

Abstract
The β2 adrenergic receptor (β2-AR) has become a model system for studying the ligand recognition process and mechanism of the G protein coupled receptors activation. In the present study stereoisomers of fenoterol and some of its derivatives (N = 94 molecules) were used as molecular probes to identify differences in stereo-recognition interactions between β2-AR and structurally similar agonists. The present study aimed at determining the 3D molecular models of the fenoterol derivative-β2-AR complexes. Molecular models of β2-AR have been developed by using the crystal structure of the human β2-AR T4 lysozyme fusion protein with bound (S)-carazolol (PDB ID: 2RH1) and more recently reported structure of a nanobody-stabilized active state of the β2-AR with the bound full agonist BI-167107 (PDB ID: 3P0G). The docking procedure allowed us to study the similarities and differences in the recognition binding site(s) for tested ligands. The agonist molecules occupied the same binding region, between TM III, TM V, TM VI and TM VII. The residues identified by us during docking procedure (Ser203, Ser207, Asp113, Lys305, Asn312, Tyr308, Asp192) were experimentally indicated in functional and biophysical studies as being very important for the agonist-receptor interactions. Moreover, the additional space, an extension of the orthosteric pocket, was identified and described. Furthermore, the molecular dynamics simulations were used to study the molecular mechanism of interaction between ligands ((R,R’)- and (S,S’)-fenoterol) and β2-AR. Our research offers new insights into the ligand stereoselective interaction with one of the most important GPCR member. This study may also facilitate the design of improved selective medications, which can be used to treat, prevent and control heart failure symptoms.
Electronic supplementary material
The online version of this article (doi:10.1007/s00894-013-1981-y) contains supplementary material, which is available to authorized users.
Keywords: β2 adrenergic receptor, GPCRs, Docking, Fenoterol, Molecular dynamics, Stereoisomers
Introduction
Fenoterol (compound 1, Table 1) is a selective full agonist of the β2-adrenergic receptor (β2-AR). The preliminary studies showed that stereoisomers of the fenoterol (Fig. 1) differ significantly in their effect on the adrenergic system [1]. Several years ago, adopting the concept of selective optimization of side activities (the SOSA approach) [2] a medicinal chemistry project was initiated for development of β2-AR selective agonist based on the template of compound 1 which, combined with selective β1-AR antagonist, could be used for the treatment of specific cardiovascular condition, namely, congestive heart failure [3]. During the course of this research more than 90 compounds were designed and synthesized with various modification at the aminoalkyl tail of the fenoterol molecule and different stereochemical configurations [1, 4–6]. These compounds, presented in Table 1, show highly diverse binding affinities (pK i exp.) and functional activities toward β2-AR, depending on the constitution and stereoconfiguration of their molecules. Generally, among the different structural analogs of 1, (R,R’)-isomers are the most active and (S,S’)-isomers are the least active while derivatives in (R,S’)- and (S,R’)- configurations show moderate activities. Apart from the study focused on the stereoselectivity, the influence of the R1 and R2 substituents (Fig. 1) on the ligand-receptor interaction was investigated. This also showed the results demonstrated that the most active among fenoterol derivatives are (R,R’)-isomers of such compounds as: 1, 2, 5, and 54, although their relative affinity/activity measures varied significantly depending on the assay used [1, 4]. In our previous computational studies, the ligand–receptor interactions were modeled using the 3D-QSAR approach. The CoMFA (comparative molecular field analysis) models achieved very good statistics (for details see ref [4]) and, thus, were further used to design a series of new molecules and to predict their pK i values in silico [1, 4]. Detailed experimental characterization of these newly designed compounds, revealed that one of these compounds, (R,R’)-54 was one of the most β2 selective agonists ever reported with its selectivity ratio K i (β1-AR)/K i(β2-AR) = 573 [4].
Table 1.
The MolDock score values obtained during docking of fenoterol stereoisomers and its derivatives to In_β2-AR and Ac_β2-AR
Fig. 1.

The structures of fenoterol stereoisomers (compound 1) and the molecular template representing the fenoterol derivatives
In the current study we use the molecular modeling approach based on the knowledge of the molecular structure of the β2 adrenergic receptor. The target, β2-AR, belongs to the class A of G-protein-coupled receptors (GPCRs) family and is one of the best characterized in terms of molecular structure and function [7]. In recent years a series of crystal structures of β2-AR were obtained. At first, the human β2-AR was crystallized in complex with an inverse agonist, (S)-carazolol (PDB ID: 2RH1) [8]. This structure was followed by the reports of the β2-AR co-crystallized with other inverse agonists or antagonists including alprenolol and ICI-118,551 [10] or timolol [9]. All these structures reveal conserved mode of binding of antagonist and inverse agonists and are thought to represent the inactive state of the receptor. β2-AR co-crystalized in the complex with covalently bound irreversible agonist resulted in a structure showing significant similarities to an inactive conformation stabilized by an antagonist. An important breakthrough in the understanding of receptor activation was accomplished with the report by Rasmussen et al. [11] with the use of a camelid Nb80 nanobody mimicking G-protein interactions with the intracellular interface of β2-AR and crystallization of the receptor in significantly altered conformation (PDB ID: 3P0G). This structure shows outward movement (∼11 Å) of the cytoplasmic end of the sixth transmembrane domain (TM VI) and rearrangement of TM V and TM VII. Moreover, this structure of β2-AR highly resembles the one reported for opsin, an active form of a rhodopsin [11]. The latest crystal structure of β2-AR crystallized in the complex with Gs protein provides insights into the process of agonist binding and receptor activation process [12].
In the current work, the two crystal models of β2-AR were employed to investigate molecular interactions between β2-AR and fenoterol analogues. Molecules of fenoterol and its derivatives, in all possible stereochemical configurations (94 structures, Table 1) were docked to the receptor into two forms: (i) a high resolution structure of carazolol-bound receptor (an inactive form of β2-AR, In_β2-AR, PDB: 2RH1) and (ii) a structure stabilized with Nb80 nanobody (an active form of β2-AR; Ac_β2-AR, PDB: 3P0G). The structure of β2-AR co-crystalized with Nb80 nanobody was selected because no significant changes in the binding pocket were observed in comparison with the most recent one structure (PDB ID: 3SN6). In addition, both these structures represent β2-AR co-crystalized with the same agonist molecule, BI-167107 and the ligand location and conformation is nearly the same. Furthermore, the use of the 3SN6-based structure is hindered as it shows missing residues from second extracellular loop: Ala176, Thr177, His178. Moreover, some residues creating receptor binding pocket have missing atoms (or even whole side chains: Asp192, Phe193, Phe194, Thr195) [12].
