

Abstract
The population genetics and phylogenetic relationships of Culex mosquitoes inhabiting the Sonoran Desert region of North America were studied using mitochondrial DNA and microsatellite molecular markers. Phylogenetic analyses of mitochondrial cytochrome c oxidase subunit I (COI) from mosquitoes collected over a wide geographic area, including the Baja California peninsula, and mainland localities in southern Arizona, USA and Sonora, Mexico, showed several well-supported partitions corresponding to Cx. quinquefasciatus, Cx. tarsalis, and two unidentified species, Culex sp. 1 and sp. 2. Culex quinquefasciatus was found at all localities and was the most abundant species collected. Culex tarsalis was collected only at Tucson, Arizona and Guaymas, Sonora. The two unidentified species of Culex were most abundant at Navojoa in southern Sonora. Haplotype and nucleotide diversities in the COI gene segment were substantially lower in Cx. quinquefasciatus compared with the other three species. Analysis of molecular variance revealed little structure among seven populations of Cx. quinquefasciatus, whereas significant structure was found between the two populations of Cx. tarsalis. Evidence for an historical population expansion beginning in the Pleistocene was found for Cx. tarsalis. Possible explanations for the large differences in genetic diversity between Cx. quinquefasciatus and the other species of Culex are presented.
1. Introduction
The population structure, dispersal capabilities, and systematics of mosquitoes in the genus Culex (Culicidae: Culicinae: Culicini) from the Sonoran Desert of North America are poorly known. Several species reported from this region, including Cx. quinquefasciatus Say, a member of the Cx. pipiens Linnaeus complex, and Cx. tarsalis Coquillett, are important vectors of the West Nile and St. Louis encephalitis viruses that infect humans. Although presently not as serious of a health problem in Mexico as the dengue fever virus vectored by the introduced Aedes aegypti (Linnaeus), a single mortality from West Nile viral infection recorded in 2009 in the northern city of Monterrey, Nuevo León [1], and an infection reported from southern Sonora in which the patient later recovered [2], raises concern that there is a potential for this disease to emerge in northern Mexico and that it should be monitored more closely by health officials. Owing to the lack of a vaccine for the West Nile virus, vector control is the only tool presently available to combat this disease. Efficient monitoring of vector control measures, and inferring sources of reinvasion, depends on an understanding of the dispersal capability and genetic diversity of the mosquitoes, as well as on accurate taxonomic identifications. The genus Culex contains 768 described species, many of which (198) are grouped in the subgenus Culex, and important gaps still exist in our knowledge of their taxonomy and relationships [3, 4].
In the Cx. pipiens complex, the species-level taxa present in the New World are Cx. pipiens and Cx. quinquefasciatus which show an extensive zone of hybridization at mid latitudes in the USA [5–7]. Some authors, however, place Cx. quinquefasciatus as a subspecies of Cx. pipiens [8]. Here they are treated as separate species. Culex quinquefasciatus is widely distributed, found in southern USA, Mexico, Central America, and most of South America [5].
Culex quinquefasciatus and Cx. tarsalis have markedly different histories in the Sonoran Desert, as well as elsewhere in the New World, and a comparison of their population genetics is predicted to reveal patterns reflecting these differences. Culex tarsalis is native to North America, whereas both Cx. pipiens and Cx. quinquefasciatus are thought to have evolved in Africa [6]. Although the route and timing of the postulated arrival of the Cx. pipiens complex to the New World are controversial [8], if Cx. quinquefasciatus is a relatively recent arrival to the New World it may still retain the genetic signature of a founder event.
