

Abstract
Purpose
Vitamin A metabolites, such as all-trans-retinoic acid (RA) that act through the nuclear receptor; retinoic acid receptor (RAR), have been shown to polarise T cells towards Th2, and to be important in resistance to helminth infections. Co-incidentally, people harbouring intestinal parasites are often supplemented with vitamin A, as both vitamin A deficiency and parasite infections often occur in the same regions of the globe. However, the impact of vitamin A supplementation on gut inflammation caused by intestinal parasites is not yet completely understood.
Methods
Here, we use Trichuris muris, a helminth parasite that buries into the large intestine of mice causing mucosal inflammation, as a model of both human Trichuriasis and IBD, treat with an RARα/β agonist (Am80) and quantify the ensuing pathological changes in the gut.
Results
Critically, we show, for the first time, that rather than playing an anti-inflammatory role, Am80 actually exacerbates helminth-driven inflammation, demonstrated by an increased colonic crypt length and a significant CD4+ T cell infiltrate. Further, we established that the Am80-driven crypt hyperplasia and CD4+ T cell infiltrate were dependent on IL-6, as both were absent in Am80-treated IL-6 knock-out mice.
Conclusions
This study presents novel data showing a pro-inflammatory role of RAR ligands in T. muris infection, and implies an undesirable effect for the administration of vitamin A during chronic helminth infection.
Electronic supplementary material
The online version of this article (doi:10.1007/s10875-013-9936-8) contains supplementary material, which is available to authorized users.
Keywords: Trichuris muris, retinoic acid, inflammation, IL-6
Introduction
Human Trichuriasis affects millions of people across the globe, causing significant gut inflammation and morbidity. The human nematode parasite Trichuris trichiura is modelled in mice using the analogous parasite Trichuris muris. T. muris buries into the colonic and caecal epithelium causing inflammation which is characterised by morphological changes such as intestinal crypt hyperplasia, as well as an infiltration of inflammatory cells and the production of inflammatory mediators [1–3]. Furthermore, the inflammation seen in response to Trichuris muris infection, has demonstrated numerous similarities to the genetic and pathological changes that occur during human inflammatory bowel disease (IBD) [4]. Therefore, it is important to understand the mechanisms that mediate the inflammatory processes during infection with this intestinal parasite, not only from a parasitological point of view and but also because of its relevance to other diseases.
Resistance to T. muris is critically dependent on the ability of the mouse to mount a Th2 response, characterised by the secretion of cytokines such as IL-4, IL-5, IL-6, IL-9 and IL-13 [5] and is exemplified by acute infection of the BALB/c strain of mouse. Conversely, susceptible strains of mouse, such as AKR, make an inappropriate Th1 response, identified by the production of IFN-γ [6] and develop chronic patent infections [7]. Furthermore, manipulation of infection dose determines whether an acute or chronic infection prevails in C57BL/6 mice [8]. Thus, high levels of infection of 100–200 eggs are expelled, whilst low-level infections, establishing from fewer than 60 eggs, persist. Low-level infection of C57BL/6 mice is therefore a useful tool to create susceptibility to infection equivalent to that seen in certain inbred strains of mouse such as AKR, enabling chronic infection experiments to be conducted in the multitude of transgenic mouse strains that exist on a C57BL/6 background.
Vitamin A and its major metabolite all-trans-retinoic acid (RA) have recently been shown to be key regulators of immune responses. RA regulates gene expression by binding to the nuclear hormone receptors; retinoic acid receptor (RAR) and retinoic X receptor (RXR), each of which exist in three isoforms; α, β, and γ [9]. RAR proteins are expressed ubiquitously and their expression is up-regulated in the presence of retinoic acid [10]. Upon complexing with retinoic acid, the RARs heterodimerise with nuclear hormone receptors (NHRs) of the RXR family. These heterodimers then interact with retinoic acid response elements (RAREs) within the promoters of retinoic acid responsive genes to modify transcriptional rates of, for example, inflammatory mediators [9].
RA has profound effects on the immune system, including influencing the development of T cell subsets. Ex vivo stimulation of lymphocytes from vitamin A-deficient mice induces IFN-γ production, and these cells produce less IL-4 and IL-5 [11]. Conversely, supplementation with RA boosts IL-4, -5 and -10 production, while reducing IFN-γ [12, 13]. RARα-selective retinoids can also inhibit the progression of Th1-associated immunological diseases such as delayed type hypersensitivity [14] and experimental arthritis [15]. Therefore, RA favours Th2 immune responses, and has been shown to enhance the differentiation of these cells [16]. Recent work on RA has focussed on its role in regulatory T cell (Treg) induction. Studies have shown that RA is able to induce the conversion of naïve FoxP3−CD4+ T cells into FoxP3+ Tregs in both mice and humans in vitro [17], and that FoxP3 expression is increased following RA treatment in colitis [18].
The role of RA in the regulation of chronic inflammation is largely unexplored, despite its ability to regulate gene transcription through NHRs. Further, its role in the induction of regulatory T cells might imply that vitamin A supplementation is well placed to reduce inflammatory responses. Indeed, vitamin A supplementation has been shown to reduce re-infection rates and increase faecal IL-4 protein in Ascaris lumbricoides-infected children [19, 20]. Beneficial effects of RXR (RAR’s heterodimeric partner) agonists have been reported in chemical models of IBD, and correlate with reduced TNF-α production [21], but the mechanisms have not been fully described. Here, we supplement chronically infected mice with an RARα/β agonist (Am80) and show that Am80 exacerbates helminth-driven inflammation, described by an exaggerated crypt hyperplasia and an increased cellular infiltrate. Furthermore, we provide evidence that the Am80-driven increase in mucosal inflammation occurs via an IL-6-dependent mechanism. Since vitamin A is widely used to lower morbidity in children and adults who harbour chronic parasitic infections [19], our findings are a critical addition to the understanding of potential therapies for chronically-infected individuals, as well as our knowledge of vitamin A-mediated immune responses in the gut mucosa.
Methods
Animals, T. muris and Compounds
Male AKR mice of 6–8 weeks of age were purchased from Harlan (UK). Breeding pairs of IL-6−/− mice [22] and their wild-type controls (IL-6+/+), back-crossed onto a C57BL/6 background were bred by the Biological Services Facility (BSF, University of Manchester, UK). All mice were housed at the BSF (University of Manchester) in sterile conditions, within individually-ventilated cages. This investigation was carried out in accordance with the UK Scientific Procedures (Animals) Act 1986.
