

Abstract
Rickets/osteomalacia is an important problem in a tropical country. Many cases are due to poor vitamin D intake or calcium deficient diets and can be corrected by administration of calcium and vitamin D. However, some cases are refractory to vitamin D therapy and are related to renal defects. These include rickets of renal tubular acidosis (RTA), hypophosphatemic rickets, and vitamin D dependent rickets (VDDR). The latter is due to impaired action of 1α-hydroxylase in renal tubule. These varieties need proper diagnosis and specific treatment.
Keywords: Hypophosphatemic rickets, osteomalacia, renal rickets, rickets, renal tubular acidosis, vitamin D rickets
INTRODUCTION
Normal calcium and phosphate are needed for bone mineralization. Deficient mineralization leads to rickets and/or osteomalacia. Rickets is characterized by deficient mineralization at the growth plate. Osteomalacia is characterized by impaired mineralization of bone matrix. Rickets and osteomalacia usually occur together as long as the growth plates are open as in the growing period; only osteomalacia occurs after the growth plates have fused.[1]
Rickets can be classified broadly as calcipenic or phosphopenic. Calcipenic rickets is often associated with low serum calcium levels, while phosphopenic rickets is characterized by low serum levels of phosphorus. Calcipenic rickets is caused by calcium deficiency, which usually is due to insufficient intake or metabolism of vitamin D, or in some cases due to inadequate intake or absorption of calcium.[2,3,4,5] Phosphopenic rickets usually is caused by renal phosphate wasting.[5]
CLINICAL FEATURES
Rickets initially manifest at the distal forearm, knee, and costochondral junctions as these are the sites of rapid bone growth and mineralization.[2] The important clinical features include:
Delayed closure of the fontanelles
Parietal and frontal bossing with caput quadratum
Craniotabes (soft skull bones)
Enlargement of the costochondral junction along the anterolateral aspects of chest (“rachitic rosary”)
Harrison sulcus groove at lower margin of thorax caused by muscular pull of diaphragmatic attachments to lower ribs
Sternum may be pulled into a pigeon-breast deformity
Widening of wrist
Bowing of distal radius and ulna (coxa vera)
Lateral bowing of femur and tibia (genu valgum and varum)
Double malleoli (Marfan’ sign)
Lower extremities tend to be predominantly affected in heritable forms of phosphopenic rickets.
Kyphoscoliosis
Pot belly and visceroptosis.
The type of deformity and the limb involved depends upon the age of child and weight-bearing in the limbs. Thus, deformities of the forearms and posterior bowing of the distal tibia are more common in infants, whereas marked bowing of the legs (genu varum) is a characteristic finding in a child who has started to walk. In the older child, valgus deformities of the legs or a windswept deformity (valgus deformity of one leg and varus deformity of the other) may be seen. Hypoplasia of the dental enamel is a typical finding of calcipenic rickets, whereas dental abscesses occur more often in phosphopenic rickets. Calcipenic rickets is characterized by decreased muscle tone and delayed motor milestones. Hypocalcemic seizures may occur in the 1st year of life. Children with calcipenic rickets are prone to recurrent infections and may manifest with hyperhidrosis due to bone pain.[4]
Radiographic Findings
The changes of rickets are seen best at growth plate of rapidly growing bones. Thus, in the upper limbs, the early signs of impaired mineralization are seen in the ulna. In the lower limbs metaphyses above and below the knees are the most useful sites. The best single radiographic view for infants and children younger than 3 years is an anterior view of the knee. There is widening of the epiphyseal plate and loss zone of provisional calcification at the epiphyseal/metaphyseal junction. Later, there is cupping, splaying, formation of cortical spurs, and stippling. The appearance of the epiphyseal bone centers is delayed, or they may be small. The diaphysis is osteopenic and the cortex becomes thin. The trabecular pattern is reduced and becomes coarse. Deformities of the shafts of the long bones are present. Pathological fractures and Looser's zones (also known as Milkman pseudofractures) may be noted. Looser's zones are radiolucent pseudofractures 2-5 mm wide and have sclerotic borders. They are seen characteristically in osteomalacia. They are usually bilateral and symmetric, and are perpendicular to the cortical margins of bones. Common sites for pseudofractures include femoral neck, medial part of femoral shaft, immediately under lesser trochanter, on pubic, and ischial rami. They may also be seen in ulna, scapula, clavicle, rib, and metatarsal bones. Pseudofractures also can be seen as hot spots on bone scans.
Evaluation
The evaluation of a child with clinical signs of rickets should include a dietary history with particular attention given to calcium and vitamin D intake along with a medication history and a history of sun exposure. Radiographic evaluation of a child with rickets should include, at least, plain films of wrist and hand or knees. The causes of rickets include conditions that lead to hypocalcemia and/or hypophosphatemia as a result of decreased intake; malabsorption; and/or increased excretion of calcium, phosphate, or vitamin D. To determine the optimal treatment, the common nutritional causes of rickets must be distinguished from those caused by a gastrointestinal or renal disease.
Laboratory investigations [Figure 1]
Figure 1.
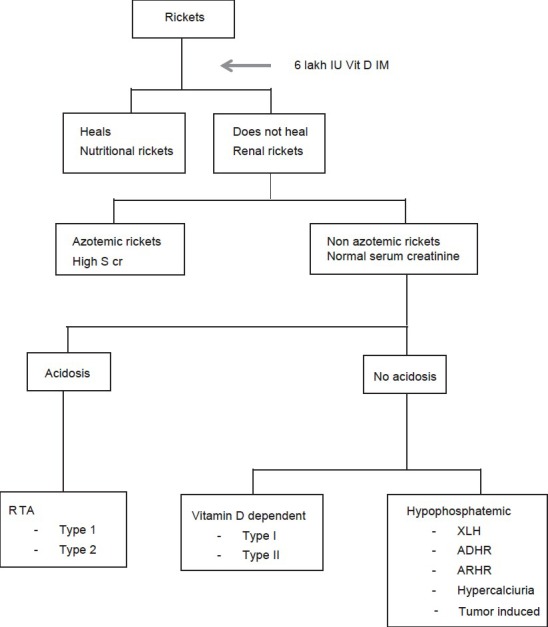
Approach to rickets
Alkaline phosphatase is an excellent marker of disease activity. In X-linked hypophosphatemic rickets (XLH), the serum alkaline phosphatase activity is moderately elevated (400-800 international units per liter (IU/L)) whereas in calcipenic rickets, values often reach greater levels (>1,500IU/L).
