

Abstract
Cornelia de Lange syndrome (CdLS) is a genetic disorder linked to mutations in cohesin and its regulators. To date, it is unclear which function of cohesin is more relevant to the pathology of the syndrome. A mouse heterozygous for the gene encoding the cohesin loader Nipbl recapitulates many features of CdLS. We have carefully examined Nipbl deficient cells and here report that they have robust cohesion all along the chromosome. DNA replication, DNA repair and chromosome segregation are carried out efficiently in these cells. While bulk cohesin loading is unperturbed, binding to certain promoters such as the Protocadherin genes in brain is notably affected and alters gene expression. These results provide further support for the idea that developmental defects in CdLS are caused by deregulated transcription and not by malfunction of cohesion-related processes.
Keywords: Cornelia de Lange Syndrome, Nibpl, cohesin, transcription, mouse model, cohesin, telomere fragility, DNA repair, mouse model, Cornelia de Lange syndrome
Introduction
Cornelia de Lange Syndrome (CdLS) is a developmental disorder affecting 1:30,000 newborns, that is characterized by mental retardation, reduced body size, dysmorphic face and upper limb defects among additional organ abnormalities [1]. This human syndrome has been linked to dysfunction of cohesin, a four-subunit protein complex (Smc1, Smc3, Rad21 and either SA1 or SA2) initially identified for its role in sister chromatid cohesion [2-5]. Cohesion is essential for accurate chromosome segregation and for homologous recombination (HR)-mediated DNA repair [6]. More recent studies have shown the ability of cohesin to embrace two DNA segments not only in trans but also in cis, leading to the formation of chromatin loops that allow efficient firing of DNA replication origins [7], promote recombination-mediated locus rearrangement [8, 9] and are the basis of transcriptional regulation of a number of loci [10-13]. To date, it is still unclear which of these functions is most directly related to the pathology of CdLS.
In almost half of CdLS patients, the syndrome is caused by heterozygous mutations in the cohesin loader Nipbl [14, 15]. Metaphase spreads from CdLS cells do not show consistently cohesion defects whereas microarray studies reveal altered patterns of gene expression [16, 17]. A mouse model that recapitulates CdLS has been generated through deletion of a NIPBL allele. Embryonic fibroblasts from these animals lack overt centromeric cohesion defects and instead display altered transcriptional profiles [18]. However, defects in telomere and arm cohesion, which cannot be scored in metaphase spreads, have not been analyzed in these cells and they have been shown to impact DNA replication, DNA repair and chromosome segregation. Indeed, we have recently reported that cells deficient in cohesin-SA1 show robust centromeric cohesion, a task performed by cohesin-SA2, but lack proper telomere cohesion. As a consequence, telomere replication is not efficient and leads to chromosome segregation defects and aneuploidy [19]. It is also not known whether limiting amounts of the cohesin loader may affect differentially cohesin-SA1 and cohesin-SA2. Thus, we decided to evaluate arm/telomere cohesion, chromosome segregation, DNA damage repair and cohesin loading in Nipbl deficient mouse embryonic fibroblasts (MEFs) in order to understand the contribution of these aspects of cohesin behavior to the pathogenesis of CdLS.
Material and Methods
Mouse handling and MEFs culture
Mice were housed in a pathogen-free animal facility according to the institution standards for animal care. MEFs were isolated from E14.5 embryos as described [18] and cultured in DMEM/10% FBS.
FISH (Fluorescence In Situ Hybridization)
MEFs in culture were arrested in 0.1 μg/ml colcemide for 4 h and harvested by trypsinization. Metaphases for FISH were prepared and hybridized with a telomeric probe as previously described [19]. Images were acquired using a Leica DM6000 microscope. In the indicated cases, cells were cultured in 0.5 μM aphidicolin for 24 h.
mRNA isolation and quantitative RT-PCR analysis
Total RNA was isolated using RNeasy Kit (Qiagen) and cDNA was synthesized with SuperScript™ II reverse transcriptase (Invitrogen) using random hexamer primers. An Applied Biosystems 7900HT Fast qRT-PCR was used to determine mRNA levels. GAPDH was used for normalization. Primers used for mRNA amplification are listed in Supplementary Table S1.