All experiments used the Edinburgh (E) strain of T. muris, which was maintained as previously described [23]. T. muris Excretory/Secretory (E/S) antigen was collected by incubating adult worms in RPMI 1640 medium for 4 or 24 h. AKR mice were infected with ~200 eggs, while WT C57BL/6 and IL-6 KO mice were infected with 40–60 eggs to establish a chronic infection. Worm burden analysis was conducted as previously described [24]. Mice were treated orally with vehicle or an RARα/β agonist (Am80; 20 mg/kg (dose taken from [21]), obtained from Hiroyuki Kagechika, Tokyo Medical and Dental University, Tokyo, Japan) daily from day 21 to day 35 post-infection. Am80 was dissolved in 2 % DMSO, 40 % PEG, 58 % water and 2 drops of Tween 20. Vehicle treatment consisted of this mixture without the active compound.
Histology
Proximal colon tissue was fixed in neutral-buffered formalin and embedded in paraffin wax. Sections of 5 μm thickness were de-waxed with Citroclear and rehydrated through decreasing concentrations of ethanol. Haematoxylin and eosin staining was used to study the gut morphology. ImageJ analysis software (National Institutes of Health) was used to measure proximal colon crypt length. Twenty measurements from each tissue section were taken randomly.
Immunohistochemistry
Tissue samples from the proximal colon were frozen in Optimal Cutting Temperature (OCT) embedding medium (R.A. Lamb, Eastbourne, U.K.) and cut into 5 μm sections (Micron HM560 cryostat). CD4+ cells and FoxP3+ cells were identified by immunohistochemistry. Briefly, tissue samples were fixed in 4 % paraformaldehyde in PBS. Endogenous peroxidase activity was quenched using glucose/azide solution with glucose oxidase (Sigma). Samples were blocked with 7 % rat serum followed by an avidin/biotin blocking step. Biotin-conjugated rat anti-mouse CD4 (eBioscience) was used at 5 μg/ml in PBS. Rat anti-mouse FoxP3 antibody (eBioscience) was used at 2.5 μg/ml in PBS followed by goat anti-rat secondary (Santa Cruz) at 1:400. The avidin-biotin complex (ABC Kit, Vector Laboratories), 3,3′-diaminobenzidine (DAB Kit, Vector Laboratories), and haematoxylin (haematoxylin QS, Vector Laboratories) steps were then conducted. CD4+ cells and FoxP3+ cells were assessed by counting the number of positively-stained cells per 20 crypt units, in three sections per mouse.
Cell Culture and Cytokine Analysis
MLNs were removed from mice at day 35 post-infection and the cells were resuspended in medium (RPMI 1640, Invitrogen) supplemented with 10 % FCS, 1 % L-glutamine and 100U/ml penicillin/100μg/ml streptomycin (all from PAA Laboratories, GmbH, UK) at a concentration of 5 × 106/ml. Cell cultures were stimulated with 50 μg/ml of T. muris excretory/secretory antigen at 37 °C. For the T cell stimulation assay, 1 × 106 MLN cells in 1 ml were added to a 48-well plate pre-coated with anti-CD3 antibody (3μg/ml) (eBioscience). Anti-CD28 antibody (BD Biosciences) was added at 5μl/well. Cells were left alone or treated with either 100 nM Am80, 1μM Am80 or vehicle (0.4 % DMSO). Plates were then incubated at 37 °C/5 % CO2 in 95 % humidity for 48 h. Bone marrow-derived macrophages (BMDM) were isolated from C57BL/6 wild-type mice by flushing femurs with Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) containing 10 % FCS, 1 % L-glutamine and 100U/ml penicillin/100μg/ml streptomycin (all from PAA Laboratories, GmbH, UK) (complete DMEM). Cells were washed and cultured at 1 × 106/ml in complete DMEM containg 30 ng/ml M-CSF (eBioscience) for 7–10 days, with the media being replaced after 4–5 days. Macrophage purity was assessed by staining with 7AAD viability stain (eBioscience), anti-CD11b-APC-Cy7, anti-F4/80-APC and anti-CD11c-PE-Cy7 (eBiosciences) and aquirng on a LSR II flow cytometer (BD Biosciences). In all cultures used, the percentage of 7AAD−CD11b+F480+ cells were found to be over 72 %. The BMDMs (0.5 × 106) were adhered to 48 well plates for 48 h in the presence of complete DMEM media alone or with T. muris E/S (50 ng/ml). Two differing concentrations of Am80 (1μM or 100 nM) were also added to the appropriate wells. CMT93, a mouse rectal epithelial cell line was grown in complete DMEM in 48 well plates. When confluent, cells were left for 48 h in the presence of complete DMEM media alone or with T. muris E/S (50 ng/ml). Again Am80 (1μM or 100 nM) was also added to the appropriate wells. All supernatants were stored at −20 °C until analyzed for cytokine content. Supernatants were assessed for IL-4, IL-5, IL-6, IL-9, IL-10, IL-13, IL-17a, IFN-γ, TNF-α and CCL2 using a customised cytometric bead array (CBA) kit (BD Biosciences).
Statistical Analyses
Statisical analysis was performed using either the Student’s unpaired t-test, or one way ANOVA (with Tukey’s multiple comparison post-hoc test) as appropriate with the statistical package GraphPad Prism (Version 3). A p value of less than 0.05 was considered significant.
Results
Activating RAR Increases Intestinal Inflammation During Chronic T. muris Infection Through an IL-6 Dependent Mechanism
Given previous work showing a beneficial effect for retinoic acid and activation of RAR in gut inflammation [18, 25] we hypothesised that supplementing with a synthetic analogue of retinoic acid during worm persistence may reduce intestinal inflammation. Am80 is widely used in vivo in the mouse as an agonist of RAR [15, 26, 27]. However, a single preliminary experiment delivering Am80 to high-dose infected susceptible AKR mice (Fig S1A) from day 21 onwards, revealed significantly exacerbated gut pathology. Thus, comparing Am80-treated chronically infected mice with vehicle controls revealed a significant increase in crypt lengths (Fig S1B and C) and exacerbated CD4+ T cell infiltrate into the colon (Fig S1D and E) in the former. Furthermore, we identified significantly elevated levels of the pro-inflammatory cytokine IL-6 in both the MLN and the gut (Fig. S1F and G). To dissect these observations further, we chose to use the low dose C57BL/6 chronic infection model [8], and explored the role of IL-6 in the Am80-driven pathology through the use of IL-6 KO mice on a C57BL/6 background. As both C57BL/6 and IL-6 KO mice are resistant to a high-dose infection (Hurst et al., unpublished observations), C57BL/6 mice (wild-type:WT, IL-6+/+) and IL-6 KO (IL-6−/−) mice were infected with a low dose (60 T. muris eggs) infection and treated from day 21 to day 35 post-infection with vehicle or an RARα/β agonist (Am80), during the development of a persistent infection and chronic inflammation. A low dose infection allows the development of adult worms in all mouse strains [8].