The serum calcium is usually decreased in calcipenic rickets (nutritional, vitamin D dependent rickets (VDDR) or renal tubular acidosis (RTA) and renal failure rickets; discussed below), while it is normal in phosphopenic rickets.
Serum phosphorus concentrations usually are low in both calcipenic and phosphopenic rickets. The total reabsorption of phosphorus (TRP) and the maximal tubular reabsorption of phosphorus per glomerular filtration rate (TmP/GFR) usually are decreased in both calcipenic and phosphopenic rickets, but decrease is severe in renal phosphate wasting type of rickets. These values are quite elevated in the setting of nutritional phosphate deprivation. In rickets due to renal failure there is high serum phosphorus.
Serum creatinine: Elevated in renal failure rickets.
Arterial blood gas (ABG): In rickets due to RTA there is normal anion gap (hyperchloremic metabolic acidosis. In renal failure rickets there is high anion gap metabolic acidosis. In all other varieties the ABG is normal.
Urine pH is >5.5 in distal RTA (DRTA) while it is <5.5 in proximal RTA (PRTA). Acid load test are the other tests done in RTA (discussed below).
A generalized aminoaciduria occurs from hyperparathyroidism. However, aminoaciduria does not occur in XLH.
Glycosuria and bicarbonaturia is seen in Fanconi's syndrome.
The serum concentration of parathyroid hormone (PTH) typically is quite elevated in calcipenic rickets. In contrast, PTH concentrations are usually normal or modestly elevated in phosphopenic rickets. Elevated PTH levels may also be seen in X-linked hypophosphatemia (XLH). Therefore, if calcipenic rickets is diagnosed, it is mandatory to observe appropriate healing during therapy, and if predicted response does not occur, XLH should be considered.
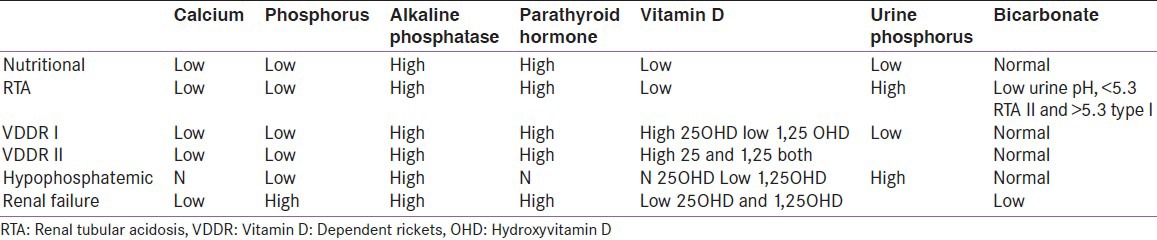
Serum concentrations of 25-hydroxyvitamin D (25OHD) and 1,25-dihydroxyvitamin D (1,25(OH)2 D)-25 OHD reflects body's vitamin D stores, and consequently, is low in nutritional rickets due to vitamin D deficiency; but it may be normal if there is associated calcium deficiency. In extremely severe liver disease or in intestinal disorders such as celiac disease, vitamin D levels may be low due to defective absorption or metabolism. Anticonvulsants may interfere with vitamin D metabolism leading to low levels. In type I VDDR 25OHD levels are high while 1,25(OH)2 D levels are low while in type II VDDR, 25OHD levels are normal and 1,25(OH)2 D levels are high. In some forms of phosphopenic rickets (XLH, tumor-induced osteomalacia (TIO)), serum concentrations of 1,25(OH)2 D may be low or inappropriately normal (despite hypophosphatemia). In other forms of phosphopenic rickets (hereditary hypophosphatemic rickets with hypercalcuria (HHRH)), the serum concentration of 1,25(OH)2 D may be elevated [Table 1].
Table 1.
Laboratory diagnosis: Etiology of rickets
TYPES OF RICKETS
Nonrenal rickets
Nutritional rickets
It occurs due to deficient intake or impaired absorption of calcium, phosphorus, or vitamin D. Mother's milk is poor in vitamin D. Sunlight exposure also influences vitamin D stores. Treatment for rickets may be administered gradually over several months or in a single day dose of 15,000 μg (600,000 U) of vitamin D. In gradual method, 125-250 μg (5,000-10,000 U) is given daily for 2-3 months until healing is well established and the alkaline phosphatase concentration becomes normal. Compliance to the daily regime is essential. If the vitamin D dose is administered in a single day, it is usually divided into four or six oral doses. An intramuscular injection is also available. Vitamin D (cholecalciferol) is well stored in the body and is gradually released over many weeks. Because both calcitriol and calcidiol have short half-lives, these agents are unsuitable for treatment. In addition they bypass the natural physiologic controls of vitamin D synthesis and may cause toxicity. The single day therapy avoids problems with compliance and may be helpful in differentiating nutritional rickets from nonnutritional rickets. In nutritional rickets, the phosphorus level rises in 96 h and radiographic healing is visible in 6-7 days. Neither happens with renal rickets. If severe deformities have occurred, orthopedic correction may be required after healing. Most of the deformities correct with growth.[6,7,8,9]
Malabsorption
Malabsorption syndromes may be associated with rickets.
Liver disease may be associated with inadequate conversion of vitamin D; 25 hydroxyvitamin D.
Anticonvulsants interfere with metabolism of vitamin D and cause calcipenic rickets or osteomalacia.
Renal Rickets
Renal tubular acidosis
Acid base balance of the body is maintained by lungs and kidneys. The lungs remove carbondioxide. Acid base balance by kidneys occurs by two processes:
Bicarbonate reabsorption and
Acid excretion.
Most of the bicarbonate that is filtered by the glomerulus is predominantly resorbed as a result of Na-H exchange by the proximal tubules. Approximately, 85-90% of the filtered load is reclaimed at this site. By comparison, 10% is reclaimed in the distal nephron, primarily via hydrogen secretion by (H-adenosine triphosphatase (ATPase) proton pump. Under normal conditions, there is no bicarbonate in urine. In addition the proximal tubule contributes to bicarbonate regeneration during ammoniagenesis (described below).