Extract preparation, immunoblotting and immunofluorescence
For whole-cell extracts, cells were collected by trypsinization, washed once in PBS, resuspended in SDS-PAGE loading buffer and sonicated. Equal amounts of protein were run in 7.5% Bis/Tris gels followed by western blotting. Chromatin fractionation was performed as previously described [20]. For immunofluorescence, cells were cultured on polylysine-coated coverslips, fixed in 2% formaldehyde for 10 min at room temperature, permeabilized in 0.1% Sodium Citrate / 0.1% Triton-X100 for 5 min at room temperature and subjected to antibody incubation. Images were acquired using a Leica DM6000 microscope. Antibodies used in this study were: SA1, SA2, Sororin [19], Smc1 and Smc3 [21], Rad21 [4], Nipbl (Bethyl, A301-779A), 53BP1 (Novus Biologicals, NB100-304), Histone 3 (phospho-Ser10) (Abcam, ab14955), α-tubulin (Sigma, DM1A), Histone 3 (Abcam, AB1791), Mek2 (BD, M24520). Antibodies against Wapl and Scc4 were a kind gift from J.M. Peters (IMP, Vienna) and E. Watrin (U. Rennes).
Survival upon damage induction
Exponentially growing cells were trypsinized and 50,000 cells per well were seeded onto 6-well plates. The following day, cells were subjected to irradiation (8 Gy) or treated for 24 h with aphidicolin, hydroxyurea or mitomycin C at different concentrations. Cells were collected by trypsinization and counted on a Countess™ Automated Cell Counter device (Invitrogen) 5 days after drug withdrawal. Cell survival is represented as percentage of survival relative to untreated cells.
ChIP-qPCR in embryonic brain
Brains form E17.5 embryos were dissected and minced in cold PBS with protease inhibitors cocktail (Roche #11873580001). Small tissue pieces were cross-linked for 20 min at room temperature in 1% formaldehyde and fixation was stopped by adding 1/20 volume of 2.5 M Glycine for 5 min at room temperature. Tissue pieces were lysed, sonicated and further processed for ChIP analysis as we previously described [21]. Primers used for ChIP-qPCR are listed in Supplementary Table S2.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5 software. A two-tailed Student’s t-test was applied. Data are shown as mean ± s.e.m (standard error of the mean). P < 0.05 was considered significant.
Results and Discussion
Robust sister chromatid cohesion in Nipbl heterozygous MEFs
We prepared metaphase spreads from heterozygous Nipbl primary MEFs. As previously reported, no centromeric cohesion defects can be observed in these chromosomes (Figure 1A; [18]). Cohesion mediated by cohesin is important for the restart of stalled replication forks at regions difficult to replicate like telomeres and fragile sites [19]. In the absence of cohesin-SA1, telomere replication is impaired and mitotic chromosomes display an irregular telomeric structure, a phenotype that has been called telomere fragility [22]. Telomere fragility can be observed also at telomeres of wild-type cells treated with low doses of the replication inhibitor aphidicolin. As readout of telomere cohesion defects, we determined the frequency of fragile telomeres by fluorescence in situ hybridization (FISH) analysis of mitotic chromosomes with a telomeric repeat probe. We observed no difference in the percentage of fragile telomeres in Nipbl deficient MEFs in comparison to wild-type controls, and a similar increase in its incidence upon treatment with aphidicolin (Figure 1B). Thus, telomere cohesion is not impaired in Nipbl heterozygous cells. To examine arm cohesion, we measured the frequency of breaks along the arms in mitotic chromosomes from cells either untreated or treated with a low dose of aphidicolin. No differences were found between the two genotypes (Figure 1C) suggesting that arm cohesion is also properly maintained in the Nipbl deficient MEFs. Thus, the limited amount of Nipbl present in these MEFs (Figure S1A and B) is sufficient to maintain the fraction of cohesin in charge of assuring robust sister chromatid cohesion at centromeres, telomeres and along chromosome arms. Consistent with the absence of cohesion defects, we observed no chromosome segregation anomalies upon careful examination of mitotic progression (Figure 1D) and no reduction in the proliferative capability of Nipbl deficient MEFs (Figure 1E). Therefore, we discard the contribution of cohesion, chromosome segregation and proliferation defects to the developmental delay and CdLS phenotypes observed in the Nipbl heterozygous mice.
