

Abstract
Abnormal expression of Aurora-A and epidermal growth factor receptor (EGFR) is observed in different kinds of cancer and associated with poor prognosis in cancer patients. However, the relationship between Aurora-A and EGFR in tumour development was not clear. In previous reports, we found that EGFR translocates to nucleus to activate Aurora-A expression after EGF treatment in EGFR-overexpressed cells. However, we also observed that not all the EGFR-overexpressed cells have the nuclear EGFR pathway to mediate the Aurora-A expression. In this study, we demonstrated that EGF signalling increased the Aurora-A protein expression in EGFR-overexpressed colorectal cancer cell lines via increasing the translational efficiency. In addition, the overexpression of EGFR was also associated with higher expression of Aurora-A in clinical colorectal samples. Activation of the PI3K/Akt/mTOR and MEK/ERK pathways mediated the effect of EGF-induced translational up-regulation. Besides, only the splicing variants containing exon 2 of Aurora-A mRNA showed increased interaction with the translational complex to synthesize Aurora-A protein under EGF stimulus. Besides, the exon 2 containing splicing variants were the major Aurora-A splicing forms expressed in human colorectal cancers. Taken together, our results propose a novel regulatory mechanism for the abnormal expression of Aurora-A in EGFR-overexpressed cancers, and highlight the importance of alternative 5′-UTR splicing variants in regulating Aurora-A expression. Furthermore, the specific expression of exon 2 containing splicing variants in cancer tissues may serve as a potential target for cancer therapy in the future.
Keywords: Aurora-A, translation, EGF, 5′UTR, alternative splicing, colorectal cancer
Introduction
Aurora-A is a centrosomal protein kinase that is involved in centrosome maturation and spindle formation. The protein, mRNA expression levels and kinase activity of Aurora-A increased at the G2-M transition and completely disappear at the G1 phase [1]. Overexpression of Aurora-A may lead to genomic instability and neoplastic transformation, suggesting that Aurora-A is a bona fide oncogene [2]. In addition, overexpression of Aurora-A is also associated with a poor prognosis in cancer patients, such as breast, colon, pancreatic, ovarian and stomach cancers [3, 4].
The epidermal growth factor receptor (EGFR) is a type of receptor tyrosine kinase that regulates cell proliferation, growth, migration, invasion and inhibition of apoptosis. Aberrant EGFR signalling has been associated with many cancers, and the development of new therapeutic agents targeting EGFR has attracted attention [5, 6]. In colorectal cancer, EGFR overexpressed in 65–70% of tumours, and the extent of EGFR expression showed a positive correlation with poor prognosis [7]. Interestingly, it was reported that the Aurora-A protein occurred in approximately 50–70% of patients with colorectal cancer. There was also a trend that patients with negative expression of Aurora-A protein showed slightly better survival rates than those with positive protein expression in stage III colorectal cancer [8, 9]. Because EGFR and Aurora-A are all important prognosis indicators in cancer development, it would be interesting to analyse the relationship between EGF signalling and the activation of Aurora-A expression in cancers.
Controlling the translation of mRNA is a fundamental event in many aspects of cell metabolism. It organizes a critical step for the control of gene expression, and hence cell growth, proliferation and differentiation [10]. It was reported that the PI3K/Akt/mTOR (mammalian target of rapamycin [mTOR]) signalling pathway deeply affects mRNA translation through the phosphorylation of downstream targets such as eukaryotic initiation factor 4E (eIF4E)-binding protein (4E-BP) and ribosomal protein S6 kinase (S6K) [11]. Because the activation of mTOR can mediate the translation of mRNAs which are related to cell cycle progression, cell survival, metastasis and angiogenesis, the deregulation of PI3K/Akt/mTOR may result in tumorigenesis and metastasis [12].
Two kinds of EGFR signalling pathways were proposed to mediate the EGF effects. One is the traditional EGFR signalling pathway and the other is the nuclear EGFR signalling pathway. In our previous report, the treatment of EGF can increase Aurora-A gene expression through the nuclear EGFR signalling pathway, where the EGFR translocated into the nucleus to act as a transcriptional activator [13]. However, it is still interesting to check whether the activation of traditional EGFR signalling pathway possesses the ability to transactivate the Aurora-A gene. In the present study, we demonstrate that traditional EGFR signalling pathway increased the protein expression level of Aurora-A through activation of the translational machinery by the ERK and Akt pathways. A specific splicing variant of Aurora-A mRNA is responsible for increasing translational efficiency under EGF stimulation. Overall, our studies provide new insights into the regulation of Aurora-A overexpression in cancers.
Materials and methods
Cell culture and drug treatment
LS174T cells were cultured in minimum essential medium α (MEM-α, GIBCO, Invitrogen, Carlsbad, CA, USA). SW480 and SW620 cells were cultured in Leibovitz’s L-15 medium (L-15, GIBCO). HT29 and HCT116 cells were cultured in RPMI medium 1640. All media were supplemented with 10% foetal bovine serum (FBS), 100 μg/ml streptomycin and 100 U/ml penicillin. In this series of experiments, cells were treated with 60 ng/ml (10 nM) EGF in optimal serum-free conditions.
Quantitative reverse-transcription polymerase chain reaction (qRT-PCR) and RT-PCR
The qRT-PCR and RT-PCR reactions were performed as described previously [13]. The sequences of primers used for the PCR are described as follows: Aurora-A forward, 5′-ATGGACCGATCTAAAGAAAAC-3′, and reverse, 5′-CGATTCCTAAGACTGTTTGC-3′; and GAPDH forward, 5′-CCATCACCATCTTCCAGGAG-3′ and reverse, 5′-CCTGCTTCACCACCTTCTTG-3′; Aurora-A 5′UTR forward F1, 5′-GTCAACCAATCACAAGGCAGC-3′, forward F2, 5′-GCTGAGCTCTTGGAAGAC-3′, and reverse R1, 5′-CAGTTTTCTTTAGATCGGTCC-3′, reverse R2, 5′-GAAAATGCTGGGATTACGGG-3′. For the qRT-PCR, first-strand cDNA was synthesized at 50°C for 60 min., followed by a 10 min. denaturation at 95°C. PCRs were then perfumed in the same tubes using the following conditions for 35 cycles: 95°C for 15 sec., 60°C for 15 sec. and 68°C for 30 sec. The sequences of primers used for the qRT-PCR were as follows: human Aurora-A forward, 5′-AATGCCCTGTCTTACTGTCATTC-3′ and reverse, 5′-TCCAGAGATCCACCTTCTCATC-3′; and human RPL13A forward, 5′-CCTGGAGGAGAAGAGGAAAGAGA-3′ and reverse, 5′-TTGAGGACCTCTGTGTATTTGTCAA-3′. Real-time fluorescence monitoring and a melting curve analysis were performed with LightCycler according to the manufacturer’s recommendations (Roche Molecular Diagnostics, Lewes, East Sussex, UK). Data were analysed by LightCycler software version 3.5 (Roche Molecular Diagnostics) to determine the threshold cycle (Cp) above the background for each reaction. The relative transcript amount of the target gene, calculated using standard curves of serial RNA dilutions, was normalized to that of RPL13A of the same RNA.