Hydrogen ion excretion is carried out by collecting tubules. The body produces an average daily acid load of 1 mEq/kg. This acid combines with buffers in the tubular lumen. Buffers enable the urine pH to be maintained above 4. Ammonia (measured as ammonium) and phosphate (titratable acidity) are the two main buffers in the urine. Ammonia is generated in the proximal tubule cells from glutamic acid. Two molecules of bicarbonate are generated in the process. NH3 diffuses freely across the tubular membranes and combines with H+ in the tubular lumen and forms ammonium. NH4+ cannot diffuse across membranes and thus leads to trapping of NH3 in the lumen. Daily approximately 40 mEq of NH4 is excreted in urine. NH3 generation is stimulated by intracellular acidosis and hypokalemia. Defects in the ability to secrete either ammonium or hydrogen ions can cause metabolic acidosis.
RTA occurs due to tubular defect leading to systemic acidosis with preserved GFR. Proximal (type 2) RTA is due to decreased ability of the proximal tubule to reclaim filtered bicarbonate with consequent increase in delivery of bicarbonate to the distal nephron with bicarbonaturia, fall in serum bicarbonate, and metabolic acidosis. DRTA results from impaired hydrogen ion secretion, while defective ammoniagenesis is the primary defect in renal failure.
All types of RTA present with a normal anion gap (hyperchloremic) metabolic acidosis. The degree of acidosis varies with the type of RTA: In distal (type 1) RTA, there is hydrogen ion retention and fall of plasma bicarbonate to even less than 10 mEq/L. The urine pH is 5.5 or higher, while patients with metabolic acidosis not due to RTA have a urine pH that is 5.3 or less. In proximal (type 2) RTA, whenever the plasma bicarbonate is above threshold, that is, 15 mEq/l there is bicarbonate wasting. The distal segments can absorb up to 15% bicarbonate, hence some bicarbonate which is not absorbed in the proximal tubule in PRTA is absorbed by the distal tubule; however, if plasma bicarbonate is above threshold, that is, 15 mEq/l there is bicabonaturia. Plasma bicarbonate concentration is usually between 15 ± 3 meq/L in untreated patients. The urine pH is appropriately low (5.3 or less) in untreated patients.[10]
Promimal (Type 2) RTA
Proximal (type 2) RTA occurs as an isolated defect or along with generalized dysfunction of proximal tubule (Fanconi syndrome). It is characterized by bicarbonaturia, glucosuria, phosphaturia, uricosuria aminoaciduria, and tubular proteinuria. Hypophosphatemia and hypouricemia are seen. The most common cause of PRTA with or without Fanconi syndrome in adults is monoclonal gammopathy. In children, inherited diseases are most often responsible for PRTA. Isolated RTA is rare in children and Fanconi's is the commonest presentation.[11]
If the acidosis is severe (serum bicarbonate < 15 mEq/l), urine pH is < 5.3 in PRTA. Urine infection with urea splitting organisms causes a falsely elevated urine pH, hence urine infection must always be excluded before interpreting urine pH. If the plasma bicarbonate is >15 mEq/l, then acid load test is done before interpreting urine pH. Acid load test consists of administration of 0.1 g/kg of ammonium chloride after a baseline blood gas analysis and testing the urine pH every hour for 5 h or till the urine pH falls below 5.3 (if pH stays above 5.3, it indicates presence of DRTA). An arterial blood gas is performed at 2 h after acid load to confirm that serum bicarbonate is <15. In patients with liver disease, arginine hydrochloride can be used. Other test is furosemide test with measurement of H and K excretion after 1 mg/kg of furosemide.
The diagnosis of PRTA can also be confirmed by measurement of the urine pH and fractional bicarbonate excretion during a bicarbonate infusion. The urine pH is above 7.5 and fractional excretion of bicarbonate is >15% when the serum bicarbonate concentration is raised to normal. Patients with PRTA should be evaluated for renal glycosuria, hypophosphatemia, and hypouricemia to rule out Fanconi's syndrome. Most children require 10-15 mEq/kg of bicarbonate per day to maintain a normal pH. Treatment of mild acidosis is not necessary in adults. Many bicarbonate preparations are available: Shohl's solution (citric acid and sodium citrate) 1 ml contains 1 mEq/l of bicarbonate, Bicitra-K, and Polycitra. Potassium citrate solution contains 2 mEq/l each of bicarbonate and potassium. Bicarbonate tablets 325 mg contains 4 mEq of bicarbonate. Bicarbonate powder can also be used (15 mEq/g). Other additional therapies include magnesium, carnitine, and thiazide diuretics to reduce polyuria and vitamin D supplements. Some cases of Fanconi's need phosphorus supplementation.
Distal (Type 1) RTA
Distal (type 1) RTA is characterized by an impaired capacity for hydrogen ion excretion. Hence, there is a decrease in ammonium secretion in the collecting tubules. There is a high urine pH (5.3 or higher) even during systemic acidosis with plasma bicarbonate <15 mEq/l. DRTA results from one of several defects in distal hydrogen ion secretion. Decreased proton pump (H-ATPase) activity causes: (i) Increased luminal membrane permeability with backleak of hydrogen ions, (ii) voltage defect, and (iii) rate defect. Voltage defect is often associated with hyperkalemia. The most common causes of DRTA in adults are autoimmune disorders and other conditions associated with chronic hyperglobulinemia. In children, DRTA is most often a primary, hereditary condition.
Patients with DRTA have a normal anion gap (hyperchloremic) metabolic acidosis and an inappropriately high urine pH (5.3 or higher). Hypercalciuria occurs due to bone resorption as a result of chronic acidosis and decreased renal tubular reabsorption of calcium. Hypercalciuria and hypocitraturia may cause nephrolithiasis and nephrocalcinosis, which is generally not seen in PRTA.[12] Hypokalemia, sometimes severe, is often seen in DRTA and may produce muscle weakness. Some patients may present with quadriparesis. Potassium wasting occurs due to defect in H+ -K + ATPase pump in the distal tubule.[13]
Correction of acidosis with alkali therapy (usually potassium citrate) generally improves both calcium and potassium balance, and prevents stones and nephrocalcinosis. In contrast to the high alkali requirement in PRTA, the daily bicarbonate requirement is only 1-2 mEq/kg per day to replace the daily acid load.