As expected, at day 35 p.i. all mice harboured a low-dose chronic infection with a spread of patent adult worm burdens which is not atypical of this type of infection (Fig. 1a). WT mice displayed increased crypt lengths following treatment with Am80 during T. muris infection (p < 0.05 compared to vehicle controls) (Fig. 2b and c). Interestingly, this crypt cell hyperplasia was not observed in IL-6 KO mice treated with Am80: indeed crypt lengths were significantly lower compared to Am80-treated wild-type mice (p < 0.05) (Fig. 1b and c). Crypt lengths in vehicle-treated wild-type and vehicle-treated IL-6 KO mice post-infection were similar, indicating that the decrease in mucosal inflammation seen in Am80-treated IL-6 KO mice does not simply reflect the absence of the pro-inflammatory mediator IL-6. There was no effect of Am80 in uninfected animals (Fig. 1b and c).
Fig. 1.

Activation of the retinoic acid receptor (RAR) increases pathology during T. muris infection but not in the absence of IL-6. a Wild-type or IL-6 KO mice were infected with 60 T. muris eggs to establish a chronic infection and treated with either vehicle or Am80. At day 35 p.i. worm burdens were determined. b Paraffin-embedded proximal colon sections were stained with haemotoxylin and eosin (H&E) and crypt lengths measured using Image J software. c Representative images of T. muris-infected proximal colon sections stained with H&E. The arrows demonstrate examples of the measurements which were taken. d Immunohistochemical staining of CD4+ cells was conducted on proximal colon sections from wild-type and IL-6 knock-out animals at day 35 p.i. Quantitative analysis of immunohistochemistry was carried out by counting the number of positively-stained CD4+ cells. e A representative image of each infected treatment group is shown, where positively stained cells are brown. Scale bars indicate 200 μm. Graphs show means + SEM (n = 5). Data are representative of two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001
Fig. 2.

Am80 treatment of T. muris-infected mice increases the production of IL-6. In the absence of IL-6, Am80 can still imprint gut homing on MLN T cells. Wild-type or IL-6 KO mice were infected with a low-dose of T. muris eggs to induce a chronic infection and treated with either vehicle or Am80 between days 21 and 35p.i.. At day 35p.i. MLN cells were isolated. a 5 × 106 MLN cells were cultured with T. muris Excretory/Secretory (E/S) antigen at 50μg/ml for 48 h. Levels of IL-9, IL-13, IFN-γ, TNF-α, IL-6, IL-10 and IL-17A were measured by CBA. Data are representative of two independent experiments. b Flow cytometric analysis of MLN cells. Graph shows the percentage of 7AAD−CD4+ cells that are α4β7+. All graphs show means + SEM (n = 4–8). *p < 0.05, **p < 0.01, ***p < 0.001, ns = no significance
Previously, retinoic acid has been shown to boost T cell proliferation through RAR [28], as well as their migration to the gut via up-regulation of gut homing receptors [29]. Therefore, we quantified the CD4+ infiltrate during T. muris infection of the proximal colon by immunohistochemical staining in chronically infected wild-type and IL-6 KO mice. In both wild-type and IL-6 KO mice, the numbers of CD4+ T cells in the proximal colon increased post-infection (Fig. 1d and e). Am80-treatment lead to a further increase in the number of CD4+ T cells in the wild-type animals (p < 0.01), however, this did not occur in IL-6 KO mice (Fig 1d and e). Indeed in IL-6 KO mice treated with Am80, the number of CD4+ T cells in the gut was significantly lower than in Am80-treated wild-type mice (p < 0.05) (Fig. 1d and e).
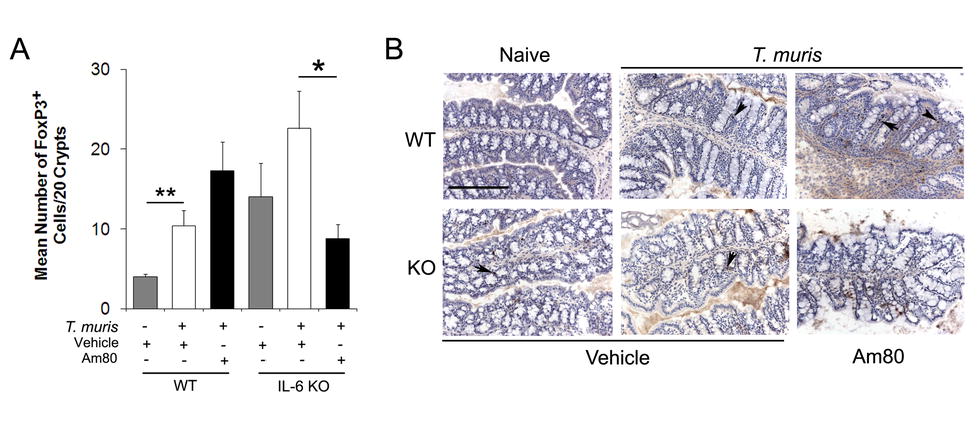
In order to further determine the identity of these CD4+ T cells infiltrating the gut following Am80 treatment of T. muris-infected mice, FoxP3+ regulatory T cells (Tregs) were quantified in the proximal colon of WT and IL-6 KO mice. The pooled results of two independent experiments suggest that in WT mice, the numbers of FoxP3+ cells in the proximal colon increased post-infection, however, there was no significant affect on FoxP3+ cell numbers with Am80 treatment of WT mice (Fig. S2), suggesting that they do not account for the additional CD4+ infiltrate driven by Am80 treatment of WT mice. Furthermore, Am80 treatment of IL-6 KO mice significantly decreased the numbers of FoxP3+ cell numbers in the gut compared to vehicle controls, which would not help to explain the decreased inflammation observed in this group (Fig. S2). Overall, these data suggest that the Am80-driven CD4+ T cell infiltrate seen during chronic T. muris infection is dependent on the production of IL-6, but does not involve the generation of Tregs.
Parasite-specific cytokine responses of MLN cells were analysed in order to explore any correlations between elevations in pro-inflammatory or regulatory cytokines and the exaggerated pathology seen with Am80 treatment. In wild-type (WT) mice, Am80 treatment had no significant effect on the levels of the pro-inflammatory cytokines IFN-γ, IL-17A and TNF-α compared to vehicle controls (Fig. 2a). IL-10 and the Th2 cytokines; IL-9 and IL-13, were raised in Am80-treated infected mice compared to vehicle controls, although, this failed to reach statistical significance (Fig. 2a). However, a dramatic rise in IL-6 production was seen in MLNs from Am80-treated infected mice compared to vehicle controls (Fig. 2a), suggesting a role for IL-6 in Am80-driven pathology. No effects of Am80 were observed in naïve wild-type mice (Fig. 2a).
