

Abstract
Recent advances have led to several systems to study transcription from defined loci in living cells. It has now become possible to address long-standing questions regarding the interplay between the processes of DNA damage repair and transcription—two disparate processes that can occur on the same stretch of chromatin and which both lead to extensive chromatin change. Here we describe the development of a system to create enzymatically induced DNA double-strand breaks (DSBs) at a site of inducible transcription and methods to study the interplay between these processes.
Keywords: Double-strand breaks, Transcription, ATM, Ubiquitin
1 Introduction
The repair of damaged DNA requires the timely and coordinated recruitment of multiple DNA damage repair (DDR) proteins to the lesion. These events have been especially well studied at DNA double-strand breaks (DSBs). The signal amplification that leads to the recruitment of many molecules of a given DDR protein to a site of damage results in a spreading of these repair factors along chromatin near a break and also makes this recruitment amenable to visualization by immunofluorescence microscopy. A common method of inducing damage to initiate these recruitment events is the use of ionizing radiation, which leads to the formation of multiple ionizing radiation-induced foci (IRIF) of repair factors throughout the irradiated nuclear volume. Alternatively, an ultraviolet laser can be used to create a stripe across the nucleus of a cell that has been first sensitized to DSB induction by treatment with bromodeoxyuridine. Subsequent laser microirradiation leads to the creation of a large and easily visualized swath of DNA DSBs across the nucleus [1, 2]. Methods such as these have been useful in the study of multiple repair factors and chromatin modifications that occur at DSBs, as well as the kinetics and relative requirements for accumulation of many of these marks at sites of damage [2–4]. In addition to these methods, which create damage at somewhat random sites within the genome, several groups have created methods for enzymatically induced breaks at defined locations in the genome. Some of these involve the use of restriction enzymes which target endogenous loci [5–7], while others have integrated exogenous restriction enzyme target sites at defined genomic loci to study the DDR [8–10].
A long-standing question in the field of DNA damage repair has been the interplay between the creation and repair of DSBs, and transcription occurring on nearby chromatin. Though a variety of DNA lesions are known to interfere with transcription [11, 12], the question has remained largely unanswered for DSBs. To address this question, varying groups have used ionizing radiation, followed by nuclear run-on assays [13, 14], as well as enzyme- induced DSBs coupled with chromatin immunoprecipitation and quantitative reverse-transcription PCR at nearby endogenous or reporter genes [6, 7, 15].
In this chapter, we describe the experimental procedures to efficiently create multiple nuclease-induced DSBs at a known distance away from an inducible and visualizable transcriptional reporter and methods to study both the DDR and its effects on local transcription. To do this, we utilize a previously described genomically integrated transcriptional reporter system in which genomic DNA, transcribed RNA, and translated protein can be visualized simultaneously in real-time or in fixed cells [16, 17]. The genomic locus where the reporter is integrated is visualized by the tandem lac operator present in the reporter. Expression of a mCherry-fluorescent-tagged lac-repressor protein (mCherry-LacI) leads to a collection of fluorescent proteins at the reporter, which is easily visualized by fluorescence microscopy. Depending on the specific cell line used, transcription can be activated by expressing a “tet-off” transactivator that translocates to the nucleus upon treatment with 4-hydroxytamoxifen (2-6-3 cells) [16, 17], or by doxycycline treatment of a variant cell line that stably expresses YFP-MS2, as well as a “tet-on” transactivator (2-6-3 rtTA + YFP-MS2 cells) [16]. The 2-6-3 cells do not stably express any fluorescent proteins and therefore are amenable to the study of fluorescence-tagged or immunostained repair factors of interest. The 2-6-3 rtTA + YFP-MS2 cells stably express a YFP-MS2 fusion protein and are therefore useful in studying transcription from the reporter. Transcription is visualized due to the 24 repeats of the MS2 sequence, which are integrated in the 3′-UTR of the reporter gene. The sequence, from the MS2 bacteriophage, forms a stem–loop structure in the transcribed RNA, which is then bound strongly and specifically by dimers of the YFP-tagged MS2 coat protein [17–19]. Finally, after induction of transcription, the reporter protein—a CFP-tagged peroxisome-targeting SKL peptide—can be visualized collecting in the cytoplasmic puncta of peroxisomes. RNA and protein production in this system can also be measured through qRT-PCR and immunoblotting, respectively.