Our main goal was to investigate the structural aspects of the β2-AR-fenoterol derivative complexes with the particular emphasis put on the stereoselective effects of binding. Four stereoisomers of fenoterol and some of its derivatives were used as a molecular probe to identify differences in stereo-recognition of structurally similar agonists. Moreover, we interpreted the molecular modeling results in terms of the binding affinities of studied compounds determined in [3H]CGP-12177 displacement experiments reported earlier [1, 4]. To address this, a series of docking studies followed by molecular dynamics (MD) simulations were carried out. MD studies were restricted to two stereoisomers of fenoterol (compound 1) ((R,R’)-1 and (S,S’)-1, respectively) and aimed at providing additional insights into stereochemical aspects of 1 bound to β2-AR.
Methods
Docking methodology
The molecules of fenoterol derivatives and their stereoisomers presented in Table 1 were prepared using HyperChem 6.03 (HyperCube Inc., Gainesville, FL) by using Model Build procedure and optimized using the AM1 method. Ligands were protonated according to the physiologal pH. Their positive charge (+1) was manually added in Molegro Virtual Docker software (MVD v. 2010.4.0.0) employed for docking procedures [14]. The flexible, optimized ligands were docked into the binding pocket of two high resolution X-ray crystal structures of β2-AR (PDB IDs: 2RH1 and 3P0G). Receptor models were prepared according to the procedure described in the Modeling β2 adrenergic receptors section. The receptor structures were edited in order to remove ligands and other non-protein molecules. The procedure of docking was performed using the binding cavity within the sphere of a radius 11 Å, covering the ligands originally co-crystallized with β2-AR ((S)-carazolol and BI-167107) and the closest residues, e.g., Asp113, Ser203, Ser204, Ser207, Asn293, Asn312, Tyr308 and Cys191, Asp192, Phe193 [8, 11, 15–22]. The MolDock SE search algorithm was used, and the number of searching runs was set to 100. The following parameters were set during docking simulation: population size = 50, maximum iteration = 1500, energy threshold = 100.00, max steps = 300, the maximum number of poses to generate was increased to 10 from a default value of 5. The estimation of the ligand-protein interactions was described by the MVD implemented scoring function (MolDock Score) [23]. The predicted positions of the ligands in the β2-AR cavity were characterized by a simultaneous lowering of the scoring function values; this corresponds to the high values of the ligand binding energy. The docking algorithm was initially validated with docking simulations of the molecules originally co-crystallized with these models (the (S)- and (R)-carazolol molecules were docked to In_β2-AR and agonist BI-167107 was docked to Ac_β2-AR).
Molecular dynamics
Modeling β2 adrenergic receptors
Modeling of the complete structure of β2-AR in its inactive and active states was done on the basis of the crystal structure of human β2-AR-T4 lysozyme fusion protein with bound (S)-carazolol [8] and the structure of a nanobody-stabilized active state of the β2-AR with the bound full agonist BI-167107 [11], respectively. Conformations of N-/C- termini and second intracellular loop connecting TM V and TM VI, not seen in the crystal structures, were predicted using the ab initio approach. The human β2-AR amino acid sequence was obtained from the Swiss-Prot database (code P07550). Modeling of N- and C-terminal domains of the receptor (residues Met1 to Glu30 and Cys341 to Leu413 respectively) was conducted using I-TASSER server [23–27]. The structure of the longest second intercellular loop of the receptor (residues Leu230 to Leu266) was predicted using CABS program [28]. The rest of the receptor structure was assumed to be rigid and provided restraints for the modeled loop. The resulting structure was refined by applying a simulated annealing routine implemented in GROMACS (v. 3.3) [29]. The partial atomics charges for all receptor protein atoms used during MD calculations, were assigned applying PDB2GMX procedure available from GROMACS (v. 3.3) program package [29]. The total atomic charge of the receptor protein was +2. Charges were assigned to be in agreement with the parameterization of modified GROMOS96 force field [32].
Simulating β2 adrenergic receptor models in the membrane
Two β2-AR models (In_β2-AR and Ac_β2-AR model) were inserted into equilibrated palmitoyl-oleoyl-phosphatidylcholine (POPC) cell membrane model applying Inflategro procedure [30]. Models of β2-AR embedded in POPC lipid bilayer were solvated with water molecules and ions were added. Two systems under consideration consisted of receptor protein, 125 POPC lipid molecules, 16271 water molecules (including 16 water molecules seen in the β2-AR crystal structure; PDB ID: 2RH1 in the case of In_β2-AR model) and two sodium ions. Energy minimization was conducted applying 2000 steps of steepest decent algorithm followed by 2000 steps of l-bfgs algorithm. Then, the four step molecular dynamics (MD) simulation was performed. At first step each system was simulated for 100 ps with no pressure coupling and all the protein atoms were constrained to its initial positions with the “freeze” option. Second step included MD simulation lasting 1 ns with position restraints imposed on all backbone atoms of the receptor model and Berendsen method [31] for pressure coupling. During the third step of MD simulations, lasting 2 ns, position restraints were removed from N- and C-terminal domain of the β2-AR model and from loops connecting seven trans-membrane helices. Finally, production run was performed lasting 40 ns with no restraints. All MD simulations were conducted using modified GROMOS96 force field (ffG53a6 parameters set) [32] with additional parameters for POPC molecules taken from Kukol [33]. SPC water model [34] was used and the PME method [35] was applied for treatment of the long-range electrostatic interactions. All bonds with hydrogen atoms were constrained by the LINCS algorithm [36]. MD was performed at the temperature of 310 K, pressure of 1013 hPa, and simulation time step was 1 fs. All calculations and data analysis were conducted using GROMACS (v. 3.3) program [29]. For comparison, MD simulation of the crystal structure of a human β2-AR T4 lysozyme fusion protein bound to the partial inverse agonist (S)-carazolol [8] was also preformed according to the previously described simulation scheme.
