated in the colocalization of NK cells and DC subsets in pathologic peripheral tissues (46). Lipoxin A4 is present in asthmatic lung (47), and when administered to asthmatic subjects via nebulization, lipoxin A4 prevents LTC4-mediated bronchoconstriction (48). In addition, treatment of the allergic condition infantile eczema with topical 15(R/S)-methyl-lipoxin A4 significantly decreases eczema severity and duration and improved patients’ quality of life, with similar efficacy as topical corticosteroids (49). Bioactive lipoxin A4 stable analogs have been prepared that in experimental asthma, block both airway hyper-responsiveness and allergic inflammation (14). Eosinophil trafficking and tissue accumulation are markedly inhibited by lipoxins (14, 50, 51), and lipoxin A4 decreases NK cell cytotoxicity in vitro (52, 53). Severe asthma is characterized by a defect in lipoxin biosynthesis (4, 5), suggesting that the chronic inflammatory airway responses in asthma may stem, in part, from inadequate counter-regulation. NK cells and ILC2 expressed the pro-resolving receptors ALX/FPR2 and CMKLR1, and NK cells from subjects with severe asthma had increased ALX/FPR2 expression. In nanomolar concentrations and in an ALX/FPR2-dependent manner, lipoxin A4 decreased release of IL-13 by ILC2 and increased NK cell mediated eosinophil apoptosis. These properties of lipoxin A4 are consistent with potent anti-inflammatory (ILC2) and pro-resolving (NK cell) actions on ILCs and highlight a putative mechanism for the pathogenesis of severe asthma that links defective lipoxin A4 generation to the increased eosinophilia and chronic airway inflammation that characterize the disease.
The peripheral blood characteristics of ILCs (NK cells, ILC2s) identified here may serve as useful biomarkers of asthma control and severity; however, there are potential limitations to note. Because type 2 ILCs are a rare cell population, an enrichment step was required that limited their accurate enumeration and characterization and restricted the cell numbers available for functional assay. While type 2 ILCs were detectable in peripheral blood and lung tissue, they were not found in aliquots of BALF and it was not feasible to obtain lung samples from asthmatic subjects. NK cells were more abundant, but the pool of peripheral blood NK cells is heterogeneous and dynamically regulated by several environmental triggers, most notably viral infection. In addition, in this mechanistic study there was in depth analysis of a relatively small sample size. Because of asthma’s syndromic nature with individual variation in severity and medication use, the differences identified in ILCs between severe and mild asthma will require validation in larger clinical cohorts.
In conclusion, NK cells and ILC2 were present in human peripheral blood and lung from healthy and asthmatic subjects. In subjects with severe asthma, the circulating NK cells were highly activated and associated with peripheral eosinophilia. During coincubations simulating cell-cell interactions in inflamed tissue, NK cells interacted with autologous eosinophils and neutrophils to promote their apoptosis. In addition to the airway epithelial cytokines IL-25 and IL-33, PGD2 induced the release of IL-13 by CRTH2+ILC2. Both NK cells and ILC2 expressed ALX/FPR2 receptors, which were increased in severe asthma, and the natural ALX/FPR2 ligand lipoxin A4 displayed potent anti-inflammatory and pro-resolving actions; decreasing PGD2 and cytokine mediated IL-13 release by ILC2 and increasing NK cell mediated eosinophil apoptosis. Together, these findings assign critical roles to ILCs in asthma pathobiology and identify new cellular targets and mechanisms for endogenous counter-regulatory mediators. The anti-inflammatory and pro-resolving actions for lipoxin A4 also suggest a potential new therapeutic strategy in asthma and allergic diseases that emphasizes this natural resolution pathway and ILCs.
Material and Methods
Patients and samples
After approval by the Partners Healthcare institutional review board and written informed consent was obtained, subjects with severe or mild asthma were recruited by the Brigham and Women’s Hospital Asthma Research Center using criteria defined by the Severe Asthma Research Program (23). Healthy control subjects were in good health, denied a history of atopy, asthma or other chronic medical illness and had taken no prescription or over-the-counter medications within two weeks prior to enrollment. Heparinized peripheral whole blood was collected by venipuncture from severe asthmatics (n=11), mild asthmatics (n=11) and healthy subjects (n=12). Paired samples of peripheral blood and bronchoalveolar lavage fluid (BALF) were obtained from 5 severe asthmatics. Peripheral blood mononuclear cells (PBMC) and granulocytes were isolated by density-gradient centrifugation over Histopaque 1077 (Sigma Aldrich) and Histopaque 1119 (Sigma Aldrich). Residual erythrocytes were lysed by incubating the cell pellet with a small volume of distilled water for 20 s followed by quenching with a large volume of isotonic PBS (as in (54)).