In infected IL-6 KO mice, levels of IFN-γ, IL-17A, IL-10 and TNF-α were increased with Am80 treatment (p < 0.05, p < 0.05, p < 0.01 and p < 0.01, respectively, compared to vehicle controls) (Fig. 2a), although, importantly, levels were not significantly different to those seen in WT mice treated with Am80, suggesting that the Am80-driven increases are independent of IL-6, unlike the pathological changes shown in Fig. 1. Levels of IL-9 and IL-13 did not change significantly (p > 0.05) in IL-6 KO mice with Am80-treatment compared to IL-6 KO vehicle controls. Although levels of IL-9 and IL-13 produced by MLNs from IL-6 KO mice treated with Am80 were reduced compared to MLNs from WT mice treated with Am80, this difference was not statistically significant in both independent experiments (Fig. 2a). Therefore, it is unlikely that changes in these cytokines can help to explain the decreased pathology observed in IL-6 KO mice treated with Am80 compared to WT mice. By definition, the Am80-driven increase in IL-6 seen in WT mice was not observed in IL-6 KO mice (p < 0.001), both validating the KO and supporting the role for IL-6 in Am80-driven pathology. No effects of Am80 were observed in naïve IL-6 KO mice (Fig. 2a).
Am80 Imprints Gut Homing on T Cells, But This is Not IL-6 Dependent
To determine whether the CD4+ infiltrate seen with Am80-treatment of T. muris-infected mice was due to enhanced gut homing we isolated MLN cells from Am80-treated wild-type and IL-6 KO mice and stained them with antibodies for α4β7. Flow cytometric analysis revealed an increase in the percentage of α4β7-positive cells in the MLNs of both Am80-treated wild-type and IL-6 KO mice (p < 0.001 compared to vehicle controls). This Am80-driven increase occurred in both uninfected and infected mice (p < 0.001 in wild-type mice compared to vehicle controls) (Fig. 2b).
To determine whether the Am80-driven CD4+ infiltrate observed in T. muris-infected mice was due to increases in gut chemokines attracting T cells to the site of infection, qPCRs were carried out on gut tissue from Am80-treated wild-type and IL-6 KO mice infected with T. muris to day 35 p.i. The expression of the T cell chemokines CCL3, CCL5, CXCL10, CXCL11, CCL20 and CCL28 was analysed. No differences were observed at this time point that could account for the Am80-driven CD4+ infiltrate (data not shown). Overall, these results suggest that neither an increase in CD4+ T cell gut homing nor enhanced chemotaxis underlie the IL-6 dependent Am80-driven T cell infiltrate observed in T. muris-infected mice.
Am80 Drives IL-6 Production from T Cells During T. muris Infection
As the Am80-dependent increase in pathology was dependent on IL-6, we went on to determine a possible cellular source of the Am80-driven increase in IL-6 production during T. muris infection. MLNs were taken from naïve and infected, wild-type mice at day 21 p.i. and T cells were stimulated using anti-CD3 and anti-CD28 antibodies, and cultured with vehicle, Am80 (1μM) or Am80 (100 nM) in vitro. T cells from naïve, wild-type mice produced low levels of IL-6 and the addition of Am80 at either concentration had no affect on IL-6 production (Fig. 3a). T cells from T. muris-infected mice however, produced higher levels of IL-6 than naïve T cells (Fig. 3a). Addition of Am80 (1μM) led to a significant increase in IL-6 levels compared to both untreated cells (p < 0.05) and vehicle treated cells (p < 0.001) (Fig. 3a). Levels of IL-6 produced by T cells cultured with 100nM Am80 were not significantly different from vehicle-treated controls, indicating a dose-dependent effect (Fig. 3a).
Fig. 3.

T cells, but not macrophages or epithelial cells, display enhanced IL-6 production following Am80-treatment in vitro. a Wild-type C57BL/6 mice were infected with a high dose of T. muris eggs and MLN cells extracted at day 21 p.i.. 1 × 106 T cells were stimulated using anti-CD3 (3μg/ml) and anti-CD28 (5μl/well) antibodies, and treated with vehicle, Am80 (1μM) or Am80 (100 nM) in vitro for 48 h. Similar data was obtained in an independent experiment sourcing T cells from a low dose infection. b Bone marrow-derived macrophages were grown from wild-type C57BL/6 mice and 0.5 × 106 were stimulated in vitro with T. muris Excretory-Secretory antigens (E/S) with or without Am80 (1 μM) or Am80 (100 nM) for 48 h. c The mouse rectal epithelial cell line CMT93 was grown and 0.5 × 106 cells were stimulated in vitro with T. muris Excretory-Secretory antigens (E/S) with or without Am80 (1 μM) or Am80 (100 nM) for 48 h. Values are means + SEM. The data are representative of two independent experiments. (n = 3–4). *p < 0.05, ***p < 0.001, ns = no significance
Bone marrow-derived macrophages were cultured from wild-type mice and stimulated in vitro with T. muris E/S antigens. This increased the production of IL-6 by the macrophages compared to media only controls (p < 0.001) (Fig. 3b), however, the addition of 1μM or 100 nM Am80 had no effect on the production of IL-6 and the levels were not significantly altered from control values (Fig. 3b).
The mouse rectal epithelial cell line CMT93 was used to investigate whether Am80 alters the production of IL-6 by epithelial cells. Stimulating these cells with T. muris E/S antigens increased the production of IL-6 by the epithelial cells (p < 0.01) (Fig. 3c) but no change in IL-6 production was observed with the addition of 1μM or 100nM Am80 (Fig. 3c). These results suggest that T cells, but not macrophages or epithelial cells are able to respond to Am80 treatment by increasing their production of IL-6, which may drive pro-inflammatory response in the infected gut. Thus, T cells may be the cellular source of the enhanced IL-6 production observed following the treatment of T. muris-infected mice with Am80.
Discussion
Vitamin A and its metabolites are biologically active agents that can mediate immunological effects through modulation of their nuclear hormone receptor RAR. Previously, retinoic acid and activation of RAR have been shown to protect against colitis, rheumatoid arthritis, and Trichinella spiralis infection [15, 18, 30]. However, there are still many unanswered questions regarding the immunomodulatory effects of retinoic acid in chronic mucosal inflammation and parasitic infections. Here we present novel data showing a role for retinoic acid in T. muris infection, which gives further insight into the function of RAR in a biologically relevant model of gut inflammation.
