

Abstract
Primary endothelial cells are fully resistant to TNF-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis. Here, we demonstrate that certain environmental conditions, such as exposure to the widespread allergen nickel, can dramatically increase the susceptibility of naturally resistant primary endothelial cells or keratinocytes to TRAIL-induced apoptosis. While nickel treatment increased surface expression of the apoptosis-inducing TRAIL receptors TRAIL-R1 and TRAIL-R2, it also up-regulated the apoptosis-deficient TRAIL-R4, suggesting that modulation of TRAIL receptor expression alone is unlikely to fully account for the dramatic sensitization effect of nickel. Further analysis of candidate mediators revealed that nickel strongly repressed c-FLIP at mRNA and protein levels. Accordingly, increased activation of Caspase-8 and Caspase-3 following nickel treatment was observed. Importantly, depletion of c-FLIP by RNA interference could largely recapitulate the effect of nickel and sensitize endothelial cells to TRAIL-dependent apoptosis in the absence of nickel pre-treatment. Conversely, ectopic expression of c-FLIPL largely protected nickel-treated cells from TRAIL-mediated apoptosis. Our data demonstrate that one key mechanism of sensitization of primary human endothelial cells or keratinocytes is transcriptional down-regulation of c-FLIP. We hypothesize that environmental factors, exemplified by the contact allergen nickel, strongly modulate death ligand sensitivity of endothelial cells and keratinocytes thus influencing vascular and epidermal function and integrity under physiological and pathophysiological conditions.
Keywords: nickel ions, contact hypersensitivity, endothelial cells, apoptosis, c-FLIP, TRAIL receptor, DISC
Introduction
Nickel (Ni2+) is an important inducer of contact allergy reactions, widely distributed in numerous industrial products, including bimetallic coins, cardiovascular stents and orthopaedic and dental biomedical alloys [1]. Ni2+ ions are able to penetrate the skin, although the absolute concentration of Ni2+ in epidermis and dermis has not yet been determined in vivo[2]. Ni2+ hypersensitivity responses primarily manifest as contact dermatitis, a frequent cause of professional incapability, but also have been implicated with in-stent restenosis, a severe complication of patients with cardiovascular disease [3]. Besides eliciting antigen-specific signals in such T-cell-prevailing conditions, Ni2+ also induces a non-specific pro-inflammatory signal in endothelial cells (ECs) and keratinocytes, which initiates and propagates the inflammatory response by induction of cytokines and cell-surface molecules required for recruitment of antigen-specific lymphocytes to the exposed tissue [4]. Several signal transduction pathways such as the IKK2/NFκB cascade, hypoxia-inducible factor 1α (HIF-1α) and the p38 mitogen-activated protein kinase (MAPK) pathway [5–7] coordinate this inflammatory response in ECs, resulting in a characteristic pattern of gene products that guide attraction and interaction with leukocytes or platelets, affect vascular permeability and finally control the course and outcome of inflammatory reactions. Surprisingly, own micro-array studies in EC revealed that Ni2+ can also regulate components of the TNF-related apoptosis-inducing ligand (TRAIL) death receptor pathway [6], suggesting that Ni2+ might also affect blood vessel integrity and remodelling.
The TRAIL system consists of soluble or cell surface-bound TRAIL protein and four membrane-bound receptors [8]. Only TRAIL receptor 1 (TRAIL-R1) and TRAIL-R2 contain functional intracellular death domains. In contrast, TRAIL-R3 and 4 are incapable of apoptosis induction and either completely lack a cytoplasmic domain (TRAIL-R3) or only contain an incomplete death domain (TRAIL-R4) unable to transduce a death signal [9, 10]. The cross-linking of the two apoptosis-inducing TRAIL receptors results in the recruitment of FADD (MORT1) and Procaspase-8 to the death-inducing signalling complex (DISC) [8], resulting in proteolytic cleavage of Caspase-8 and initiation of the apoptotic process via activation of downstream effector caspases.
In many primary cells, death receptor-mediated apoptosis is effectively inhibited by high expression of cellular Fas-associated death domain-like interleukin-1-converting enzyme (FLICE) inhibitory protein (c-FLIP), an intracellular homologue of Caspase-8 [11]. c-FLIP isoforms are recruited to the TRAIL DISC and inhibit full cleavage and release of active Caspase-8 and Caspase-10 [11], allowing survival of these cells in the presence of receptor ligation, a finding that has merited great attention since many tumour cells are highly sensitive to TRAIL-mediated apoptosis [12]. However, the insensitivity of primary cells to TRAIL-mediated apoptosis may underlie plasticity under distinct physiological or pathophysiological conditions [13].
In this report, we have studied the impact of Ni2+ on TRAIL apoptosis sensitivity in primary ECs in detail. We found that Ni2+ strongly sensitizes naturally resistant ECs to TRAIL-induced apoptosis. This sensitization could only partially be explained by TRAIL-R regulation since Ni2+ simultaneously up-regulated apoptosis-proficient and -deficient members of the TRAIL-R family. Instead, we demonstrate that transcriptional repression of c-FLIP provides a functionally relevant mechanism by which Ni2+ sensitizes ECs for death ligand-mediated apoptosis. Similar results could be obtained with primary keratinocytes, another important type of effector cells in contact eczema. Our data show for the first time that environmental conditions, such as exposure to the contact allergen Ni2+, can greatly influence apoptosis resistance to death ligands and implicate continuous c-FLIP transcription as an essential determinant sustaining vascular and epidermal integrity.
Materials and methods
Cell culture and materials
Human umbilical vein endothelial cells (HUVEC) and human primary keratinocytes (KCs) were generated and cultured as described [6, 14]. Cells were exposed to 1.5 mM NiCl2.6H2O (Merck, Darmstadt, Germany; subsequently referred to as Ni2+) or medium as control and subsequently stimulated with Leucine-Zipper-TRAIL (kindly provided by H. Walczak) [15] at 100 ng/ml unless indicated otherwise. The following antibodies and reagents were used: z-Val-Ala-Asp-fluoromethyl ketone (zVAD-fmk; Bachem, Heidelberg, Germany), mouse monoclonal antibodies (Abs) against FLICE/Caspase-8 (C-15) and c-FLIP (NF-6) (generously provided by P. H. Krammer), total Caspase-3 Ab (MF #393; kindly provided by D. W. Nicholson, Merck Frost, Quebec, Canada) and rabbit polyclonal Ab against cleaved Caspase-3 (Cell Signaling, Danvers, MA, USA), Abs to tubulin and actin (Sigma, St. Louis, MO, USA), monoclonal Abs against FADD (Becton Dickinson (BD), Heidelberg, Germany), Ab against Erk2 (C-14, Santa Cruz Technology, Santa Cruz, CA), monoclonal Abs against Poly-ADP-ribose-polymerase (PARP; clone C2–10; Biomol, Hamburg, Germany). Polyclonal Abs for Western blot detection of TRAIL-R1 (ab8414) and TRAIL-R2 (ab8416) were from Abcam (Cambridge, UK), and rabbit polyclonal Abs to TRAIL-R4 were from Santa Cruz (Santa Cruz, CA, USA; C-20; sc-7550). Brefeldin A was obtained from Applichem GmbH, Darmstadt, Germany. For detection of human interleukin-8 (IL-8) protein in cell supernatants, a commercial ELISA kit from BD Pharmingen (BD OptEIA human IL8 Elisa Set) was used according to the manufacturer’s instructions.
Apoptosis and cytotoxicity assays
Crystal violet staining of surviving attached cells was performed 6 hrs after addition of FLAG-TRAIL in 96-well plates as described [16]. For analysis of apoptosis, cells were harvested 8 hrs after addition of TRAIL, fixed and examined by DNA-profiling employing propidium iodide staining and flow cytometric analysis of subdiploid DNA content. Internucleosomal degradation of genomic DNA was detected using the Cell Death Detection ELISAPLUS assay (Roche Molecular Diagnostics, Mannheim, Germany).
Western blot analysis
Total cellular proteins were lysed as described [6, 17]. Five to 75 μg of protein were electrophoresed on SDS-PAGE gels and Western blot analysis using the indicated primary and appropriate horseradish peroxidase-conjugated secondary Abs was performed as described [6, 16].
Analysis of the death-inducing signalling complex (DISC)
For precipitation of the native TRAIL-DISC, HUVEC were washed with RPMI medium and subsequently incubated for the indicated time periods in the presence of 1 μg/ml FLAG-TRAIL (kindly provided by H. Walczak) pre-complexed with 2 μg/ml anti-FLAG M2 Ab (Sigma) for 30 min., or, for non-stimulated controls, in the absence of FLAG-TRAIL as described for other cells [16]. DISC formation was stopped by washing the monolayer with ice-cold PBS. Cells were lysed by addition of lysis buffer (30 mM Tris-HCl pH 7.5 at 21°C, 120 mM NaCl, 10% Glycerol, 1% Triton X-100, Complete® protease inhibitor cocktail [Roche Molecular Diagnostics]). After 15 min. of incubation, lysates were centrifuged to pellet cellular debris. DISC complexes were precipitated from the lysates by co-incubation with 20 μl protein G beads (Roche Molecular Diagnostics) for 12 hrs on an end-over-end shaker at 4°C. For precipitation of non-stimulated receptors, 200 ng FLAG-TRAIL and 400 ng mouse-anti-FLAG M2 antibody (Sigma) were added to the lysates prepared from non-stimulated cells to control for protein association with non-stimulated receptor(s). Ligand affinity precipitates were washed before the protein complexes were eluted from the beads by addition of 2× standard reducing sample buffer. Subsequently, proteins were separated by SDS-PAGE on 4–12% NuPage Bis-Tris gradient gels (Invitrogen, Karlsruhe, Germany) and DISC components detected by Western blot analysis.
Retroviral modulation of c-FLIP expression in ECs
For stable expression of c-FLIP, HUVEC were infected with the retroviral vector PINCO containing FLAME1/c-FLIPL cDNA (kind gift of G. Stassi) as described [6]. For RNA interference studies, HUVEC were infected with retroviral expression vectors encoding either small hairpin (shRNA) against the long and the short isoform of human c-FLIP (pRS-c-FLIP; targeting sequence: GGAGCAGGGACAAGTTACA) or an empty pRS-retrovirus, respectively. To obtain optimal knock-down efficiencies, pRS-derived retroviruses were produced employing a high titre-producing packaging cell line [18]. Seventy-two hours after infection, cells were selected for puromycin resistance (2 μg/ml puromycin, 24 hrs) and incubated overnight in puromycin-free medium before stimulation. Knock-down efficiencies were routinely monitored by Western blot.