Figure 1. Reduced Nipbl levels do not affect cohesion and progression through the cell cycle.

(A) Metaphase spreads from wild-type and Nipbl heterozygous MEFs showing robust centromere cohesion. (B) Telomere fragility measured in two clones each of wild-type and Nipbl heterozygous MEFs untreated or treated with 0.5 μM aphidicolin for 24 h. White arrows on the images indicate the aberrant telomeres displaying two instead of a single dot. The number of chromosomes examined is indicated above each bar. (C) Quantification of breaks along the chromosome arms (white arrows) in cells treated as in (A). (D) Frequency of normal anaphases and aberrant anaphases showing lagging chromosomes or bridges in wild-type and Nipbl heterozygous MEFs (n ≥ 50 cells per clone from two independent clones per genotype). (E) Growth curves of wild-type and Nipbl heterozygous MEFs (n=2 clones per genotype).
DNA repair pathways work efficiently in Nipbl deficient MEFs
Next, we examined whether limiting amounts of Nipbl confers sensitivity to DNA damaging agents. Short-term viability assays were used to measure the effect of gamma irradiation as well as treatment with three different drugs on wild-type and Nipbl deficient primary MEFs: aphidicolin, hydroxyurea (both DNA replication inhibitors) and mitomycin C (MMC, a DNA interstrand cross-linker). Nipbl deficient cells showed dose-response survival curves similar to the wild-type controls in the four different treatments (Figure 2A). These results contrast with a previous report of increased sensitivity to MMC in fibroblasts and B cells from CdLS patients [23]. At present, we cannot discard the possibility that different cell types display slightly different sensitivity to DNA damage and/or that this sensitivity depends on the fraction of functional Nipbl present in the cell, which may be different depending on the causative mutation.
Figure 2. Nipbl deficiency does not increase DNA damage sensitivity in MEFs.

(A) Survival of wild-type and Nipbl heterozygous cells after exposure to the DNA damaging agents aphidicolin, hydroxyurea, mitomycin C and gamma irradiation at the indicated doses (n=2 clones per genotype). (B) Left: 53BP1 staining of G2 irradiated (8Gy) cells, identified as pH3(S10) positive, was categorized in three different classes: homogenous nuclear staining (class I), few foci with diffuse staining (class II) and many strong foci without diffuse staining (class III). Right: Quantification of these phenotypes before, immediately after and 3 hours after irradiation of wild-type and Nipbl heterozygous MEFs. (n ≥ 50 G2-cells per condition and per clone from two independent clones per genotype).
Cohesin is also required for the DNA damage-induced G2/M-checkpoint and for efficient recruitment of 53BP1 to double strand breaks [24]. Thus, we also tested the effect of reduced amounts of Nipbl to this function. Cells in G2, whose heterochromatin regions appear labeled by phospho histone H3, were scored for foci formation by 53BP1 before, right after and three hours after irradiation (8 Gy). We did not observe any delay or impairment in recruitment of 53BP1 in Nipbl heterozygous cells (Figure 2B). Therefore, it seems unlikely that the CdLS-like phenotypes of the Nipbl mouse model derive from defective HR-mediated DNA repair.
Bulk cohesin loading remains unchanged upon reduction of Nipbl
Embryos lacking cohesin-SA1 display phenotypes reminiscent of CdLS and altered transcription patterns that coincide with those observed in the Nipbl mouse model [21]. Thus, we asked whether the loading of the two cohesin complexes, cohesin-SA1 and cohesin-SA2, could be differentially affected by the reduction in the amount of loader. Chromatin fractionation of two clones each of wild-type and Nipbl heterozygous MEFs showed no significant differences in the amount of either cohesin complex present on chromatin (Figure 3). In contrast, the amount of Nipbl itself, as well as its partner Scc4/Mau-2, was clearly diminished both in total cell extract and in the chromatin-enriched fraction from Nipbl heterozygous MEFs. Importantly, the amount of cohesive complexes, marked by the presence of Sororin [25], was identical among the four clones. The levels of another factor regulating cohesin dynamics, Wapl, were unaffected in Nipbl deficient cells. Thus, bulk loading of both cohesin-SA1 and cohesin-SA2 is achieved normally with lower levels of Nipbl, being these complexes competent for cohesion establishment and maintenance all along the chromosome
Figure 3. Nipbl deficiency does not affect bulk cohesin loading.