Many reports have shown that retinoic acid modulates the Th1/Th2 balance in favour of Th2 responses [16, 31]. The ability to expel T. muris is directly related to the development of a Th2 immune response [32]. IL-9 and IL-13 are important cytokines in immunity to intestinal nematodes [33–35]. However, in the current study, only a non-significant increase in Th2 cytokines was observed in the MLNs of mice treated with the RAR agonist Am80 and levels were not sufficient to induce resistance to the low-dose infection as worms were still present at day 35 p.i.. In contrast, previous studies have shown that vitamin A deficiency in mice causes increased susceptibility to acute infections with Nippostrongylus [36], increased IFN-γ production and delayed expulsion following acute Trichinella spiralis infection [30], as well as higher parasite burdens and reduced Th2 cytokine responses in Schistosoma mansoni-infected rats and mice [37, 38]. Although it is currently unclear why, there was a trend towards lower cytokine levels in the vehicle-treated IL-6KO mice compared to WT controls. These levels are amplified in the presence of Am80. Furthermore, we have unpublished data showing that mice which lack the IL-6 receptor on T cells (CD4-Cre GP130flf/mice) are still able to make Th1, Th2, Treg and Th17 derived cytokines (Hurst & Else, unpublished findings). Thus, it is unlikely that T cells are unable to respond to parasite antigen in the absence of IL-6. Equally, it is unlikely that low cytokine levels reflect low worms numbers, as the majority of mice in the WT vehicle-treated group harbor the same number of adult worms as seen in the IL-6 KO vehicle-treated group.
Importantly, our experiments showed that Am80, an RARα/β agonist, exaggerated the pathology caused by chronic T. muris infection of mice, demonstrated by a crypt hyperplasia and CD4+ T cell infiltrate in this treatment group. This is surprising, given previous reports showing the beneficial effects of Am80 in DSS colitis [25], EAE [27] and GvH disease [39]. Although, we identify IL-6 as a key component of the mechanism underlying this Am80-driven increase in mucosal inflammation, the downstream events remains unclear. Retinoic acid is well-known for its ability to imprint gut homing on CD4+ T cells via an up-regulation of the gut homing receptor α4β7 [29], and we also observed this in MLNs of mice treated with Am80. However, we found this not to be dependent on the presence of T. muris infection or IL-6, suggesting that this is also not the mechanism behind the IL-6 dependent Am80-driven increase in CD4+ T cells in the colon. The expression of CD4+ T cell chemoattractants in the gut (CCL3, CCL5, CXCL10, CXCL11, CCL20 and CCL28) also failed to account for the accumulation of CD4+ T cells in Am80-treated wild-type mice post T. muris infection, however, this analysis was carried out on gut tissue from day 35 p.i. which may have been too late to identify chemokines responsible for recruiting CD4+ T cells into the gut during the infection period. IL-6 is able to increase leukocyte adhesion via L-selectin [40], and blocking RXR can reduce the expression of ICAM-1 and VCAM-1 on the endothelium [41]. Therefore it is possible that local changes in adhesion molecule expression may, at least in part, explain the increased leukocyte presence in the gut following Am80 treatment and its reduction in the absence of IL-6, although further work is needed to confirm this.
Despite the established role played by RA in the induction of Tregs [17] and the role played by IL-6 in driving Th17 development [42], we have no evidence to suggest that the increased inflammation caused by activation of RAR in WT mice is due to increased Th17 cell numbers or that the decrease in inflammation in the absence of IL-6 is due to elevated Treg numbers. Thus, IL-17 and IL-10 levels in the MLN cells of WT mice were not significantly changed following Am80-treatment and intestinal Treg numbers in Am80-treated mice were also unaltered.
From these findings, we suggest that Am80 promotes IL-6 transcription and production leading to increased gut inflammation. The mouse IL-6 gene is reported to have a putative RARE binding site [43], however we currently cannot distinguish whether the Am80-driven increase in IL-6 in our model of gut inflammation is due to a direct or indirect effect on the transcription of this gene. Indeed, previous work has shown that Am80 treatment can decrease IL-6 production in an in vivo model of collagen-induced arthritis and in a human osteosarcoma cell line [15] which may suggest that other factors are involved in the regulation of IL-6 in our model. Alternatively, differing concentrations of Am80 or lengths of treatments may determine the transcriptional regulation of inflammatory genes, including IL-6. This is supported by a report which shows that Am80 treatment is beneficial in acute EAE by inhibiting the induction of Th17 cells, but not in chronic EAE where a prolonged treatment with Am80 caused suppression of IL-10 production by T cells [27].
CD4+ T cells are a major producer of IL-6 [44] and we have shown that administration of Am80 to stimulated CD4+ T cells can result in the increased production of IL-6 in vitro. Therefore, CD4+ T cells may be the major source of IL-6, controlled directly or indirectly by the RAR agonist Am80, in our experiments. Furthermore, it has been shown that naive CD4+ T cells express RARα, and that TCR stimulation is required for their response to RA [45]. Therefore, CD4+ T cells certainly have the ability to respond to retinoic acid and Am80 during infection with T. muris. We have also used bone marrow-derived macrophages and the mouse rectal epithelial cell line CMT93 to show that macrophages and epithelial cells do not respond to Am80 in vitro to produce elevated levels of IL-6. However, other investigators have shown that intestinal pig epithelial cells predominantly produce IL-10 and IL-6 following stimulation with T. suis E/S, and so these cells may also be responsible for at least some of the increased IL-6 post Am80-treatment [46].