Hypophosphatemic rickets
Most of these cases are caused by renal phosphate wasting. This disorder initially called vitamin D resistant rickets, is now called hereditary hypophosphatemic rickets because the primary problem is phosphate wasting rather than true vitamin D resistance. Hereditary forms of hypophosphatemic rickets include X-linked, autosomal dominant, and autosomal recessive disease, as well as hypophosphatemic rickets with hypercalciuria. The X-linked form is most common; the other forms are rare. An acquired disorder, tumor-induced (or oncogenic) osteomalacia, has similar clinical manifestations to the familial syndromes. In addition to hypophosphatemia, these disorders all have normal serum levels of calcium and PTH. Most of these disorders also have high circulating levels of fibroblast growth factor 23 (FGF23), a circulating hormone that causes renal phosphate wasting and is a common final pathway.
X-linked hypophosphatemic rickets
It is a dominant disorder with a prevalence of approximately one case per 20,000 live births. The renal tubular abnormality in XLH, is caused by one or more circulating factors.[14] These circulating factors promote phosphate excretion and impair bone mineralization and are called “phosphatonins”. The gene for XLH is located on chromosome Xp22.1 and is named PHEX (Phosphate regulating Endopeptidase on the X chromosome). PHEX is expressed mainly in bone and teeth.[15] The principal phosphatonin involved in the pathogenesis of XLH is FGF23. Mutations in PHEX (in bone tissue) indirectly affect the degradation and generation of FGF23, causing increased circulating levels of phosphatonin. FGF23 acts as a counter-regulatory hormone to inhibit phosphate reabsorption by sodium/phosphate cotransporter in the kidney, acting through FGF receptors with cofactor klotho.[16,17] Elevated levels of FGF23 are also seen in other forms of hereditary hypophosphatemic rickets, as well as TIO, although the mechanisms vary. FGF23 causes renal phosphate-wasting, and the hypophosphatemia contributes to rickets. However, FGF23 does not mediate all of the clinical features of XLH. Enthesopathy and dental abnormalities are mediated by mechanisms other than FGF23. XLH is characterized by inappropriately low level of calcitriol (1,25-(OH) 2 vitamin D3).[18]
Most common clinical features include poor growth, rickets, or osteomalacia. An outstanding feature of familial hypophosphatemic rickets is short stature. The short stature associated with this condition is disproportionate, resulting from deformity and growth retardation of the lower extremities. Low serum phosphorus is seen soon after birth. At the time of weight bearing leg deformities (e.g., bowing) are seen. Lower limb involvement is more common. Many adults have enthesopathy (calcification of tendons, ligaments, and joint capsules). Younger siblings of affected patients should be screened to diagnose the disorder before these complications develop. Screening can be accomplished by measuring fasting serum phosphorus, and if necessary, renal phosphorus excretion. Dental abscesses and early tooth decay occurs in children due to defective dentin. Hypertension, left ventricular hypertrophy, and craniosynostosis are some other features seen. Hyperparathyroidism may develop due to oral phosphate supplementation. Muscle weakness is rare. Tetany is absent. The severity of XLH varies even among family members. There is no gender difference in disease severity.
Patients with XLH have hypophosphatemia, decreased tubular reabsorption threshold for phosphorus, normal serum levels of calcium, normal-to-high PTH levels, increased (or normal) alkaline phosphatase activity, normal plasma calcidiol concentration, and normal or slightly reduced plasma calcitriol concentration.[18] Calcitriol synthesis is abnormal in XLH because calcitriol is expected to be elevated in the presence of hypophosphatemia. The defect is in translation of the 25(OH) D-1alpha-hydroxylase messenger ribonucleic acid (mRNA). Determination of the PHEX mutation can confirm the diagnosis. Early diagnosis in newborns may be facilitated by knowing which mutation runs in the family, but it is not clear whether this information improves clinical care.
Therapy for XLH in children consists of the oral administration of phosphate and calcitriol. Phosphate increases the plasma phosphate concentration, which lowers the plasma ionized calcium, and decreases the plasma calcitriol concentration (by correcting hypophosphatemia). Hypocalcemia and low calcitriol causes secondary hyperparathyroidism which aggravates the bone disease and increase urinary phosphate excretion. Calcitriol should be given along with oral phosphorus. Calcitriol is administered in two doses per day (10-20 ng/kg per dose). Phosphorus is administered in four to five doses per day; the starting dose is 40 mg of elemental phosphorus/kg per day. The daily phosphorus dose should be increased in steps of 250 mg up to a maximum of 3500 mg/day till normal growth is achieved. Various forms of phosphate tablets are available in western countries (e.g. sodium and potassium phosphate tablets) which contain 250 mg elemental phosphorus per tablet. Children can be given phosphorus supplementation in the form of Joulie's solution (155 g of dibasic anhydrous sodium phosphate and 64 g of phosphoric acid 85% per liter solution, corresponding to 50 mg/mL of elemental phosphorus. Oral phosphorus causes diarrhea. If diarrhea occurs, the dose of phosphorus should be decreased then gradually reincreased. The goals of therapy include acceptable height velocity and improvement in skeletal deformities and not normalization of phosphorus. Therapy is continued as long as the growth plates are open. Addition of a calcimimetic (cinacalcet) to the regimen may prevent secondary hyperparathyroidism.[19] However, long-term studies in children with XLH are necessary before calcimimetics can be generally recommended. Newer therapeutic strategies include neutralizing antibody to FGF23.[20]
Most of the adults are asymptomatic and may not benefit from therapy. Some may have calcifications of tendons and ligaments. Unnecessary treatment may cause complications. Hence, treatment is only indicated in symptomatic patients for bone pain or non-uniting fractures or those who are to undergo orthopedic surgery in next 3-6 months as this helps in recovery. Calcitriol is administered in two doses per day (10-20 ng/kg per dose). Phosphate is administered in a dose of 1-4 g/day in three to four divided doses. Adults should be monitored at least annually for serum phosphorus, calcium, creatinine, and PTH. Treatment can be discontinued once symptoms subside. Pregnancy is another indication where treatment should be considered to maintain serum phosphorus concentrations.