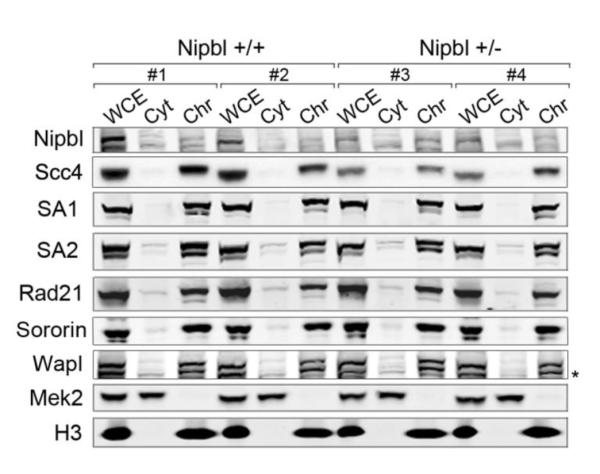
Asynchronous cultures of two clones per genotype were subjected to chromatin fractionation followed by immunoblotting with the indicated antibodies. Mek2 cytoplasmatic kinase and histone H3 are used as control for the fractionation procedure. WCE whole-cell extract; Cyt, cytoplasm; Chr, chromatin-enriched fraction. The soluble nucleoplasmic fraction was not loaded in this gel. In the immunoblot of Wapl, the lower band indicated by the asterisk correspond to SA2, since the membrane was reused.
Local effect on cohesin loading upon reduction of Nipbl levels affects transcription
Next, we look at specific cohesin binding sites. We had previously shown that the presence of cohesin at the promoters of Myc and the protocadherins (Pcdhs) is required for their expression since we observed a significant downregulation of such genes in the brains of SA1 null embryos [21]. Similar transcriptional changes are also detected in the brains of the Nipbl heterozygous embryos (Figure 4A, grey bars and [18]). The reduced gene expression is most likely the consequence of the decrease in the amount of cohesin present at all the promoters examined (Figure 4B). Further support for this hypothesis comes from the observation that the expression of Myc and Pcdhs is unaltered in the brains from SA1 heterozygous E17.5 embryos (Figure 4A, white bars), consistent with the fact that SA1 heterozygous mice do not present CdLS-like phenotypes [19]. The changes in cohesin binding at the corresponding gene promoters in the brains from SA1 heterozygous embryos are much milder and not always result in a net decrease in cohesin (Figure 4C).
Figure 4. Nipbl deficiency alters cohesin binding at certain promoters and affects gene expression.

(A) mRNA levels of the indicated genes in the brains from E17.5 Nipbl +/− (grey bars) and SA1 +/− embryos (white bars). Three embryos per genotype were tested and three independent qPCR reactions per condition were performed. Values are represented as log2 of FC versus its respective wild-type controls. * P <0.05, ** P <0.01, n.s = not significant. (B) In vivo ChIP-qPCR of SA1 and SMC1 binding at the promoter of the indicated genes in E17.5 brains from Nipbl +/− (n=5) and their wild-type littermates (n=5). (C) Same analysis by ChIP-qPCR of SA1 and SMC1 binding in E17.5 brains from SA1 +/− (n=6) and their wild-type littermates (n=4).