In conclusion, we show that delivery of the RAR agonist Am80 during a chronic T. muris infection leads to increased Th2 responses, but these are insufficient to induce resistance to the parasite. Further, Am80 treatment leads to an augmented inflammatory response in the gut mucosa demonstrated by increased CD4+ T cell infiltration and exaggerated crypt hyperplasia. We find that these parameters are dependent on IL-6. These data are in opposition to previous reports where retinoic acid and activation of both RAR and RXR have been shown to down-regulate inflammatory processes in both human ulcerative and murine colitis [18, 21]. Vitamin A supplementation has been widely used to lower morbidity in children and adults harbouring parasitic infections [47]. However, our findings add caution to the practice of supplementing parasite-infected individuals with vitamin A, due to the increased production of the pro-inflammatory cytokine IL-6 following RAR activation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
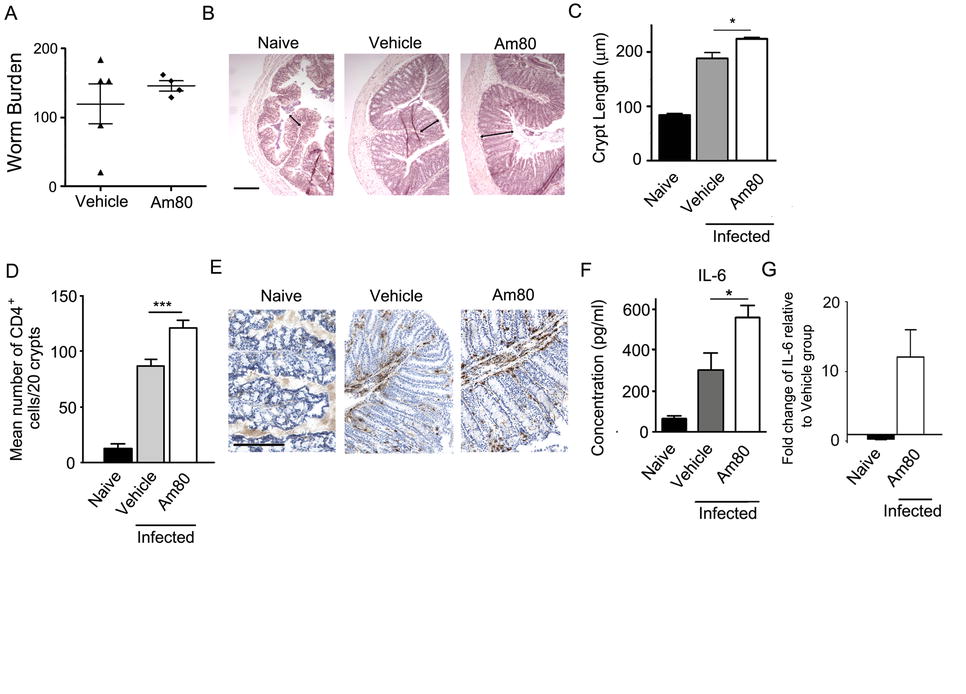
Activation of the retinoic acid receptor (RAR) increases pathology during T. muris infection. (A) AKR mice, treated with either vehicle or an RAR agonist (Am80), were infected with T. muris for 35 days and worm burdens were determined. (B) Paraffin-embedded proximal colon sections were stained with haemotoxylin and eosin (H&E) and images of H&E-stained proximal colon sections were taken. Scale bar indicates 200 μm. (C) Crypt lengths were measured using Image J software as indicated by arrows in B. (D) Immunohistochemical staining of CD4+ cells was conducted on proximal colon sections at day 35 p.i. Quantitative analysis of immunohistochemistry was carried out by counting the number of positively-stained cells per 20 crypts. (E) A representative image of each treatment group is shown, where positively stained cells are brown. (F) MLN cells were isolated 35 days p.i. and 5 × 106 were cultured with T. muris Excretory/Secretory (E/S) antigen at 50 μg/ml for 24 hours. Levels of IL-6 were measured by CBA. (G) Total RNA from whole proximal colon tissue was recovered from uninfected (naive) mice and T. muris-infected mice treated with Am80 or vehicle. Measurements of IL-6 by quantitative-RT-PCR are expressed as fold changes over infected mice treated with vehicle, after normalisation to 18s. Graphs show means+SEM (n = 4 or 5). Scale bars indicate 200μm.*p < 0.05, ***p < 0.001. (JPEG 74 kb)
{kind=link}
Am80 treatment of T. muris-infected mice does not cause an increase in the number of FoxP3+ cells in the gut. (A) Proximal colon sections from wild-type and IL-6 knock-out animals at day 35 p.i. were stained by immunohistochemistry for FoxP3+ cells. Quantitative analysis was carried out by counting the number of positively-stained FoxP3+ cells. (B) A representative image of each infected treatment group is shown, where positively stained cells are brown. Scale bars indicate 200 μm. Graphs show means + SEM (n = 9–13). Data is representative of the pooled results from two independent experiments. *p < 0.05, **p < 0.01. (JPEG 72 kb)
Acknowledgments
Work was supported by the Wellcome trust (Grant number: 091815) and BBSRC.
Conflicts of Interest
The authors declare that they have no conflict of interest.
Abbreviations
- IBD
Inflammatory bowel disease
- NHR
Nuclear hormone receptor
- RA
All-trans-retinoic acid
- RAR
Retinoic acid receptor
- RARE
Retinoic acid response element
- RXR
Retinoid X receptor
- Treg
Regulatory T cell
References
- 1.Bundy DA, Cooper ES. Trichuris and trichuriasis in humans. Adv Parasitol. 1989;28:107–173. doi: 10.1016/S0065-308X(08)60332-2. [DOI] [PubMed] [Google Scholar]
- 2.Cliffe LJ, Grencis RK. The Trichuris muris system: a paradigm of resistance and susceptibility to intestinal nematode infection. Adv Parasitol. 2004;57:255–307. doi: 10.1016/S0065-308X(04)57004-5. [DOI] [PubMed] [Google Scholar]
- 3.Genta RM. Diarrhea in helminthic infections. Clin Infect Dis. 1993;16(Suppl 2):S122–S129. doi: 10.1093/clinids/16.Supplement_2.S122. [DOI] [PubMed] [Google Scholar]
- 4.Levison SE, McLaughlin JT, Zeef LA, Fisher P, Grencis RK, Pennock JL. Colonic transcriptional profiling in resistance and susceptibility to trichuriasis: phenotyping a chronic colitis and lessons for iatrogenic helminthosis. Inflamm Bowel Dis. 2010;16(12):2065–2079. doi: 10.1002/ibd.21326. [DOI] [PubMed] [Google Scholar]
- 5.Else KJ. deSchoolmeester ML. Immunity to Trichuris muris in the laboratory mouse. J Helminthol. 2003;77(2):95–98. doi: 10.1079/JOH2002162. [DOI] [PubMed] [Google Scholar]
- 6.Else KJ, Grencis RK. Cellular immune responses to the murine nematode parasite Trichuris muris. I. Differential cytokine production during acute or chronic infection. Immunology. 1991;72(4):508–513. [PMC free article] [PubMed] [Google Scholar]
- 7.Blackwell NM, Else KJ. A comparison of local and peripheral parasite-specific antibody production in different strains of mice infected with Trichuris muris. Parasite Immunol. 2002;24(4):203–211. doi: 10.1046/j.1365-3024.2002.00452.x. [DOI] [PubMed] [Google Scholar]
- 8.Bancroft AJ, Else KJ, Grencis RK. Low-level infection with Trichuris muris significantly affects the polarization of the CD4 response. Eur J Immunol. 1994;24(12):3113–3118. doi: 10.1002/eji.1830241230. [DOI] [PubMed] [Google Scholar]
- 9.Mora JR, Iwata M, von Andrian UH. Vitamin effects on the immune system: vitamins A and D take centre stage. Nat Rev. 2008;8(9):685–98. [DOI] [PMC free article] [PubMed]
- 10.Blomhoff R, Blomhoff HK. Overview of retinoid metabolism and function. J Neurobiol. 2006;66(7):606–630. doi: 10.1002/neu.20242. [DOI] [PubMed] [Google Scholar]