Quantitative real-time reverse transcriptase-PCR (qRT-PCR)
qRT-PCR was performed using the TaqMan Universal PCR Mastermix (Applied Biosystems, Darmstadt, Germany) and an ABI PRISM 7000 lightcycler (Applied Biosystems). Primers recognizing both c-FLIPL and c-FLIPS or IL-8 were purchased from Applied Biosystems. Gene expression was normalized to the endogenous housekeeping control gene glyceraldehyde phosphate dehydrogenase (GAPDH) and relative expression of c-FLIP and IL-8 was calculated using the comparative threshold cycle (CT) method [19].
Flow cytometric analysis
Surface staining of TRAIL-R1-4 using monoclonal Abs against TRAIL-R1-4 that are available from Alexis Corp. (Lansen, Switzerland) [20, 21], or control mIgG1 at 10 μg/ml was essentially performed and analysed by flow cytometry as described [5].
Results
Ni2+ sensitizes primary ECs to TRAIL-induced apoptosis
The resistance of ECs to death ligand-mediated apoptosis is crucial for the maintenance of vascular integrity. We previously analysed the effect of the pro-inflammatory agent and potent contact allergen Ni2+ on endothelial gene expression and observed up-regulation of several transcripts involved in death receptor signalling. Most prominently, we found a marked up-regulation of TRAIL-R2 within 5 hrs of Ni2+ treatment, suggesting a potential effect of Ni2+ on death ligand sensitivity [6]. We thus compared the apoptotic response of untreated HUVEC and overnight Ni2+-pretreated cells to increasing concentrations of recombinant TRAIL protein. Figure 1A illustrates that even concentrations of 3000 ng/ml of recombinant TRAIL failed to induce cell death in these cells. In contrast, pre-incubation with the bivalent transition metal Ni2+ dramatically sensitized HUVEC to TRAIL-mediated death. After pre-incubation with Ni2+ for 16 hrs, TRAIL efficiently induced cell death of HUVEC with an IC50 of 5–10 ng/ml (Fig. 1A). The sensitization to TRAIL was dose-dependent with an effective range between 0.25 mM to 1.5 mM Ni2+ (Fig. S1A). Cell death occurred by apoptosis as evident by the presence of cells with subdiploid DNA content in DNA profile analysis (Fig. 1B) and the appearance of fragmented DNA in internucleosomal DNA fragmentation assays (Fig. 1C). TRAIL-mediated cell death after Ni2+ pre-incubation was strictly caspase-dependent, because the pancaspase inhibitor zVAD-fmk fully protected Ni2+-treated cells from TRAIL-induced apoptosis in internucleosomal DNA fragmentation assays (Fig. 1C) and promoted cell survival as determined by crystal violet staining (data not shown). Taken together, these data demonstrate that Ni2+ is capable of sensitizing ECs to TRAIL-mediated apoptosis.
Fig 1.
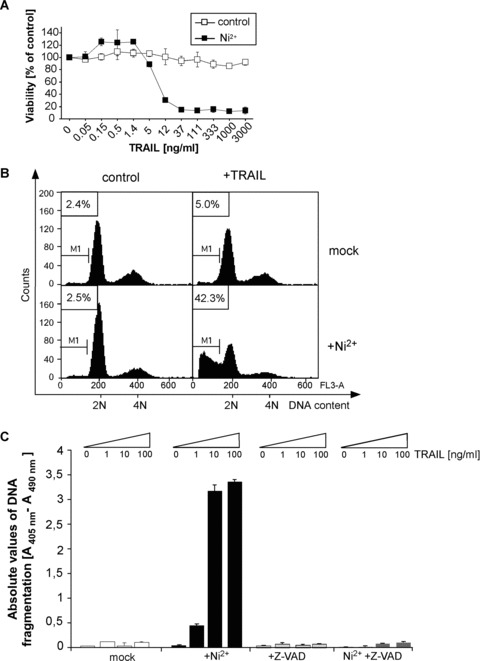
Ni2+ sensitizes HUVEC to TRAIL-induced apoptosis. (A) Cell viability was assessed by crystal violet staining. HUVEC were left untreated or stimulated with Ni2+ for 16 hrs. Subsequently, cells were cultured for another 6 hrs in Ni2+-free medium containing the indicated TRAIL concentrations. Experimental values were normalized to control values of diluent- or Ni2+-treated controls to facilitate direct comparison. (B) Quantification of apoptosis by flow cytometric analysis of hypodiploid DNA content (M1) of propidium iodide-stained cells. HUVEC were pre-stimulated as in (A) and subsequently exposed to 100 ng/ml TRAIL for 8 hrs. (C) Quantification of apoptosis by measuring internucleosomal DNA fragmentation. HUVEC were treated as in (A) and additionally exposed to 10 μM of the pan-caspase inhibitor zVAD-fmk as indicated. Data in (A) and (C) show mean values ± S.D. from one representative experiment out of 2–3 independent experiments performed in triplicate.
TRAIL receptor cell surface expression does not fully account for the increased TRAIL sensitivity in Ni2+-treated HUVEC
In order to elucidate whether Ni2+-mediated sensitization can be accounted for by modification of TRAIL receptor surface expression in HUVEC, we characterized the four membrane-bound TRAIL receptors in HUVEC by flow cytometry. While control HUVEC showed significant TRAIL-R2 and TRAIL-R3 surface expression and low TRAIL-R1 levels, membrane-bound TRAIL-R4 was undetectable (Fig. 2A). Incubation with Ni2+ for 16 hrs led to a minor up-regulation of TRAIL-R1, moderate up-regulation of TRAIL-R2, and a strong induction of TRAIL-R4. In contrast, TRAIL-R3 surface expression was slightly repressed (Fig. 2A). Very similar findings were obtained using primary human KC, indicative of the broader relevance of our findings (compare Fig. 7B). In order to investigate if the induction of TRAIL-R is the critical event required for sensitization of EC to TRAIL-mediated apoptosis we performed experiments with Brefeldin A that disturbs the Golgi apparatus required for protein secretion and externalization of receptors [22]. However, pre-incubation of Ni2+-treated EC with 2 μg/ml Brefeldin A did not block Ni2+-induced sensitization to TRAIL (Fig. 2C) despite effective interference with Ni2+-induced TRAIL-R surface expression (Supplemental Fig. 2A) and IL-8 secretion (Fig. S2B). Taken together, our data of coincident up-regulation of TRAIL-R1, TRAIL-R2 and TRAIL-R4 together with the data using Brefeldin A did not sufficiently explain the dramatic sensitization of HUVEC to TRAIL following Ni2+ exposure and suggested the existence of additional intracellular regulatory mechanisms.
Fig 2.
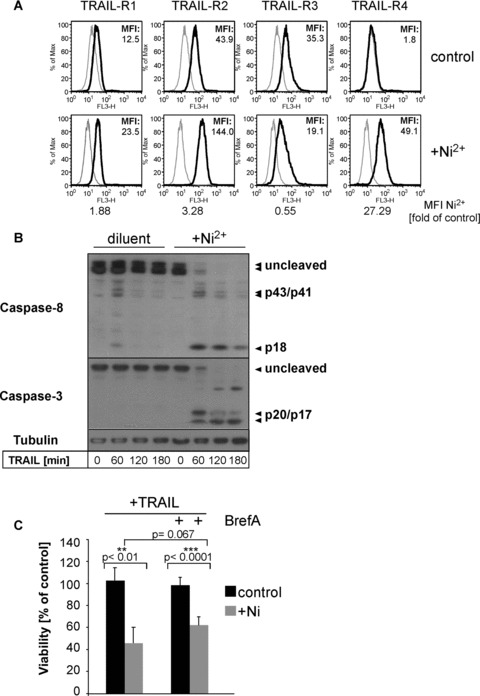
Ni2+ leads to altered TRAIL-receptor expression and rapid Caspase-8 and -3 cleavage upon TRAIL stimulation. (A) Flow cytometric analysis of TRAIL-R1, -R2, -R3 and -R4 cell surface expression of non-stimulated or Ni2+-stimulated (16 hrs) HUVEC using specific antibodies to TRAIL-R1-R4 (filled lines). Grey lines indicate staining with the respective isotype-specific control antibodies. One of four independent experiments is shown with the respective background-subtracted median fluorescence intensity (MFI). Additionally, MFI ratios of Ni2+-treated samples and its respective controls are shown. (B) HUVEC were left untreated or pre-incubated with Ni2+ for 16 hrs and subsequently incubated with TRAIL as indicated. Lysates were analysed for cleavage of Caspase-8 (p43/p41/p18), and Caspase-3 (p20/p17) by Western blot. Membranes were rehybridized with an Ab to tubulin to control for equal protein loading. (C) EC were pre-treated for 16 hrs with Brefeldin A (2 μg/ml) in the presence or absence of Ni2+ (1.5 mM) and subsequently treated with TRAIL (100 ng/ml) for 6 hrs. The viability was subsequently determined by crystal violet assay. Statistical significance of the detected effects was evaluated by Student’s t-test. Statistically significant changes (P < 0.05) are marked by two, highly significant changes (P < 0.005) by three asterisks. Data are derived from a total of five experiments each performed in triplicate wells.
Fig 7.