Modeling of the ligand-receptor complexes
The PRODRG server [37] was used to obtain ligand structures ((R,R’)- and (S,S’)-fenoterol) and force field parameters for MD simulation. Geometry optimization of two ligands in their protonated-nitrogen forms was completed using Hartree–Fock procedure employing the 6–31G* basis set in Gaussian (v.03 rev. C.02, Gaussian Inc.) [38]. Partial charges for two molecules were obtained using R.E.D.III procedure [39]. The ligand-receptor complexes were built based on the low-energy structures obtained during the docking simulations. Ligands were inserted in the middle of the binding sites of In_β2-AR model and Ac_β2-AR model to preserve the interaction between Asp113 and the protonated amine nitrogen of the ligands (i.e. the two meta-OH groups located at the 3,5-dihydroxyphenyl moiety positioned in direction of TM V and the 3,5-dihydroxyphenyl group located in the vicinity of loop covering receptor binding site, connecting TM IV and TM V). The similar starting structures of all four receptor-ligand complexes were generated during restrained MD simulation lasting 200 ps. At that point, protein backbone atoms were constrained to its initial positions using “freeze” option and weak harmonic distance restraints (applying the force constant 1000 kJ mol-1 nm-1 and the distance parameters: r0 = 0.0 nm, r1 = 0.3 nm and r2 = 0.5 nm) were imposed on three receptor-ligand atom pairs (pair 1: oxygen atom of hydroxyl group of Ser203 and the oxygen atom of the first meta-OH group of 3,5-dihydroxyphenyl moiety; pair 2: the oxygen atom of hydroxyl group of Ser207 and the oxygen atom of the second meta-OH group of 3,5-dihydroxyphenyl moiety; pair 3: Cγ atom of Asp113 and the protonated nitrogen atom of the ligand) in order to preserve similar starting position of two ligands inside the binding cavity. Finally, two step MD simulation of the receptor-ligand complexes was preformed. During the first step, lasting 2 ns, weak harmonic position restraints were imposed on backbone atoms of trans-membrane helices of the receptor only and ligand-receptor restraints were released. Next, second step included 5 ns production run with no restraints. Described two step MD simulation scheme was repeated 44 times applying random starting velocities for every atom, 11 times for the In_β2-AR-(S,S’)-fenoterol complex, 11 times for the In_β2-AR-(R,R’)-fenoterol complex, 11 times for the Ac_β2-AR-(S,S’)-fenoterol complex and 11 times for the Ac_β2-AR-(R,R’)-fenoterol complex, respectively. Simulation parameters were identical to those used previously during MD simulations of the β2-AR models.
Results and discussion
Ligand binding sites found by docking simulations using the active and inactive β2-AR model
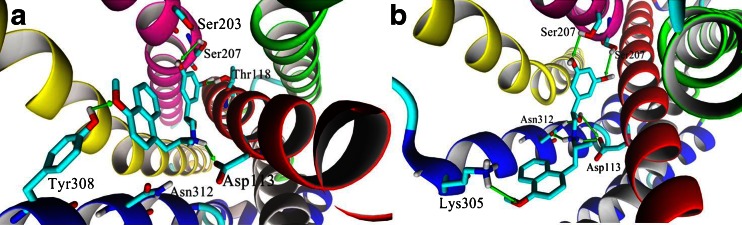
The interactions of the β2-AR and its ligands are well defined as the receptor was co-crystallized with a series of ligands including an inverse agonist, (S)-carazolol and an agonist, BI-167107. All docked molecules were located according to the similar pattern in the binding cavity in comparison to the positions of (S)-carazolol and BI-167107 in the crystal β2-AR structures (Fig. 2). The results of docking simulations clearly show the differences in the binding modes of fenoterol interacting with the active and inactive states of β2-AR. The main findings obtained in the docking study are summarized in Table 1. In the case of both β2-AR models, the 3,5-dihydroxyphenyl and amine moieties of docked ligands lie in the orthosteric site, located between TM III, TM V, TM VI and TM VII. The positions of all docked compounds are very similar, especially in the orthosteric site, but the binding modes are slightly different depending on the receptor model. This concerns the meta-OH groups and amine moiety of the ligands mainly. The schemes of the possible interaction between β2-AR and fenoterol derivatives are shown in Figs. 3 and 4. Two meta-OH groups of the 3,5-dihyroxyphenyl moiety of all docked compounds can create hydrogen bonds (HBs) with Ser203 and Ser207 (TM V). Moreover, only in the case of In_β2-AR model, one of the meta-OH group can interact with Thr118 (TM III) (Figs. 1 and 3). The latter type of interactions is not observed in the case of Ac_β2-AR model. This is probably caused by slight translation of ligands toward Asn312 (TM VII) and the second extracellular loop (ECL2). In the case of both β2-AR models the Asp113 residue creates the salt bridge with the positively charged amine of the ligand. This confirms that the protonated (positively charged) nitrogen atom of the ligand plays a significant role in binding to β2-AR. Additionally, β-OH moiety of the ligand, especially in the (R,*’)-stereoisomers, form HB involving Asp113. The protonated nitrogen atom and β-OH located at the chiral center of the ligand creates HBs with Asn312 but only in the case of Ac_β2-AR. The results obtained for fenoterol derivatives suggest that simultaneous formation of HBs with serines on TM V and Asn312 (TM VII) is not possible in the case of In_β2-AR, it is caused by increased distance between serines on the 5th domain and Asn312 (∼2 Å). Nevertheless, for Ac_β2-AR model, tilting of TM V toward the center of the receptor causes a more intensive polar receptor-ligand interactions for all fenoterol analogues, by enabling optimal engagement of agonists with two experimentally identified anchor sites, formed by Asp113/Asn312 and Ser203/Ser207 side chains (Fig. 4). The part of the ligand molecule containing methyl moiety located at the α’ carbon atom of the compound (the second chiral center) and phenyl/naphthyl rings is located in the extension of the orthosteric site. In the case of In_β2-AR model the 4'-hydroxy-/4'-methoxy-/4'-amino-phenyl and methoxynaphthyl/naphthyl moieties of fenoterol derivatives can potentially assume three possible positions in an extension of the orthosteric site (Fig. 3). The 4'-OH, 4'-NH2 and 4'-OCH3 groups of the fenoterol derivatives can create HB with Cys191 or Asp192 (ECL2) (IIA site) or Thr110 (TM III) (IIB site) or Tyr308 (TM VII) (IIC site). Furthermore, the ligand positions are stabilized by a favorable edge-to-face interaction with Phe193 residue (ECL2). It is depicted in Fig. 3. The schemes of the possible interactions created between β2-AR in its inactive (Fig. 3) and active (Fig. 4) state with fenoterol derivatives were presented below.