Nephrocalcinosis and hyperparathyroidism are important complications. Nephrocalcinosis can be demonstrated in up to 80% of patients with XLH. The renal calcifications are located in the tubules and are composed of calcium phosphate. Nephrocalcinosis correlates with the phosphate dose, but not with the dose of calcitriol or the duration of therapy. Nephrocalcinosis may result in renal insufficiency. Nonhypercalcemic analogues of calcitriol, such as 22-oxacalcitriol, may prevent nephrocalcinosis.
Calcium with phosphate complexes result in intermittent hypocalcemia and persistent stimulation of PTH release despite the administration of calcitriol. In some cases autonomous (tertiary) hyperparathyroidism occurs, and parathyroidectomy may be required. Other adjuvants to therapy include recombinant growth hormone in children to improve stature.[21] Hydrochlorothiazide or amiloride decreases urinary calcium excretion and prevents nephrocalcinosis.[22] Addition of 24,25-dihydroxyvitamin D may improve hyperparathyroidism.[23]
Autosomal dominant hypophosphatemic rickets
Results from activating mutations in the FGF23 gene on chromosome 12p13. The mutant protein is resistant to proteases, thus serum FGF23 is increased and this causes phosphaturia.[24] The clinical features are similar to X-linked disease. Patients with early onset disease have phosphate wasting, rickets, and lower extremity deformities in childhood; but may lose the phosphate-wasting defect after puberty. Those who present after growth; plate closure have bone pain and fractures, but no deformities. Many females present after pregnancy. Pseudofractures are common. Vitamin D is inappropriately normal. Treatment is similar to that of X-linked hypophosphatemic rickets.
Autosomal recessive hypophosphatemic rickets
Patients exhibit isolated renal phosphate wasting. The disorder is caused by inactivating mutations in the DMP1 gene, which encodes dentin matrix protein-1. DMP suppresses FGF23 production from bone.[25]
Hypophosphatemic rickets with hypercalciuria
HHRH is inherited in an autosomal recessive manner and is associated with high levels of vitamin D. HHRH is caused by genetic mutations of the renal type 2c sodium-phosphate cotransporter. The gene is located on chromosome 9q34, which contains the gene SLC34A3. Rickets and/or osteomalacia is the presenting feature like XLH. Mild forms may present with hypercalciuria and nephrolithiasis without bone disease. Patients should be treated only with phosphorus supplementation. Calcitriol should not be used. Plasma calcitriol levels and urinary calcium excretion should be monitored.[26]
Dent's disease
Dent's disease results from mutations in the CLCN5 gene encoding a voltage-gated chloride transporter in about 60% of patients, and with the OCRL1 gene in another 15%. It is an X-linked recessive syndrome in which a primary defect in the cells of the proximal renal tubule results in a phenotype of proximal tubular solute-wasting, hypercalciuria, nephrocalcinosis, kidney stones, renal failure, and in some cases rickets. The most consistent feature is low molecular weight (LMW) proteinuria; other evidence of proximal solute reabsorptive failure includes glycosuria, aminoaciduria, and phosphaturia; though not bicarbonaturia. Rickets occurs only in 25% of patients and it is seen in early childhood. In a hypophosphatemic child with rickets, if other features suggest Dent's disease, urinary LMW proteins (β2 microglobulin and retinol-binding protein) should be measured.[27]
Tumor induced osteomalacia (oncogenic osteomalacia)
Usually seen in adults where tumor leads to severe hypophosphatemia with osteomalacia. Rarley in children when growth plates are open, tumor may cause rickets. The tumors associated with rickets or osteomalacia are benign, small, and originate from the mesenchyma (e.g., sclerosing type of hemangiopericytoma).[28] The tumor produces PTH related peptide (PTH rP). Unlike PTH, PTH rP affects only phosphate transport and has no effect on calcium metabolism. PTH antagonists do not block PTH rP and and its effects are not mediated by cyclic adenosine monophosphate (AMP). The phosphaturic hormone implicated in TIO is FGF23.[29,30] There may be a possible pathogenetic role for FGF23 in some patients with TIO. Vitamin D metabolism is abnormal in TIO and plasma calcitriol is reduced despite hypophosphatemia. TIO is a differential of XLH and ADRH and is characterized by hypophosphatemia, osteomalacia or rickets, renal phosphate wasting, no other proximal tubular defects, and an inappropriately low plasma calcitriol. The family history is negative unlike the other two disorders. Tumor localization is a major challenge. Despite total body magnetic resonance imaging (MRI), scintigraphy using octreotide labeled with indium-111 (because the tumors typically express somatostatin receptors) or scintigraphy combined with positron emission tomography/computerized tomography (PET/CT) which are often employed; identification of the tumor is often difficult as it is generally small and intraosteal.[31,32] Systemic venous sampling for FGF23 levels is one of the newer methods used for tumor detection. Functional imaging studies included whole body[11] In-octreotide single photon emission computed tomography (SPECT) and, if necessary, whole body[18] fluorodeoxyglucose PET/CT and anatomic imaging (CT, MRI).[33] Treatment is similar to those of X-linked disease. Cinacalcet may be useful.[34] Therapy is continued until the causative tumor can be removed or indefinitely if tumor removal is not possible. Treatment with octreotide may be considered when the tumor cannot be found.[35]
Vitamin D dependent rickets
Vitamin D dependent rickets type 1
VDDR-I is also called pseudovitamin D deficiency rickets. It is caused due to 1-alpha-hydroxylase deficiency. VDDR-I is characterized by early onset of rickets (within the 1st year of life) and severe hypocalcemia (sometimes with tetany), with accompanying moderate hypophosphatemia. Enamel hypoplasia may occur. These features distinguish VDDR-I from XLH (previously known as X-linked vitamin D-resistant rickets).