Our primary goal in the present study was to utilize mitochondrial DNA (mtDNA) sequences from a segment of the cytochrome c oxidase subunit I (COI) gene, known as the barcode segment [9], to examine population genetics of Culex mosquitoes collected from widely separated localities in the Sonoran Desert region, including the Baja California peninsula, mainland Sonora, Mexico and southeastern Arizona, USA, to test the prediction that Cx. quinquefasciatus and Cx. tarsalis will show evidence of different demographic histories. Because we found extremely low genetic variability in the COI gene in Cx. quinquefasciatus, we also used a small set of nuclear microsatellite loci to obtain preliminary estimates of population structure in this species. Also, because it is well known that taxonomic identifications based on morphological examination of females of certain species of Culex can be especially problematic [5, 10], COI barcodes, which can reliably distinguish many nominal species of Culex [11, 12], were used to estimate the overall biodiversity and phylogenetic relationships of Culex obtained in our survey.
2. Materials and Methods
2.1. Sampling
Adult female mosquitoes were collected at seven localities, including the states of Sonora (Hermosillo, Guaymas, Ciudad Obregón, and Navojoa) and Baja California Sur (Bahía Tortugas and Santa Rosalía) in Mexico and southeastern Arizona (Tucson) in the USA (Figure 1). Ciudad Obregón and Navojoa are located in a thornscrub biome south of the Sonoran Desert, but are included here as part of the Sonoran Desert region. Carbon dioxide traps (BioQuip, Rancho Dominguez, CA, USA) were placed at collection sites from 1700 until 0900 when mosquitoes were collected from the traps. Mosquitoes were provisionally identified by examination under a dissecting microscope and then either frozen in liquid nitrogen, placed on dry ice, or immediately used for DNA extractions.
Figure 1.
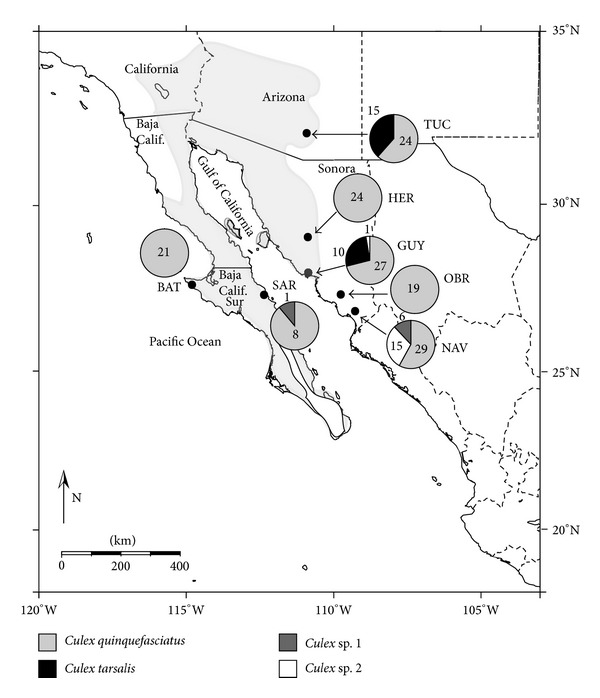
Map showing collection localities for Culex spp. in northwestern Mexico and southern Arizona, USA. Light gray shading on map shows approximate boundaries of the Sonoran Desert. Pie charts show numbers of individuals of each species analyzed from each locality. TUC: Tucson; HER: Hermosillo; GUY: Guaymas; OBR: Ciudad Obregón; NAV: Navojoa; SAR: Santa Rosalía; BAT: Bahía Tortugas.