We found that ablation of one of the two alleles of Nipbl results in a significant decrease in mRNA levels both in MEFs (around 50% left) and embryonary brains (only 25% left; Figure S1). As a consequence, the amount of Nipbl protein bound to chromatin is also significantly decreased, but nevertheless sufficient to load the bulk of both cohesin-SA1 and cohesin-SA2 (Figure 3). We have also shown here that no significant defects in sister chromatid cohesion along the chromosomes (centromeres, arms and telomeres), in chromosome segregation or in DNA repair can be observed in cells partially deficient for Nipbl. However, we found reduced amounts of cohesin at specific genomic locations that correlate with altered gene expression. It is known that binding of cohesin to chromatin is dynamic and thus the amount of cohesin at a given position at any given time depends on two antagonistic actions, namely the loading by Nipbl and the unloading by Wapl [26]. It is likely that cohesin, and in particular cohesin-SA1, is more dynamic at certain locations depending on additional factors such as active transcription. In such scenario, cohesin may need to be constantly reloaded by Nipbl to deal with the passage of the transcriptional machinery and/or to maintain a higher-order chromatin structure compatible with the dynamics of transcription factories. In the SA1 heterozygous brains, despite the reduction in cohesin-SA1 levels (Figure S2), Nipbl maintains a fraction of cohesin-SA1 at gene promoters that is sufficient to assure their proper gene expression. In contrast, in the Nipbl heterozygous brains, the reduced levels of the loader produce modest changes in the binding of cohesin at gene promoters that trigger dramatic transcriptional changes. In an attempt to most likely compensate for its defective loading, SA1 is upregulated in Nipbl deficient brains (Supplementary Figure S1C). How may increased levels of cohesin at a given promoter favor transcription? We speculate that they may promote interaction with an enhancer through stabilization of a chromatin loop between the two elements. They could also enhance the stability of RNA polymerase binding and thus the efficiency of transcription, as recently shown in Drosophila [27].
The combinatorial expression of Pdch genes appears to provide a sort of barcode for each neuron that allows self-recognition and is essential for neural circuit assembly [28]. Cohesin and CCCTC-binding factor (CTCF) contribute to specify these barcodes [29]. Mental retardation associated to CdLS could be therefore related to anomalous expression of protocadherins. Defects in myc expression could also contribute to the growth retardation in CdLS [30]. The beta globin locus is another example of a locus whose expression depends on the amount of cohesin and is very sensitive to the partial reduction on Nipbl [13]. A detailed knowledge of cohesin binding sites in different tissues and how they are affected upon reduction of Nipbl levels will therefore be of particular importance to further understand the mechanisms that underlie CdLS pathogenesis.
Conclusions
Our careful examination of cohesion-dependent functions in cells from Nipbl deficient mice, which recapitulate human CdLS, suggests that these functions have little contribution to the etiology of the syndrome. While bulk loading of cohesin and its interactors on chromatin is not noticeably changed, we did observe decreased binding to the promoters of Myc and Protocadherin genes, whose expression is downregulated in the brains of Nipbl deficient mice. Thus, our results provide further support for the idea that developmental defects in CdLS are caused by deregulated transcription of a subset of genes highly sensitive to small variations in the amount of cohesin present at their promoters.
Supplementary Material
Highlights.
- Reduced Nipbl levels in a CdLS mouse model does not affect bulk cohesin loading.
- Cohesin binding to genes relevant to CdLS is reduced and affects their expression.
- Nipbl deficient cells present robust cohesion all along the chromosome.
- Impairment of cohesion-dependent functions is unlikely to contribute to CdLS.
Acknowledgements
We are grateful to J. M. Peters and E. Watrin for the Wapl and Scc4 antibodies, L. Ström for sending the Nipbl +/− frozen sperm, R. Santos and S. Qurashi for assisting with MEF preparation and M. Rodríguez-Corsino for additional technical assistance. This research has been supported by the Spanish Ministry of Economy and Competitiveness (SAF-2010-21517 and CSD2007-00015 Inesgen to AL; “Ramón y Cajal” grant to AC) and NIH (grant P01-HD052860 to ALC and ADL). SR was the recipient of a La Caixa predoctoral fellowship.