VDDR-I is an autosomal recessive disorder. It is characterized by defective conversion of 25OHD-1,25(OH)2 D.[36] The characteristic biochemical findings of VDDR-I are normal serum levels of 25OHD and low levels of 1,25(OH)2 D. Patients with VDDR-I have inactivating mutations in the CYP27B1 gene which encodes the enzyme 1-alpha hydroxylase responsible for the conversion of 25OHD to 1,25 (OH)2 D. The treatment for VDDR-I is 1,25 (OH)2 D (calcitriol). The initial dose for rickets is 1 μg/day till bone healing occurs. Later the dose is reduced to 0.25 and 1 μg/day. Adequate intake of dietary calcium (30-75 mg/kg/day of elemental calcium) should be maintained. Possible side effects of 1,25(OH)2 D therapy include hypercalcemia, hypercalciuria, nephrocalcinosis, and intraocular calcifications. Therefore, it is important to check the urinary calcium/creatinine ratio and kidney function (e.g. serum creatinine) at each visit. Renal ultrasound and ophthalmologic consultation (slit lamp examination) should be performed once per year.
Vitamin D dependent rickets type II
Is a very rare form of rickets. It is an autosomal recessive disorder. It is associated with end-organ resistance to 1,25(OH)2 D, usually caused by mutations in the gene encoding the vitamin D receptor. The defect in the receptor interferes with the function of the hormone-receptor complex, thereby preventing 1,25(OH)2 D action.[37,38,39] The clinical spectrum of VDDR II varies widely with the type of mutation. Affected children usually appear normal at birth, but develop rickets within the first 2 years of life. Alopecia is seen in resulting from the lack of vitamin D receptor activity within keratinocytes develops in approximately two-thirds of cases and is a marker of disease severity.[40,41,42,43,44,45] The treatment of VDDR II consists of therapeutic trial of 1,25(OH)2 D (calcitriol) and calcium. The severity of the receptor defect varies among patients. Therapy is started at daily doses of 2 μg of 1,25 (OH)2 D and 1 g of elemental calcium. However, in some cases administration of very high doses of 1,25(OH)2 D (up to 60 μg/day) and calcium (up to 3 g per day) may be necessary. Intravenous calcium infusion into central vein may be needed for resistant patients and must be continued for many months. Oral calcium therapy may be sufficient once radiographic healing has occurred.
Chronic kidney disease rickets (renal osteodystrophy)
In patients with moderate to severe chronic kidney disease, there is a high anion gap acidosis. In chronic kidney disease as the number of functioning nephrons decreases, acid excretion is initially maintained by increased ammonium excretion per nephron. However, when the GFR is below 40 ml/min, ammonium excretion begins to fall due to decrease in number of functioning nephrons. There is retention of hydrogen ions, which can lead to a progressive metabolic acidosis. The anion gap is elevated due to retention of anions such as phosphate, sulfate, urate, and hippurate due to reduced GFR. Other factors contributing to rickests include reduced formation of 1,25(OH)2 D, administration of aluminum, and secondary hyperparathyroidism. Patients who have a normal anion gap differ from those with DRTA as they have high serum creatinine and a urine pH of < 5.3. It is associated with elevated phosphorus and low calcium.[46]
In our study we found that in children with renal rickets; 20% had azotemic rickets, 64.3% had DRTA, 7.1% had PRTA, and 6.4% had hypophosphatemic rickets, while 1.7% had VDDR.[47] Cystinosis is an important cause of PRTA in children and can be diagnosed by slit lamp.[48]
Monitoring
During initial phase, patients should be evaluated at least once per week with serum calcium, phosphorus, alkaline phosphatase, creatinine, 1,25(OH)2 D, PTH, and urinary calcium/creatinine ratio. If the biochemical parameters do not respond, the dose of 1,25(OH)2 D should be gradually increased to reach serum concentrations of up to 100 times the normal mean value. Failure of therapy should be considered if no biochemical response occurs after 3-5 months of treatment. Radiographs should show clear improvement after 4 weeks of therapy and should be repeated after 3 months; when the growth plates should have regained a normal appearance. Patients may be evaluated at 3-month intervals during maintenance therapy. Hand radiographs are performed once per year to check for the reappearance of rachitic changes.
Differential diagnosis
Rare metabolic bone diseases, including hypophosphatasia, have been confused with rickets in infancy. Jansen syndrome is a rare autosomal dominant form of short-limbed dwarfism in which infants present with metaphyseal chondroplasia. Hypophosphatasia is a rare genetic disorder of alkaline phosphatase activity. Like rickets, it is characterized by bone demineralization. In contrast to rickets, serum alkaline phosphatase activity is very low.[49] Skeletal dysplasia (e.g. achondroplasia, pseudoachondroplasia, and metaphyseal chondrodysplasia) is another cause of bilateral, symmetric bowed legs. The radiographic features can be similar to those of rickets. However, serum inorganic phosphorus and PTH concentrations usually are normal in children with skeletal dysplasia.