Media and reagents
RPMI-1640 (Lonza) supplemented with 2 mM L-glutamine, and 10% heat-inactivated fetal calf serum (FCS) (all from Gibco BRL) was used as media for NK cell culture with granulocytes (4h, 37°C, 5% CO2). For longer (24h) incubations, this medium was supplemented with 100 IU/mL penicillin and 100 μg/ml streptomycin. Lipoxin A4, PGD2, 15(R)-methyl-PGD2, BW 245C, BW A868C and BAY-u3405 were purchased from Cayman Chemical. WRW4 was purchased from Tocris Bioscience. Recombinant human (rh) IL-2, rhIL-25 and rhIL-33 were purchased from R&D Systems. Phorbol 12-myristate 13-acetate (PMA) and the divalent cation ionophore A23187 were obtained from Sigma.
Flow cytometry analysis and sorting
The following antibodies to human proteins were used: anti-CD3 (SK7), anti-CD56 (CMSSB), anti-CD16 (CB16), anti-CD69 (FN50), anti-CD161 (HP-3G10), anti-CD11c (3.9), anti-human-CD123 (6H6), anti-CD19 (HIB19), anti-CD14 (61D3), anti-alpha beta TCR (IP26), anti-gamma delta TCR (B1.1), mouse IgG1 kappa isotype control (P3.6.2.8.1) (all from eBioscience; anti-CD34 (581), Annexin V and 7 AAD (from BD Pharmingen; anti-CD335 (NKp46) (9E2), anti)NKG2D (1D11), anti-CD117 (104D2), anti-CD127 (A019D5), anti-CD294 (CRTH2) (BM16), mouse IgG1 kappa isotype control (MOPC 21), Rat IgG2a kappa isotype control (RTK2758) (from Biolegend); anti-ChemR23 (84939), anti-FPRL1 (304405), mouse IgG3 isotype control (133319) and mouse IgG2b isotype control (133303) (from R & D Systems).
For analyses of cell phenotype, data were acquired on a Canto II flow cytometer (Becton Dickinson Bioscience) and were analyzed with FlowJo software (Tree Star). NK cells were identified in a PBMC suspension or in BALF samples as a cell population with a lymphoid morphology based on forward and side scatter characteristics expressing NKp46 and lacking CD3. 20,000 events of NK cells were acquired and results were expressed as a percentage of the total lymphocyte population. If required, cells were preincubated with blocking mAb (Purified Human IgG, R & D Systems) before specific staining. For ILC phenotyping, PBMCs were first depleted of most T and B lymphocytes and monocytes by labeling with FITC conjugated anti-CD3, anti-CD14 and anti-CD19 (eBioscience) plus anti-FITC magnetic beads (Easy Sep, StemCell Technology) and further identified among a cell population with a lymphoid morphology based on forward and side scatter characteristics. For compensation control, BD CompBeads (BD Pharmingen) were used and appropriate isotype negative controls were used to define background staining.
For flow cytometry sorting, peripheral blood mononuclear cells were depleted of most of T cells, B cells and monocytes by labeling with FITC conjugated anti-CD3, anti-CD14 and anti-CD19 (eBioscience) plus anti-FITC magnetic beads (Easy Sep, StemCell Technology). Remaining cells were labeled with FITC-conjugated anti-CD3, TCR-αβ, TCR-γδ, CD14, CD19, CD16, CD123, CD11c, CD34, CD56 and CD127. Lin−(CD3−TCR-αβ−TCR-γδ−CD14−CD19−CD16−CD123−CD11c−CD34−)CD127+ ILCs, Lin−CD56bright NK cells and Lin−CD56dim NK cells were sorted with an Aria SORP flow cytometer (Becton Dickinson Bioscience) to a purity of ≥96%.