Footnotes
Author Contributions: AL designed and supervised the study. SR designed, performed, analyzed and interpreted most experiments with contributions from AC. SK, ALC and ADL provided the Nipbl heterozygous MEFs. AL and SR wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Liu J, Krantz ID. Cornelia de Lange syndrome, cohesin, and beyond. Clin Genet. 2009;76:303–314. doi: 10.1111/j.1399-0004.2009.01271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Guacci V, Koshland D, Strunnikov A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell. 1997;91:47–57. doi: 10.1016/s0092-8674(01)80008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Michaelis C, Ciosk R, Nasmyth K. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell. 1997;91:35–45. doi: 10.1016/s0092-8674(01)80007-6. [DOI] [PubMed] [Google Scholar]
- [4].Losada A, Hirano M, Hirano T. Identification of Xenopus SMC protein complexes required for sister chromatid cohesion. Genes Dev. 1998;12:1986–1997. doi: 10.1101/gad.12.13.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Losada A, Yokochi T, Kobayashi R, Hirano T. Identification and characterization of SA/Scc3p subunits in the Xenopus and human cohesin complexes. J Cell Biol. 2000;150:405–416. doi: 10.1083/jcb.150.3.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nasmyth K, Haering CH. Cohesin: its roles and mechanisms. Annu Rev Genet. 2009;43:525–558. doi: 10.1146/annurev-genet-102108-134233. [DOI] [PubMed] [Google Scholar]
- [7].Guillou E, Ibarra A, Coulon V, Casado-Vela J, Rico D, Casal I, Schwob E, Losada A, Mendez J. Cohesin organizes chromatin loops at DNA replication factories. Genes Dev. 2010;24:2812–2822. doi: 10.1101/gad.608210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Seitan VC, Hao B, Tachibana-Konwalski K, Lavagnolli T, Mira-Bontenbal H, Brown KE, Teng G, Carroll T, Terry A, Horan K, Marks H, Adams DJ, Schatz DG, Aragon L, Fisher AG, Krangel MS, Nasmyth K, Merkenschlager M. A role for cohesin in T-cell-receptor rearrangement and thymocyte differentiation. Nature. 2011;476:467–471. doi: 10.1038/nature10312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Degner SC, Verma-Gaur J, Wong TP, Bossen C, Iverson GM, Torkamani A, Vettermann C, Lin YC, Ju Z, Schulz D, Murre CS, Birshtein BK, Schork NJ, Schlissel MS, Riblet R, Murre C, Feeney AJ. CCCTC-binding factor (CTCF) and cohesin influence the genomic architecture of the Igh locus and antisense transcription in pro-B cells. Proc Natl Acad Sci U S A. 2011;108:9566–9571. doi: 10.1073/pnas.1019391108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nativio R, Wendt KS, Ito Y, Huddleston JE, Uribe-Lewis S, Woodfine K, Krueger C, Reik W, Peters JM, Murrell A. Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet. 2009;5:e1000739. doi: 10.1371/journal.pgen.1000739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hadjur S, Williams LM, Ryan NK, Cobb BS, Sexton T, Fraser P, Fisher AG, Merkenschlager M. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature. 2009;460:410–413. doi: 10.1038/nature08079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mishiro T, Ishihara K, Hino S, Tsutsumi S, Aburatani H, Shirahige K, Kinoshita Y, Nakao M. Architectural roles of multiple chromatin insulators at the human apolipoprotein gene cluster. Embo J. 2009;28:1234–1245. doi: 10.1038/emboj.2009.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chien R, Zeng W, Kawauchi S, Bender MA, Santos R, Gregson HC, Schmiesing JA, Newkirk DA, Kong X, Ball AR, Jr., Calof AL, Lander AD, Groudine MT, Yokomori K. Cohesin mediates chromatin interactions that regulate mammalian beta-globin expression. J Biol Chem. 2011;286:17870–17878. doi: 10.1074/jbc.M110.207365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Krantz ID, McCallum J, DeScipio C, Kaur M, Gillis LA, Yaeger D, Jukofsky L, Wasserman N, Bottani A, Morris CA, Nowaczyk MJ, Toriello H, Bamshad MJ, Carey JC, Rappaport E, Kawauchi S, Lander AD, Calof AL, Li HH, Devoto M, Jackson LG. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet. 2004;36:631–635. doi: 10.1038/ng1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tonkin ET, Wang TJ, Lisgo S, Bamshad MJ, Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat Genet. 2004;36:636–641. doi: 10.1038/ng1363. [DOI] [PubMed] [Google Scholar]