2.2. Molecular Protocol and Sequence Analysis
Total genomic DNA was extracted from each mosquito using the DNeasy (QIAGEN Inc., Valencia, CA, USA) protocol. Samples not analyzed immediately were stored at −20°C. The polymerase chain reaction (PCR) was used to amplify a segment of the COI gene using the primer pair LCO1490f/HCO2198r and standard assay conditions [13]. Sequencing reactions were performed on an Applied Biosystems (Foster City, CA, USA) ABI 3730XL DNA sequencer at the Genomic Analysis and Technology Core Facility, University of Arizona, Tucson, USA, using the amplifying primers. Sequences were proofread and aligned in either Sequencher 4.1 (GeneCodes Corp., Ann Arbor, MI, USA) or ClustalX 1.81 [14] followed by manual editing. Sequences were trimmed to remove ambiguous sites, resulting in a final segment of 624 bp in 23 of the 25 Cx. tarsalis (see Table 1) and 611 bp in Cx. quinquefasciatus and Culex sp. 1 and sp. 2. The first nucleotide in the 624 bp segment of Cx. tarsalis corresponds to position no. 1527 in the complete mitochondrial genome of Drosophila yakuba (GenBank Accession no. NC001322). The first nucleotide position in Cx. quinquefasciatus and Culex sp. 1 and sp. 2 corresponds to position no. 1515 in D. yakuba. GenBank accession numbers for the new Culex COI sequences obtained here are JX297260–JX297304.
Table 1.
Summary of genetic diversity indices and results of neutrality tests (Tajima's D and Fu's F S) in the mitochondrial COI gene segment of Culex spp.
| Species | N | L | k | K | h (±SD) | π (±SD) | Tajima's D | Fu's F S |
|---|---|---|---|---|---|---|---|---|
| Cx. quinquefasciatus | 28 | 611 | 1 | 2 | 0.071 ± 0.065 | 0.00012 ± 0.00011 | −1.15 | −1.15* |
| Cx. tarsalis | 23** | 624 | 15 | 14 | 0.921 ± 0.042 | 0.00460 ± 0.00069 | −1.05 | −7.54* |
| Culex sp. 1 | 7 | 611 | 5 | 4 | 0.857 ± 0.102 | 0.00405 ± 0.00075 | 1.06 | 0.28 |
| Culex sp. 2 | 16 | 611 | 8 | 6 | 0.675 ± 0.117 | 0.00218 ± 0.00066 | −1.61 | –1.81 |
N: number of sequences; L: sequence length (bp); k: number of variable sites; K: number of haplotypes; h: haplotype diversity; π: nucleotide diversity; *significant at the 0.05 level; **two of the 25 sequences obtained for Cx. tarsalis (GUY1 and GUY3) contained 558 bp and were omitted.
With one exception, all individuals of Cx. quinquefasciatus possessed the same COI haplotype. To obtain a preliminary estimate of population structure in Cx. quinquefasciatus, therefore, we also analyzed four microsatellite loci (CQ16, CQ26, CQ29, and CQ41) as described by Fonseca et al. [15]. Most of the 134 specimens of Cx. quinquefasciatus analyzed for microsatellites were not the same as those used for COI analyses. Several individuals from Hermosillo (N = 6), Guaymas (N = 2), and Santa Rosalía (N = 2), however, were analyzed for both molecular markers. Genetic diversity for each locus in each of the seven populations (Figure 1), as well as over all loci and populations, was quantified using Microsatellite Analyser (MSA) version 4.00 [16] and ARLEQUIN version 3.5.1.3 [17]. Deviations from Hardy-Weinberg equilibrium (HWE) were tested for each locus and over all loci in ARLEQUIN using a Markov chain approximation [18]. All estimates were assessed for significance using a test analogous to Fisher's exact test, with 100,000 steps in the Markov chain and 5000 dememorization steps. Significance for all estimates was placed at the 0.05 level. Other details on the microsatellite protocol are given elsewhere [19].
Calculations of Kimura's [20] 2-parameter genetic distances (d) were carried out in MEGA version 5.0.5 [21]. Genetic diversity indices were calculated in DnaSP version 5.00.07 [22]. Neutrality tests (Tajima's D [23] and Fu's F S [24]) were carried out in ARLEQUIN. Fu's F S test is also useful for detecting signatures of population expansions, which lead to large negative values in the test statistic [24, 25]. The significance of F S at the 0.05 level is indicated when P values are <0.02 [17]. Networks for COI haplotypes were constructed using statistical parsimony implemented in TCS version 1.21 [26]. The connection limit among haplotypes was set to the default value of 95%, unless indicated otherwise.