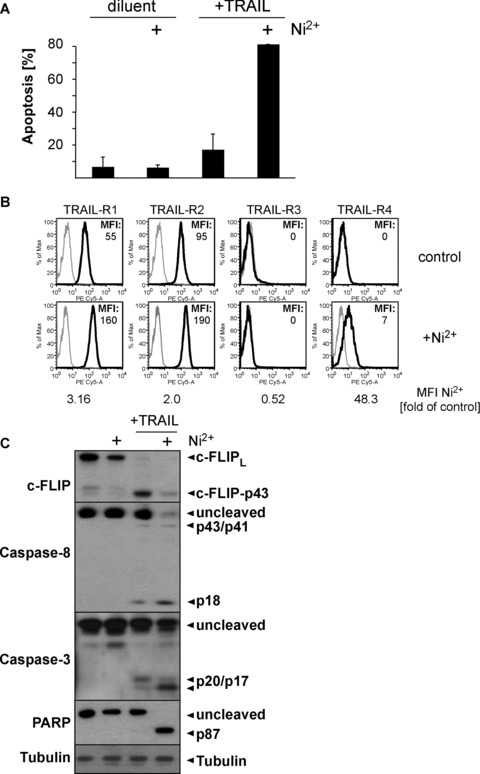
Ni2+ sensitizes human primary keratinocytes for TRAIL-induced apoptosis. Human primary keratinocytes were stimulated with 1.5 mM Ni2+ for 16 hrs or diluent and either directly processed for flow cytometric assessment of TRAIL-R1-4 surface expression (B), or subsequently exposed to 25 ng/ml TRAIL or diluent alone for 6 hrs (A, C). (A) Apoptosis induction as determined by flow cytometric subdiploidy analysis. Average values of two independent experiments using KCs from two different donors ± S.D. are shown. (B) Cell surface expression of TRAIL-R1-4 as determined by flow cytometry. Isotype controls are shown in grey, expression of the respective TRAIL-Rs is depicted in black. Background-corrected MFI values of the stainings are indicated. (C) Western blots showing Ni2+-dependent repression of c-FLIP protein and sensitization of TRAIL-induced apoptosis as evident by induction of Caspase-8, Caspase-3 and PARP cleavage. Tubulin served as a loading control.
Ni2+ promotes TRAIL-mediated Caspase-8 processing in HUVEC
We next analysed the expression and TRAIL-induced activation of initiator and effector caspases in HUVEC upon Ni2+ exposure. Ni2+ did not modify basal expression of initiator Caspase-8 and effector Caspase-3. Neither did we observe prominent processing of Caspase-8 and Caspase-3 in control cells that were stimulated with TRAIL alone for a time period of up to 3 hrs (Fig. 2B, lanes 1–4). In contrast, pre-incubation of HUVEC with Ni2+ resulted in rapid and complete activation of Caspase-8 within 1 hr after TRAIL addition, as indicated by the detection of the p43/41 fragments as well as the fully cleaved p18 fragment of Caspase-8 (Fig. 2B, lanes 5–8). Moreover, cleavage of Caspase-3 to its active p17 fragments was detected within 1–3 hrs after TRAIL stimulation (Fig. 2B, lanes 5–8). Thus, our biochemical data suggested that pre-incubation with Ni2+ allows for efficient initiator and effector caspase activation following TRAIL stimulation in ECs.
Ni2+ affects TRAIL-induced apoptosis via down-regulation of c-FLIP
TRAIL-mediated apoptotic signalling is crucially counteracted by c-FLIP expression in primary cells [23]. Moreover, mouse embryonic fibroblasts from c-FLIP-deficient animals were shown to be highly sensitive to TRAIL-induced apoptosis [24]. We thus explored if Ni2+ affected c-FLIP protein expression or processing in ECs. Ni2+ dose-dependently repressed cFLIPL protein expression (Fig. S1B). In unstimulated HUVEC, c-FLIP protein was largely expressed in its long isoform, c-FLIPL, with barely detectable expression of c-FLIPS. In agreement with previous data from primary human keratinocytes [14], addition of TRAIL to unstimulated HUVEC resulted in a time-dependent cleavage of c-FLIPL to p43 but observed no prominent Caspase-8 cleavage (Fig. 3A) or apoptosis induction (compare Fig. 1B and C). Interestingly, we detected a 25-kD fragment reactive with the cFLIP antibody in all TRAIL-stimulated cells, which may reflect NF-κB-induced cFLIPS expression as reported [25, 26]. Alternatively, this fragment may represent the cleaved prodomain (about 25 kD) of cFLIPL, although the fact that we also detect this fragment in Ni2+-treated cells in the absence of TRAIL favours induction of cFLIPS by TRAIL or Ni2+ (Fig. 3C). Pre-incubation with Ni2+ for 16 hrs led to repression of basal c-FLIP protein (Fig. 3A and B, compare lanes 1 and 2) but likewise triggered no obvious Caspase-8 activation. However, when TRAIL was added to Ni2+-pre-exposed cells a rapid Caspase-8 cleavage to its p43/41 and p18 cleavage products was detected (Fig. 3B). This Caspase-8 cleavage correlated with a very rapid cleavage of c-FLIPL to p43 within 15–30 min. (Fig. 3B, lane 3) and subsequent total loss of detectable c-FLIP proteins (Fig. 3B). We next investigated the dynamics of Ni2+-mediated repression of c-FLIP protein. Interestingly, we first observed up-regulation of c-FLIPS at 1–4 hrs after Ni2+ stimulation while c-FLIPL protein was unchanged at these early time-points. At later time-points, however, protein expression of both c-FLIPL and c-FLIPs was strongly repressed (Fig. 3C). c-FLIP levels are regulated at the transcriptional as well as post-transcriptional level [27]. Considering the late kinetics of c-FLIP protein repression we suspected that Ni2+ might affect c-FLIP expression at the RNA level. We thus performed qRT-PCR for c-FLIP with primers detecting both c-FLIPL and c-FLIPS. Consistent with the protein data, Ni2+ regulated c-FLIP mRNA in a biphasic manner (Fig. 3D). Within 2–4 hrs after Ni2+ treatment, mRNA for c-FLIP was induced 3–5 fold (Fig. 3D). In marked contrast, down-regulation of c-FLIP mRNA was detected at later time-points between 8–16 hrs to roughly 20% of baseline mRNA levels (Fig. 3D). This late repression was not a general phenomenon of Ni2+-induced gene regulation because in the same samples mRNA expression of CXCL-8/IL-8, an established Ni2+-regulated gene [6], was still induced (Fig. 3D). In summary, our data demonstrate that Ni2+-dependent modulation of c-FLIP proteins is at least partially regulated at the mRNA level.
Fig 3.
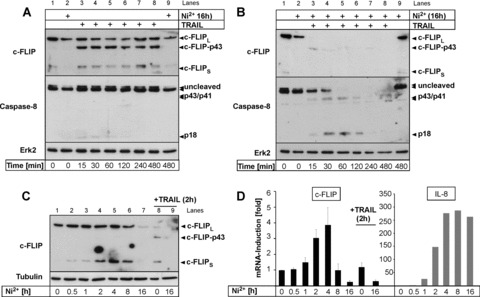
c-FLIP is down-regulated by Ni2+ at the mRNA and protein level. (A, B) Untreated HUVEC (A) or HUVEC pre-incubated with Ni2+ for 16 hrs (B) were stimulated with 100 ng/ml TRAIL as indicated and analysed for expression and cleavage of c-FLIP and Caspase-8 by Western blot. Membranes were rehybridized with an Ab to Erk2 to control for even protein loading. Lanes 1, 2 and 9 in (A) and (B) contain equal amounts of identical samples to control for similar exposures of the different membranes. (C, D) HUVEC were treated for the indicated times with Ni2+ and expression of c-FLIP protein (C) or mRNA (D; left panel) was assayed by Western blotting or qRT-PCR, respectively. For comparison, HUVEC were also treated with TRAIL for 2 hrs (C, lanes 8, 9; D, left panel). Data in (D; left panel) represent mean values of two experiments each performed in triplicates ± S.D. In (D; right panel) additional qRT-PCR analysis of IL-8 mRNA for the indicated times is shown.
Ectopic c-FLIPL expression can counteract Ni2+-dependent sensitization to TRAIL-mediated apoptosis in ECs
Our observation that TRAIL stimulation triggers cleavage of c-FLIP while Ni2+ simultaneously decreases its mRNA levels suggests that Ni2+ might exert its sensitizing effect on TRAIL-induced apoptosis by preventing resynthesis of c-FLIP proteins consumed by death receptor stimulation. To test this hypothesis we expressed c-FLIPL from an ectopic promoter that should no longer be responsive to Ni2+. We predicted that ectopic c-FLIPL expression should blunt the sensitizing effect of Ni2+ on TRAIL-mediated apoptosis and protect Ni2+-stimulated ECs from ligand-induced cell death. Indeed, retroviral expression of c-FLIPL strongly reduced Caspase-8 cleavage (Fig. 4A) and considerably protected Ni2+-exposed cells from TRAIL-mediated apoptosis even at high TRAIL concentrations (Fig. 4B). At the same time, it had no protective effect on paclitaxel- or doxorubicin-induced apoptosis (Fig. S3), which is largely independent of death receptor-mediated signals [28]. HUVEC were not fully protected from TRAIL-mediated apoptosis, which may possibly be explained by the well-known effect of high levels of cFLIP overexpression that may result in the generation of pro-apoptotic signals [29, 30]. Nevertheless, these data indicate that Ni2+-dependent depletion of c-FLIP mRNA critically contributes to its sensitizing effect on TRAIL-induced apoptosis in primary ECs.
Fig 4.
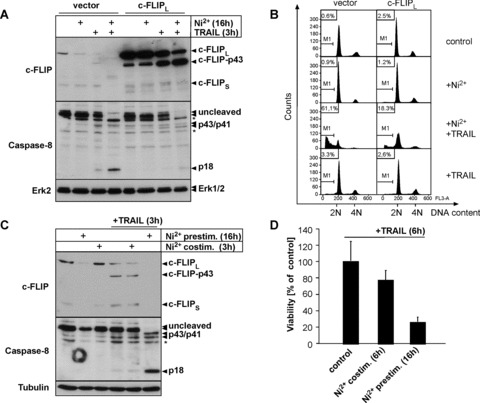
Transcriptional repression of c-FLIP expression is a functionally relevant mechanism by which Ni2+ sensitizes ECs to TRAIL-induced apoptosis (A, B) Ectopic expression of cFLIPL protects Ni2+-exposed HUVEC against TRAIL-mediated apoptosis. HUVEC were infected with the respective retroviruses and pre-treated with Ni2+ or diluent for 16 hrs. Subsequently, cells were cultured for 3 (A) or 8 hrs (B) in Ni2+-free medium in the presence or absence of TRAIL, respectively. (A) Western blots showing protein expression of c-FLIP and Caspase-8. Molecular weights of full-length proteins and cleavage products are indicated. Asterisks denote unspecific bands consistently obtained with the Caspase-8 antibody. Erk2 served as a loading control. (B) Apoptosis induction as determined by subdiploid DNA analysis. (C, D) Short-term Ni2+-stimulation is insufficient to sensitize ECs to TRAIL-induced apoptosis. HUVEC were either pre-incubated with Ni2+ and subsequently exposed to 100 ng/ml TRAIL or co-stimulated with Ni2+ and TRAIL for 3 (C) or 6 hrs (D). (C) Western blots demonstrate that TRAIL-treated EC maintain c-FLIP protein expression and fail to initiate full Caspase-8 cleavage upon co-treatment of Ni2+ and TRAIL. Tubulin staining served as loading control. (D) Crystal violet viability assays showing strongly decreased capacity of a short-term (6 hrs) Ni2+ co-treatment to sensitize ECs to TRAIL-mediated apoptosis. Data are derived from three independent experiments each performed in triplicate and are presented as percentage of viable cells ± S.D. related to the respective non-Ni2+-stimulated controls that arbitrarily were set to 100%.