Reporter assay
Cells were transfected with the human Aurora-A promoter (–968 to +124 nt) luciferase construct (pGL2-Aurora-A promoter) by LipofectAMINE 2000 (Invitrogen) according to the manufacturer’s instructions. Promoter activities of the Aurora-A gene under EGF treatment at various time-points were calculated as described previously [13]. Briefly, 24 hrs following transfection, cells were serum-starved for another 24 hrs, then stimulated with 60 ng/ml (10 nM) EGF under serum-free condition for various times, harvested and subjected to the luciferase assay using a dual-luciferase reporter assay kit (Promega, Madison, WI, USA). After normalization with Renilla luciferase activity, mean luciferase activities and standard deviations were derived from three independent experiments.
Preparation of cell lysates and immunoblot analysis
For total cell lysates, cells from 10 cm plastic dishes were washed twice with PBS and lysed with RIPA buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.1% NP-40, 0.5% sodium deoxycholate, 1 mM ethylenediaminetetraacetic acid (EDTA) and 2 mM EGTA) containing 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin and 10 μg/ml aprotinin. The lysates were centrifuged at 12,000 ×g for 5 min. at 4°C. The supernatants were collected and stored at −70°C until used. The immunoprecipitation complexes were resolved by SDS-PAGE and subjected to immunoblotting analysis as indicated in the figure legends. Anti-EGFR (sc-03) and anti-phospho-ERK (sc-7383) antibodies were purchased from Santa Cruz (Santa Cruz, CA, USA), anti-ERK1/2 (06–182) was purchased from Upstate (Charlottesville, VA, USA), anti-Aurora-A (ab13824) was purchased from Abcam (Cambridge, UK) and anti-α-tubulin (DM1A) was purchased from Sigma (St. Louis, MO, USA), whereas anti-Akt (#9272), anti-phospho-Akt/Ser473 (#9271), anti-phospho-4E-BP/Thr70 (#9455), anti-phospho-S6K/Thr389 (#9206) and anti-phospho-mTOR/Ser2448 (#2971) were all from Cell Signaling (Beverly, MA, USA).
Pulse-chase assay
Serum-depleted cells were cultured in methionine-free medium for 2 hrs, and then treated with 10 nM EGF for 1.5 hrs, followed by addition of 300 μCi 35S-methionine for 30 min. Cells were then harvested and lysed in immunoprecipitation buffer (50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 5 mM EDTA, 0.5% Triton X-100, 0.1% NP-40 and 2 mM EGTA) containing 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin and 10 μg/ml aprotinin. Non-specific binding was pre-cleared by incubating with protein A agarose beads at 4°C for 1 hr, and then the clear supernatant was collected for the immunoprecipitation assay using anti-Aurora-A polyclonal antibodies (ab1287, Abcam) or normal rabbit immunoglobulin G (IgG) (Santa Cruz). The immunoprecipitated protein was analysed by 10% SDS-PAGE and autoradiography.
S6 immunoprecipitation assay
After serum-starvation, cells were treated with 10 nM EGF for 2 hrs, and then cells were harvested and lysed in RNA-IP buffer (100 mM KCl, 10 mM HEPES, 5 mM MgCl2, 1% Triton X-100, 0.5% Na-deoxycholate and 100 unit/ml RNaseOUT (Invitrogen) and 100 μg/ml cycloheximide). One milligram of total lysate was pre-cleaned, and then the immunoprecipitation assay was performed by anti-ribosomal protein S6 polyclonal antibodies (sc-20085, Santa Cruz). The RNA in the immunoprecipitated complex was purified with the Trizol reagent as the manufacturer’s instructions. The amounts of Aurora-A and GAPDH mRNA were evaluated by RT-PCR using the primers described above.
In vivo and in vitro translation assay
The in vivo translation assay was performed as described previously [14]. The Aurora-A mRNA 5′UTR isoforms were cloned into the pGL3 promoter vector, which contains a SV40 promoter and a luciferase reporter. These constructs were transient transfected into cells by LipofectAMINE 2000 (Invitrogen) according to the manufacturer’s instructions. Promoter activities of the pGL3 promoter carrying the different Aurora-A 5′UTR isoforms under EGF treatment at various time-points were calculated as described previously [14].
The in vitro translation assay was performed as described previously [14]. Briefly, the fragments contained Aurora-A mRNA 5′UTR and luciferase gene were subcloned into pGEM-7Z vector. After linearizing, these constructs were used to perform the in vitro transcription assay using the SP6 polymerase according to the manufacturer’s instructions (Promega). Equal amount of RNA was incubated with 10 μg EGF-treated or untreated LS174T lysate, respectively. Then in vitro translation assay in rabbit reticulocyte lysates (Promega) was performed as described previously [14].
Immunohistochemistry
An immunohistochemical assay was performed as previously described [15]. After dewaxing and rehydration, formalin-fixed and paraffin-embedded sections were boiled in 0.01 M sodium citrated buffer (pH 6.0) to retrieve the antigen. Expressions of the EGFR and Aurora-A were detected by the ABC method. Briefly, after blocking endogenous peroxidase and biotin, sections were incubated with anti-EGFR (sc-03, Santa Cruz) or anti-Aurora-A antibodies (ab1287, Abcam). Sections were counterstained with haematoxylin. The human colorectal samples were obtained according to the guidelines and were approved by the Human Experiment & Ethics Committee in National Cheng Kung University Hospital.
Results
Traditional EGF signalling pathway increases Aurora-A expression in EGFR-overexpressed cells through ERK and Akt signalling pathways
In our previous report, the EGF signaling enhanced the transcriptional activity of Aurora-A gene through the nuclear EGFR signaling pathway in EGFR-overexpressed A431 cell was demonstrated [13]. In that study, we observed that the Aurora-A mRNA increased after long-term EGF stimulus in A431 cells (at least 18 hrs) [13]; however, the protein expression of Aurora-A was induced in a bi-phase manner, which increased after 2 and 24 hrs EGF treatment (Fig. 1A). Therefore, we concluded that the nuclear EGFR signalling pathway, where the nuclear EGFR serves as a transcriptional coactivator, contributed to the increase expression of Aurora-A after EGF treatment for 24 hrs but not 2 hrs. This result prompts us to speculate whether traditional EGFR signalling pathway regulates the protein expression of Aurora-A in the early stage of EGF stimulus in EGFR-overexpressed cells.