NK cells and granulocyte incubations
Peripheral blood NK cells were isolated from PBMCs using a NK cell isolation kit (EasySep, Stem Cell Technologies). Briefly, non-NK cells (i.e., T cells, B cells, stem cells, DCs, monocytes, granulocytes, and erythroid cells) were magnetically labeled and depleted using a magnet. The purity of isolated NK cells was assessed by flow cytometric analysis of cells stained against CD3 and NKp46. The purity of isolated NK cells was > 96%.
Autologous human granulocytes were freshly isolated from peripheral whole blood, resuspended in RPMI 1640 medium, supplemented with 10% heat inactivated FCS, 2 mM L-glutamine and incubated in the absence of presence of NK cells (1:5 ratio of granulocytes:NK cells) (4h, 37°C, 5% CO2) and assayed by FACS using Annexin V and 7-AAD (BD Pharmingen) to identify and distinguish cells undergoing apoptosis from necrosis. Neutrophils and eosinophils were gated based on FSC and SSC characteristics, and eosinophils (CD16−) were also distinguished from neutrophils (CD16+) by immunostaining. In some experiments, cells were selectively exposed (15 min, 37°C) to lipoxin A4 (100 nM) or vehicle (0.1% ethanol) prior to co-incubation. To assess ALX/FPR2-mediated signaling, some NK cells were exposed to the receptor antagonist WRW4 (230 nM, IC50) prior to lipoxin A4 (100 nM).
ILC cultures and IL-13 release
In some subjects, Lin−CD127+, CD56bright NK cells and CD56dim NK cells were isolated from whole blood by flow cytometry (see above) and cultured for 24h (37°C, 5% CO2) in the presence of vehicle (0.1% ethanol) or soluble stimuli, including PMA (50 ng/ml) and calcium ionophore A23187 (500 ng/ml) (PMA/A2), IL-2 alone (2 ng/ml) (IL-2), IL-2 (2 ng/ml), IL-25 (50 ng/ml) and IL-33 (50 ng/ml) (IL-2/IL-25/IL-33), IL-2 (2 ng/ml) and PGD2 (100 nM) (IL2/PGD2), or IL-2 (2 ng/ml), IL-25 (50 ng/ml), IL-33 (50 ng/ml) and PGD2 (100 nM) (IL-2/IL-25/IL-33/PGD2). In select experiments, pharmacological agents were used to investigate receptor dependency, including BWA868C (5 nM, DP1 IC50), BAY-u3405 (100 nM, CRTH2 (DP2) IC50), BW245C (2.5 nM, DP1 EC50), 15(R)-methyl-PGD2 (2.5 nM, DP2 EC50) and WRW4 (230 nM, ALX/FPR2 IC50). IL-13 was measured in the collected cell culture supernatants by ELISA (eBioscience).
Immunofluorescent identification of ILCs and mast cells in human lung
Healthy human lungs that were not used for transplantation, and had their vasculature perfused to remove the blood, were fixed in 10% buffered formalin and paraffin embedded for immunofluorescence staining. For ckit/CD161 double staining, the 5 um sections were incubated with a ckit antibody (1:100, Anti-human CD117 polyclonal rabbit IgG, Dako cat#A4502) and a CD161 antibody (1:10, anti-human CD161 monoclonal IgG, Abcam cat#ab23624) at 4°C overnight, followed by incubation with secondary antibodies of rabbit IgG conjugated with FITC (1:50) and mouse IgG conjugated with TRITC (1:30), respectively, at 37°C for 1h. For ckit/CD161/perforin triple staining, after the ckit/CD161 staining was complete, the sections were incubated with a perforin antibody (1:20, Anti-human perforin monoclonal IgG, eBioscience cat#14-9994-80) at 37°C for 2h, following by incubation with secondary anti-mouse antibody conjugated with far-red (1:30 for 1h). For ckit/tryptase staining, after the ckit staining was complete, the sections were incubated with goat anti-human tryptase antibody (1:30, Santa Cruz sc-17039), followed by incubation with an anti-goat IgG conjugated with far-red (1:30) for 1h. Stained slides were photographed by immunofluorescence microscope or confocal microscopy.