Fig. 2.

The comparison of the positions of ligands co-crystallized with β2-AR (gray), (S)-carazolol (PDB ID: 2RH1) (a) and BI-167107 (PDB ID: 3P0G) (b) with (R,R’)-1 docked to In_β2-AR (a) and Ac_β2-AR model (b). The positions of both aromatic rings of (R,R’)-1 are very similar to those of (S)-carazolol and BI-167107 present in the crystal structures of β2-AR. Positions of (R,R’)-1, characterized by the lowest scoring function values are presented.
Fig. 3.

The scheme of the possible interactions in the (R,R’)-1-In_β2-AR complex. Red and green lines denote hydrogen bonds and hydrophobic interactions ( Π-Π interactions), respectively. The selected amino acid residues are presented in the 1-letter code [40]
Fig. 4.

The scheme of the possible interactions in the (R,R’)-1-Ac_β2-AR complex. Red and green lines denote hydrogen bonds and hydrophobic interactions (Π-Π interactions), respectively. The selected amino acid residues are presented in the 1-letter code [40]
The amine and β-OH moieties of (R,R’)-1 docked to the Ac_β2-AR model are trapped in the network of HBs involving Asp113 and Asn312. Two meta-OH groups of ligand form HBs with Ser203 and Ser207 of TM V (the orthosteric site), Fig. 4. The additional hydrogen bond was formed between 4'-OH moiety of (R,R’)-1 and residues of the second extracellular loop (ECL2) (Cys191 or Asp192) creating the IIA site or residues of TM VII creating the IIB site: Lys305, Tyr308 or Trp313 (Fig. 4). The lowest scoring function values were obtained for positions in which 4'-OH moiety of the ligand creates HB with Lys305 (TM VII) or Cys191 (ECL2). Thus, (R,R’)-1 forms a very similar pattern of interactions as BI-167107 molecule co-crystallized originally with β2-AR (Ac_β2-AR model) (Fig. 2b). In the (R,R’)-1-In_β2-AR complex two meta-OH moieties form HBs with Ser203 (TM V) (seldom with Ser207) and Thr118 (TM III). The amine and β-OH moieties of the ligand form HBs with Asp113 (TM III) (Fig. 3). In this case Asn312 is too distant to interact with the amine or β-OH group of the ligand; similarly, due to the reorientation of Lys305 in the In_β2-AR model, no direct interaction between this residue and the ligand 4'-OH moiety was observed. In the case of In_β2-AR model, the 4'-OH moiety interacts with Tyr308 (TM VII) by hydrogen bonding mainly. Note than the abovementioned residue, Tyr308, plays an important role in the selective binding of the agonists to β2-AR with respect to β1-AR [41]. The oxygen atom of the phenolic group of the ligand seems to interact preferentially with the hydroxyl group of the Tyr308 residue [42]; its interactions with Cys191 and Asp192 (ECL2) are also possible. Comparison of MolDock Score functions characterizing the two complexes including (R,R’)-1 suggests that the complex formed with Ac_β2-AR should be more stable; the difference between MolDock Score values (Table 1) is -4.5 kJ mol-1.
The analysis of the β2-AR molecular models reveals that Tyr308 is neighbored by several other aromatic residues (e.g., Trp109, Phe193, Trp286, Phe289, Phe290, His296 or Tyr316) and the aminoaryl moiety of the fenoterol analogues can form a network of π−π interactions within this region of β2-AR. Especially, the naphthyl moiety with increased π electron density can interact strong enough to overcome the lack of the hydrogen bond acceptor effect in (R,R’)-5. We observed that the number of aromatic rings influences the scoring function values. For compounds with the naphthyl moiety (compounds: 5 and 53) MolDock Score values are smaller than that for compound with the phenyl ring (compound 3). Moreover, compounds containing naphthyl moieties are characterized by higher pK i values in comparison to the compound with phenyl ring (Table 1). The naphthyl moiety can create π−π interactions with several aromatic residues distributed in the binding region instead of one preferential direction, characteristic of (R,R’)-1 or (R,R’)-2 donating their oxygen atoms for HB interaction with Tyr308. In several cases, the naphthyl moieties of the studied ligands take the same position as aromatic rings of carazolol bound to the In_β2-AR structure (Fig. 2a) in which a carbazole heterocycle ring of carazolol forms a network of π−π interactions with Phe289, Phe290 and Phe193 [8]. Docking results show that compounds containing the naphthyl group, contrary to compounds with phenyl moiety, are more likely to interact with the other aromatic residues by π−π stacking. This type of interaction is not preferred in the case of derivatives with the phenolic moiety; their rings are directed rather toward Tyr308 or residues located on ECL2 (Cys191 or Asp192) but with no significant π−π interactions.