- 11.Cantorna MT, Nashold FE, Hayes CE. In vitamin A deficiency multiple mechanisms establish a regulatory T helper cell imbalance with excess Th1 and insufficient Th2 function. J Immunol. 1994;152(4):1515–1522. [PubMed] [Google Scholar]
- 12.Cui D, Moldoveanu Z, Stephensen CB. High-level dietary vitamin A enhances T-helper type 2 cytokine production and secretory immunoglobulin A response to influenza A virus infection in BALB/c mice. J Nutr. 2000;130(5):1132–1139. doi: 10.1093/jn/130.5.1132. [DOI] [PubMed] [Google Scholar]
- 13.Racke MK, Burnett D, Pak SH, Albert PS, Cannella B, Raine CS, et al. Retinoid treatment of experimental allergic encephalomyelitis. IL-4 production correlates with improved disease course. J Immunol. 1995;154(1):450–458. [PubMed] [Google Scholar]
- 14.Niwa S, Ochi T, Hirano Y, Wang T, Inagaki N, Shudo K, et al. Effect of Am-80, a retinoid derivative, on 2, 4-dinitrofluorobenzene-induced contact dermatitis in mice. Pharmacology. 2000;60(4):208–214. doi: 10.1159/000028371. [DOI] [PubMed] [Google Scholar]
- 15.Nagai H, Matsuura S, Bouda K, Takaoka Y, Wang T, Niwa S, et al. Effect of Am-80, a synthetic derivative of retinoid, on experimental arthritis in mice. Pharmacology. 1999;58(2):101–112. doi: 10.1159/000028272. [DOI] [PubMed] [Google Scholar]
- 16.Lovett-Racke AE, Racke MK. Retinoic acid promotes the development of Th2-like human myelin basic protein-reactive T cells. Cell Immunol. 2002;215(1):54–60. doi: 10.1016/S0008-8749(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 17.Kang SG, Lim HW, Andrisani OM, Broxmeyer HE, Kim CH. Vitamin A metabolites induce gut-homing FoxP3+ regulatory T cells. J Immunol. 2007;179(6):3724–3733. doi: 10.4049/jimmunol.179.6.3724. [DOI] [PubMed] [Google Scholar]
- 18.Bai A, Lu N, Guo Y, Liu Z, Chen J, Peng Z. All-trans retinoic acid down-regulates inflammatory responses by shifting the Treg/Th17 profile in human ulcerative and murine colitis. J Leukoc Biol. 2009;86(4):959–969. doi: 10.1189/jlb.0109006. [DOI] [PubMed] [Google Scholar]
- 19.Long KZ, Estrada-Garcia T, Rosado JL, Ignacio Santos J, Haas M, Firestone M, et al. The effect of vitamin A supplementation on the intestinal immune response in Mexican children is modified by pathogen infections and diarrhea. J Nutr. 2006;136(5):1365–1370. doi: 10.1093/jn/136.5.1365. [DOI] [PubMed] [Google Scholar]
- 20.Payne LG, Koski KG, Ortega-Barria E, Scott ME. Benefit of vitamin A supplementation on ascaris reinfection is less evident in stunted children. J Nutr. 2007;137(6):1455–1459. doi: 10.1093/jn/137.6.1455. [DOI] [PubMed] [Google Scholar]
- 21.Desreumaux P, Dubuquoy L, Nutten S, Peuchmaur M, Englaro W, Schoonjans K, et al. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimer. A basis for new therapeutic strategies. J Exp Med. 2001;193(7):827–838. doi: 10.1084/jem.193.7.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, et al. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature. 1994;368(6469):339–342. doi: 10.1038/368339a0. [DOI] [PubMed] [Google Scholar]
- 23.Wakelin D. Acquired immunity to Trichuris muris in the albino laboratory mouse. Parasitology. 1967;57(3):515–524. doi: 10.1017/S0031182000072395. [DOI] [PubMed] [Google Scholar]
- 24.Else KJ, Wakelin D. Genetically-determined influences on the ability of poor responder mice to respond to immunization against Trichuris muris. Parasitology. 1990;100(Pt 3):479–489. doi: 10.1017/S0031182000078793. [DOI] [PubMed] [Google Scholar]
- 25.Wada Y, Hisamatsu T, Kamada N, Okamoto S, Hibi T. Retinoic acid contributes to the induction of IL-12-hypoproducing dendritic cells. Inflamm Bowel Dis. 2009;15(10):1548–1556. doi: 10.1002/ibd.20934. [DOI] [PubMed] [Google Scholar]
- 26.Nishimori H, Maeda Y, Teshima T, Sugiyama H, Kobayashi K, Yamasuji Y, et al. Synthetic retinoid Am80 ameliorates chronic graft-versus-host disease by down-regulating Th1 and Th17. Blood. 2012;119(1):285–295. doi: 10.1182/blood-2011-01-332478. [DOI] [PubMed] [Google Scholar]
- 27.Klemann C, Raveney BJ, Klemann AK, Ozawa T, von Horsten S, Shudo K, et al. Synthetic retinoid AM80 inhibits Th17 cells and ameliorates experimental autoimmune encephalomyelitis. Am J Pathol. 2009;174(6):2234–2245. doi: 10.2353/ajpath.2009.081084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ertesvag A, Engedal N, Naderi S, Blomhoff HK. Retinoic acid stimulates the cell cycle machinery in normal T cells: involvement of retinoic acid receptor-mediated IL-2 secretion. J Immunol. 2002;169(10):5555–5563. doi: 10.4049/jimmunol.169.10.5555. [DOI] [PubMed] [Google Scholar]
- 29.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity. 2004;21(4):527–538. doi: 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 30.Carman JA, Pond L, Nashold F, Wassom DL, Hayes CE. Immunity to Trichinella spiralis infection in vitamin A-deficient mice. J Exp Med. 1992;175(1):111–120. doi: 10.1084/jem.175.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoag KA, Nashold FE, Goverman J, Hayes CE. Retinoic acid enhances the T helper 2 cell development that is essential for robust antibody responses through its action on antigen-presenting cells. J Nutr. 2002;132(12):3736–3739. doi: 10.1093/jn/132.12.3736. [DOI] [PubMed] [Google Scholar]
- 32.Else KJ, Finkelman FD, Maliszewski CR, Grencis RK. Cytokine-mediated regulation of chronic intestinal helminth infection. J Exp Med. 1994;179(1):347–351. doi: 10.1084/jem.179.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bancroft AJ, McKenzie AN, Grencis RK. A critical role for IL-13 in resistance to intestinal nematode infection. J Immunol. 1998;160(7):3453–3461. [PubMed] [Google Scholar]