To create DSBs in this system, we employ the nonspecific nuclease domain of the FokI nuclease [20, 21], fused to the mCherryLacI protein (FokI-mCherry-LacI) (Fig. 1). Upon expression of this construct, the lac-repressor domain targets the nuclease domain to a site approximately 5 kb upstream of the inducible reporter gene, where it can dimerize and create DSBs. As a control, we use a catalytically ineffective mutant of FokI (FokI- D450A) [20]. In Subheadings 2.1 and 2.2, we describe the induction of damage at the reporter and subsequent detection of DDR proteins through immunostaining or expression of fluorophore- tagged proteins. In Subheadings 2.3 and 2.4, we describe the quantification of transcription from the reporter by immunofluorescence and by qRT-PCR, respectively. The system entails the first example of a method that enables simultaneous, real-time analysis of DNA damage responses and their impact on local transcriptional events occurring on a contiguous stretch of chromatin. The DSBs induce chromatin changes that influence transcription by preventing elongation of RNA polymerase and subsequent transcription.
Fig. 1.
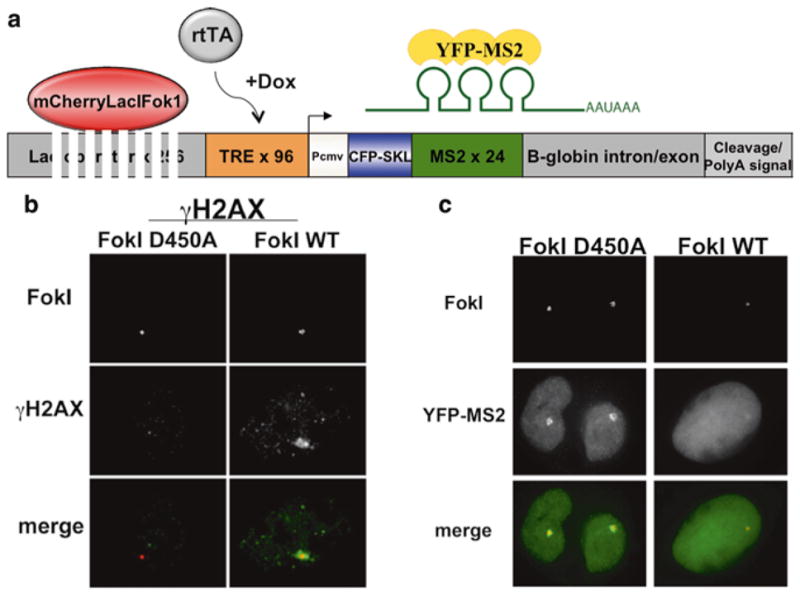
System for monitoring communication between DNA damage responses and local transcription. (a) Schematic of reporter. The mCherry-LacI-Fok1 fusion protein creates DSBs within 256 lac operator repeats. Transcription is inducible upon doxycycline (Dox) addition, which activates a CFP gene. YFP-MS2 focally accumulates at the locus by binding to stem–loop structures within the 3′-UTR of the nascent transcript. (b) Expression of wild-type mCherry-LacI-Fok1 (FokI WT), but not nuclease-inactive FokI D450A, induces DSBs as indicated by gH2AX focus formation. (c) YFP-MS2 accumulates at the locus in FokI D450A-expressing cells indicating the presence of nascent transcription. This does not occur in FokI WT cells indicating that DSB responses silence transcription
2 Materials
2.1 Expression of FokI-mCherry-LacI in Reporter Cells and Induction of Transcription
2-6-3 and 2-6-3 rtTA + YFP-MS2 reporter cell lines [15, 16].
50 mg/ml Hygromycin sterile solution.
100 mg/ml G418 filter sterilized.
FokI-mCherry-LacI plasmid: use 200 ng per transfection [15].
ERTA plasmid: use 200 ng per transfection; only for 2-6-3 cells [16].
LipoD-293 transfection reagent (Signagen Laboratories, Rockville, MD, USA).
Dulbecco’s Modified Eagle’s Medium (DMEM) + Glutamax.
Tet-system-approved fetal bovine serum.
Glass optical cover slips.
PBS for washing.
-
Formaldehyde fixative solution: 3 % paraformaldehyde/2 % sucrose in PBS.
Dissolve 15 g paraformaldehyde and 10 g sucrose in 500 ml PBS and place in a 65 °C water bath shaking occasionally until almost the PFA is in solution.
Adjust pH to 7.0–7.4.
Allow solution to cool slightly, then filter through Whatman paper and store at −20° C (see Note 1).
24-Well tissue-culture-treated plates.
2 mg/ml doxycycline in DMSO.
1 mM 4-hydroxytamoxifen in EtOH.
2.2 Immunofluorescent Detection of DNA Damage Response
Fixed coverslips of transfected cells in a 24-well plate (see Subheading 2.1).