Docking simulations: stereoselective binding of fenoterol derivatives to β2-AR
Experimental binding affinities determined for the studied compounds suggest that the stereochemistry plays a crucial role in the interactions with the receptor [1, 4–6]. Thus, it is very important to describe how docking simulations explain the stereoselective nature of ligand-receptor interactions. Figure 5 presents the binding modes of the (S,S’)-1 molecule to In_β2-AR and Ac_β2-AR, respectively. Docking of (S,S’)-1 to the In_β2-AR model reveals that the position of the molecule’s backbone is virtually the same as in (R,R’)-1 docked to the In_β2-AR model. The only difference lies in the reorientation of moieties associated with the two chiral centers, β-OH and –CH3 groups, which are in the opposite direction when comparing with (R,R’)-1-In_β2-AR complex, Fig. 5a. As a result, the β-OH moiety of (S,S’)-1 does not form a HB with Asp113 of In_β2-AR and the methyl moiety does not exhibit favorable orientation toward hydrophobic surface of the binding site formed by TM III (Trp109) and ECL2 (Phe193). The inverted positions of two moieties in (S,S’)-1-In_β2-AR complex result in significant increase of the MolDock Score value with respect to the (R,R’)-1-In_β2-AR complex; the difference is 9.0 kJ mol-1 (Table 1). This value reflects the difference in pK i values determined experimentally for (R,R’)-1 and (S,S’)-1 (∆pK i = 1.9, Table 1). Docking of (S,S’)-1 to Ac_β2-AR reveals more significant differences in the interactions pattern with respect to the (R,R’)-1-Ac_β2-AR complex. The aminoalkyl part of the (S,S’)-1 molecule is oriented in almost the same manner within the binding site as the corresponding part of the (R,R’)-1, 4'-OH moiety interacts via HB with Lys305 and both amine and β-OH groups form a network of hydrogen bonds with Asp113 and Asn312, Fig. 5b. The 3,5-dihydroxyphenol moiety of (S,S’)-1 is significantly reoriented that only one meta-OH moiety is able to form HB with Ser203. Moreover, β-OH moiety of (S,S’)-1 does not form HB with Asp113 of Ac_β2-AR. Similarly, the S’ configuration forces the methyl moiety associated with the second chiral center to reorient and point away from the surface of the TM III (Trp109), Fig. 6. Surprisingly, these significant differences between (R,R’)-1-Ac_β2-AR and (S,S’)-1-Ac_β2-AR complexes are not translated into notable differences in MolDock Score values, equal to -133.5 and -134.6 kJ mol-1, respectively (Table 1). Docking results show a good correlation with the binding affinities (pK i values) of the fenoterol derivatives. The In_β2-AR model predicts the stereoselective binding effects better than the Ac_β2-AR model, Tab. S1, Figs. S1, S2. This results from the better correlation of scoring function values with pK is determined using β2 selective antagonist, [3H]CGP-12117, in a radioligand displacement study [1, 4]. Thus, the experimental pK i values reflect the inactive state of receptor rather than the active one. Moreover, the applied docking procedure appeared not to be sensitive enough to differentiate between binding affinities of stereoisomers of the same compound (Table 1).
Fig. 5.

The structural models of the (S,S’)-fenoterol-In_β2-AR (a) and the (S,S’)-fenoterol-Ac_β2-AR complexes (b)
Fig. 6.

Structural models of the (R,R’)-52-In_β2-AR complex (a) and the (R,R’)-52-Ac_β2-AR complex (b)
Docking simulations: influence of the R1 and R2 substituents on the binding to β2-AR
Figures 6 and 7 show the differences of the docking results of the two selected fenoterol derivatives, namely (R,R’)-52 and (R,R’)-54 (Table 1) compared to the binding modes of fenoterol to inactive and active form of the receptor (Fig. 5a, b). The replacement methyl moiety for the ethyl group attached to the second chiral center (R1 substituent, Fig. 1) causes the reorientation of the 4'-hydroxyphenyl moiety of the (R,R’)-52 in the complex with Ac_β2-AR in comparison to the (R,R’)-1-Ac_β2-AR complex. The ethyl group causes the shift of the 4'-hydroxyphenyl moiety away from Lys305 and therefore lack of HB with this residue. All other interactions are analogous to those present in the (R,R’)-1-Ac_β2-AR complex, Fig. 4. The (R,R’)-52-In_β2-AR complex shown in Fig. 6a reveals a very similar pattern of the ligand-receptor interactions as that found in (R,R’)-1-In_β2-AR complex (Fig. 3). The (R,R’)-54 molecule contains 4’-methoxy-1-naphthyl moiety at the aminoalkyl tail (R2 substituent). This modification does not produce very significant changes in the position of the molecule within the Ac_β2-AR binding site in comparison to that of fenoterol. All the principal interactions observed for the (R,R’)-54-Ac_β2-AR complex are still present, Fig. 7. More significant differences can be noticed for (R,R’)-54 docked to In_β2-AR. In this case, the ligand molecule is reoriented to prevent the β-OH group from creating a HB with Asp113 and bulky 4-methoxy-1-naphthyl moiety is pushed into the center of the binding site due to the interaction with Tyr308. Both (R,R’)-52 and (R,R’)-54 showed qualitatively different behavior in a functional test in comparison to the compounds like (R,R’)-1 or (R,R’)-2. (R,R’)-52, having an ethyl moiety attached to the second chiral center, has a significantly weaker binding affinity relative to (R,R’)-1 (1,273 nM versus 345 nM) when [3H]CGP-12177 is the marker ligand, is active in the cAMP accumulation assay but is not active in the cardiomyocyte contractility test [4]. Interestingly, (R,R’)-54, which contains the 4’-methoxy-1-naphthyl moiety, is not active in the cAMP accumulation assay, but this stereoisomer is the most potent derivative, appeared to have very low K i value determined experimentally using the radioactive antagonist [3H]CGP-12177 and the exceptional subtype selectivity (the K iβ1/K iβ2 ratio = 578). This compound was further characterized in the cardiomyocyte contractility assay and its EC50 value was 3 nM which place this compound among the strongest selective agonists of the β2-AR ever described [4]. The obtained results confirm the influence of R1 and R2 substituents in the fenoterol molecule (Fig. 1) on its binding to β2-AR. This is in agreement with the experimentally determined pKi values for stereoisomers of fenoterol and its derivatives [1, 4]. Considering the R2 substituent in the template molecule (Fig. 1), the pKi values corresponding to the fenoterol derivatives exhibit the following trend: 4'-OH > 4'-OCH3 > 4'-NH2. The above trend holds also in the case of compounds containing either phenyl or naphthyl group in R2. Furthermore, the compounds containing the naphthyl rings in the R2 substituents exhibit larger affinity for β2-AR in comparison to the corresponding ligands with phenyl ring in R2 (Table S2) [unpublished data].
Fig. 7.