- 34.Bancroft AJ, Artis D, Donaldson DD, Sypek JP, Grencis RK. Gastrointestinal nematode expulsion in IL-4 knockout mice is IL-13 dependent. Eur J Immunol. 2000;30(7):2083–2091. doi: 10.1002/1521-4141(200007)30:7<2083::AID-IMMU2083>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 35.Faulkner H, Renauld JC, Van Snick J, Grencis RK. Interleukin-9 enhances resistance to the intestinal nematode Trichuris muris. Infect Immun. 1998;66(8):3832–3840. doi: 10.1128/iai.66.8.3832-3840.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.watt JY, Golden WR, Olason F, Mladinich G. The relationship of Vitamin A to resistence to nippostrongylus muris. Science (New York, NY) 1943;97(2521):381–382. doi: 10.1126/science.97.2521.381. [DOI] [PubMed] [Google Scholar]
- 37.Parent G, Rousseaux-Prevost R, Carlier Y, Capron A. Influence of vitamin A on the immune response of Schistosoma mansoni-infected rats. Trans R Soc Trop Med Hyg. 1984;78(3):380–383. doi: 10.1016/0035-9203(84)90127-5. [DOI] [PubMed] [Google Scholar]
- 38.Broadhurst MJ, Leung JM, Lim KC, Girgis NM, Gundra UM, Fallon PG, et al. Upregulation of retinal dehydrogenase 2 in alternatively activated macrophages during retinoid-dependent type-2 immunity to helminth infection in mice. PLoS Pathog. 2012;8(8):e1002883. doi: 10.1371/journal.ppat.1002883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishimori H, Maeda Y, Teshima T, Sugiyama H, Kobayashi K, Yamasuji Y, et al. Synthetic retinoid Am80 ameliorates chronic graft-versus-host disease by down-regulating Th1 and Th17. Blood. 2012;119(1):285–95. [DOI] [PubMed]
- 40.Rose-John S, Neurath MF. IL-6 trans-signaling: the heat is on. Immunity. 2004;20(1):2–4. doi: 10.1016/S1074-7613(04)00003-2. [DOI] [PubMed] [Google Scholar]
- 41.Sanz MJ, Albertos F, Otero E, Juez M, Morcillo EJ, Piqueras L. Retinoid X receptor agonists impair arterial mononuclear cell recruitment through peroxisome proliferator-activated receptor-gamma activation. J Immunol. 2012;189(1):411–424. doi: 10.4049/jimmunol.1102942. [DOI] [PubMed] [Google Scholar]
- 42.Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. 2010;40(7):1830–1835. doi: 10.1002/eji.201040391. [DOI] [PubMed] [Google Scholar]
- 43.Schug J. Using TESS to predict transcription factor binding sites in DNA sequence. Curr Protoc Bioinform. 2008;21:2.6.1–2.6.15. [DOI] [PubMed]
- 44.Zubiaga AM, Munoz E, Merrow M, Huber BT. Regulation of interleukin 6 production in T helper cells. Int Immunol. 1990;2(11):1047–1054. doi: 10.1093/intimm/2.11.1047. [DOI] [PubMed] [Google Scholar]
- 45.Ohoka Y, Yokota A, Takeuchi H, Maeda N, Iwata M. Retinoic acid-induced CCR9 expression requires transient TCR stimulation and cooperativity between NFATc2 and the retinoic acid receptor/retinoid X receptor complex. J Immunol. 2011;186(2):733–744. doi: 10.4049/jimmunol.1000913. [DOI] [PubMed] [Google Scholar]
- 46.Parthasarathy G, Mansfield LS. Trichuris suis excretory secretory products (ESP) elicit interleukin-6 (IL-6) and IL-10 secretion from intestinal epithelial cells (IPEC-1) Vet Parasitol. 2005;131(3–4):317–324. doi: 10.1016/j.vetpar.2005.03.043. [DOI] [PubMed] [Google Scholar]
- 47.Long KZ, Santos JI, Rosado JL, Lopez-Saucedo C, Thompson-Bonilla R, Abonce M, et al. Impact of vitamin A on selected gastrointestinal pathogen infections and associated diarrheal episodes among children in Mexico City, Mexico. J Infect Dis. 2006;194(9):1217–1225. doi: 10.1086/508292. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Activation of the retinoic acid receptor (RAR) increases pathology during T. muris infection. (A) AKR mice, treated with either vehicle or an RAR agonist (Am80), were infected with T. muris for 35 days and worm burdens were determined. (B) Paraffin-embedded proximal colon sections were stained with haemotoxylin and eosin (H&E) and images of H&E-stained proximal colon sections were taken. Scale bar indicates 200 μm. (C) Crypt lengths were measured using Image J software as indicated by arrows in B. (D) Immunohistochemical staining of CD4+ cells was conducted on proximal colon sections at day 35 p.i. Quantitative analysis of immunohistochemistry was carried out by counting the number of positively-stained cells per 20 crypts. (E) A representative image of each treatment group is shown, where positively stained cells are brown. (F) MLN cells were isolated 35 days p.i. and 5 × 106 were cultured with T. muris Excretory/Secretory (E/S) antigen at 50 μg/ml for 24 hours. Levels of IL-6 were measured by CBA. (G) Total RNA from whole proximal colon tissue was recovered from uninfected (naive) mice and T. muris-infected mice treated with Am80 or vehicle. Measurements of IL-6 by quantitative-RT-PCR are expressed as fold changes over infected mice treated with vehicle, after normalisation to 18s. Graphs show means+SEM (n = 4 or 5). Scale bars indicate 200μm.*p < 0.05, ***p < 0.001. (JPEG 74 kb)
Am80 treatment of T. muris-infected mice does not cause an increase in the number of FoxP3+ cells in the gut. (A) Proximal colon sections from wild-type and IL-6 knock-out animals at day 35 p.i. were stained by immunohistochemistry for FoxP3+ cells. Quantitative analysis was carried out by counting the number of positively-stained FoxP3+ cells. (B) A representative image of each infected treatment group is shown, where positively stained cells are brown. Scale bars indicate 200 μm. Graphs show means + SEM (n = 9–13). Data is representative of the pooled results from two independent experiments. *p < 0.05, **p < 0.01. (JPEG 72 kb)