To further confirm this hypothesis, we next compared the effect of Ni2+ pre-treatment versus Ni2+-co-treatment on TRAIL-induced apoptosis. The rationale behind this experiment was the observation that repression of c-FLIP protein and mRNA expression was only observed after 8–16 hrs whereas at earlier time-points c-FLIP mRNA and protein levels were still unaffected or even induced by Ni2+ (compare Fig. 3C and D). Hence, co-treatment of cells with TRAIL and Ni2+ for 3–6 hrs should not be capable to sensitize cells to TRAIL-induced apoptosis. Indeed, concomitant stimulation of ECs with both Ni2+ and TRAIL for 3 hrs failed to induce Caspase-8 cleavage (Fig. 4C) and only marginally affected cell viability after 6 hrs of co-treatment (Fig. 4D). In contrast, pre-treatment of cells with Ni2+ strongly induced Caspase-8 cleavage and cell death upon subsequent TRAIL treatment within the same time periods in parallel experiments (Fig. 4C and D). These assays further support the hypothesis that a transcriptional event is responsible for the sensitizing effect of Ni2+ on TRAIL-induced apoptosis.
Depletion of c-FLIP is sufficient to sensitize ECs to TRAIL-mediated apoptosis
To analyse whether loss of c-FLIP mRNA is sufficient to sensitize ECs for TRAIL-mediated apoptosis, we next depleted endogenous c-FLIP mRNA by retroviral expression of shRNA. As shown in Fig. 5A, both isoforms of c-FLIP were efficiently suppressed by the shRNA. TRAIL treatment of empty pRS vector-infected cells did not induce substantial Caspase-8 cleavage, while a prominent cleavage of c-FLIPL to its p43 fragment was detected. In contrast, TRAIL treatment of c-FLIP shRNA-expressing HUVEC led to substantial cleavage of Caspase-8 to its active p18 form (Fig. 5A). Accordingly, down-regulation of c-FLIP was sufficient to sensitize HUVEC to TRAIL-mediated apoptosis as determined by hypodiploidy analysis (Fig. 5B). To investigate at which level c-FLIP interferes with TRAIL-induced apoptosis in HUVEC, we next analysed the composition of the TRAIL DISC by ligand affinity precipitation using a Flag-tagged recombinant TRAIL, which has not been studied thus far in ECs (Fig. 5C). The adaptor molecules FADD and Caspase-8 p55/53 were recruited in the TRAIL DISC of control shRNA-infected ECs. Moreover, we mainly detected the DISC-cleaved p43/41 fragments in the ligand affinity precipitates. c-FLIPL was strongly detectable as p43 fragment in the DISC of control shRNA-infected cells, but not in c-FLIP-depleted cells, while full length c-FLIPL was undetectable in the DISC under both conditions. These findings indicate that c-FLIPL p43 is enriched and retained in the DISC, consistent with findings in other cellular models [14, 31]. In contrast, DISC precipitates of c-FLIP-depleted HUVEC (pRS-cFLIP-infected cells) contained larger amounts of unprocessed Procaspase-8 (p55/53) in the DISC, indicating that cleaved Caspase-8 is rapidly released from the receptor complex when c-FLIP is absent. Thus, c-FLIP is an important regulator of sensitivity to TRAIL in primary ECs at the level of the TRAIL DISC.
Fig 5.
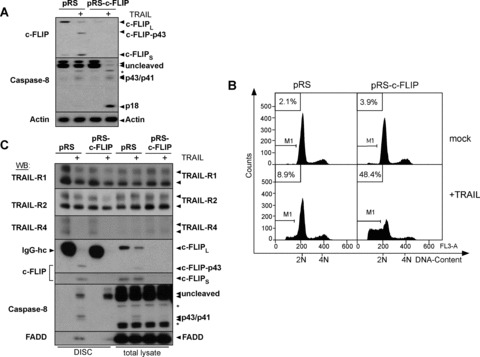
Depletion of endogenous c-FLIP by retroviral expression of c-FLIP shRNA sensitizes HUVEC for TRAIL-induced apoptosis. (A–C) HUVEC were infected with the respective constructs and stimulated with 100 ng/ml TRAIL or diluent for 3–8 hrs. (A) Western blot for c-FLIP or Caspase-8 showing TRAIL-dependent cleavage of Caspase-8 upon knock-down of c-FLIP. Molecular weight of full-length proteins and cleavage products are indicated. Cells were lysed 3 hrs after exposure to TRAIL or diluent. The asterisk denotes unspecific bands. Actin served as loading control. (B) Subdiploidy analysis of the differently infected cells harvested 8 hrs after addition of TRAIL or diluent. One representative experiment of three is shown. (C) DISC analysis of TRAIL receptor signalling complexes from the differently infected cells (for details, see Materials and Methods). Cells were cultured for 30 min. in presence of 1 μg/ml Flag-TRAIL pre-complexed with anti-Flag mAb (+) or diluent alone and equal amounts of anti-Flag immunoprecipitates (DISC) were analysed by Western blot. Protein association with non-stimulated receptors was monitored by supplementing lysates from unstimulated cells with anti-Flag-Ab and Flag-TRAIL prior to immunoprecipitation. IgG-hc indicates mouse IgG heavy chain of anti-Flag Ab detected by the respective Abs that is added to the lysates generated from non-stimulated cells as indicated in detail in Materials and Methods. Western blotting with TRAIL-R1, TRAIL-R2 and TRAIL-R4 Abs controls for excessive precipitation of unstimulated receptors as compared to stimulated (+) receptors. Asterisk denotes unspecific bands. Arrows indicate detection of cleavage fragments of the respective proteins.
Prolonged Ni2+-treatment facilitates TRAIL-dependent cleavage of Caspase-8 at the DISC by reducing the available amounts of protective c-FLIP protein. In order to test if TRAIL-mediated loss of c-FLIP protein in Ni2+ pre-exposed cells (compare Fig. 3B) correlates with the disappearance of protective c-FLIP protein from the activated receptor, we finally performed DISC analysis in Ni2+-treated ECs using Flag-tagged recombinant TRAIL (Fig. 6). Since we detected increased surface expression of TRAIL-R1/TRAIL-R2 and TRAIL-R4, we analysed the DISC composition under conditions of (a) Ni2+ pre-treatment for 16 hrs followed by TRAIL stimulation for 30 min. (Fig. 6, lane 6), (b) concomitant Ni2+- and TRAIL-treatment for 30 min. (Fig. 6, lane 5), or (c) treatment with TRAIL for 30 min. alone (Fig. 6, lane 4). Interestingly, Ni2+-pre-treatment resulted in a marked up-regulation of total (lanes 9 and 12) and ligand-bound TRAIL-R1, TRAIL-R2, and TRAIL-R4 (lanes 3 and 6), which was not seen when TRAIL and Ni2+ were co-administered (lanes 2 and 5, lanes 8 and 11). Proteolytic cleavage of c-FLIPL to c-FLIP-p43 was evident under all conditions in the TRAIL DISC (Fig. 6, lanes 4–6), suggesting the presence of functional receptor complexes in all cases. However, compared to both TRAIL-stimulated as well as Ni2+-co-stimulated cells, we observed a clearly reduced amount of c-FLIP-p43 in the TRAIL-DISC of Ni2+ pre-stimulated cells despite enhanced precipitation of the TRAIL receptors TRAIL-R1, TRAIL-R2, and TRAIL-R4 (compare Fig. 6, left panel, lane 6). Furthermore, an elevated amount of uncleaved Procaspase-8 was recruited in the TRAIL-DISC of Ni2+-pre-treated cells (Fig. 6, lane 6), while in the other TRAIL-treated samples, only partially cleaved Caspase-8 was detectable, indicative of a c-FLIPL-inhibited DISC [32, 33]. Remarkably, the pattern of Ni2+-pre-stimulated cells closely resembled the TRAIL-DISC following depletion of c-FLIP by shRNA with the exception of TRAIL-R4 that was specifically increased in the DISC of Ni2+-treated cells, but not modified by knock-down of cFLIP in the DISC (compare Fig. 5C). Thus, the enhanced Procapase-8 recruitment most likely reflects an accelerated turnover of Caspase-8 within the DISC and subsequent rapid release of the cleaved fragments from the DISC, similar to results in other cells [31, 33]. In line with this conclusion, total cellular lysates demonstrated a strong reduction of total c-FLIP protein and presence of fully cleaved Caspase-8 p18 in TRAIL-treated samples of Ni2+-pre-exposed cells (Fig. 6, lane 12). In contrast, neither fully cleaved Caspase-8 nor loss of total c-FLIP protein was observed in lysates from Ni2+-co-treated or solely TRAIL-treated cells (Fig. 6, lanes 10 and 11). These results suggest that the amount of c-FLIPL cleaved within the TRAIL-DISC was sufficient to prevent the release of active Caspase-8 p18 from the DISC in the absence of Ni2+. Collectively, these data imply that prolonged Ni2+ exposure strongly decreases the availability of protective c-FLIP protein at the receptor complex thereby critically influencing the outcome of TRAIL-R ligation in primary ECs.
Fig 6.