Statistical analysis
Data are shown as mean ± SEM. The statistical significance of differences was assessed by two-tailed Student’s t-test or one-way analysis of variance (ANOVA) followed by post-hoc tests to determine the levels of significance for each group. Correlations were evaluated by Spearman or Pearson’s correlation coefficient (r). A p-value <0.05 was considered significant.
Supplementary Material
Acknowledgments
The work has been funded in part by AI068084 (BDL), HL107166 (BDL, EI), HL109172 (BDL, EI), HL102225 (EI) and Fonds de dotation “Recherche en Santé Respiratoire” 2011 (CB).
Footnotes
Author contributions: C.B. designed the study, performed experiments, collected and analyzed data, and wrote the manuscript; M.C. provided conceptual advice and edited the manuscript, S.D., S.K., M.E.W. and E.I. provided samples from asthma patients and edited the manuscript; X.L. and M.P. performed experiments, collected and analyzed data, and edited the manuscript; B.D.L. designed the study, performed experiments, analyzed data, and wrote the manuscript.
Competing interest:
BDL is a co-inventor on patents related to lipoxin A4 and asthma that have been licensed by the Brigham and Women’s Hospital (BWH) for clinical development and he receives a share of licensing income through BWH. None of the other authors declare any financial competing interests.
References and Notes
- 1.Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8:183–192. doi: 10.1038/nri2254. [DOI] [PubMed] [Google Scholar]
- 2.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. 2007;25:101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- 4.Planaguma A, Kazani S, Marigowda G, Haworth O, Mariani TJ, Israel E, Bleecker ER, Curran-Everett D, Erzurum SC, Calhoun WJ, Castro M, Chung KF, Gaston B, Jarjour NN, Busse WW, Wenzel SE, Levy BD. Airway lipoxin A4 generation and lipoxin A4 receptor expression are decreased in severe asthma. Am J Respir Crit Care Med. 2008;178:574–582. doi: 10.1164/rccm.200801-061OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy BD, Bonnans C, Silverman ES, Palmer LJ, Marigowda G, Israel E. Diminished lipoxin biosynthesis in severe asthma. Am J Respir Crit Care Med. 2005;172:824–830. doi: 10.1164/rccm.200410-1413OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nature Immunology. 2011;12:21–27. doi: 10.1038/ni.1962. [DOI] [PubMed] [Google Scholar]
- 7.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schleinitz N, Vely F, Harle JR, Vivier E. Natural killer cells in human autoimmune diseases. Immunology. 2010;131:451–458. doi: 10.1111/j.1365-2567.2010.03360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haworth O, Cernadas M, Levy BD. NK cells are effectors for resolvin E1 in the timely resolution of allergic airway inflammation. J Immunol. 2011;186:6129–6135. doi: 10.4049/jimmunol.1004007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, Fokkens WJ, Cupedo T, Spits H. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12:1055–1062. doi: 10.1038/ni.2104. [DOI] [PubMed] [Google Scholar]
- 11.Liang HE, Reinhardt RL, Bando JK, Sullivan BM, Ho IC, Locksley RM. Divergent expression patterns of IL-4 and IL-13 define unique functions in allergic immunity. Nat Immunol. 2012;13:58–66. doi: 10.1038/ni.2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, Dekruyff RH, Umetsu DT. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12:631–638. doi: 10.1038/ni.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. 2003;3:781–790. doi: 10.1038/nri1199. [DOI] [PubMed] [Google Scholar]
- 14.Levy BD, De Sanctis GT, Devchand PR, Kim E, Ackerman K, Schmidt BA, Szczeklik W, Drazen JM, Serhan CN. Multi-pronged inhibition of airway hyper-responsiveness and inflammation by lipoxin A(4) Nat Med. 2002;8:1018–1023. doi: 10.1038/nm748. [DOI] [PubMed] [Google Scholar]