Ice-cold PBS.
Ice-cold PBS with 0.1 % Tween (PBST).
Ice-cold permeabilization buffer: PBS with 0.5 % Tween.
Parafilm.
Humidifying chamber: a small plastic lidded container big enough to hold the lid of a 24-well plate, with a damp paper towel or cheesecloth at the bottom.
37-Degree incubator.
IF-validated antibodies to targets of choice.
Fluorescent-conjugated secondary antibodies.
Glass optical cover slips.
Fluorescence mounting medium with DAPI.
2.3 Quantification of DDR Foci/YFP-MS2 with Imaging Software
Fluorescence microscope (wide-field or confocal) of choice with camera attachment. We use a QImaging RETIGA-SRV camera connected to a Nikon Eclipse 80i microscope driven by ImagePro 6.2 software.
Computer equipped with ImageJ software (NIH).
2.4 Quantification of Transcript with qRT-PCR
6-Well tissue-culture-treated dishes.
Trizol reagent or other RNA isolation reagent of choice.
High-capacity cDNA reverse transcriptase kit.
-
Control human primers. We use GAPDH.
hGAPDH_F: CTCAAGATCATCAGCAATGCC
hGAPDH_R: CATCACGCCACAGTTTCCC
-
Reporter transcript qPCR primers. We use the following two sets of primers, both of which amplify regions of the ECFP reporter gene:
ECFP_1F: GACGTAAACGGCCACAAGTT
ECFP_1R: GAACTTCAGGGTCAGCTTGC
ECFP_2F: CGACCACTACCAGCAGAACA
ECFP_2R: GAACTCCAGCAGGACCATGT
SYBR Green 2× master mix.
384-Well qPCR plates and seals.
Conventional PCR block.
qPCR machine with analysis software. We use the 7900HT real-time PCR machine from Applied Biosystems.
3 Methods
3.1 Expression of FokI-mCherry-LacI into Reporter Cells and Induction of Transcription
Grow reporter cells in DMEM with 10 % Tet-system-approved FBS and 1 % Penicillin-Streptomycin. 2-6-3 cells are also grown in the presence of 100 μg/ml hygromycin, and 2-6-3 rtTA + YFP-MS2 cells in the presence of 100 μg/ml hygromycin, as well as 400 μg/ml G418, both of which are added fresh at each passage (see Note 2).
1 day prior to transfection, seed approximately 5 × 104 cells per well in a 24-well dish, onto sterilized glass coverslips, without antibiotics. The cells should be 60–80 % confluent the day of transfection (see Note 3).
The next day, change medium on the cells before transfection.
We transfect the cells using the LipoD-293 protocol (Signagen), with some modifications. For each transfection, place 200 ng total DNA to be transfected in 1 sterilized 1.5 ml microcentrifuge tube with 10 μL serum-free DMEM. For instance, if cotransfecting 2 plasmids, add 100 ng each. Tap to mix.
After the DNA for each transfection has been added to the tubes, create a master mix of transfection reagent by adding 0.5 μL transfection reagent per transfection to 10 μL serum-free DMEM per transfection, with some extra to account for some loss upon pipetting. Tap to mix.
Incubate the transfection reactions for 15 min at room temperature.
Immediately after the 15 min incubation, add 20 μL transfection reagent/DNA mixture to each well and swirl gently to mix.
-
18–24 h after transfection, wash cells 1× in ice-cold PBS.
If using 2-6-3 rtTA + YFP-MS2 cells and transcription activation is desired, first treat cells with 1 μg/ml doxycycline in complete growth medium for 3 h before washing and fixing (see Note 4).
If using 2-6-3 cells which have been transfected with the ER-TA, a fusion of TA with estradiol receptor transactivator, and transcription is desired, first treat cells with 1 μM 4-hydroxytamoxifen in complete growth medium for 3 h before washing and fixing.
Fix in formaldehyde fixative solution for 10 min at room temperature.
Wash cells 2× in ice-cold PBS and proceed to immunofluorescence (Subheading 3.2). At this step, cells can be kept at 4 °C in the dark before staining, but mCherry fluorescence will be slowly lost over this time.
3.2 Immunofluorescent Detection of DNA Damage Response
Aspirate PBS and add 0.5 ml ice-cold permeabilization buffer to each well.
Incubate on ice 5 min.
Aspirate permeabilization buffer and wash 3× with ice-cold PBST, leaving the coverslips in PBST after the third wash.
Using tweezers, place coverslips atop a piece of parafilm that has been pressed onto the lid of a 24-well dish.