Structural models of the (R,R’)-54-In_β2-AR complex (a) and the (R,R’)-54-Ac_β2-AR complex (b)
MD simulations: binding modes of two fenoterol isomers
The radioligand binding studies showed that fenoterol stereochemistry greatly influences the binding affinity of derivatives to β2-AR with relative order (R,R’)>(R,S’)>(S,R’)>(S,S’) [1] and a similar trend was found in functional assays (ligand induced the cAMP accumulation and the cardiomyocyte contractility measurements). Two models: In_β2-AR and Ac_β2-AR were used for investigation of different binding modes of (R,R’)- and (S,S’)-fenoterol isomers of 1 using MD simulations. Both ligands were inserted in the middle of the receptor binding site and 44 MD simulations were conducted. In general, the starting positions of two fenoterol isomers inside the binding site of In_β2-AR and Ac_β2-AR are very similar (Online Resource 4). Two hydroxyl groups (meta-OH) of the ligand 3,5-dihydroxyphenyl moiety are situated in a vicinity of the Ser203 and Ser207 residues from TM V, whereas the ligand protonated nitrogen atom was in close distance to Asp113 (TM III) (Fig. 3). Slightly different binding mode was observed in the case of Ac_β2-AR-(S,S’)-fenoterol complex (Fig. 5). The phenolic group of the ligand was oriented toward TM VII, whereas its hydroxyl group was directed toward extracellular part of the receptor. During 44 MD simulations with no restraints (11 MD simulations for each fenoterol isomer–receptor complex, each lasting 5 ns) both fenoterol isomers adopted different conformations inside the receptor binding sites (Online resource 5). Inspection of the 44 MD trajectories of the receptor-ligand complexes revealed much higher conformational stability of the (R,R’)-fenoterol when compared to the (S,S’)-fenoterol isomer, Online resource 5. The flexibility of two ligands has been monitored in the form of RMSD autocorrelation function. The observed averaged RMSD values are lower for (R,R’)-fenoterol isomer when interacting with both In_β2-AR and Ac_β2-AR for all monitored time intervals (Online resource 5). The calculation of root mean square fluctuation (RMSF) parameter for position of all ligand atoms indicated specific fragments of two fenoterol isomers showing the highest difference in RMSD fluctuation (Fig. 8). In the case of In_β2-AR-fenoterol complexes atoms creating the chain connecting 3,5-dihydroxyphenyl and phenolic moieties and atoms present in the 4’-hydroxyphenyl group of (R,R’)-fenoterol display much lower averaged displacement during the simulation. Plot presenting measured distances between receptor-fenoterol atom pairs (Online resource 6) indicated that those fragments include atoms (protonated amine and 4’-OH moiety) responsible for direct contact with β2-AR binding site. Additionally, the biggest fluctuation of ligand conformations were observed for ligand-In_β2-AR complexes, Fig. 8.
Fig. 8.

The averaged RMSF values of ligand atoms during 5 ns of MD simulation of ligand-In_β2-AR (top) and ligand-Ac_β2-AR (bottom) complexes: (R,R’)-fenoterol – red, (S,S’)-fenoterol – black. Blue and green rectangles represent molecule fragment with highest difference of RMSF values
The analysis of MD trajectories indicated that (R,R’)-fenoterol in the complex with In_β2-AR creates a stable HB network with residues Asp113 and Asn312 (Online resource 6). The carboxyl group of Asp113 forms close contact with hydrogen atoms at the ligand protonated amine group and with H16 atom of β-OH (see Fig. 8 for atom numbering). The position of Asp113 sidechain is also stabilized by the interaction with Tyr316 (TM VII). The nitrogen and hydrogen atoms of the Asn312 residue form close contacts with ligands H20 and O15 atoms, respectively. The HB between 4’-hydroxyphenyl group of the ligands and Asp192 (ECL2) was also well preserved during MD simulation. Two meta-OH groups of the ligand 3,5-dihydroxyphenyl moiety were oriented toward Ser203 and Ser207 residues from TM V, but the distance was too large to form a contact. Analysis of the MD simulation trajectories of Ac_β2-AR-ligand complexes revealed that no stable connection with Ser203/207 from TM helix V was observed. During the MD simulations two ligand's hydroxyl groups occasionally created hydrogen bonds with serine residues but this event was rather rare (Fig. 6 shows the measured ligand-receptor atom pairs distances averaged over 22 MD simulations). In our opinion this is due to two main reasons: (i) fenoterol moiety containing one aromatic ring with two hydroxyl groups directed toward TM helix V occupies the same space as the much larger two ring system of the BI-167107 agonist moiety seen in 3P0G crystal structure which allows for higher mobility of this fenoterol fragment inside the receptor binding site; (ii) hydroxyl group of phenolic moiety of fenoterol molecule interacts with Asp192 from second extracellular loop which favors shifting the ligand position; this, in turn, increases the distance from TM helix V and weakens the interaction with two Ser203/207 residues. In addition, Asn293 residue, which is believed to play a crucial role in stereoselective agonist binding and receptor activation [43] did not form a stable interaction with the ligand during MD simulation. In the contrast, the (S,S’)-fenoterol isomer in complex with In_β2-AR did not maintained a stable position in the receptor binding cavity. On average, only one well preserved contact during each of 11 MD simulations was formed between ligand protonated nitrogen atom and carboxyl group of the Asp113 residue. Interaction between Asn312 and the ligand involved rotation of amino acid side chain and was frequently interrupted which is associated with high conformational flexibility of the (S,S’)-fenoterol isomer. The average distance between 4’-hydroxyphenyl group of the ligand and carboxyl group of Asp192 was too large (> 4 Å) to establish a stable hydrogen bond. Ligand movement in the binding cavity allowed two hydroxyl groups of the 3,5-dihydroxyphenyl moiety to move closer toward Ser203 and Ser207, but no stable connection was made. Analysis of MD trajectories revealed that the two fenoterol isomers of 1 are conformationally flexible and do not form common invariable interaction pattern with Ac_β2-AR. Ionic interactions between ligands protonated nitrogen and Asp113 and HB between 4’-hydroxyphenyl group of the ligand and carboxyl group of Asp192 are the most frequently observed interactions, Online resource 6. A small number of water molecules entered the receptor binding cavity during all MD simulations but no common interaction pattern was observed.
Conclusions
The main results of our studies can be summarized as follows:
All derivatives of 1 take similar positions in the binding cavity of β2-AR, especially in the orthosteric site (Figs. 3 and 4). The meta-OH groups of tested compounds create hydrogen bonds with serines on TM V (Ser203, Ser207). The protonated amine of the ligands creates a salt bridge with the Asp113 sidechain (TM III). Moreover, the β-OH moiety of (R,*’)-stereoisomers can form the HB with Asp113. The remaining part of ligands, including α’ alkyl chain (R2 substituent) and phenyl/naphthyl rings, is located in the extension of the orthosteric site. The compounds can be oriented in the extension of the orthosteric site according to diverse patterns, depending on the character of the R2 substituent in the leading structure of 1 (Fig. 1) and the β2-AR model. The observations listed above are common for the two structural models of the receptor: inactive β2-AR (In_β2-AR) and active β2-AR (Ac_β2-AR).