- 15.Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, Yang R, Petasis NA, Serhan CN. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med. 2005;201:713–722. doi: 10.1084/jem.20042031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, Ichimasa M, Sugamura K, Nakamura M, Takano S, Nagata K. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med. 2001;193:255–261. doi: 10.1084/jem.193.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, McKenzie AN. Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol. 2012;129:191–198. e191–194. doi: 10.1016/j.jaci.2011.09.041. [DOI] [PubMed] [Google Scholar]
- 18.Kim HY, Chang YJ, Subramanian S, Lee HH, Albacker LA, Matangkasombut P, Savage PB, McKenzie AN, Smith DE, Rottman JB, DeKruyff RH, Umetsu DT. Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. J Allergy Clin Immunol. 2012;129:216–227. e211–216. doi: 10.1016/j.jaci.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolterink RG, Kleinjan A, van Nimwegen M, Bergen I, de Bruijn M, Levani Y, Hendriks RW. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur J Immunol. 2012;42:1106–1116. doi: 10.1002/eji.201142018. [DOI] [PubMed] [Google Scholar]
- 20.Jira M, Antosova E, Vondra V, Strejcek J, Mazakova H, Prazakova J. Natural killer and interleukin-2 induced cytotoxicity in asthmatics. I. Effect of acute antigen-specific challenge. Allergy. 1988;43:294–298. doi: 10.1111/j.1398-9995.1988.tb00903.x. [DOI] [PubMed] [Google Scholar]
- 21.Timonen T, Stenius-Aarniala B. Natural killer cell activity in asthma. Clin Exp Immunol. 1985;59:85–90. [PMC free article] [PubMed] [Google Scholar]
- 22.Di Lorenzo G, Esposito Pellitteri M, Drago A, Di Blasi P, Candore G, Balistreri C, Listi F, Caruso C. Effects of in vitro treatment with fluticasone propionate on natural killer and lymphokine-induced killer activity in asthmatic and healthy individuals. Allergy. 2001;56:323–327. doi: 10.1034/j.1398-9995.2001.00879.x. [DOI] [PubMed] [Google Scholar]
- 23.Moore WC, Bleecker ER, Curran-Everett D, Erzurum SC, Ameredes BT, Bacharier L, Calhoun WJ, Castro M, Chung KF, Clark MP, Dweik RA, Fitzpatrick AM, Gaston B, Hew M, Hussain I, Jarjour NN, Israel E, Levy BD, Murphy JR, Peters SP, Teague WG, Meyers DA, Busse WW, Wenzel SE, L. B. I. s. S. A. R. P. National Heart Characterization of the severe asthma phenotype by the National Heart, Lung, and Blood Institute’s Severe Asthma Research Program. J Allergy Clin Immunol. 2007;119:405–413. doi: 10.1016/j.jaci.2006.11.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin SJ, Chang LY, Yan DC, Huang YJ, Lin TJ, Lin TY. Decreased intercellular adhesion molecule-1 (CD54) and L-selectin (CD62L) expression on peripheral blood natural killer cells in asthmatic children with acute exacerbation. Allergy. 2003;58:67–71. doi: 10.1034/j.1398-9995.2003.t01-1-23697.x. [DOI] [PubMed] [Google Scholar]
- 25.Fehniger TA, Cooper MA, Nuovo GJ, Cella M, Facchetti F, Colonna M, Caligiuri MA. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell-derived IL-2: a potential new link between adaptive and innate immunity. Blood. 2003;101:3052–3057. doi: 10.1182/blood-2002-09-2876. [DOI] [PubMed] [Google Scholar]
- 26.Dalbeth N, Callan MF. A subset of natural killer cells is greatly expanded within inflamed joints. Arthritis Rheum. 2002;46:1763–1772. doi: 10.1002/art.10410. [DOI] [PubMed] [Google Scholar]
- 27.Pridgeon C, Lennon GP, Pazmany L, Thompson RN, Christmas SE, Moots RJ. Natural killer cells in the synovial fluid of rheumatoid arthritis patients exhibit a CD56bright, CD94bright, CD158negative phenotype. Rheumatology (Oxford) 2003;42:870–878. doi: 10.1093/rheumatology/keg240. [DOI] [PubMed] [Google Scholar]