Dilute primary antibodies of choice in ice-cold PBST. For instance, if staining with rabbit anti-protein X and mouse anti-protein Y, both can be diluted in the same PBST.
Add 25–30 μL primary antibody solution to the top of each coverslip.
Incubate, in the humidifying chamber, at 37 °C × 20 min.
Place coverslips back in wells with cold PBST (this is the first wash).
Aspirate PBST and wash 3× with cold PBST (do not aspirate after final wash).
Remove coverslips and place on fresh piece of laboratory film pressed onto lid of 24-well plate.
Add 25–30 μL diluted secondary antibody to each coverslip (we use secondary antibodies from Jackson Immunoresearch, each diluted 1:200).
Incubate, in the humidifying chamber, at 37 °C × 20 min.
Place coverslips back in wells with cold PBST, for the first wash.
Aspirate PBST and wash 4× with cold PBST. Do not aspirate after final wash.
With fine tweezers, remove coverslips, dab edge on paper towel to remove excess liquid, and place facedown onto 4 μL mounting medium on glass microscopy slides.
Pat dry gently with paper towels or laboratory tissue.
Seal edges of coverslips with clear coverslip sealant or clear nail polish. Let dry in dark.
Store slides at −20 °C, protected from light, until ready for microscopy.
3.3 Quantification of DDR Foci/YFP-MS2 with Imaging Software
Using the microscopy settings of your choice, obtain images of your cells of interest. We obtain pictures at 60× oil-immersion magnification.
When taking pictures, all pictures for a given experiment should be taken on the same day. When comparing conditions, the same exposure settings should be used for all comparisons.
Open images of interest in the ImageJ program.
For each cell measured, measure a region of interest (e.g., a focus of 53BP1 accumulation), as well as a background fluorescence image elsewhere in the cell, using the elliptical ROI tool. Note that adjusting brightness and contrast in ImageJ will alter the appearance of the image, but not the values obtained, and can therefore be helpful in measurements. For images in which a given signal is absent (e.g., no 53BP1 accumulation at the reporter locus in a cell expressing the FokI nuclease-deficient mutant), overlay the mCherry image to locate the locus and draw an ROI at that point.
Measure 50–100 cells per experiment, then subtract each background from each signal, and calculate the mean fluorescence intensity (MFI) for a given signal.
3.4 Quantification of Transcript with qRT-PCR
-
We use a modified version of the RNA isolation protocol from Trizol (Invitrogen).
You can use any RNA isolation reagent/protocol. Be sure all reagents/equipment have been cleaned and are free of any RNAse/DNAse contamination.
After isolating RNA, measure the concentration and convert 1–2 μg to cDNA using the high-capacity cDNA kit or another cDNA kit of your choice.
After the reverse-transcription reaction is complete, dilute the cDNA with sterile water. 1 μg is diluted to 100 μl.
-
We use the following reaction mixture per well in a 384-well qPCR plate.
5 μL 2× SYBR Green master mix.
4 μL diluted cDNA.
1 μL 9 μM primer mix (a mix of both forward and reverse primers for a given amplicon, with each primer being at a final concentration of 9 μM).
Final volume—10 μL
Run the qPCR with the following conditions.
Stage 1 (×1): 50° × 2 min.
Stage 2 (×1): 95° × 10 min.
Stage 3 (×40): 95° × 15 s.
60°×1 min.
Analyze the data using the SDS and RQ manager programs (ABI), and the relative quantity method, normalizing to a given sample, and to human GAPDH within samples. For example, a given experiment may compare transcript levels after induction with doxycycline in the presence of FokI(WT)-mCherry-LacI, as compared to expression of the nuclease-deficient mutant FokI(D450A)-mCherry-LacI.
Footnotes
For formaldehyde fixative solution, perform all steps in a chemical hood to avoid fumes. Single-use aliquots can be stored at −20 °C (we use pop-cap culture tubes).
When growing and passaging the cells, we split the cells 1:2–1:4 every 2–3 days, once the cells reach 80–90 % confluence. It is also important to freeze down multiple vials of early passage cells in 10 % DMSO-containing freezing media, as later passage cells can lose the integrated reporter or inducibility.
We sometimes see problems with transfection toxicity if the cells are too sparse (less than approximately 50–60 % confluent) and problems with transgene inducibility if the cells are too dense (close to or completely confluent).
As the doxycycline is light sensitive, we store small aliquots of the 2 mg/ml stock in the dark at −20 °C.