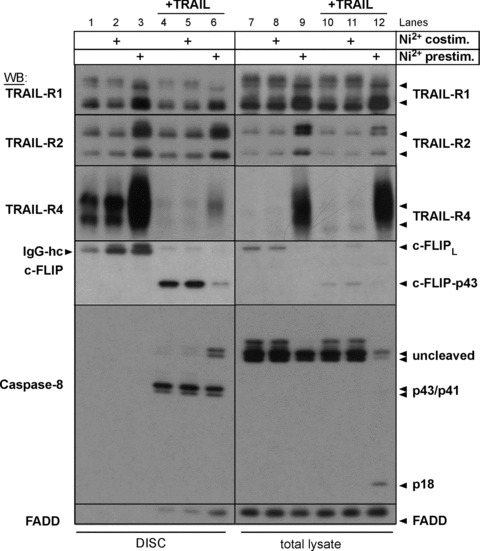
Prolonged Ni2+-treatment triggers TRAIL-dependent activation of Caspase-8 by reduced c-FLIP recruitment to the TRAIL DISC. EC were pre-treated with Ni2+ for 16 hrs (lanes 3, 6, 9, 12) or treated with diluent alone. Cells were subsequently cultured in the presence of 2.5 μg/ml Flag-TRAIL pre-complexed with anti-Flag mAb (+) or diluent alone in the absence or presence of Ni2+ for 30 min. Subsequently, equal amounts (1 mg of total cellular proteins) were subjected to anti-Flag immunoprecipitation (DISC, lanes 1–6). Total cellular lysates of all conditions were analysed in parallel (total lysate, lanes 7–12). Protein association with non-stimulated receptors was monitored by supplementing equal amounts of lysates from unstimulated cells with Flag-TRAIL that was pre-complexed with anti-Flag mAb prior to immunoprecipitation. IgG-hc indicates mouse IgG heavy chain of anti-Flag mAbs detected by the respective Abs.
Ni2+ facilitates TRAIL-induced apoptosis in human primary keratinocytes
To finally investigate whether Ni2+ can also sensitize other primary cells for TRAIL-mediated apoptosis, we employed human KCs that represent important effector cells in allergic contact der matitis and have previously been associated with death-receptor mediated apoptosis in Ni2+-induced contact eczema [34]. Under basal conditions, KCs are already more prone to TRAIL-induced apoptosis than ECs at high concentrations of the death ligand ([23] and data not shown). However, Ni2+ pre-incubation strongly increased apoptosis induction at suboptimal TRAIL concentrations (Fig. 7A). Consistent with our observation in ECs, examination of TRAIL-R surface expression revealed moderate up-regulation of both the apoptosis-proficient TRAIL-R1 and 2 and induction of the apoptosis-deficient TRAIL-R4 (Fig. 7B) in response to Ni2+. Similarly, Western analysis indicated that Ni2+ also decreased c-FLIP protein levels and allowed for efficient cleavage of Caspase-8, Caspase-3, and its downstream effector PARP upon TRAIL treatment. These data suggest that our findings in respect to Ni2+ ions are also relevant in other primary cells that are potentially exposed to death ligands.
Discussion
TRAIL has attracted wide interest due to its selective apoptosis-inducing activity for tumour cells, while most primary cells are resistant to its pro-apoptotic action in vitro and in vivo. However, emerging evidence suggests that a number of primary cells, although resistant under basal conditions, can be sensitized to the pro-apoptotic signals exerted by TRAIL under certain conditions (for review see [8]). Here we describe the bivalent cation Ni2+, a widely distributed noxious agent and inducer of contact allergy reactions [4], as novel sensitizer of ECs or KCs to TRAIL-mediated apoptosis, underscoring the plasticity of TRAIL-induced responses. Early studies in various cell systems had suggested that TRAIL resistance and conversely sensitivity for TRAIL-induced apoptosis could simply result from altered expression of the ‘decoy’ receptors TRAIL-R3 and TRAIL-R4, which lack functional death domains and thus might counteract the apoptosis-inducing capacity of TRAIL [8]. Consistent with other reports [35–37] we found TRAIL-R1–3 to be moderately expressed on unstimulated ECs while TRAIL-R4 was absent. Interestingly, Ni2+ increased surface expression of both TRAIL-R1 and TRAIL-R2, whereas TRAIL-R3 levels remained unchanged and TRAIL-R4 expression was strongly induced in both KCs as well as ECs. Current models suggest a rather complex function of TRAIL-R4 beyond a sole function as decoy receptor: TRAIL-R4 was shown to either corecruit with TRAIL-R2 [10] or may directly bind to TRAIL-R2 in a ligand-independent manner via the pre-ligand assembly domain [38]. Despite this complex picture, the overall anti-apoptotic function of TRAIL-R4 remains undisputed to date. Our experiments using Brefeldin A in order to block externalization of newly formed receptors further corroborate our hypothesis that there is a critical role for a loss of cFLIP in the presence of Ni2+ to increased sensitivity of ECs to TRAIL-mediated apoptosis. Our data rather argue against the hypothesis that TRAIL-R1/TRAIL-R2 modulation alone is responsible for Ni2+-dependent sensitization to TRAIL-induced apoptosis. They are in line with data collected in different tumour cell models, which previously revealed that in tumour cells pharmacological inhibitors such as bortezomib or depsipeptide lead to sensitization to TRAIL-mediated apoptosis also independent of an up-regulation of TRAIL-R2 [39–41]. Another level of complexity of the TRAIL/TRAIL-R system further corroborated by our data is the detection of TRAIL-R4 in the native TRAIL DISC of Ni2+-treated ECs (compare Fig. 6). However, TRAIL-R4 in the DISC would then not only block caspase-8 processing in and release of active caspase-8 from the DISC [10], but should also lead to increased resistance to TRAIL-mediated apoptosis which is clearly not the case in the presence of Ni2+.
Interestingly, neither the alterations in TRAIL receptor expression nor the sensitizing effect of Ni2+ can be attributed to Ni2+-induced NF-κB stimulation, which accounts for a prominent proportion of Ni2+-dependent gene regulation in HUVEC [6], since the expression of a kinase-dead mutant of IKK2 was unable to prevent this up-regulation and failed to rescue TRAIL-induced apoptosis of Ni2+-pre-incubated cells (Fig. S4). Furthermore, the prototypic NF-κB activator TNF did not significantly change surface expression of TRAIL-R1-R4 (Fig. S5), although a modest increase in the mean fluorescence intensity of TRAIL-R2 surface expression was detectable. Taken together, these data indicate that Ni2+ utilizes NF-κB-independent signalling pathways for TRAIL-R surface regulation. A potential candidate pathway is the p38 MAPK cascade, which has previously been shown to contribute to the Ni2+-induced transcriptome in HUVEC [6]. Such a notion would corroborate earlier studies by Guan et al.[42] who reported regulation of TRAIL-R1 by AP-1 heterodimers that are known transcriptional mediators of the stress-activated JNK and p38 MAPK pathways. It should be noted, however, that we were unable to prevent TRAIL-dependent Caspase-8 cleavage of Ni2+-pretreated ECs by co-incubation with an inhibitor of p38, SB202190 (Fig. S6). Thus, this pathway likewise appears to be irrelevant for the increased susceptibility of Ni2+-treated ECs for TRAIL-induced apoptosis.
Apart from altered TRAIL-R expression, modulation of internal regulators of the apoptotic machinery, most notably c-FLIP, represents an equally important mechanism, by which cells can be sensitized for TRAIL-induced apoptosis (reviewed in [8]). It was previously reported that the Forkhead transcription factor FOXO3a may control endothelial viability via transcriptional repression of cFLIP [43]. While our initial micro-array experiment revealed that Ni2+ could up-regulate FOXO3a expression in HUVEC [6], we failed to revert the sensitizing effect of Ni2+ by expression of a dominant-negative FOXO mutant (Fig. S7) for TRAIL-induced apoptosis excluding a role for FOXO3.
Based on our observations that Ni2+ incubation decreased c-FLIP mRNA levels (Fig. 3D) and at the same time triggered a rapid loss of protective c-FLIP proteins upon TRAIL stimulation (Fig. 3B), we propose the following model for Ni2+-induced sensitization for TRAIL-induced apoptosis (Fig. 8): Pre-incubation with Ni2+ results in an overall decrease of c-FLIP protein due to Ni2+-mediated repression of c-FLIP mRNA via an as yet unidentified mechanism. In the absence of TRAIL-R stimulation this has no consequence for the viability of the respective cells, since the low amount of remaining c-FLIP protein is still sufficient to counteract basal TRAIL-R activity and high levels of Caspase-8 activation in the cell, in particular because the affinity of cFLIP isoforms for the DISC is high as compared to caspase-8 [14, 27]. Thus maintenance of cFLIP levels in the absence of Ni2+ is critical to protect from excessive caspase-8 activation necessary for cell death induction. Of note, our data depicted in Fig. 3A clearly indicate that there is sufficient TRAIL DISC-associated caspase-8 activity needed for cFLIPL cleavage in the absence of Ni2+. Upon TRAIL-R stimulation, however, c-FLIP is increasingly recruited to the receptor, where it is required to limit the DISC turnover. Thus, further caspase-8 recruitment to, cleavage within, and subsequent release of active Caspase-8 from the DISC is blocked. Normally, a constant supply of resynthesized c-FLIP protein can sustain the anti-apoptotic action of c-FLIP upon further receptor stimulation and compensate the greater demand of protective c-FLIP proteins under these conditions (Fig. 8A). Due to Ni2+-dependent repression of c-FLIP mRNA the resupply of c-FLIP protein is shut off (Fig. 8B). The available c-FLIP protein at the DISC now becomes rate-limiting and is no longer able to block subsequent release of active caspase-8. This effect may also be intensified by an increased need for c-FLIP at the receptors due to an elevated surface expression of TRAIL-R1 and -R2 as indicated by our studies. As a result, Caspase-8 is increasingly recruited to the DISC and cleaved to its active form that is rapidly released from the DISC into the cytoplasm, thereby exerting its pro-apoptotic function. Of note our proposed model does not necessarily reflect the full complexity of the TRAIL/TRAIL-R system. The failure of c-FLIP overexpression to completely revert the sensitizing effect of Ni2+ on TRAIL-induced apoptosis suggests that Ni2+ may interfere with the apoptotic machinery at multiple levels and potentially modulate expression of additional regulators of apoptosis, a hypothesis to be investigated in the future. It is currently unclear if and to what extent other regulators of the apoptotic machinery are involved but recent micro-array analysis revealed that Ni2+ can also down-regulate other anti-apoptotic genes such as Mcl-1 at the mRNA level [6]. Interestingly, we detect a partial loss of caspase-8 in total cellular lysates in the presence of Ni2+ in some of our experiments. One explanation could be a transcriptional or translational repression of caspase-8 in addition to cFLIP, as both genes appear to be similarly regulated at the transcriptional level [44]. However, since loss of caspase-8 would rather favour apoptosis protection, this additional effect does not likely explain the increased sensitivity to TRAIL-mediated cell death in the presence of Ni2+ and requires additional investigations in the future. Irrespective of these issues, our data clearly demonstrate that Ni2+-dependent down-regulation of c-FLIP constitutes a relevant mechanism by which it can sensitize different primary cells such as ECs for TRAIL-induced apoptosis.