- 28.Dalbeth N, Gundle R, Davies RJ, Lee YC, McMichael AJ, Callan MF. CD56bright NK cells are enriched at inflammatory sites and can engage with monocytes in a reciprocal program of activation. J Immunol. 2004;173:6418–6426. doi: 10.4049/jimmunol.173.10.6418. [DOI] [PubMed] [Google Scholar]
- 29.Robinson BW, Pinkston P, Crystal RG. Natural killer cells are present in the normal human lung but are functionally impotent. J Clin Invest. 1984;74:942–950. doi: 10.1172/JCI111513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bordignon C, Villa F, Vecchi A, Giavazzi R, Introna M, Avallone R, Mantovani A. Natural cytotoxic activity in human lungs. Clin Exp Immunol. 1982;47:437–444. [PMC free article] [PubMed] [Google Scholar]
- 31.Katchar K, Soderstrom K, Wahlstrom J, Eklund A, Grunewald J. Characterisation of natural killer cells and CD56+ T-cells in sarcoidosis patients. Eur Respir J. 2005;26:77–85. doi: 10.1183/09031936.05.00030805. [DOI] [PubMed] [Google Scholar]
- 32.Campbell JJ, Qin S, Unutmaz D, Soler D, Murphy KE, Hodge MR, Wu L, Butcher EC. Unique subpopulations of CD56+ NK and NK-T peripheral blood lymphocytes identified by chemokine receptor expression repertoire. J Immunol. 2001;166:6477–6482. doi: 10.4049/jimmunol.166.11.6477. [DOI] [PubMed] [Google Scholar]
- 33.Frey M, Packianathan NB, Fehniger TA, Ross ME, Wang WC, Stewart CC, Caligiuri MA, Evans SS. Differential expression and function of L-selectin on CD56bright and CD56dim natural killer cell subsets. J Immunol. 1998;161:400–408. [PubMed] [Google Scholar]
- 34.Schuster M, Tschernig T, Krug N, Pabst R. Lymphocytes migrate from the blood into the bronchoalveolar lavage and lung parenchyma in the asthma model of the brown Norway rat. Am J Respir Crit Care Med. 2000;161:558–566. doi: 10.1164/ajrccm.161.2.9812021. [DOI] [PubMed] [Google Scholar]
- 35.Fanta CH. Asthma. N Engl J Med. 2009;360:1002–1014. doi: 10.1056/NEJMra0804579. [DOI] [PubMed] [Google Scholar]
- 36.Matsubara S, Takeda K, Kodama T, Joetham A, Miyahara N, Koya T, Swasey CH, Okamoto M, Dakhama A, Gelfand EW. IL-2 and IL-18 attenuation of airway hyperresponsiveness requires STAT4, IFN-gamma, and natural killer cells. Am J Respir Cell Mol Biol. 2007;36:324–332. doi: 10.1165/rcmb.2006-0231OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA, Caldicott A, Martinez-Losa M, Walker TR, Duffin R, Gray M, Crescenzi E, Martin MC, Brady HJ, Savill JS, Dransfield I, Haslett C. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med. 2006;12:1056–1064. doi: 10.1038/nm1468. [DOI] [PubMed] [Google Scholar]
- 38.Thoren FB, Riise RE, Ousback J, Della Chiesa M, Alsterholm M, Marcenaro E, Pesce S, Prato C, Cantoni C, Bylund J, Moretta L, Moretta A. Human NK Cells induce neutrophil apoptosis via an NKp46- and Fas-dependent mechanism. J Immunol. 2012;188:1668–1674. doi: 10.4049/jimmunol.1102002. [DOI] [PubMed] [Google Scholar]
- 39.Papakosta D, Manika K, Kyriazis G, Kontakiotis T, Gioulekas D, Polyzoni T, Bouros D, Patakas D. Bronchoalveolar lavage fluid eosinophils are correlated to natural killer cells in eosinophilic pneumonias. Respiration. 2009;78:177–184. doi: 10.1159/000203989. [DOI] [PubMed] [Google Scholar]
- 40.Haworth O, Cernadas M, Yang R, Serhan CN, Levy BD. Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation. Nat Immunol. 2008;9:873–879. doi: 10.1038/ni.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonnans C, Fukunaga K, Levy MA, Levy BD. Lipoxin A(4) regulates bronchial epithelial cell responses to acid injury. American Journal of Pathology. 2006;168:1064–1072. doi: 10.2353/ajpath.2006.051056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Filep JG, Zouki C, Petasis NA, Hachicha M, Serhan CN. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 modulate adhesion molecule expression on human leukocytes in whole blood and inhibit neutrophil-endothelial cell adhesion. Advances in Experimental Medicine & Biology. 2002;507:223–228. doi: 10.1007/978-1-4615-0193-0_34. [DOI] [PubMed] [Google Scholar]