Fig 1.
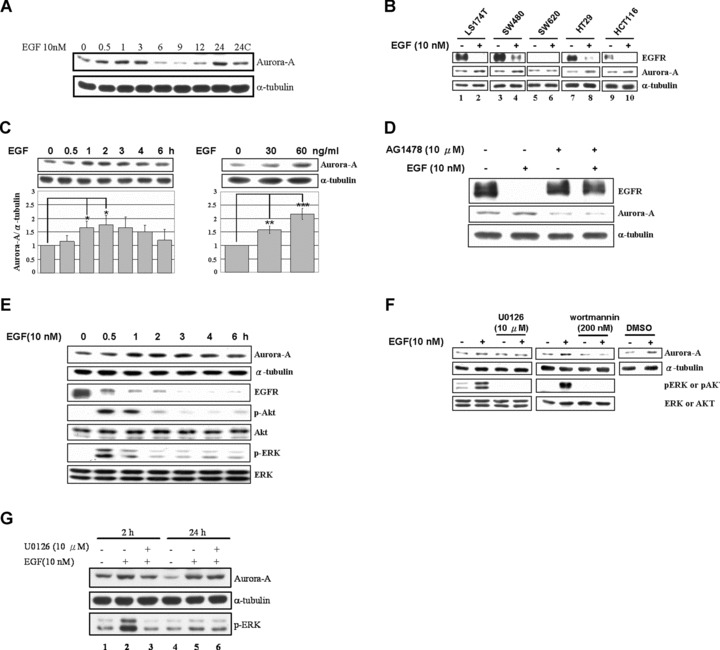
EGF increases the expression level of Aurora-A through the activation of ERK and Akt signalling pathways in human colorectal cancer cell lines. (A) The expression of Aurora-A was enhanced by EGF treatment in a bi-phase manner. Total lysates from EGF treated A431 cells were harvested for performing the Western blot analysis to demonstrate the expression of Aurora-A. The time-points indicated the duration of EGF treatment. α-Tubulin was used as a loading control. (B) Five colorectal cancer cell lines were treated with EGF for 2 (LS174T) or 3 hrs (SW480, SW620, HT29 and HCT116), and then Western Blot analysis was performed. (C) LS174T cells were treated with EGF for various times as indicated (left) or treated with 0, 30 or 60 ng/ml EGF for 2 hrs (right). The lysates were analysed as in (B). The quantitative results are shown below. Statistic results from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (D) Cells were pre-treated with AG1478 for 30 min., followed by treatment with EGF for 2 hrs. The expressions of Aurora-A and EGFR were analysed as in (B). (E) EGF-stimulated cells were harvested at the indicated times, and the expressions of EGFR, Aurora-A, p-Akt, Akt, p-ERK, ERK and α-tubulin were analysed. (F) Cells were pre-treated with U0126 or wortmannin for 30 min., followed by EGF treatment for 2 hrs (DMSO, vehicle control). α-Tubulin was used as the loading control. (G) The induction of Aurora-A by short-term (2 hrs) EGF treatment was inhibited by U0126. Serum-starved A431 cells were pre-treated without (lanes 2, 5) or with (lanes 3, 6) U0126 (10 μM) for 30 min. and then treated without (lanes 1, 4) or with EGF (10 nM) for 2 (lanes 2–3) or 24 hrs (lanes 5–6). Total lysates were harvested for performing the Western blotting analysis by using the indicated antibodies.
To address it, Western blot analysis was performed to examine the expression levels of five cultured human colorectal cell lines, which are all mediating the EGF signal through the traditional EGFR signalling pathway (Fig. 1B). The Aurora-A expression was observed in all of these colorectal cell lines, and the EGFR was overexpressed in four of these cell lines (Fig. 1B). Interestingly, the expression level of Aurora-A was enhanced by EGF stimulation only in those EGFR-overexpressed cell lines (Fig. 1B). To further characterize this phenomenon, LS174T cells were treated with EGF, and the cell lysate was collected for Western blot analysis. It showed that the expression of Aurora-A appears to increase via time- and dose-dependent manners (Fig. 1C). This phenomenon was abolished by pre-treating cells with an EGFR inhibitor, AG1478, to block the EGF signalling pathway (Fig. 1D). Moreover, it is interesting to examine which signalling pathways mediated the EGF-enhanced Aurora-A expression. As shown in Fig. 1E, the Akt and ERK signalling pathways were activated after 30 min. of EGF treatment, and then returned to an inactivation state after 1 hr of stimulation. In the same circumstance, the protein expression level of Aurora-A reached a peak after 2 hrs of EGF treatment. When cells were pre-treated with U0126, a MEK inhibitor, or wortmannin, a PI3K inhibitor, the expression level of Aurora-A was blocked under EGF treatment, but not the control with DMSO treatment (Fig. 1F). Furthermore, pre-treating the U0126 to inhibit the ERK signalling pathway in A431 cells, the expression of Aurora-A only decreased in the early stage (2 hrs), not in the late stage (24 hrs), of EGF treatment (Fig. 1G). This result strengthens our previous hypothesis that the traditional EGF signalling pathway is also involved in the regulation of Aurora-A expression under EGF treatment in EGFR-overexpressed cells, and that the Akt and ERK signalling pathways play important roles in mediating this signal.
Co-expression of the EGFR and Aurora-A in colorectal cancers
The result above indicates that the expression of EGFR is highly important for the Aurora-A expression under EGF signal. Therefore, it would be interesting to study the expression patterns of Aurora-A and EGFR in clinical samples. To address it, the expression levels of EGFR and Aurora-A were detected in 13 colorectal cancer specimens. From Fig. 2A, the co-expression of EGFR and Aurora-A was found in 61.54% (8/13) of colorectal cancer samples. And only 15.38% (2/13) exhibited a high expression level of Aurora-A, but no EGFR expression. Furthermore, the activated statuses of PI3K/AKT and MEK/ERK signal pathways were also verified. As shown in Fig. 2B, the Akt signal pathway activation was noted in 87.5% (7/8) of colorectal cancers with EGFR and Aurora-A co-expression; the ERK pathway was activated in 50% (4/8) of samples and both the activated Akt and ERK were detected in four samples (50%). Among them, 23.08% (3/13) of colorectal cancers with elevated phosph-AKT activity did not express the EGFR and the Aurora-A protein. It indicates that other signaling pathways may contribute to the activation of Akt, but not through the EGFR signal in those clinical samples. These results showed a strong correlation between EGFR and Aurora-A expression in colorectal cancer. It also implied that the mechanism, which EGF signaling enhances the expression of Aurora-A, underlies the development of colorectal cancer, and the Akt and ERK pathways may also be involved in this regulation.