Fig 8.
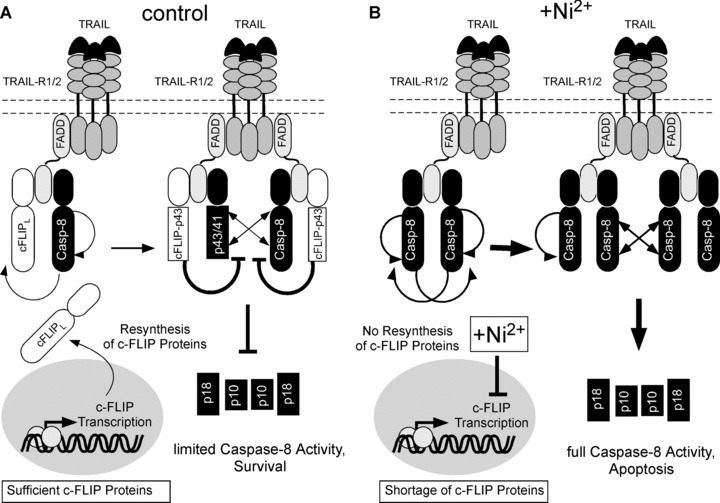
Model for Ni2+-dependent regulation of TRAIL resistance in primary human ECs. (A) In the absence of Ni2+, TRAIL stimulation leads to efficient recruitment of FADD, Caspase-8 and c-FLIP to the DISC. c-FLIPL and Caspase-8 are cleaved within the DISC (see arrows), but c-FLIP-p43 prevents further cleavage within and release of Caspase-8 from the DISC. Continuous transcription of c-FLIP proteins maintains blockade of TRAIL death receptors and thus inhibits the DISC turnover over time. (B) In the presence of Ni2+, re-supply of c-FLIP protein is shut off due to repression of c-FLIP transcription. TRAIL stimulation under these conditions leads to an increased caspase-8 recruitment to, cleavage of, and release from the TRAIL DISC thus leading to rapid consumption of remnant c-FLIP protein. Once the cellular pool of c-FLIP is consumed by continuous receptor triggering, Caspase-8 is cleaved in large amounts at the DISC and rapidly released in large quantities, thereby inducing cellular apoptosis. Ni2+-dependent up-regulation of TRAIL-R1 and 2 may further increase the requirement of protective c-FLIP protein at the DISC.
The data presented here strongly suggest that the cellular responses to TRAIL are subject to substantial modulation by environmental factors, as exemplified by the bivalent cation Ni2+. In particular the susceptibility of primary human ECs and KCs to TRAIL-mediated apoptosis may strongly depend on the micro-environmental conditions that dictate the outcome of ligand-induced death receptor activation by regulation of anti-apoptotic proteins such as c-FLIP. Similar conclusions were previously drawn concerning sensitivity of ECs to CD95-mediated apoptosis [45, 46]. Interestingly, in the T-cell prevailing acute phase of nickel-induced contact dermatitis death-receptor-mediated apoptosis of KCs is a well-documented phenomenon [34]. In addition, recent evidence indicates that cFLIP protects the basal cell layer of the interfollicular epidermis from death ligand-mediated cell death and may be responsible for the maintenance of the stem cell pool in the basal layer of the epidermis whenever activated T cells expressing death ligands are invading the epidermis [47, 48]. It is tempting to speculate that the apoptosis-sensitizing effect of Ni2+ on KCs and ECs contributes to the frequency of contact allergy to Ni2+ altogether. A scenario could well be in place that increased apoptosis of KCs may contribute to epidermal spongiosis upon Ni2+ exposure. This further facilitates the penetration of Ni2+ to the dermis or may activate apoptosis-dependent signals in the skin [49]. Subsequently, pro-inflammatory properties [6] as well as apoptosis-sensitizing effects of Ni2+ may recruit additional immune cells to the skin. It will be interesting to analyse whether this scenario holds true for ECs and KCs during acute eczema in situ. Considering the close contact of the endothelium with TRAIL-positive cells such as vascular smooth muscle cells [50], NK cells, monocytes, or T-cells which potentially trigger cell death of ECs and KCs we conclude that a high degree of resistance to TRAIL-induced apoptosis is crucial for sustaining vascular and epidermal integrity under physiological conditions. However, conditions such as inflammation, viral infection or tumour formation may also require the capability of primary cells to locally permit receptor-mediated apoptosis to enable the organism to adequately respond to environmental cues. Future in vivo studies are mandatory to elucidate this hypothesis in greater detail.
Acknowledgments
We thank Peter H. Krammer and H. Walczak for numerous invaluable reagents, H. Mehmet for CPP32 antiserum, L.M. Martins and M. MacFarlane for retroviral constructs, O. Micheau for helpful suggestions, and E. Horn and R. Alt for expert technical assistance. Parts of this study were funded by grants of the Wilhelm-Sander-Stiftung (2008.072.1), Deutsche Krebshilfe (106849), Exzellenzförderung Sachsen-Anhalt (N2_OGU, TP6) and the Deutsche Forschungsgemeinschaft (DFG) (grant Le 953/5-1) to Martin Leverkus as well as by grants of the DFG (Go 811/1-5) and the Exzellenzzentrum Dermatologie Baden-Württemberg to Matthias Goebeler. M. Hupe is a member of GRK1167 of the Deutsche Forschungsgemeinschaft.
Supporting Information
Fig. S1 Ni2+ sensitizes human ECs toTRAIL-induced apoptosis in a concentration-dependent manner. (A) Cell viability as assessed by crystal violet staining.HUVEC were pre-treated with the indicatedNi2+-doses or medium alone for 16 hrs andsubsequently exposed to the indicated concentrations of TRAIL for 6hrs prior to analysis by crystal violet assay. Experimental valueswere normalized to control values of diluent- orNi2+-treated controls to facilitate directcomparison. Error bars represent standard deviations derived fromone representative experiment performed in triplicate wells.(B) Concentration-dependent repression of cFLIP protein byNi2+ as determined by Western blot. HUVEC cellswere treated with the indicated Ni2+ doses for16 hrs and cFLIP protein expression in total cellular lysates wascompared by immunoblot analysis. Effectiveness of the respectiveNi2+ concentration was monitored by measuringprotein levels of the NF-κB target IL8 in the supernatants ofthe Ni2+-treated cells as previously described[1]. Immunoblotting with Abs to Tubulin was usedas loading control.
Fig. S2 Brefeldin A (BrefA) treatment blocksNi2+-dependent surface expression of TRAIL-Rsin ECs. (A) HUVEC were incubated with either diluent,Ni2+ or 2 μg/ml BrefA alone, or thecombination of both compounds for 16 hrs, and surface expression ofthe indicated TRAIL-Rs was analysed by flow cytometry. FACShistograms representing overlays of the individual TRAIL-R-stainedsamples (black, thick line) and their respective IgG1control-stained counterparts (grey, thin line) are shown. Thevalues of MFI in the upper right corner of each FACS histogramrepresent background-corrected MFI values for each individualTRAIL-R staining obtained after MFI subtraction of thecorresponding IgG1-control. Values in the table are calculated asfold MFI in relation to the respective untreated control for eachTRAIL-R. (B) HUVEC cells were either treated with theindicated BrefA concentrations alone, or the combination withNi2+ [1.5 mM] for 16 hrs. The effectiveness of BrefA on the IL-8 release from differentially treated cells was assessed by quantification of IL-8 protein in the supernatants by ELISA.
Fig. S3 cFLIPL overexpression does not affectDoxorubicin- or Paclitaxel-induced apoptosis. Primary ECs wereinfected with the indicated retroviruses and subsequently treatedwith 1 μM Paclitaxel (Tax) or 1 μg/ml Doxorubicin (Dox) for30 or 24 hrs, respectively. Subsequently, DNA fragmentation wasassessed by subdiploidy analysis in the FACS.
Fig. S4 Expression of dominant-negative IKK2 does notaffect Ni2+-dependent TRAIL-R surfaceregulation or TRAIL-induced Caspase-8 cleavage. HUVEC were infectedwith either empty retrovirus, or a retrovirus for dominant-negativeIKK2 (IKK2kd). Ni2+-induced modulation ofTRAIL-R surface expression (A) or TRAIL-induced Caspase-8and Caspase-3 cleavage (B) was assessed by flow cytometry,or Western blot analysis, respectively. (A) FACS profilesrepresenting overlays of the differently infected and treated cellsstained with specific antibodies for the indicated TRAIL-Rs (black,thick line) or an unspecific control IgG1-antibody (grey, thinline) are shown. Values in the upper right panel of each FACSprofile represent differential MFI values of the individual TRAIL-Rstainings after background subtraction of the corresponding IgG1control MFI value. For better comparison, additionallybackground-corrected MFI ratios of theNi2+-treated versus control-treatedsamples are shown as fold MFI. Incubation time withNi2+ was 16 hrs in all cases. (B) Afterinfection with the indicated retroviruses, cells were eithermock-treated or pre-treated with Ni2+ for 16 hrs. Subsequently, cells were stimulated with either TRAIL or equivalent amounts of diluent for 3 hrs. Expression and cleavage of cFLIP, Caspase-8 and Caspase-3 protein in the lysates of the differently treated cells was then analysed by immunoblot using appropriate antibodies for the indicated proteins. An immunoblot for Tubulin is shown as loading control; immunoblot for IKK2 is included as expression control for the IKK2 mutant. For better visualization both long and short exposures of the Caspase-8 immunoblot are shown; asterisks denote unspecific bands consistently seen with the employed Caspase-8 antibody.
Fig. S5 TNF treatment does not change the overall pattern of TRAIL-R surface expression. Human primary ECs were treated with 2 ng/ml TNF for 16 hrs and surface expression of the individual TRAIL-Rs was monitored by flow cytometry. Overlays of FACS profiles from the individual TRAIL-R-stained (black, thick lines) and IgG1-control-stained (grey, thin lines) samples are shown. Background-corrected MFI are plotted in the upper right corner of each FACS profile. For better comparison, MFI ratios of TNF-stimulated and unstimulated control cells for each TRAIL-R are included on the right side.