- 43.Godson C, Mitchell S, Harvey K, Petasis NA, Hogg N, Brady HR. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. Journal of Immunology. 2000;164:1663–1667. doi: 10.4049/jimmunol.164.4.1663. [DOI] [PubMed] [Google Scholar]
- 44.Morris T, Stables M, Colville-Nash P, Newson J, Bellingan G, de Souza PM, Gilroy DW. Dichotomy in duration and severity of acute inflammatory responses in humans arising from differentially expressed proresolution pathways. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:8842–8847. doi: 10.1073/pnas.1000373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wittamer V, Franssen JD, Vulcano M, Mirjolet JF, Le Poul E, Migeotte I, Brezillon S, Tyldesley R, Blanpain C, Detheux M, Mantovani A, Sozzani S, Vassart G, Parmentier M, Communi D. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J Exp Med. 2003;198:977–985. doi: 10.1084/jem.20030382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parolini S, Santoro A, Marcenaro E, Luini W, Massardi L, Facchetti F, Communi D, Parmentier M, Majorana A, Sironi M, Tabellini G, Moretta A, Sozzani S. The role of chemerin in the colocalization of NK and dendritic cell subsets into inflamed tissues. Blood. 2007;109:3625–3632. doi: 10.1182/blood-2006-08-038844. [DOI] [PubMed] [Google Scholar]
- 47.Lee TH, Crea AE, Gant V, Spur BW, Marron BE, Nicolaou KC, Reardon E, Brezinski M, Serhan CN. Identification of lipoxin A4 and its relationship to the sulfidopeptide leukotrienes C4, D4, and E4 in the bronchoalveolar lavage fluids obtained from patients with selected pulmonary diseases. American Review of Respiratory Disease. 1990;141:1453–1458. doi: 10.1164/ajrccm/141.6.1453. [DOI] [PubMed] [Google Scholar]
- 48.Christie PE, Spur BW, Lee TH. The effects of lipoxin A4 on airway responses in asthmatic subjects. Am Rev Respir Dis. 1992;145:1281–1284. doi: 10.1164/ajrccm/145.6.1281. [DOI] [PubMed] [Google Scholar]
- 49.Wu SH, Chen XQ, Liu B, Wu HJ, Dong L. Efficacy and Safety of 15(R/S)-Methyl-Lipoxin A4 in Topical Treatment of Infantile Eczema. British Journal of Dermatology. 2012 doi: 10.1111/j.1365-2133.2012.11177.x. [DOI] [PubMed] [Google Scholar]
- 50.Bandeira-Melo C, Bozza PT, Diaz BL, Cordeiro RS, Jose PJ, Martins MA, Serhan CN. Cutting edge: lipoxin (LX) A4 and aspirin-triggered 15-epi-LXA4 block allergen-induced eosinophil trafficking. J Immunol. 2000;164:2267–2271. doi: 10.4049/jimmunol.164.5.2267. [DOI] [PubMed] [Google Scholar]
- 51.Soyombo O, Spur BW, Lee TH. Effects of lipoxin A4 on chemotaxis and degranulation of human eosinophils stimulated by platelet-activating factor and N-formyl-L-methionyl-L-leucyl-L-phenylalanine. Allergy. 1994;49:230–234. doi: 10.1111/j.1398-9995.1994.tb02654.x. [DOI] [PubMed] [Google Scholar]
- 52.Ramstedt U, Serhan CN, Nicolaou KC, Webber SE, Wigzell H, Samuelsson B. Lipoxin A-induced inhibition of human natural killer cell cytotoxicity: studies on stereospecificity of inhibition and mode of action. J Immunol. 1987;138:266–270. [PubMed] [Google Scholar]
- 53.Ramstedt U, Ng J, Wigzell H, Serhan CN, Samuelsson B. Action of novel eicosanoids lipoxin A and B on human natural killer cell cytotoxicity: effects on intracellular cAMP and target cell binding. J Immunol. 1985;135:3434–3438. [PubMed] [Google Scholar]
- 54.Munoz NM, Leff AR. Highly purified selective isolation of eosinophils from human peripheral blood by negative immunomagnetic selection. Nat Protoc. 2006;1:2613–2620. doi: 10.1038/nprot.2006.340. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.