Fig 2.
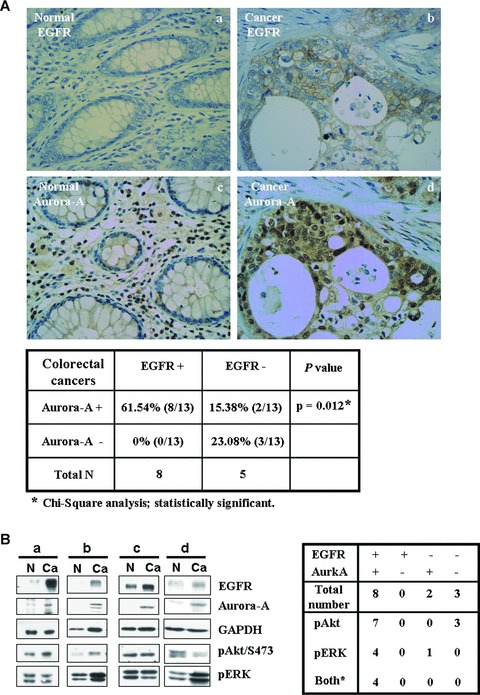
Co-expression of EGFR and Aurroa-A, and activation of Akt and ERK pathways in human colorectal tissues. (A) The expression of the EGFR and Aurora-A in normal colon tissue (a and c) and colorectal cancer (b and d). Representative data are shown. The statistical result of the immunohistochemical staining of 13 human colorectal specimens is shown below. (B) The expression levels of the EGFR and Aurora-A and the activation statuses of Akt (Akt/pS473) and ERK (pERK) in human normal colon tissues (N) and colorectal cancers (Ca) were analysed by Western Blot. The statistical result is shown in right panel.
Traditional EGF signalling pathway did not activate the Aurora-A mRNA expression in colorectal cancer cells
Transcriptional regulation of Aurora-A was reported in non-EGFR-overexpressed H-1299 cells via Akt signal pathway [16]. To further demonstrate whether the same mechanism is functional in EGFR-overexpressed colorectal cancer cells, the expression level of Aurora-A mRNA was examined after EGF stimulation. The result showed that the expression of Aurora-A mRNA was maintained at a constant level regardless of whether EGF was stimulated or not (Fig. 3A). Blocking the ERK and Akt signal pathways, which regulated the Aurora-A protein expression upon EGF treatment (Fig. 1F), showed no effects on Aurora-A mRNA expression (Fig. 3B). In addition, the promoter activity of the Aurora-A gene was also not activated in response to EGF stimulus (Fig. 3C). Furthermore, cells were pre-treated with actinomycin D to block the transcriptional activity, and the protein expression level of Aurora-A under EGF treatment was further increased (Fig. 3D). Overall, these results indicated that transcriptional regulation was not involved in the increase of Aurora-A protein in EGFR-overexpressed cells.
Fig 3.
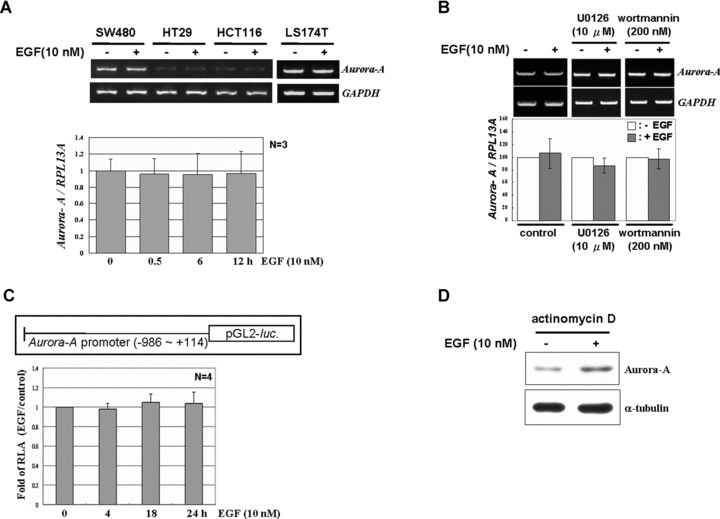
EGF does not alter the Aurora-A mRNA level in human colorectal cancer cell lines. (A) Serum-starved LS174T cells were treated with EGF for 3 hrs, and total RNA was harvested. The expression levels of Aurora-A and GAPDH mRNA were detected by RT-PCR. The lower panel, serum-starved cells were treated with EGF for different time-points, and the expression level of Aurora-A mRNA was quantified by real-time RT-PCR and normalized with RPL13A (ribosomal protein L 13A). The mean ± S.D. (error bars) was obtained from three independent experiments. (B) Neither U0126 nor wortmannin changed the expression level of Aurora A mRNA under EGF treatment. The expression of Aurora-A mRNA was analysed as in (A). (C) Cells were transiently transfected with the Aurora-A promoter-luciferase construct. After starvation, cells were treated with EGF for 0, 4, 18 and 24 hrs, and the reporter assay was performed with a dual luciferase assay kit. The mean ± S.D. was obtained from three independent experiments. (D) Serum-starved cells were pre-treated with actinomycin D for 30 min., followed by EGF for another 2 hrs. The expressions of Aurora-A and α-tubulin were analysed by Western Blot.
Traditional EGF signalling pathway activates Aurora-A expression through the translational up-regulatory mechanism
Enhanced protein stability or protein synthesis rate are two major regulatory mechanisms to increase protein expression. It was reported that Aurora-A may be degraded by APC/CCdh1 complex during mitotic exit [17]. To address whether elevated Aurora-A is associated with increased protein stability, cells were treated with EGF for 2 hrs prior to addition of cyclohexamide to block subsequent protein synthesis, and then the Aurora-A protein degradation rate with or without EGF treatment was measured. From Fig. 4A, the protein stability did not change under EGF stimulation. In addition, the expression of the Cdh1, an E3 ubiquitin ligase, was also kept at a constant level during EGF stimulus (Fig. 4B). It indicated that the increased expression of Aurora-A was not because of the decrease of protein degradation rate. Therefore, whether the EGF signal enhances the translation efficiency of Aurora-A mRNA was examined. First, the pulse-chase assay was performed to examine the de novo protein synthesis of Aurora-A mRNA upon EGF treatment. From Fig. 4C, the newly synthesized Aurora-A protein increased under EGF treatment. Furthermore, using the S6 immunoprecipitation assay, we demonstrated that the association of Aurora-A mRNA with the ribosomal S6 subunit increased under EGF treatment, indicating that Aurora-A mRNA engaged in an active translation status (Fig. 4D). This phenomenon was also observed in other colorectal cancer cell lines, such as HT29, HCT116 and SW480, and found in the EGFR-overexpressed cervical cancer cell line, A431 (Fig. 4E). Overall, these results suggested that a translational regulatory mechanism might play an important role in controlling Aurora-A expression under EGF signalling.