2.3. Phylogenetic Analyses
Relationships among COI haplotypes in Sonoran Desert Culex were examined using maximum parsimony (MP) and Bayesian inference. For all phylogenetic analyses, sequences for Cx. tarsalis were trimmed from 624 to 611 bp to correspond to the sequence length of the other samples (Table 1). We also incorporated GenBank sequences for several different species of Culex into the data matrix, including Cx. (Neoculex) territans Walker and Cx. (Culiciomyia) nigropunctatus Edwards. All other Culex species treated here are presently assigned to the subgenus Culex [4]. Culiseta inornata (Williston) from the tribe Culisetini was used as the outgroup based on results of previous molecular studies of Culicidae [11, 27]. Maximum parsimony analyses were carried out in MEGA using the CNI heuristic search option and 100 random additions of sequences. Relative support for tree topology was obtained by bootstrapping [28] using 1000 pseudoreplicates. Bayesian analyses were implemented in MrBayes version 3.1 [29]. The model of nucleotide substitution that best fitted the data set, determined with jModelTest 0.1.1 [30] using the Akaike Information Criterion was, TVM + G. The substitution model was set to nst = “2” and rates = “gamma”, and the analysis was run for 1,000,000 generations, sampled every 250th generation (4,000 trees sampled), using the default random tree option to begin the analysis. We also conducted an analysis with nst = “6,” used for the more highly parameterized GTR substitution model, and obtained the same tree topology and similar clade support values. Clade support, expressed as posterior probabilities, was estimated utilizing a Markov chain Monte Carlo (MCMC) algorithm.
2.4. Population Structure and Historical Demography
Analysis of molecular variance (AMOVA) [31], performed in ARLEQUIN, was used to test for population structure among populations of Cx. quinquefasciatus and Cx. tarsalis. The significance of population pairwise comparisons of the fixation indices, ΦST for COI and F ST for microsatellites, was based on 10,000 permutations of the data matrix and assessed at α = 0.05 (Cx. tarsalis) or using a sequential Bonferroni correction [32] for multiple comparisons of Cx. quinquefasciatus. Estimates of the number of migrants per generation (N m) among populations were also calculated in ARLEQUIN.
The demographic history of Cx. tarsalis from the Sonoran Desert was inferred by performing three different tests of the sequence data. For all demographic tests, we chose a value of 2.3% pairwise sequence divergence per million years for COI [33]. This resulted in a neutral mutation rate per site per generation (μ) of 1.15 × 10−8 assuming a single generation per year (see Section 4). A mismatch distribution analysis [34, 35] of COI sequence data was performed in ARLEQUIN. The significance of the estimated parameters of the sudden expansion model of the mismatch distribution is obtained from the sum of square deviations (SSD) statistic and the raggedness statistic (rg) and their corresponding P values. The sudden expansion model is rejected at P < 0.05. A Bayesian skyline analysis, which provides an estimate of changes in effective population size through time utilizing MCMC sampling of sequence data, was conducted in BEAST version 1.3 [36]. Because the TVM substitution model is not available in BEAST, analyses were run using both the HKY + G and GTR + G substitution models (four gamma categories) for five million iterations sampled every 1000 iterations. Bayesian skyline plots generated with TRACER version 1.5 [36] were essentially identical in the two analyses. A maximum-likelihood estimate of the exponential population growth parameter (g) and the mutation parameter θ in Cx. tarsalis was obtained with the program FLUCTUATE version 1.4 [37] using the program settings described previously [38].
3. Results
3.1. Sequence Analysis
Culex COI sequences were translated in MEGA. No frameshifts or stop codons were found. Base composition showed little variation among sequences, with CG content averaging 31%. Together these results suggest that our sequences represent mtDNA and are not nuclear mitochondrial pseudogenes (numts) which have been reported for the COI gene in insects [39].