Contributor Information
Niraj M. Shanbhag, Department of Cancer Biology, Basser Research Center for BRCA1/2, Abramson Family Cancer Research Institute, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA
Roger A. Greenberg, Department of Pathology, Basser Research Center for BRCA1/2, Abramson Family Cancer Research Institute, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, 19104-6160, USA
References
- 1.Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat Cell Biol. 2003;5(3):255–260. doi: 10.1038/ncb945. [DOI] [PubMed] [Google Scholar]
- 2.Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, et al. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol. 2006;173(2):195. doi: 10.1083/jcb.200510130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118(6):699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 4.Lisby M, Mortensen UH, Rothstein R. Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat Cell Biol. 2003;5(6):572–577. doi: 10.1038/ncb997. [DOI] [PubMed] [Google Scholar]
- 5.Berkovich E, Monnat RJ, Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007;9(6):683–690. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- 6.Pankotai T, Bonhomme C, Chen D, Soutoglou E. DNAPKcs-dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat Struct Mol Biol. 2012;19(3):276–282. doi: 10.1038/nsmb.2224. [DOI] [PubMed] [Google Scholar]
- 7.Iacovoni JS, Caron P, Lassadi I, Nicolas E, Massip L, Trouche D, et al. High- resolution profiling of |[gamma]|H2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010;29(8):1446–1457. doi: 10.1038/emboj.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soutoglou E, Dorn JF, Sengupta K, Jasin M, Nussenzweig A, Ried T, et al. Positional stability of single double-strand breaks in mammalian cells. Nat Cell Biol. 2007;9(6):675–682. doi: 10.1038/ncb1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Honma M, Izumi M, Sakuraba M, Tadokoro S, Sakamoto H, Wang W, et al. Deletion, rearrangement, and gene conversion; genetic consequences of chromosomal double-strand breaks in human cells. Environ Mol Mutagen. 2003;42(4):288–298. doi: 10.1002/em.10201. [DOI] [PubMed] [Google Scholar]
- 10.Rouet P, Smih F, Jasin M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endo-nuclease. Mol Cell Biol. 1994;14(12):8096–8106. doi: 10.1128/mcb.14.12.8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anindya R, Aygün O, Svejstrup JQ. Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Mol Cell. 2007;28(3):386–397. doi: 10.1016/j.molcel.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 12.Svejstrup JQ. Contending with transcriptional arrest during RNAPII transcript elongation. Trends Biochem Sci. 2007;32(4):165–171. doi: 10.1016/j.tibs.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 13.Solovjeva LV, Svetlova MP, Chagin VO, Tomilin NV. Inhibition of transcription at radiation-induced nuclear foci of phosphorylated histone H2AX in mammalian cells. Chromosome Res. 2007;15(6):787–797. doi: 10.1007/s10577-007-1162-x. [DOI] [PubMed] [Google Scholar]
- 14.Kruhlak M, Crouch EE, Orlov M, Montano C, Gorski SA, Nussenzweig A, et al. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature. 2007;447(7145):730–734. doi: 10.1038/nature05842. [DOI] [PubMed] [Google Scholar]
- 15.Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell. 2010;141(6):970–981. doi: 10.1016/j.cell.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, et al. From silencing to gene expression: real-time analysis in single cells. Cell. 2004;116(5):683–698. doi: 10.1016/s0092-8674(04)00171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rafalska-Metcalf IU, Janicki SM. Show and tell: visualizing gene expression in living cells. J Cell Sci. 2007;120(14):2301–2308. doi: 10.1242/jcs.008664. [DOI] [PubMed] [Google Scholar]
- 18.Bertrand E, Chartrand P, Schaefer M, Shenoy SM, Singer RH, Long RM. Localization of ASH1 mRNA particles in living yeast. Mol Cell. 1998;2(4):437–445. doi: 10.1016/s1097-2765(00)80143-4. [DOI] [PubMed] [Google Scholar]
- 19.Yunger S, Shav-Tal Y. Imaging mRNAs in living mammalian cells. Methods Mol Biol. 2011;714:249–263. doi: 10.1007/978-1-61779-005-8_16. [DOI] [PubMed] [Google Scholar]
- 20.Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I. FokI dimerization is required for DNA cleavage. Proc Natl Acad Sci USA. 1998;95(18):10570–10575. doi: 10.1073/pnas.95.18.10570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wah DA, Bitinaite J, Schildkraut I, Aggarwal AK. Structure of foki has implications for DNA cleavage. Proc Natl Acad Sci USA. 1998;95(18):10564–10569. doi: 10.1073/pnas.95.18.10564. [DOI] [PMC free article] [PubMed] [Google Scholar]