- [16].Liu J, Zhang Z, Bando M, Itoh T, Deardorff MA, Clark D, Kaur M, Tandy S, Kondoh T, Rappaport E, Spinner NB, Vega H, Jackson LG, Shirahige K, Krantz ID. Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol. 2009;7:e1000119. doi: 10.1371/journal.pbio.1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, Cole KE, De Baere E, Decroos C, Di Donato N, Ernst S, Francey LJ, Gyftodimou Y, Hirashima K, Hullings M, Ishikawa Y, Jaulin C, Kaur M, Kiyono T, Lombardi PM, Magnaghi-Jaulin L, Mortier GR, Nozaki N, Petersen MB, Seimiya H, Siu VM, Suzuki Y, Takagaki K, Wilde JJ, Willems PJ, Prigent C, Gillessen-Kaesbach G, Christianson DW, Kaiser FJ, Jackson LG, Hirota T, Krantz ID, Shirahige K. HDAC8 mutations in Cornelia de Lange Syndrome affect the cohesin acetylation cycle. Nature. 2012;489:313–317. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kawauchi S, Calof AL, Santos R, Lopez-Burks ME, Young CM, Hoang MP, Chua A, Lao T, Lechner MS, Daniel JA, Nussenzweig A, Kitzes L, Yokomori K, Hallgrimsson B, Lander AD. Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/−) mouse, a model of Cornelia de Lange Syndrome. PLoS Genet. 2009;5:e1000650. doi: 10.1371/journal.pgen.1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Remeseiro S, Cuadrado A, Carretero M, Martínez P, Drosopoulos WC, Cañamero M, Schildkraut CL, Blasco MA, Losada A. Cohesin-SA1 deficiency drives aneuploidy and tumourigenesis in mice due to impaired replication of telomeres. Embo J. 2012;31:2076–2089. doi: 10.1038/emboj.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mendez J, Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol. 2000;20:8602–8612. doi: 10.1128/mcb.20.22.8602-8612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Remeseiro S, Cuadrado A, Gómez-López G, Pisano DG, Losada A. A unique role of cohesin-SA1 in gene regulation and development. EMBO J. 2012;31:2090–2102. doi: 10.1038/emboj.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vrouwe MG, Elghalbzouri-Maghrani E, Meijers M, Schouten P, Godthelp BC, Bhuiyan ZA, Redeker EJ, Mannens MM, Mullenders LH, Pastink A, Darroudi F. Increased DNA damage sensitivity of Cornelia de Lange syndrome cells: evidence for impaired recombinational repair. Hum Mol Genet. 2007;16:1478–1487. doi: 10.1093/hmg/ddm098. [DOI] [PubMed] [Google Scholar]
- [24].Watrin E, Peters JM. The cohesin complex is required for the DNA damage-induced G2/M checkpoint in mammalian cells. Embo J. 2009;28:2625–2635. doi: 10.1038/emboj.2009.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nishiyama T, Ladurner R, Schmitz J, Kreidl E, Schleiffer A, Bhaskara V, Bando M, Shirahige K, Hyman AA, Mechtler K, Peters JM. Sororin mediates sister chromatid cohesion by antagonizing Wapl. Cell. 2010;143:737–749. doi: 10.1016/j.cell.2010.10.031. [DOI] [PubMed] [Google Scholar]
- [26].Nasmyth K. Cohesin: a catenase with separate entry and exit gates? Nat Cell Biol. 2011;13:1170–1177. doi: 10.1038/ncb2349. [DOI] [PubMed] [Google Scholar]
- [27].Schaaf CA, Kwak H, Koenig A, Misulovin Z, Gohara DW, Watson A, Zhou Y, Lis JT, Dorsett D. Genome-Wide Control of RNA Polymerase II Activity by Cohesin. PLoS Genet. 2013;9:e1003382. doi: 10.1371/journal.pgen.1003382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zipursky SL, Sanes JR. Chemoaffinity revisited: dscams, protocadherins, and neural circuit assembly. Cell. 2010;143:343–353. doi: 10.1016/j.cell.2010.10.009. [DOI] [PubMed] [Google Scholar]
- [29].Monahan K, Rudnick ND, Kehayova PD, Pauli F, Newberry KM, Myers RM, Maniatis T. Role of CCCTC binding factor (CTCF) and cohesin in the generation of single-cell diversity of Protocadherin-alpha gene expression. Proc Natl Acad Sci U S A. 2012;109:9125–9130. doi: 10.1073/pnas.1205074109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rhodes JM, Bentley FK, Print CG, Dorsett D, Misulovin Z, Dickinson EJ, Crosier KE, Crosier PS, Horsfield JA. Positive regulation of c-Myc by cohesin is direct, and evolutionarily conserved. Dev Biol. 2010;344:637–649. doi: 10.1016/j.ydbio.2010.05.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.