The comparison of In_β2-AR and Ac_β2-AR reveals several substantial differences between binding modes characteristic of them. Unlike Ac_β2-AR, in the case of In_β2-AR model, only the Thr118 and Thr110 residues interact with one meta-OH and 4’-OH groups of ligand respectively. Docking results indicate that in the case of Ac_β2-AR the amine group of ligand interacts also with Asn312 (TM VII). This interaction does not exist in the case of In_β2-AR, as the location of Asn312 is too distant. In the In_β2-AR model ligands can take three possible positions in the extension of the orthosteric site (IIA, IIB and IIC site). In the case of Ac_β2-AR only two positions in the extension of the orthosteric site were observed (Figs. 3 and 4). The most significant difference between fenoterol derivatives bound to In_β2-AR and to Ac_β2-AR lies in the hydrogen bonding contacts with Lys305 (TM VII) created only in the case of Ac_β2-AR model.
Interestingly, docking studies reveal different binding modes in the extension of the orthosteric site of compounds containing naphthyl or phenyl moieties. Compounds containing the 4’-hydroxy-/4’-methoxy-/4-‘amino-phenyl group interact mainly with Cys191 or Asp192 (ECL2, IIA site). Additionally, the interactions with Lys305 (TM VII) in the case of Ac_β2-AR are preferred. In the case of compounds containing the methoxynaphthyl/naphthyl moieties, the position of these groups is usually close to Tyr308. The naphthyl moiety of derivatives 5, 53 and 54 can form π−π interactions with aromatic ring of Tyr308 and/or several aromatic residues neighboring Tyr308. These hydrophobic interactions cause the compounds 5, 53 and 54 to interact strong enough to overcome the lack of the HB acceptor/donor effect.
Molecular dynamics simulations indicate that the β-OH and (–NH2 +–) functional groups interact with Asp113 and Asn312. These interactions are more stable in the case of the receptor active state (Ac_β2-AR) than in the inactive one (In_β2-AR). The distances between the protonated amine of ligands and both Asp113 and Asn312 residues are larger for In_β2-AR. It causes the β-OH moiety of (R,R’)-fenoterol to lose HB with Asn312. In comparison to the docking results, MD simulations do not reveal the presence of HBs between Ser203/Ser207 and meta-OH moieties of ligands (Online resource 6). During the MD simulations ligand drifts away from TM V and looses the possibility of direct interaction with the serine residues. The indirect interaction is possible, though, according to the scenario water molecules form the ‘bridges’ between the meta-OH moiety (ligand) and Ser203/Ser207.
The results obtained by us are in accordance with the experimental data and indicate that the most potent compounds are those with (R,*’)-configuration. Docking studies indicate that the hydroxyl group at the first chiral center of ligand creates HB with Asp113 or/and Asn312 in the case of (R,*’)-stereoisomers mainly. Further, molecular dynamics simulations confirm the existence of the stereoselective effects accompanying the ligand-receptor interactions; namely, different stereoisomers exhibit diverse conformational behaviors and distances between characteristic ligand-receptor atom-atom pairs, Fig. S6.
Molecular dynamics simulations reveal that the available conformational phase space is larger in the case of (S,S’)-fenoterol while in the case of (R,R’)-fenoterol it is significantly reduced (Online resource 5). The largest differences between (R,R’)- and (S,S’)-fenoterol are observed in the region containing the β-OH, (–NH2 +–) and 4’-OH functional groups, which interact with the receptor (Fig. 8).
Electronic supplementary material
Below is the link to the electronic supplementary material.
(DOC 2437 kb)
Acknowledgments
The work was supported by the Polish Ministry of Science and Higher Education (Grant no. N N405 617238), by the Foundation for Polish Science (TEAM 2009-4/5 and FOCUS 4/2006 programmes), and using the equipment purchased within the Project "The equipment of innovative laboratories doing research on new medicines used in the therapy of civilization and neoplastic diseases" within the Operational Program Development of Eastern Poland 2007- 2013, Priority Axis I Modern Economy, Operations I.3 Innovation Promotion. The research was also supported in part by the Intramural Research Program of the National Institutes of Health National Institute on Aging; by the National Institutes of Health National Institute on Aging [Contract N01-AG31009].
References
- 1.Jozwiak K, Khalid C, Tanga MJ, Berzetei-Gurske I, Jimenez L, Kozocas JA, Woo A, Zhu W, Xiao RP, Abernethy DR, Wainer IW. Comparative molecular field analysis of the binding of the stereoisomers of fenoterol and fenoterol derivatives to the beta2 adrenergic receptor. J Med Chem. 2007;50:2903–2915. doi: 10.1021/jm070030d. [DOI] [PubMed] [Google Scholar]
- 2.Wermuth CG. Selective optimization of side activities: the SOSA approach. Drug Discov Today. 2006;11:160–164. doi: 10.1016/S1359-6446(05)03686-X. [DOI] [PubMed] [Google Scholar]
- 3.Woo AY, Xiao RP. β-Adrenergic receptor subtype signaling in heart: from bench to bedside. Acta Pharmacol Sin. 2012;33:335–341. doi: 10.1038/aps.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jozwiak K, Woo AY, Tanga MJ, Toll L, Jimenez L, Kozocas JA, Plazinska A, Xiao RP, Wainer IW. Comparative molecular field analysis of fenoterol derivatives: a platform towards highly selective and effective beta(2)-adrenergic receptor agonists. Bioorg Med Chem. 2010;18:728–736. doi: 10.1016/j.bmc.2009.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jozwiak K, Toll L, Jimenez L, Woo AY, Xiao RP, Wainer IW. The effect of stereochemistry on the thermodynamic characteristics of the binding of fenoterol stereoisomers to the beta(2)-adrenoceptor. Biochem Pharmacol. 2010;79:1610–1615. doi: 10.1016/j.bcp.2010.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jozwiak K, Plazinska A, Toll L, Jimenez L, Woo AY, Xiao RP, Wainer IW. Effect of fenoterol stereochemistry on the β2 adrenergic receptor system: ligand-directed chiral recognition. Chirality. 2011;23:E1–E6. doi: 10.1002/chir.20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobilka BK. G protein coupled receptor structure and activation. Biochim Biophys Acta. 2007;1768:794–807. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, Stevens RC. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wacker D, Fenalti G, Brown MA, Katritch V, Abagyan R, Cherezov V, Stevens RC. Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J Am Chem Soc. 2010;132:11443–11445. doi: 10.1021/ja105108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rasmussen SGF, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, DeVree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Graaf C, Rognan D. Selective structure-based virtual screening for full and partial agonists of the beta2 adrenergic receptor. J Med Chem. 2008;51:4978–4985. doi: 10.1021/jm800710x. [DOI] [PubMed] [Google Scholar]