Genetic diversity indices and results of neutrality tests for COI are shown in Table 1. The very low haplotype (h) and nucleotide (π) diversities found in Cx. quinquefasciatus contrast markedly with the high values seen in Cx. tarsalis and the two unidentified species. Tajima's D was not significant in any of the Culex species. A relatively large and significant Fu's F S, however, was found in Cx. tarsalis.
3.2. Phylogenetic Relationships
Phylogenetic relationships among COI haplotypes from 76 individuals of Culex collected from six Sonoran Desert localities (mosquitoes from Ciudad Obregón were analyzed only for microsatellites) revealed four well-resolved clades (Figure 2). One clade found at all six localities (N = 28) clustered with the closely related Cx. pipiens and Cx. quinquefasciatus (Cx. pipiens complex). Of these 28 individuals, 27 possessed the same haplotype, which was identical to the corresponding 611 bp COI segment reported for Cx. pipiens pallens from Japan (GenBank Accession no. FN395206). Another clade found only at Tucson and Guaymas comprised Cx. tarsalis (N = 25). The remaining two clades, found mainly at Navojoa and provisionally assigned to Culex sp. 1 and sp. 2 (Figure 1), did not cluster with any of the available GenBank sequences and remained unidentified (see Section 4). Culex nigripalpus from the Dominican Republic (GenBank Accession no. JX259910), however, resolved in a basal position to the two unidentified Culex and was closely related to them (d = 2.2 − 3.9%), supporting their assignment to the genus Culex. Mean genetic distance between Culex sp. 1 and sp. 2 was d = 2.1%. Culex sp. 1 and sp. 2 also differed from all other species of Culex analyzed here by showing a nonsynonymous first codon nucleotide substitution (G to A) at position 68 of the COI gene segment which resulted in an alanine to threonine substitution in the COI protein.
Figure 2.
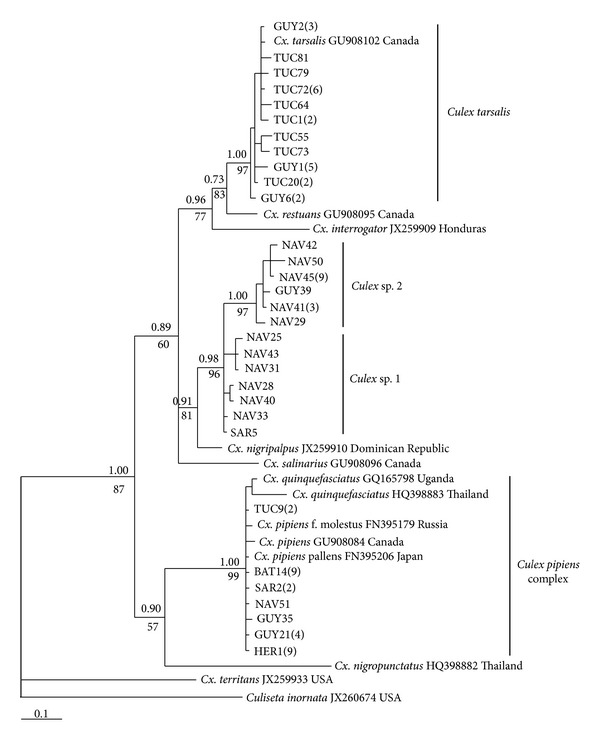
Bayesian 50% majority rule consensus tree based on COI sequences (611 bp) showing relationships among haplotypes of Culex spp. from the Sonoran Desert region and GenBank sequences from several recognized species worldwide. Clade support expressed as posterior probabilities is shown above the branches. Bootstrap support values for the maximum parsimony (MP) tree (length = 235; CI = 0.698; RI = 0.677; 154 variable sites; 90 parsimony informative sites) are shown below the branches. Branch terminals are labeled with locality abbreviation and sample identification number (see Figure 1). The total number of identical haplotypes from each region is shown in parentheses following the listed haplotype. The scale shows substitutions per site.