CONCLUSIONS
Rickets and osteomalacia are commonly encountered clinical problems. Many cases of rickets are nutritional and respond to vitamin D therapy. A single dose of 6 lakh IU intramuscular (IM) is the most practical way to treat nutritional rickets along with calcium. The serum phosphorus rises within 96 h and healing is seen on X-ray within 4 weeks. In case of nonresponse, the renal causes should be considered. A normal serum creatinine excludes renal osteodystrophy. The presence of acidosis points towards RTA. Further differentiation between types 1 and 2 is by urine pH. The absence of acidosis indicates either hypophosphatemic rickets or VDDR. Hypophosphatemic rickets shows renal phosphate wasting, while VDDR can be identified by measuring serum vitamin D. The treatment depends on etiology, hence a proper evaluation is needed.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
REFERENCES
- 1.Rauch F. The rachitic bone. Endocr Dev. 2003;6:69–79. doi: 10.1159/000072770. [DOI] [PubMed] [Google Scholar]
- 2.Misra M, Pacaud D, Petryk A, Collett-Solberg PF, Kappy M. Drug andTherapeutics Committee of the Lawson Wilkins Pediatric Endocrine Society. Vitamin D deficiency in children and its management: Review of current knowledge and recommendations. Pediatrics. 2008;122:398–417. doi: 10.1542/peds.2007-1894. [DOI] [PubMed] [Google Scholar]
- 3.William's Textbook of Endocrinology. 11th ed Rickets and Osteomalacia. [Google Scholar]
- 4.Sahay M, Sahay R. Rickets-Vitamin D deficiency and dependency. Indian J Endocrinol Metab. 2012;16:164–76. doi: 10.4103/2230-8210.93732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sahay M. Homeostasis and disorders of calcium, phosphorus and magnesium principles and practice of pediatric nephrology. In: Vijaykumar M, Nammalwar BR, editors. 2nd ed. p. 82. [Google Scholar]
- 6.Robinson PD, Högler W, Craig ME, Verge CF, Walker JL, Piper AC, et al. The re-emerging burden of rickets: A decade of experience from Sydney. Arch Dis Child. 2006;91:564–8. doi: 10.1136/adc.2004.069575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Najada AS, Habashneh MS, Khader M. The frequency of nutritional rickets among hospitalized infants and its relation to respiratory diseases. J Trop Pediatr. 2004;50:364–8. doi: 10.1093/tropej/50.6.364. [DOI] [PubMed] [Google Scholar]
- 8.Holick MF. Vitamin D. In: Shils ME, Olson J, Shike M, Ross CA, editors. Modern Nutrition in Health and Disease. 9th ed. Baltimore: Williams and Williams; 1999. pp. 329–45. [Google Scholar]
- 9.Washington: National Academy Press; 1997. Standing Committee on the Scientific Evaluation of Dietary ReferenceIntakes. Dietary reference intakes for calcium, phosphorous, magnesium, vitamin D and fluoride. [Google Scholar]
- 10.Rodríguez Soriano J. Renal tubular acidosis: The clinical entity. J Am Soc Nephrol. 2002;13:2160–70. doi: 10.1097/01.asn.0000023430.92674.e5. [DOI] [PubMed] [Google Scholar]
- 11.Sebastian A, McSherry E, Morris RC., Jr Renal potassium wasting in renal tubular acidosis (RTA): Its occurrence in types 1 and 2 RTA despite sustained correction of systemic acidosis. J Clin Invest. 1971;50:667–78. doi: 10.1172/JCI106537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brenner RJ, Spring DB, Sebastian A, McSherry EM, Genant HK, Palubinskas AJ, et al. Incidence of radiographically evident bone disease, nephrocalcinosis, and nephrolithiasis in various types of renal tubular acidosis. N Engl J Med. 1982;307:217–21. doi: 10.1056/NEJM198207223070403. [DOI] [PubMed] [Google Scholar]
- 13.Batlle D, Moorthi KM, Schlueter W, Kurtzman N. Distal renal tubular acidosis and the potassium enigma. Semin Nephrol. 2006;26:471–8. doi: 10.1016/j.semnephrol.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 14.Alizadeh Naderi AS, Reilly RF. Hereditary disorders of renal phosphate wasting. Nat Rev Nephrol. 2010;6:657–65. doi: 10.1038/nrneph.2010.121. [DOI] [PubMed] [Google Scholar]
- 15.A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet. 1995;11:130–6. doi: 10.1038/ng1095-130. [DOI] [PubMed] [Google Scholar]
- 16.Razzaque MS, Lanske B. The emerging role of the fibroblast growth factor-23-klotho axis in renal regulation of phosphate homeostasis. J Endocrinol. 2007;194:1–10. doi: 10.1677/JOE-07-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng JQ, Clinkenbeard EL, Yuan B, White KE, Drezner MK. Osteocyte regulation of phosphate homeostasis and bone mineralization underlies the pathophysiology of the heritable disorders of rickets and osteomalacia. Bone. 2013;54:213–21. doi: 10.1016/j.bone.2013.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan B, Xing Y, Horst RL, Drezner MK. Evidence for abnormal translational regulation of renal 25-hydroxyvitamin D-1alpha-hydroxylase activity in the hyp-mouse. Endocrinology. 2004;145:3804–12. doi: 10.1210/en.2004-0192. [DOI] [PubMed] [Google Scholar]
- 19.Alon US, Levy-Olomucki R, Moore WV, Stubbs J, Liu S, Quarles LD. Calcimimetics as an adjuvant treatment for familial hypophosphatemic rickets. Clin J Am Soc Nephrol. 2008;3:658–64. doi: 10.2215/CJN.04981107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aono Y, Yamazaki Y, Yasutake J, Kawata T, Hasegawa H, Urakawa I, et al. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J Bone Miner Res. 2009;24:1879–88. doi: 10.1359/jbmr.090509. [DOI] [PubMed] [Google Scholar]
- 21.Haffner D, Nissel R, Wühl E, Mehls O. Effects of growth hormone treatment on body proportions and final height among small children with X-linked hypophosphatemic rickets. Pediatrics. 2004;113:e593–6. doi: 10.1542/peds.113.6.e593. [DOI] [PubMed] [Google Scholar]
- 22.Seikaly MG, Baum M. Thiazide diuretics arrest the progression of nephrocalcinosis in children with X-linked hypophosphatemia. Pediatrics. 2001;108:E6. doi: 10.1542/peds.108.1.e6. [DOI] [PubMed] [Google Scholar]
- 23.Carpenter TO, Keller M, Schwartz D, Mitnick M, Smith C, Ellison A, et al. 24,25 Dihydroxyvitamin D supplementation corrects hyperparathyroidism and improves skeletal abnormalities in X-linked hypophosphatemic rickets--a clinical research center study. J Clin Endocrinol Metab. 1996;81:2381–8. doi: 10.1210/jcem.81.6.8964881. [DOI] [PubMed] [Google Scholar]