Fig 4.
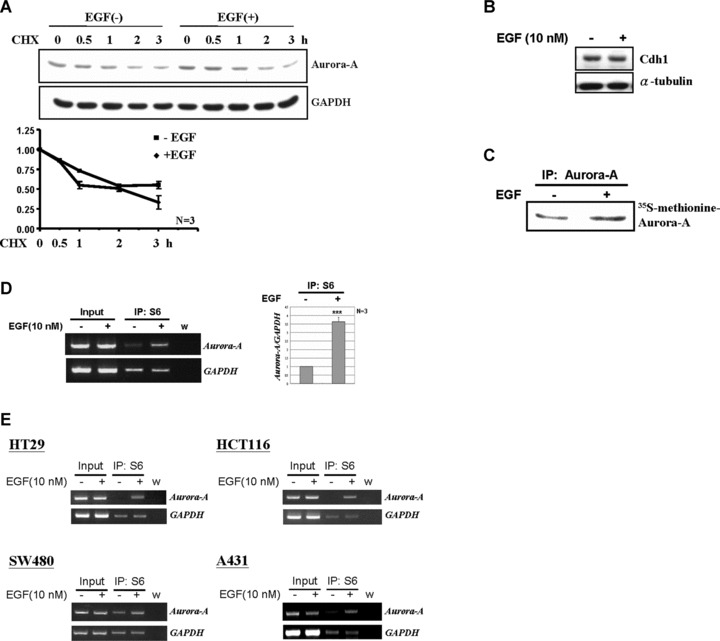
EGF increases the translational efficiency of Aurora-A mRNA. (A) LS174T cells were treated with EGF (10 nM) for 2 hrs and then further incubated with the translational inhibitor, cyclohexamide (CHX, 0.2 μg/ml), for additional 0, 0.5, 1, 2 and 3 hrs. The protein level of Aurora-A was measured by Western blot analysis. Relative protein levels of Aurora-A were normalized to GAPDH. And the quantitative results from three independent experiments was shown below. (B) The expression level of Cdh1 under EGF treated (+) or not (–) was examined by Western blot analysis. (C) 35S-methionine labelled Aurora-A was immunoprecipitated from LS174T cell lysates by anti-Aurora-A antibodies. The immunoprecipitated product was separated by 10% SDS-PAGE and analysed by radiography. (D) Equal amounts of EGF-stimulated cell lysates were used for the ribosomal protein S6-immunoprecipitation assay. The amount of Aurora-A mRNA in the S6-IP complex was analysed by RT-PCR. Quantitative results are shown in the right panel. The mean ± S.D. was obtained from three independent experiments. (w, water control; ***, P < 0.001). (E) Amounts of Aurora A mRNA in the S6-IP complexes of SW480, HT29, HCT116 and A431 cells were analysed as described in (C). (w, water control).
The PI3K/Akt/mTOR and MEK/ERK pathways regulate the EGF-induced Aurora-A protein translation
It was well established that the activated mTOR pathway regulates the mRNA translation through the phosphorylation of its down-stream targets such as eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) and the ribosomal protein S6 kinase (S6K) [11]. To further demonstrate that the EGF signal increases the Aurora-A expression via translational regulation, the mTOR signal pathway was analysed under EGF stimulus. From Fig. 5A, EGF signal augmented the expression of Aurora-A and the phosphorylation forms of Akt, mTOR, p70S6K and 4E-BP1 were increased (compare lanes 1 and 2). It indicated that the addition of EGF might turn on the mTOR pathway to activate the translational machine. To address it, the EGF-mediated activation of mTOR pathway was blocked by rapamycin treatment. The result showed that the EGF-induced Aurora-A protein increase was abolished (Fig. 5A, compare lanes 5 and 6). On the other hand, we had demonstrated that the Akt and ERK signalling pathways were involved in the expression of Aurora-A under EGF treatment (Fig. 1F). By treating cells with U0126 and wortmannin to inhibit the Akt and ERK pathways respectively, the recruitment of Aurora-A mRNA to ribosome complex was abolished (Fig. 5B). This result indicated that both Akt and ERK pathways regulate Aurora-A mRNA translation under EGF stimulus. In addition, it would be interesting to check whether these two pathways are interfering the phosphorylation status of mTOR to increase the Aurora-A mRNA translation. Addition of wortmannin inhibited the Akt pathway to block the mTOR phosphorylation (Fig. 5C, compare lanes 7 and 8) and then stopped the translation as expected (Fig. 5B). Interestingly, the ERK inhibitor, U0126, showed no effects on mTOR pathway (Fig. 5C, lanes 3 and 4). This result implied that the activated ERK might work on different targets, which are not through the mTOR signalling pathway, to regulate the Aurora-A protein synthesis.
Fig 5.
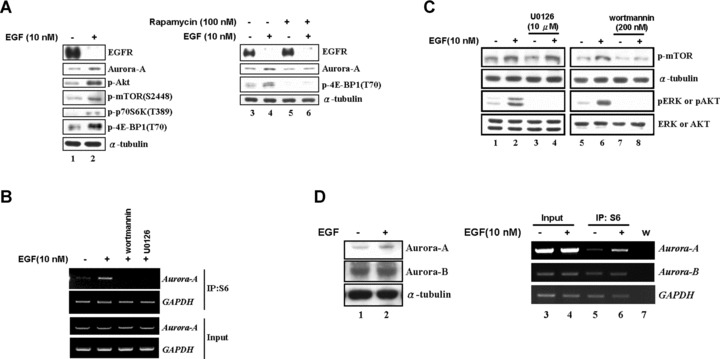
Activation of the ERK and Akt signalling pathways is important for EGF-increased Aurora-A overexpression. (A) In LS174T cells, the expression of Aurora-A, the activation of mTOR (p-mTOR/S2448) and its substrates, 70S6K (p-p70S6K/T389) and 4E-BP1 (p-4E-BP1/T70), were activated by EGF treatment (lanes 1–2). The addition of the mTOR inhibitor, rapamycin, blocked EGF effect on Aurora-A protein expression (lanes 3–6). (B) The amount of Aurora-A mRNA in the S6-IP complexes of U0126- or wortmannin pre-treated cells was analysed by RT-PCR as described in Fig. 4D. (C) Serum-starved cells were pre-treated with U0126 or wortmannin, followed by EGF treatment as described above. The phosphorylation status of mTOR (p-mTOR), ERK (pERK) and Akt (pAkt) was analysed. (D) The expression levels of Aurora-A and Aurora-B were analysed by the Western blot analysis in cells which are EGF-treated (+) or not (–) (lanes 1–2). And the amount of Aurora-A or Aurora-B mRNA in the S6-IP complexes were examined (lanes 3–6). w: H2O negative control.