The TCS analyses showed that COI haplotypes for Cx. tarsalis and the two unidentified Culex species resolved in separate networks at the 95% connection limit (Figure 3), consistent with the presence of at least two species-level taxa. The clustering of haplotypes for Culex sp. 1 and sp. 2 in a network separate from Cx. tarsalis indicates that the unidentified species are closely related, although separated by nine mutational steps. When the connection limit was increased from 95 to 97% in the TCS analysis, Culex sp. 1 and sp. 2 formed separate networks (not shown) supporting the view that they represent separate species.
Figure 3.
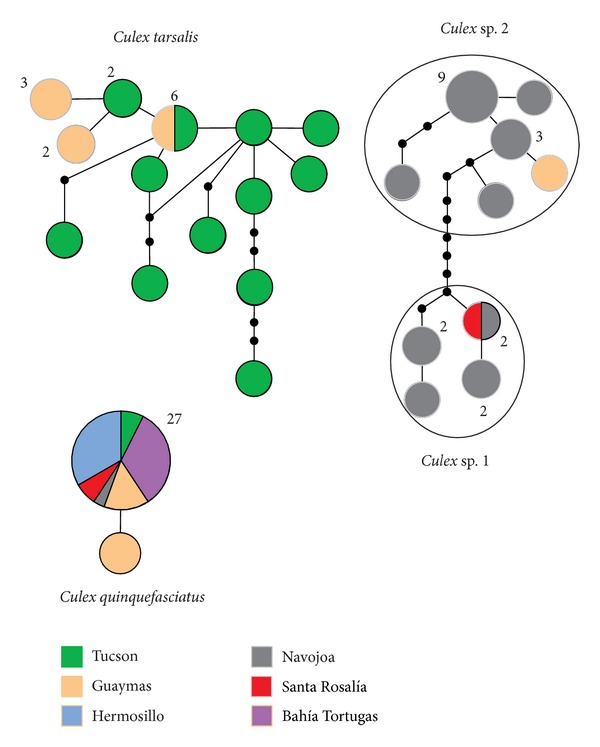
TCS haplotype network for the COI gene segment in Culex tarsalis (N = 23; see Table 1), Cx. quinquefasciatus (N = 28), Culex sp. 1 (N = 7), and Culex sp. 2 (N = 16) based on a 95% connection limit. Each line segment represents a single mutation. Inferred intermediate haplotypes that were not sampled are shown as small black dots on the line segments. Size of the circles is proportional to haplotype frequency. Numbers next to the circles represent number of individuals with that haplotype, if greater than one. Geographical localities are color-coded.
Of the fourteen haplotypes seen in Cx. tarsalis (Table 1), ten were singletons, all of which were found at Tucson (Figure 3). The abundance of singleton haplotypes suggests an expanding population, consistent with results from the demographic tests (see Section 3.4). Although the common haplotype in Cx. tarsalis was present at both Guaymas and Tucson, the geographic partitioning shown in Figure 3 also is consistent with the results from the AMOVA showing significant structure between Guaymas and Tucson populations (see Section 3.3).
3.3. Population Structure
A summary of results obtained for the four microsatellite loci in Cx. quinquefasciatus averaged over the seven Sonoran Desert populations is shown in Table 2. All four loci showed significant deviations (P < 0.01) from HWE. In all cases we found H obs< H exp indicating an excess of homozygotes. When each population was analyzed separately, however, no significant deviations from HWE were found at the CQ26 locus, and most populations showed no significant deviations at the CQ16 and CQ29 loci (not shown). For the CQ41 locus, HWE was found only in populations at Ciudad Obregón, Santa Rosalía, and Tucson. An excess of homozygotes has also been reported in other microsatellite studies of the Cx. pipiens complex and has been attributed to the presence of null alleles [40] or the Wahlund effect [7].
Table 2.