- 14.Thomsen R, Christensen MH. MolDock: a new technique for high-accuracy molecular docking. J Med Chem. 2006;49:3315–3321. doi: 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- 15.Strader CD, Sigal IS, Candelore MR, Rands E, Hill WS, et al. Conserved aspartic acid residues 79 and 113 of the beta-adrenergic receptor have different roles in receptor function. J Biol Chem. 1988;263:10267–10271. [PubMed] [Google Scholar]
- 16.Strader CD, Candelore MR, Hill WS, Sigal IS, Dixon RA. Identification of two serine residues involved in agonist activation of the beta-adrenergic receptor. J Biol Chem. 1989;264:13572–13578. [PubMed] [Google Scholar]
- 17.Sato T, Kobayashi H, Nagao T, Kurose H. Ser203 as well as Ser204 and Ser207 in fifth transmembrane domain of the human beta2-adrenoceptor contributes to agonist binding and receptor activation. Br J Pharmacol. 1999;128:272–274. doi: 10.1038/sj.bjp.0702813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katritch V, Reynolds KA, Cherezov V, Hanson MA, Roth CB, Yeager M, Abagyan R. Analysis of full and partial agonists binding to beta2-adrenergic receptor suggests a role of transmembrane helix V in agonist-specific conformational changes. J Mol Recognit. 2009;22:307–318. doi: 10.1002/jmr.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liggett SB. Molecular and genetic basis of beta2-adrenergic receptor function. J Allergy Clin Immunol. 1999;104:S42–S46. doi: 10.1016/S0091-6749(99)70272-1. [DOI] [PubMed] [Google Scholar]
- 20.Suryanarayana S, Kobilka BK. Amino acid substitutions at position 312 in the seventh hydrophobic segment of the beta 2-adrenergic receptor modify ligand-binding specificity. Mol Pharmacol. 1993;44:111–114. [PubMed] [Google Scholar]
- 21.Isogaya M, Yamagiwa Y, Fujita S, Sugimoto Y, Nagao T, Kurose H. Identification of a key amino acid of the beta2-adrenergic receptor for high affinity binding of salmeterol. Mol Pharmacol. 1998;54:616–622. [PubMed] [Google Scholar]
- 22.Wang T, Duan Y. Ligand entry and exit pathways in the beta2-adrenergic receptor. J Mol Biol. 2009;392:1102–1115. doi: 10.1016/j.jmb.2009.07.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2011;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu S, Zhang Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007;35:3375–3382. doi: 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, Skolnick J. Automated structure prediction of weakly homologous proteins on a genomic scale. Proc Natl Acad Sci U S A. 2004;101:7594–7599. doi: 10.1073/pnas.0305695101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu S, Skolnick J, Zhang Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 2007;5:17. doi: 10.1186/1741-7007-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 2007;69:108–117. doi: 10.1002/prot.21702. [DOI] [PubMed] [Google Scholar]
- 28.Kolinski A. Protein modeling and structure prediction with a reduced representation. Acta Biochim Pol. 2004;51:349–371. [PubMed] [Google Scholar]
- 29.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 30.Kandt C, Ash WL, Tieleman DP. Setting up and running molecular dynamics simulations of membrane proteins. Methods. 2007;41:475–488. doi: 10.1016/j.ymeth.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 31.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 32.Oostenbrink C, Villa A, Mark AE, van Gunsteren WF. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem. 2004;25:1656–1676. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- 33.Kukol A. Lipid Models for United-Atom Molecular Dynamics Simulations of Proteins. J Chem Theory Comput. 2009;5:615–626. doi: 10.1021/ct8003468. [DOI] [PubMed] [Google Scholar]
- 34.Van Der Spoel D, Van Maaren PJ, Berendsen HJC. A systematic study of water models for molecular simulation: derivation of water models optimized for use with a reaction field. J Chem Phys. 1998;108:10220–10230. doi: 10.1063/1.476482. [DOI] [Google Scholar]
- 35.Darden T, York D, Pedersen L. Particle mesh Ewald: An N•log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 36.Hess B, Berendsen HJC, Fraaije JGEM. LINCS: a linear constraint solver for molecular simulations. J Comput Chem. 1997;18:1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- 37.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D: Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 38.Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussion 03 Program. Wallingford: Gaussian Inc.; 2004. [Google Scholar]
- 39.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for determining atom-centered charges: the RESP Model. J Phys Chem. 1993;97:10269–10280. doi: 10.1021/j100142a004. [DOI] [Google Scholar]
- 40.Ballesteros JA, Weinstein H. Integrated methods for the construction of three dimensional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci. 1995;25:366–428. doi: 10.1016/S1043-9471(05)80049-7. [DOI] [Google Scholar]
- 41.Isogaya M, Sugimoto Y, Tanimura R, Tanaka R, Kikkawa H, Nagao T, Kurose H. Binding pockets of the beta(1)- and beta(2)-adrenergic receptors for subtype-selective agonists. Mol Pharmacol. 1999;56:875–885. doi: 10.1124/mol.56.5.875. [DOI] [PubMed] [Google Scholar]
- 42.Kikkawa H, Isogaya M, Nagao T, Kurose H. The role of the seventh transmembrane region in high affinity binding of a beta 2-selective agonist TA-2005. Mol Pharmacol. 1998;53:128–134. doi: 10.1124/mol.53.1.128. [DOI] [PubMed] [Google Scholar]
- 43.Wieland K, Zuurmond HM, Krasel C, Ijzerman AP, Lohse MJ. Involvement of Asn-293 in stereospecific agonist recognition and in activation of the beta 2-adrenergic receptor. PNAS. 1996;93:9276–9281. doi: 10.1073/pnas.93.17.9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 2437 kb)