Fig. S6 Inhibition of p38 does not modulate TRAIL-inducedCaspase 8 cleavage in the presence or absence ofNi2+. HUVEC were incubated with diluent,Ni2+or 10 μM of the p38 inhibitor SB202190alone, or were co-treated with SB202190 andNi2+ for 16 hrs. Subsequently, cells were stimulated with TRAIL or medium for 90 min. prior to lysis. Protein expression and cleavage of Caspase-8 and c-FLIP was characterized by Western blot analysis. Even loading was controlled by immunoblot analysis of Tubulin.
Fig. S7 Expression of a dominant negative FoxO4 does notaffect Ni2+-dependent sensitization toTRAIL-induced apoptosis. HUVEC were infected with a control retrovirus, or a retrovirusencoding HA-tagged dominant-negative FoxO4 (dnFOXO) andsubsequently incubated with Ni2+ or diluent for 16 hrs. Cells were further cultured in presence or absence of TRAIL for 6 hrs and cytotoxicity was analysed by crystal violet assay. Error bars denote standard deviations of one representative experiment performed in triplicates. An immunoblot using an antibody for HA demonstrates expression of HA-tagged dnFOXO in the presented experiment (inset).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Krob HA, Fleischer AB, Jr, D’Agostino R, Jr, et al. Prevalence and relevance of contact dermatitis allergens: a meta-analysis of 15 years of published T.R.U.E. test data. J Am Acad Dermatol. 2004;51:349–53. doi: 10.1016/j.jaad.2003.11.069. [DOI] [PubMed] [Google Scholar]
- 2.Larese F, Gianpietro A, Venier M, et al. In vitro percutaneous absorption of metal compounds. Toxicol Lett. 2007;170:49–56. doi: 10.1016/j.toxlet.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Koster R, Vieluf D, Kiehn M, et al. Nickel and molybdenum contact allergies in patients with coronary in-stent restenosis. Lancet. 2000;356:1895–7. doi: 10.1016/S0140-6736(00)03262-1. [DOI] [PubMed] [Google Scholar]
- 4.Hostynek JJ. Sensitization to nickel: etiology, epidemiology, immune reactions, prevention, and therapy. Rev Environ Health. 2006;21:253–80. doi: 10.1515/reveh.2006.21.4.253. [DOI] [PubMed] [Google Scholar]
- 5.Goebeler M, Roth J, Brocker EB, et al. Activation of nuclear factor-kappa B and gene expression in human endothelial cells by the common haptens nickel and cobalt. J Immunol. 1995;155:2459–67. [PubMed] [Google Scholar]
- 6.Viemann D, Schmidt M, Tenbrock K, et al. The contact allergen nickel triggers a unique inflammatory and proangiogenic gene expression pattern via activation of NF-kappaB and hypoxia-inducible factor-1alpha. J Immunol. 2007;178:3198–207. doi: 10.4049/jimmunol.178.5.3198. [DOI] [PubMed] [Google Scholar]
- 7.Goebeler M, Gillitzer R, Kilian K, et al. Multiple signaling pathways regulate NF-kappaB-dependent transcription of the monocyte chemoattractant protein-1 gene in primary endothelial cells. Blood. 2001;97:46–55. doi: 10.1182/blood.v97.1.46. [DOI] [PubMed] [Google Scholar]
- 8.Kimberley FC, Screaton GR. Following a TRAIL: update on a ligand and its five receptors. Cell Res. 2004;14:359–72. doi: 10.1038/sj.cr.7290236. [DOI] [PubMed] [Google Scholar]
- 9.Wajant H. TRAIL and NFkappaB signaling–a complex relationship. Vitam Horm. 2004;67:101–32. doi: 10.1016/S0083-6729(04)67007-5. [DOI] [PubMed] [Google Scholar]
- 10.Merino D, Lalaoui N, Morizot A, et al. Differential inhibition of TRAIL-mediated DR5-DISC formation by decoy receptors 1 and 2. Mol Cell Biol. 2006;26:7046–55. doi: 10.1128/MCB.00520-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Budd RC, Yeh WC, Tschopp J. cFLIP regulation of lymphocyte activation and development. Nat Rev Immunol. 2006;6:196–204. doi: 10.1038/nri1787. [DOI] [PubMed] [Google Scholar]
- 12.Johnstone RW, Frew AJ, Smyth MJ. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer. 2008;8:782–98. doi: 10.1038/nrc2465. [DOI] [PubMed] [Google Scholar]
- 13.Leverkus M, Diessenbacher P, Geserick P. FLIP ing the coin? Death receptor-mediated signals during skin tumorigenesis. Exp Dermatol. 2008;17:614–22. doi: 10.1111/j.1600-0625.2008.00728.x. [DOI] [PubMed] [Google Scholar]
- 14.Leverkus M, Sprick MR, Wachter T, et al. Proteasome inhibition results in TRAIL sensitization of primary keratinocytes by removing the resistance-mediating block of effector caspase maturation. Mol Cell Biol. 2003;23:777–90. doi: 10.1128/MCB.23.3.777-790.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walczak H, Miller RE, Ariail K, et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med. 1999;5:157–63. doi: 10.1038/5517. [DOI] [PubMed] [Google Scholar]
- 16.Wachter T, Sprick M, Hausmann D, et al. cFLIPL inhibits tumor necrosis factor-related apoptosis-inducing ligand-mediated NF-kappaB activation at the death-inducing signaling complex in human keratinocytes. J Biol Chem. 2004;279:52824–34. doi: 10.1074/jbc.M409554200. [DOI] [PubMed] [Google Scholar]
- 17.Leverkus M, McLellan AD, Heldmann M, et al. MHC class II-mediated apoptosis in dendritic cells: a role for membrane-associated and mitochondrial signaling pathways. Int Immunol. 2003;15:993–1006. doi: 10.1093/intimm/dxg099. [DOI] [PubMed] [Google Scholar]
- 18.Gerin PA, Gilligan MG, Searle PF, et al. Improved titers of retroviral vectors from the human FLYRD18 packaging cell line in serum- and protein-free medium. Hum Gene Ther. 1999;10:1965–74. doi: 10.1089/10430349950017329. [DOI] [PubMed] [Google Scholar]
- 19.Liu W, Saint DA. A new quantitative method of real time reverse transcription polymerase chain reaction assay based on simulation of polymerase chain reaction kinetics. Anal Biochem. 2002;302:52–9. doi: 10.1006/abio.2001.5530. [DOI] [PubMed] [Google Scholar]
- 20.Ganten TM, Haas TL, Sykora J, et al. Enhanced caspase-8 recruitment to and activation at the DISC is critical for sensitisation of human hepatocellular carcinoma cells to TRAIL-induced apoptosis by chemotherapeutic drugs. Cell Death Differ. 2004;11:S86–S96. doi: 10.1038/sj.cdd.4401437. [DOI] [PubMed] [Google Scholar]
- 21.Washburn B, Weigand MA, Grosse-Wilde A, et al. TNF-related apoptosis-inducing ligand mediates tumoricidal activity of human monocytes stimulated by Newcastle disease virus. J Immunol. 2003;170:1814–21. doi: 10.4049/jimmunol.170.4.1814. [DOI] [PubMed] [Google Scholar]
- 22.Dinter A, Berger EG. Golgi-disturbing agents. Histochem Cell Biol. 1998;109:571–90. doi: 10.1007/s004180050256. [DOI] [PubMed] [Google Scholar]
- 23.Leverkus M, Neumann M, Mengling T, et al. Regulation of tumor necrosis factor-related apoptosis-inducing ligand sensitivity in primary and transformed human keratinocytes. Cancer Res. 2000;60:553–9. [PubMed] [Google Scholar]
- 24.Yeh WC, Itie A, Elia AJ, et al. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 2000;12:633–42. doi: 10.1016/s1074-7613(00)80214-9. [DOI] [PubMed] [Google Scholar]
- 25.Kreuz S, Siegmund D, Scheurich P, et al. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol. 2001;21:3964–73. doi: 10.1128/MCB.21.12.3964-3973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leverkus M, Sprick MR, Wachter T, et al. TRAIL-induced apoptosis and gene induction in HaCaT keratinocytes: differential contribution of TRAIL receptors 1 and 2. J Invest Dermatol. 2003;121:149–55. doi: 10.1046/j.1523-1747.2003.12332.x. [DOI] [PubMed] [Google Scholar]
- 27.Kataoka T. The caspase-8 modulator c-FLIP. Crit Rev Immunol. 2005;25:31–58. doi: 10.1615/critrevimmunol.v25.i1.30. [DOI] [PubMed] [Google Scholar]
- 28.Debatin KM, Krammer PH. Death receptors in chemotherapy and cancer. Oncogene. 2004;23:2950–66. doi: 10.1038/sj.onc.1207558. [DOI] [PubMed] [Google Scholar]
- 29.Chang DW, Xing Z, Pan Y, et al. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002;21:3704–14. doi: 10.1093/emboj/cdf356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Micheau O, Thome M, Schneider P, et al. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J Biol Chem. 2002;277:45162–71. doi: 10.1074/jbc.M206882200. [DOI] [PubMed] [Google Scholar]
- 31.Krueger A, Baumann S, Krammer PH, et al. FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol Cell Biol. 2001;21:8247–54. doi: 10.1128/MCB.21.24.8247-8254.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krueger A, Schmitz I, Baumann S, et al. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J Biol Chem. 2001;276:20633–40. doi: 10.1074/jbc.M101780200. [DOI] [PubMed] [Google Scholar]
- 33.Geserick P, Drewniok C, Hupe M, et al. Suppression of cFLIP is sufficient to sensitize human melanoma cells to TRAIL- or CD95L-mediated apoptosis. Oncogene. 2008;27:3211–20. doi: 10.1038/sj.onc.1210985. [DOI] [PubMed] [Google Scholar]
- 34.Trautmann A, Akdis M, Kleemann D, et al. T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J Clin Invest. 2000;106:25–35. doi: 10.1172/JCI9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Secchiero P, Gonelli A, Carnevale E, et al. TRAIL promotes the survival and proliferation of primary human vascular endothelial cells by activating the Akt and ERK pathways. Circulation. 2003;107:2250–6. doi: 10.1161/01.CIR.0000062702.60708.C4. [DOI] [PubMed] [Google Scholar]
- 36.Alladina SJ, Song JH, Davidge ST, et al. TRAIL-induced apoptosis in human vascular endothelium is regulated by phosphatidylinositol 3-kinase/Akt through the short form of cellular FLIP and Bcl-2. J Vasc Res. 2005;42:337–47. doi: 10.1159/000086599. [DOI] [PubMed] [Google Scholar]