A specific 5′-UTR splicing variant of Aurora-A mRNA is recruited to ribosome complex after EGF treatment
The Akt/mTOR pathway targets the 4E-BP1 and S6K to regulate a common translation mechanism. However, the ribosome complex recruits a particular group of mRNAs, but not all, to translate their proteins under EGF stimulation. In order to further demonstrate whether the EGF-induced translational up-regulation is specific on Aurora-A mRNA but not the other Aurora kinases family, the levels of Aurora-B protein and Aurora-B mRNA that interacted with ribosome complex were examined (Fig. 5D). The data showed that the expression level of Aurora-B is maintained constant after EGF treatment. This result indicated that other factors might control the selection function to make sure the translational machine synthesizes the ‘right’ mRNA. In addition, our previous result also indicated that blocking the ERK pathway to inhibit the Aurora-A protein expression was not mediated by mTOR pathway (Fig. 5C, compare lanes 3 and 4). Therefore, it is interesting to study what kind of cis- or trans- factors determine the selective function. By searching the NCBI database, the 5′UTR of Aurora-A mRNA is shown to contain six types of alternative splicing variants (Fig. 6A). By using the specific primers to amplify the 5′UTR of Aurora-A mRNA, we observed that four splicing variants (133/147 and 243/257 bps) dominantly expressed in LS174T cells, and the expression level remained constant with or without EGF treatment (Fig. 6B). However, only the splicing variants – 243/257 bps (contained the exon 2) – were enhanced to bind with the translational complex after EGF treatment (Fig. 6C). To further demonstrate the exon 2 fragment is responsible for EGF-induced translational activation, the in vivo and in vitro translation assay were performed. From Fig. 6D, the specific exon 2-contained fragments increased the translation efficiency under EGF stimulation. Moreover, EGF enhanced the report gene expression only in the construct containing exon 2 splicing form in LS174T cells (Fig. 6E). The S6-IP assay further showed the increased association of the exon 2 containing construct with ribosomal complex after EGF stimulation (Fig. 6E). Besides, by examining the Aurora-A mRNA splicing variants that existed in human colorectal cancers, the exon 2 containing variants (243/257 bps) were dominantly expressed in these samples (Fig. 6F). Combining these results, they imply that the exon 2 region of Aurora-A mRNA may play a crucial role in modulating the translational efficiency under EGF treatment. Moreover, the unique expression pattern of exon 2 containing 5′UTR in cancer tissues may provide it a role as a potential target for future pharmaceutical purposes.
Fig 6.
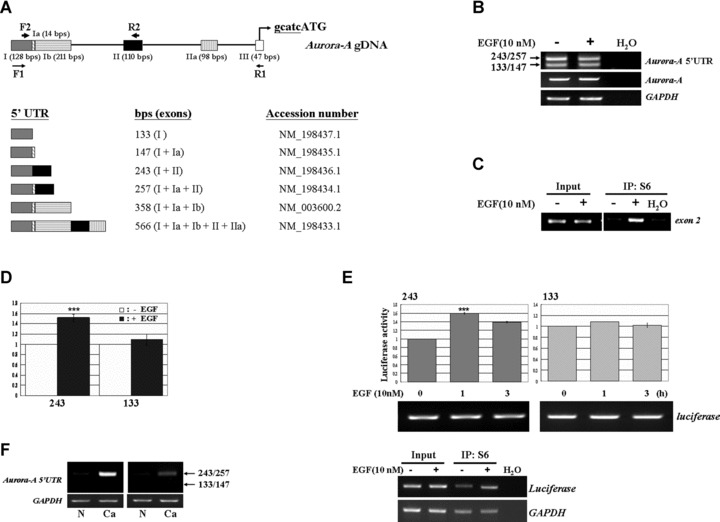
The exon 2 containing Aurora-A mRNA plays an important role in response to EGF treatment. (A) Schematic of Aurora-A mRNA 5′-UTR isoforms, and the location of primer pairs used for identifying the expression pattern of Aurora-A 5′UTR (F1 and R1) and S6-IP followed PCR (F2 and R2) are indicated. (B) The 133/147 bps and 243/257 bps 5′UTRs were dominantly expressed in LS174T cells, and the expression levels of Aurora-A mRNA and its 5′UTR isoforms were not changed under EGF stimulus. (C) The S6 ribosomal protein-associated Aurora-A mRNA was detected by primer pair F2/R2 located on the 5′UTR as indicated in (A). (D) Equal amount of RNA from different splicing variant, 133 bps and 243 bps, were incubated with EGF-treated or untreated lysates and then performed the in vitro translation assay. (E) The constructs of pGL3-promoter contained different Aurora-A 5′UTR splicing forms (243 bps and 133 bps) were transient transfected into cells, and then the reporter assays were performed. The expression level of luciferase mRNA was used for indicating the similar amount of mRNA. In the lower panel, the S6-IP assays were also conducted to exam the association of splicing form 243 bps with translational complex under EGF stimulation. (***, P < 0.001) (F) The expression pattern of Aurora-A 5′UTRs in human normal colon tissues (N) and colorectal cancers (Ca). GAPDH as an internal control.
Discussion
Deregulation of Aurora-A is highly correlated with tumour progression and the poor prognosis [3, 4]. Known instances of Aurora-A overexpression in cancers are through gene amplification, RNA transcriptional up-regulation and prolonged protein stability [18, 19]. No literature has reported whether translational regulation exists for increased expression of Aurora-A in cancer cells. This study is the first report to demonstrate that translational activation may up-regulate the expression of Aurora-A in cancers. In addition, two pathways, MEK/ERK and the PI3K/Akt/mTOR, were identified which regulate the translation of the Aurora-A protein (Fig. 1).