- 24.Shimada T, Muto T, Urakawa I, Yoneya T, Yamazaki Y, Okawa K, et al. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology. 2002;143:3179–82. doi: 10.1210/endo.143.8.8795. [DOI] [PubMed] [Google Scholar]
- 25.Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006;38:1310–5. doi: 10.1038/ng1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet. 2006;78:179–92. doi: 10.1086/499409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scheinman SJ. X-linked hypercalciuric nephrolithiasis: Clinical syndromes and chloride channel mutations. Kidney Int. 1998;53:3–17. doi: 10.1046/j.1523-1755.1998.00718.x. [DOI] [PubMed] [Google Scholar]
- 28.Drezner MK. Tumor-induced Osteomalacia. In: Favus MJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 4th ed. Philadelphia: Lippincott Williams and Wilkins; 1999. p. 331. [Google Scholar]
- 29.Wilkins GE, Granleese S, Hegele RG, Holden J, Anderson DW, Bondy GP. Oncogenic osteomalacia: Evidence for a humoral phosphaturic factor. J Clin Endocrinol Metab. 1995;80:1628–34. doi: 10.1210/jcem.80.5.7745010. [DOI] [PubMed] [Google Scholar]
- 30.Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348:1656–63. doi: 10.1056/NEJMoa020881. [DOI] [PubMed] [Google Scholar]
- 31.Chong WH, Yavuz S, Patel SM, Chen CC, Collins MT. The importance of whole body imaging in tumor-induced osteomalacia. J Clin Endocrinol Metab. 2011;96:3599–600. doi: 10.1210/jc.2011-1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clifton-Bligh RJ, Hofman MS, Duncan E, Sim IeW, Darnell D, Clarkson A, et al. Improving diagnosis of tumor-induced osteomalacia with Gallium-68 DOTATATE PET/CT. J Clin Endocrinol Metab. 2013;98:687–94. doi: 10.1210/jc.2012-3642. [DOI] [PubMed] [Google Scholar]
- 33.Chong WH, Andreopoulou P, Chen CC, Reynolds J, Guthrie L, Kelly M, et al. Tumor localization and biochemical response to cure in tumor-induced osteomalacia. J Bone Miner Res. 2013;28:1386–98. doi: 10.1002/jbmr.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geller JL, Khosravi A, Kelly MH, Riminucci M, Adams JS, Collins MT, et al. Cinacalcet in the management of tumor-induced osteomalacia. J Bone Miner Res. 2007;22:931–7. doi: 10.1359/jbmr.070304. [DOI] [PubMed] [Google Scholar]
- 35.Paglia F, Dionisi S, Minisola S. Octreotide for tumor-induced osteomalacia. N Engl J Med. 2002;346:1748–9. doi: 10.1056/NEJM200205303462215. [DOI] [PubMed] [Google Scholar]
- 36.Kitanaka S, Takeyama K, Murayama A, Kato S. The molecular basis of vitamin D-dependent rickets type I. Endocr J. 2001;48:427–32. doi: 10.1507/endocrj.48.427. [DOI] [PubMed] [Google Scholar]
- 37.Brooks MH, Bell NH, Love L, Stern PH, Orfei E, Queener SF, et al. Vitamin-D-dependent rickets type II. Resistance of target organs to 1,25-dihydroxyvitamin D. N Engl J Med. 1978;298:996–9. doi: 10.1056/NEJM197805042981804. [DOI] [PubMed] [Google Scholar]
- 38.Rut AR, Hewison M, Kristjansson K, Luisi B, Hughes MR, O›Riordan JL. Two mutations causing vitamin D resistant rickets: Modelling on the basis of steroid hormone receptor DNA-binding domain crystal structures. Clin Endocrinol (Oxf) 1994;41:581–90. doi: 10.1111/j.1365-2265.1994.tb01822.x. [DOI] [PubMed] [Google Scholar]
- 39.Malloy PJ, Eccleshall TR, Gross C, Van Maldergem L, Bouillon R, Feldman D. Hereditary vitamin D resistant rickets caused by a novel mutation in the vitamin D receptor that results in decreased affinity for hormone and cellular hyporesponsiveness. J Clin Invest. 1997;99:297–304. doi: 10.1172/JCI119158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hewison M, Rut AR, Kristjansson K, Walker RE, Dillon MJ, Hughes MR, et al. Tissue resistance to 1,25-dihydroxyvitamin D without a mutation of the vitamin D receptor gene. Clin Endocrinol (Oxf) 1993;39:663–70. doi: 10.1111/j.1365-2265.1993.tb02424.x. [DOI] [PubMed] [Google Scholar]
- 41.Li YC, Pirro AE, Amling M, Delling G, Baron R, Bronson R, et al. Targeted ablation of the vitamin D receptor: An animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci U S A. 1997;94:9831–5. doi: 10.1073/pnas.94.18.9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakai Y, Kishimoto J, Demay MB. Metabolic and cellular analysis of alopecia in vitamin D receptor knockout mice. J Clin Invest. 2001;107:961–6. doi: 10.1172/JCI11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen CH, Sakai Y, Demay MB. Targeting expression of the human vitamin D receptor to the keratinocytes of vitamin D receptor null mice prevents alopecia. Endocrinology. 2001;142:5386–9. doi: 10.1210/endo.142.12.8650. [DOI] [PubMed] [Google Scholar]
- 44.Malloy PJ, Wang J, Srivastava T, Feldman D. Hereditary 1,25-dihydroxyvitamin D-resistant rickets with alopecia resulting from a novel missense mutation in the DNA-binding domain of the vitamin D receptor. Mol Genet Metab. 2010;99:72–9. doi: 10.1016/j.ymgme.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Forghani N, Lum C, Krishnan S, Wang J, Wilson DM, Blackett PR, et al. Two new unrelated cases of hereditary 1,25-dihydroxyvitamin D-resistant rickets with alopecia resulting from the same novel nonsense mutation in the vitamin D receptor gene. J Pediatr Endocrinol Metab. 2010;23:843–50. doi: 10.1515/jpem.2010.136. [DOI] [PubMed] [Google Scholar]
- 46.Widmer B, Gerhardt RE, Harrington JT, Cohen JJ. Serum electrolyte and acid base composition. The influence of graded degrees of chronic renal failure. Arch Intern Med. 1979;139:1099–102. [PubMed] [Google Scholar]
- 47.Sahay M, Sahay R. Rickets in tropics. Int J Endocrinol Metab. 2010;23:1–5. doi: 10.1515/jpem.2010.098. [DOI] [PubMed] [Google Scholar]
- 48.Whyte MP. Physiological role of alkaline phosphatase explored in hypophosphatasia. Ann N Y Acad Sci. 2010;1192:190–200. doi: 10.1111/j.1749-6632.2010.05387.x. [DOI] [PubMed] [Google Scholar]
- 49.Sahay M, Vali SP, Ramesh VD. The Case. A child with metabolic acidosis and growth retardation. Kidney Int. 2009;75:1121–2. doi: 10.1038/ki.2009.25. [DOI] [PubMed] [Google Scholar]