- 37.Zhang XD, Nguyen T, Thomas WD, et al. Mechanisms of resistance of normal cells to TRAIL induced apoptosis vary between different cell types. FEBS Lett. 2000;482:193–9. doi: 10.1016/s0014-5793(00)02042-1. [DOI] [PubMed] [Google Scholar]
- 38.Clancy L, Mruk K, Archer K, et al. Preligand assembly domain-mediated ligand-independent association between TRAIL receptor 4 (TR4) and TR2 regulates TRAIL-induced apoptosis. Proc Natl Acad Sci USA. 2005;102:18099–104. doi: 10.1073/pnas.0507329102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koschny R, Sykora J, Walczak H, et al. Bortezomib-mediated up-regulation of TRAIL-R1 and TRAIL-R2 is not necessary for but contributes to sensitization of primary human glioma cells to TRAIL. Clin Cancer Res. 2007;13:6541–2. [Google Scholar]
- 40.Inoue S, Twiddy D, Dyer MJ, et al. Upregulation of TRAIL-R2 is not involved in HDACi mediated sensitization to TRAIL-induced apoptosis. Cell Death Differ. 2006;13:2160–2. doi: 10.1038/sj.cdd.4401977. [DOI] [PubMed] [Google Scholar]
- 41.Inoue S, Macfarlane M, Harper N, et al. Histone deacetylase inhibitors potentiate TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in lymphoid malignancies. Cell Death Differ. 2004;11:S193–S206. doi: 10.1038/sj.cdd.4401535. [DOI] [PubMed] [Google Scholar]
- 42.Guan B, Yue P, Lotan R, et al. Evidence that the human death receptor 4 is regulated by activator protein 1. Oncogene. 2002;21:3121–9. doi: 10.1038/sj.onc.1205430. [DOI] [PubMed] [Google Scholar]
- 43.Skurk C, Maatz H, Kim HS, et al. The Akt-regulated forkhead transcription factor FOXO3a controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP. J Biol Chem. 2004;279:1513–25. doi: 10.1074/jbc.M304736200. [DOI] [PubMed] [Google Scholar]
- 44.Borrelli S, Candi E, Alotto D, et al. p63 regulates the caspase-8-FLIP apoptotic pathway in epidermis. Cell Death Differ. 2009;16:253–63. doi: 10.1038/cdd.2008.147. [DOI] [PubMed] [Google Scholar]
- 45.Aoudjit F, Vuori K. Matrix attachment regulates Fas-induced apoptosis in endothelial cells: a role for c-flip and implications for anoikis. J Cell Biol. 2001;152:633–43. doi: 10.1083/jcb.152.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suhara T, Mano T, Oliveira BE, et al. Phosphatidylinositol 3-kinase/Akt signaling controls endothelial cell sensitivity to Fas-mediated apoptosis via regulation of FLICE-inhibitory protein (FLIP) Circ Res. 2001;89:13–9. doi: 10.1161/hh1301.092506. [DOI] [PubMed] [Google Scholar]
- 47.Armbruster N, Trautmann A, Brocker EB, et al. Suprabasal spongiosis in acute eczematous dermatitis: cFLIP maintains resistance of basal keratinocytes to t-cell-mediated apoptosis. J Invest Dermatol. 2009 doi: 10.1038/jid.2008.438. Doi: 10.1038/jid.2008.438. [DOI] [PubMed] [Google Scholar]
- 48.Kerstan A, Leverkus M, Trautmann A. Effector pathways during eczematous dermatitis: where inflammation meets cell death. Exp Dermatol. 2009;18:893–9. doi: 10.1111/j.1600-0625.2009.00919.x. [DOI] [PubMed] [Google Scholar]
- 49.Farley SM, Dotson AD, Purdy DE, et al. Fas ligand elicits a caspase-independent proinflammatory response in human keratinocytes: implications for dermatitis. J Invest Dermatol. 2006;126:2438–51. doi: 10.1038/sj.jid.5700477. [DOI] [PubMed] [Google Scholar]
- 50.Gochuico BR, Zhang J, Ma BY, et al. TRAIL expression in vascular smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2000;278:L1045–50. doi: 10.1152/ajplung.2000.278.5.L1045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Ni2+ sensitizes human ECs toTRAIL-induced apoptosis in a concentration-dependent manner. (A) Cell viability as assessed by crystal violet staining.HUVEC were pre-treated with the indicatedNi2+-doses or medium alone for 16 hrs andsubsequently exposed to the indicated concentrations of TRAIL for 6hrs prior to analysis by crystal violet assay. Experimental valueswere normalized to control values of diluent- orNi2+-treated controls to facilitate directcomparison. Error bars represent standard deviations derived fromone representative experiment performed in triplicate wells.(B) Concentration-dependent repression of cFLIP protein byNi2+ as determined by Western blot. HUVEC cellswere treated with the indicated Ni2+ doses for16 hrs and cFLIP protein expression in total cellular lysates wascompared by immunoblot analysis. Effectiveness of the respectiveNi2+ concentration was monitored by measuringprotein levels of the NF-κB target IL8 in the supernatants ofthe Ni2+-treated cells as previously described[1]. Immunoblotting with Abs to Tubulin was usedas loading control.
Fig. S2 Brefeldin A (BrefA) treatment blocksNi2+-dependent surface expression of TRAIL-Rsin ECs. (A) HUVEC were incubated with either diluent,Ni2+ or 2 μg/ml BrefA alone, or thecombination of both compounds for 16 hrs, and surface expression ofthe indicated TRAIL-Rs was analysed by flow cytometry. FACShistograms representing overlays of the individual TRAIL-R-stainedsamples (black, thick line) and their respective IgG1control-stained counterparts (grey, thin line) are shown. Thevalues of MFI in the upper right corner of each FACS histogramrepresent background-corrected MFI values for each individualTRAIL-R staining obtained after MFI subtraction of thecorresponding IgG1-control. Values in the table are calculated asfold MFI in relation to the respective untreated control for eachTRAIL-R. (B) HUVEC cells were either treated with theindicated BrefA concentrations alone, or the combination withNi2+ [1.5 mM] for 16 hrs. The effectiveness of BrefA on the IL-8 release from differentially treated cells was assessed by quantification of IL-8 protein in the supernatants by ELISA.
Fig. S3 cFLIPL overexpression does not affectDoxorubicin- or Paclitaxel-induced apoptosis. Primary ECs wereinfected with the indicated retroviruses and subsequently treatedwith 1 μM Paclitaxel (Tax) or 1 μg/ml Doxorubicin (Dox) for30 or 24 hrs, respectively. Subsequently, DNA fragmentation wasassessed by subdiploidy analysis in the FACS.
Fig. S4 Expression of dominant-negative IKK2 does notaffect Ni2+-dependent TRAIL-R surfaceregulation or TRAIL-induced Caspase-8 cleavage. HUVEC were infectedwith either empty retrovirus, or a retrovirus for dominant-negativeIKK2 (IKK2kd). Ni2+-induced modulation ofTRAIL-R surface expression (A) or TRAIL-induced Caspase-8and Caspase-3 cleavage (B) was assessed by flow cytometry,or Western blot analysis, respectively. (A) FACS profilesrepresenting overlays of the differently infected and treated cellsstained with specific antibodies for the indicated TRAIL-Rs (black,thick line) or an unspecific control IgG1-antibody (grey, thinline) are shown. Values in the upper right panel of each FACSprofile represent differential MFI values of the individual TRAIL-Rstainings after background subtraction of the corresponding IgG1control MFI value. For better comparison, additionallybackground-corrected MFI ratios of theNi2+-treated versus control-treatedsamples are shown as fold MFI. Incubation time withNi2+ was 16 hrs in all cases. (B) Afterinfection with the indicated retroviruses, cells were eithermock-treated or pre-treated with Ni2+ for 16 hrs. Subsequently, cells were stimulated with either TRAIL or equivalent amounts of diluent for 3 hrs. Expression and cleavage of cFLIP, Caspase-8 and Caspase-3 protein in the lysates of the differently treated cells was then analysed by immunoblot using appropriate antibodies for the indicated proteins. An immunoblot for Tubulin is shown as loading control; immunoblot for IKK2 is included as expression control for the IKK2 mutant. For better visualization both long and short exposures of the Caspase-8 immunoblot are shown; asterisks denote unspecific bands consistently seen with the employed Caspase-8 antibody.
Fig. S5 TNF treatment does not change the overall pattern of TRAIL-R surface expression. Human primary ECs were treated with 2 ng/ml TNF for 16 hrs and surface expression of the individual TRAIL-Rs was monitored by flow cytometry. Overlays of FACS profiles from the individual TRAIL-R-stained (black, thick lines) and IgG1-control-stained (grey, thin lines) samples are shown. Background-corrected MFI are plotted in the upper right corner of each FACS profile. For better comparison, MFI ratios of TNF-stimulated and unstimulated control cells for each TRAIL-R are included on the right side.
Fig. S6 Inhibition of p38 does not modulate TRAIL-inducedCaspase 8 cleavage in the presence or absence ofNi2+. HUVEC were incubated with diluent,Ni2+or 10 μM of the p38 inhibitor SB202190alone, or were co-treated with SB202190 andNi2+ for 16 hrs. Subsequently, cells were stimulated with TRAIL or medium for 90 min. prior to lysis. Protein expression and cleavage of Caspase-8 and c-FLIP was characterized by Western blot analysis. Even loading was controlled by immunoblot analysis of Tubulin.
Fig. S7 Expression of a dominant negative FoxO4 does notaffect Ni2+-dependent sensitization toTRAIL-induced apoptosis. HUVEC were infected with a control retrovirus, or a retrovirusencoding HA-tagged dominant-negative FoxO4 (dnFOXO) andsubsequently incubated with Ni2+ or diluent for 16 hrs. Cells were further cultured in presence or absence of TRAIL for 6 hrs and cytotoxicity was analysed by crystal violet assay. Error bars denote standard deviations of one representative experiment performed in triplicates. An immunoblot using an antibody for HA demonstrates expression of HA-tagged dnFOXO in the presented experiment (inset).