The constitutive activation of EGFR is proposed to play a role in chromosome instability [20, 21]. This phenomenon was also observed in cancers in which the Aurora-A protein was overexpressed [22]. Our study was designed to investigate the relationship between the overexpression of EGFR and Aurora-A in tumour cells. There are two routes for the EGF/EGFR signalling pathway: the traditional/cytoplasmic signalling pathway and a novel nuclear translocation pathway [23]. Previously, we demonstrated that EGF increased the expression of Aurora-A mRNA through the nuclear EGFR pathway. When cells were treated with EGF, the activated EGFR is translocated into the nucleus and acts as a transcription activator to interact with STAT5 to increase the expression of the Aurora-A gene in EGFR-overexpressing tumours or cells [13]. Besides, we observed a bi-phase increase in Aurora-A protein expression in A431 cells after treatment with EGF for 2 and 24 hrs, respectively (Fig. 1A). Using U0126 to inhibit the ERK signalling pathway, the expression of Aurora-A only decreased in the early stage, not the late stage, of EGF treatment (Fig. 1G). This indicates that another mechanism may exist, in addition to the nuclear EGFR pathway, which regulates Aurora-A expression under EGF treatment. To address this hypothesis, LS174T cells, which overexpress the EGFR but not exhibit the nuclear EGFR pathway (Fig. 1B, the expression of EGFR decreased after EGF treatment), were used to dissect the regulatory mechanism under the cytoplasmic/traditional EGF/EGFR signalling pathway. The result indicated that EGF increased Aurora-A protein translation (Fig. 1), but not the amount of Aurora-A mRNA (Fig. 3). Furthermore, the ribosomal S6 protein immunoprecipitation assay showed increased binding of Aurora-A mRNA with the ribosome complex which activates its translation under EGF treatment (Fig. 4D). Combining these results, we concluded that the short-term treatment of EGF might result in the translational up-regulation of Aurora-A mRNA, whereas the transcriptional regulation of Aurora-A might involve in the long-term EGF treatment. How cells determine the fate of EGFR routes, to translocate into nucleus or to undergo the cytoplasmic signalling, remain unclear. In this report, the novel control mechanism of Aurora-A expression via translational regulation was identified. This mechanism provides evidence that cells can regulate important cell cycle-related genes, such as Aurora-A, through multiple pathways to ensure their proper expression. It also provides strong evidence to support the idea that the Aurora-A protein is involved in cancer development in EGFR-overexpressing cancer cells.
Activation of Akt and Ras pathways was reported that can cooperative control translation in cancer cells [24]. The PI3K/Akt signalling pathway occurs at an early stage of cancer formation [11], and it is activated by stimulation of extracellular signals, including the EGFR, insulin-like growth factor-1 receptor, integrin and the G-protein-coupled receptor [25]. One of the well-known pathways promoted the survival of cancer cells by activating mTOR/S6K is through the increasing protein translation, and then lead to the effects on the cell cycle progression [11, 25, 26]. This study provides a linkage between the activated EGF/EGFR-PI3K/Akt/mTOR pathways with the overexpressed Aurora-A in cancer cells, and therefore confirms that complex multiple factors are involved in cancer formation. In addition, another effort of this study was to characterize the activated MEK/ERK pathway that may also participate in the overexpression of Aurora-A in cancer cells. ERK is another notable downstream target of the activated EGFR, and aberrant activation of the ERK pathway may lead cells to tumorigenesis [27]. Interestingly, it was reported that the activated MAPK pathway may directly up-regulate the Aurora-A gene in pancreatic cancer [28]. However, we demonstrated that EGF-induced activation of the ERK pathway did not activate the Aurora-A promoter, but indeed participated in increasing the protein expression of Aurora-A in colorectal cancers and cervical cancer cells (Figs 3, 4C and D). The discrepancy between these results may have been because of the different microenvironments among the different cell types (pancreatic cancer cells vs. colorectal and cervical cancer cells).
Although both the PI3K/Akt and MEK/ERK pathways are critical in regulating Aurora-A protein translation, the regulatory mechanism may different. The PI3K/Akt pathway increases the phosphorylation mTOR and enhances the translation efficiency. Nevertheless, the treatment with the MEK inhibitor, U0126, did not inhibit the phosphorylation status of mTOR (Fig. 5C), indicating that the activated ERK pathway might not work through the activation of mTOR to increase the translational efficiency. Furthermore, it was reported that the EGF-induced activation of the PI3K/Akt/mTOR pathway can inhibit lactogenic differentiation in HC11 cells [29]. This implies that cells can selectively activate the translation of certain proteins and inhibit others. This regulation may occur through an interaction between selective protein(s) with its target mRNA, and then the translational complex was recruited to the target mRNA via this selective protein. Therefore, we postulate that the ERK pathway may regulate a selective protein to enhance the formation of translational initiation complex on the Aurora-A mRNA.
The 5′-untranslated region (5′UTR) of mRNA may be the target site where the selective proteins interact and thus plays an important role in its translational regulation [30]. Alternative pre-mRNA splicing plays a crucial role in the production of proteome complexity and influences development and disease. Numerous genome-wide analyses estimated that 35–94% of all human genes appear to undergo alterative splicing, and lots of cancer-specific transcript variants were identified [31, 32]. Currently, the analyses of splice variants that are found predominantly in tumour have clear diagnostic value and may provide potential drug targets [33]. Interestingly, Aurora-A mRNA has six kinds of 5′-UTR variants (according to the NCBI databank). The expression of different 5′UTR alterative splicing variants of Aurora-A in breast cancer cell lines and primary tumours had been reported [34]. However, the functional role of these variants was unclear. Here we found the exon 2 variant contributes to the protein expression of Aurora-A under EGF treatment. Moreover, the exon 2 containing variants – 243/257 bps is dominantly expressed in tested human cancer tissues (Fig. 6F). It would be interesting to study the correlation between exon 2 variant and colorectal cancer development in the future.
In summary, this report provides a novel regulatory mechanism for Aurora-A protein expression, and highlights the relationship between EGF signalling and Aurora-A expression. Most importantly, the recruitment of specific splicing variants of Aurora-A mRNA in regulating Aurora-A protein expression provides a new approach in studying the gene expression under tumour development.
Acknowledgments
We thank Mr. Dan Chamberlin for English editing. This work was supported by grants (NSC94–2314-B-006–121, NSC95–2320-B-006–084 and NSC95–2320-B006–066-MY3) from the National Science Council, and Project of Promoting Academic Excellent and Developing World Class Research Centers, and Center for Gene Regulation and Signal Transduction Research, National Cheng Kung University.
References
- 1.Bischoff JR, Anderson L, Zhu Y, et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998;17:3052–65. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell. 2003;3:51–62. doi: 10.1016/s1535-6108(02)00235-0. [DOI] [PubMed] [Google Scholar]
- 3.Katayama H, Brinkley WR, Sen S. The Aurora kinases: role in cell transformation and tumorigenesis. Cancer Metastasis Rev. 2003;22:451–64. doi: 10.1023/a:1023789416385. [DOI] [PubMed] [Google Scholar]
- 4.Landen CN, Jr, Lin YG, Immaneni A, et al. Overexpression of the centrosomal protein Aurora-A kinase is associated with poor prognosis in epithelial ovarian cancer patients. Clin Cancer Res. 2007;13:4098–104. doi: 10.1158/1078-0432.CCR-07-0431. [DOI] [PubMed] [Google Scholar]
- 5.Burgess AW. EGFR family: structure physiology signalling and therapeutic targets. Growth Factors. 2008;26:263–74. doi: 10.1080/08977190802312844. [DOI] [PubMed] [Google Scholar]
- 6.Normanno N, Bianco C, De Luca A, et al. Target-based agents against ErbB receptors and their ligands: a novel approach to cancer treatment. Endocr Relat Cancer. 2003;10:1–21. doi: 10.1677/erc.0.0100001. [DOI] [PubMed] [Google Scholar]
- 7.Lockhart AC, Berlin JD. The epidermal growth factor receptor as a target for colorectal cancer therapy. Semin Oncol. 2005;32:52–60. doi: 10.1053/j.seminoncol.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 8.Lam AK, Ong K, Ho YH. Aurora kinase expression in colorectal adenocarcinoma: correlations with clinicopathological features, p16 expression, and telomerase activity. Hum Pathol. 2008;39:599–604. doi: 10.1016/j.humpath.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi T, Futamura M, Yoshimi N, et al. Centrosomal kinases, HsAIRK1 and HsAIRK3, are overexpressed in primary colorectal cancers. Jpn J Cancer Res. 2000;91:1007–14. doi: 10.1111/j.1349-7006.2000.tb00878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mamane Y, Petroulakis E, LeBacquer O, et al. mTOR, translation initiation and cancer. Oncogene. 2006;25:6416–22. doi: 10.1038/sj.onc.1209888. [DOI] [PubMed] [Google Scholar]
- 11.Granville CA, Memmott RM, Gills JJ, et al. Handicapping the race to develop inhibitors of the phosphoinositide 3-kinase/Akt/mammalian target of rapamycin pathway. Clin Cancer Res. 2006;12:679–89. doi: 10.1158/1078-0432.CCR-05-1654. [DOI] [PubMed] [Google Scholar]
- 12.Jiang BH, Liu LZ. Role of mTOR in anticancer drug resistance: perspectives for improved drug treatment. Drug Resist Updat. 2008;11:63–76. doi: 10.1016/j.drup.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hung LY, Tseng JT, Lee YC, et al. Nuclear epidermal growth factor receptor (EGFR) interacts with signal transducer and activator of transcription 5 (STAT5) in activating Aurora-A gene expression. Nucleic Acids Res. 2008;36:4337–51. doi: 10.1093/nar/gkn417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeh CH, Hung LY, Hsu C, et al. RNA-binding protein HuR interacts with thrombomodulin 5’untranslated region and represses internal ribosome entry site-mediated translation under IL-1 beta treatment. Mol Biol Cell. 2008;19:3812–22. doi: 10.1091/mbc.E07-09-0962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gritsko TM, Coppola D, Paciga JE, et al. Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clin Cancer Res. 2003;9:1420–6. [PubMed] [Google Scholar]
- 16.Liu X, Shi Y, Woods KW, et al. Akt inhibitor a-443654 interferes with mitotic progression by regulating aurora a kinase expression. Neoplasia. 2008;10:828–37. doi: 10.1593/neo.08408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Littlepage LE, Ruderman JV. Identification of a new APC/C recognition domain, the A box, which is required for the Cdh1-dependent destruction of the kinase Aurora-A during mitotic exit. Genes Dev. 2002;16:2274–85. doi: 10.1101/gad.1007302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marumoto T, Zhang D, Saya H. Aurora-A – a guardian of poles. Nat Rev Cancer. 2005;5:42–50. doi: 10.1038/nrc1526. [DOI] [PubMed] [Google Scholar]
- 19.He L, Yang H, Ma Y, et al. Identification of Aurora-A as a direct target of E2F3 during G2/M cell cycle progression. J Biol Chem. 2008;283:31012–20. doi: 10.1074/jbc.M803547200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Li L, Dutra A, Pak E, et al. EGFRvIII expression and PTEN loss synergistically induce chromosomal instability and glial tumors. Neuro-oncology. 2009;11:9–21. doi: 10.1215/15228517-2008-081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin ES, Tonon G, Sinha R, et al. Common and distinct genomic events in sporadic colorectal cancer and diverse cancer types. Cancer Res. 2007;67:10736–43. doi: 10.1158/0008-5472.CAN-07-2742. [DOI] [PubMed] [Google Scholar]
- 22.Zhou H, Kuang J, Zhong L, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20:189–93. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]
- 23.Lo HW, Hung MC. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br J Cancer. 2006;94:184–8. doi: 10.1038/sj.bjc.6602941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parsa AT, Holland EC. Cooperative translational control of gene expression by Ras and Akt in cancer. Trends Mol Med. 2004;10:607–13. doi: 10.1016/j.molmed.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 25.LoPiccolo J, Granville CA, Gills JJ, et al. Targeting Akt in cancer therapy. Anticancer Drugs. 2007;18:861–74. doi: 10.1097/CAD.0b013e3280cc2c6f. [DOI] [PubMed] [Google Scholar]
- 26.Jiang BH, Liu LZ. PI3K/PTEN signaling in tumorigenesis and angiogenesis. Biochim Biophys Acta. 2008;1784:150–8. doi: 10.1016/j.bbapap.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 27.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 28.Furukawa T, Kanai N, Shiwaku HO, et al. AURKA is one of the downstream targets of MAPK1/ERK2 in pancreatic cancer. Oncogene. 2006;25:4831–9. doi: 10.1038/sj.onc.1209494. [DOI] [PubMed] [Google Scholar]
- 29.Galbaugh T, Cerrito MG, Jose CC, et al. EGF-induced activation of Akt results in mTOR-dependent p70S6 kinase phosphorylation and inhibition of HC11 cell lactogenic differentiation. BMC Cell Biol. 2006;7:34. doi: 10.1186/1471-2121-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilkie GS, Dickson KS, Gray NK. Regulation of mRNA translation by 5’- and 3’-UTR-binding factors. Trends Biochem Sci. 2003;28:182–8. doi: 10.1016/S0968-0004(03)00051-3. [DOI] [PubMed] [Google Scholar]
- 31.He C, Zhou F, Zuo Z, et al. A global view of cancer-specific transcript variants by subtractive transcriptome-wide analysis. PLoS ONE. 2009;4:e4732. doi: 10.1371/journal.pone.0004732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–6. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brinkman BM. Splice variants as cancer biomarkers. Clin Biochem. 2004;37:584–94. doi: 10.1016/j.clinbiochem.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 34.Shin SO, Lee KH, Kim JH, et al. Alternative splicing in 5’-untranslational region of STK-15 gene, encoding centrosome associated kinase, in breast cancer cell lines. Exp Mol Med. 2000;32:193–6. doi: 10.1038/emm.2000.31. [DOI] [PubMed] [Google Scholar]