Summary information of the four microsatellite loci averaged over the seven populations of Culex quinquefasciatus. The number of individuals genotyped (N), observed and expected heterozygosities (H obs and H exp), fragment size range (bp), and number of alleles are shown for each locus.
| Locus | N | H obs | H exp | Size (bp) | No. of alleles |
|---|---|---|---|---|---|
| CQ16 | 101 | 0.77228* | 0.86813 | 210–260 | 17 |
| CQ26 | 132 | 0.62879* | 0.68879 | 208–220 | 7 |
| CQ29 | 131 | 0.30534* | 0.41628 | 168–180 | 5 |
| CQ41 | 133 | 0.51128* | 0.68035 | 136–154 | 8 |
*Significant deviation (P < 0.01) from Hardy-Weinberg equilibrium (HWE).
AMOVA of the microsatellite data set for Cx. quinquefasciatus from seven localities revealed that only five of the 21 pairwise comparisons of F ST were significant after Bonferroni correction for multiple comparisons (Table 3).
Table 3.
Pairwise comparisons of F ST for populations of Culex quinquefasciatus from the Sonoran Desert region based on analyses of four microsatellite loci. Sample sizes from each locality are shown in parentheses. Locality abbreviations are given in the legend of Figure 1.
| TUC | HER | GUY | OBR | NAV | SAR | BAT | |
|---|---|---|---|---|---|---|---|
| (22) | (21) | (24) | (19) | (28) | (8) | (12) | |
| TUC | — | ||||||
| HER | 0.007 | — | |||||
| GUY | 0.042* | −0.000 | — | ||||
| OBR | 0.006 | 0.011 | 0.074* | — | |||
| NAV | 0.029 | −0.003 | 0.022 | 0.015 | — | ||
| SAR | 0.019 | −0.015 | −0.059 | 0.017 | −0.033 | — | |
| BAT | 0.068* | 0.064 | 0.047 | 0.126* | 0.099* | 0.017 | — |
*Statistically significant values using a Bonferroni correction (P < 0.003).
The AMOVA of the COI data set in Cx. tarsalis collected from Tucson (N = 15) and Guaymas (N = 8) showed that 85% of the genetic variation was found within populations, but significant structure was found between populations from the two localities (ΦST = 0.15; P = 0.007). The estimated number of migrants per generation (N m) between Tucson and Guaymas, however, was 2.8. Therefore, for demographic analyses (Section 3.4), the two populations were combined.
3.4. Historical Demography
FLUCTUATE showed that the population growth parameter (g ± 95% confidence interval) in Cx. tarsalis, expressed in units of 1/μ generations, was positive and significantly different from zero (g = 664 ± 165), consistent with population growth. The maximum-likelihood estimate for the mutation parameter θ was 0.027363 ± 0.00773. Effective female population size (N ef) in Cx. tarsalis, estimated using the equation θ = 2N ef μ, was 1.19 × 106.
The mismatch distribution of COI sequences in Cx. tarsalis is shown in Figure 4. The observed distribution of pairwise differences among haplotypes showed relatively good agreement with the expected unimodal distribution for a population that has undergone an expansion [35]. The test statistics SSD (0.0092; P = 0.37) and rg (0.047; P = 0.44) were small and not statistically significant, indicating that the sudden expansion model could not be rejected. The value found for τ, the time to the population expansion, where τ = 2ut, u is the mutation rate for the entire gene segment, and t is the number of generations since the expansion [34], was 3.029 (95% confidence intervals: 1.023, 4.896). Assuming 2.3% pairwise divergence per million years in the COI gene in insects [33], the mean mutation rate per site per generation in the 624 bp segment for a single lineage is (624) × (1.15 × 10−8) or 7.176 × 10−6. Based on these values, the estimated time to the population expansion in Cx. tarsalis (with 95% confidence intervals) was 211,050 (71,279–341,140) generations ago.
Figure 4.
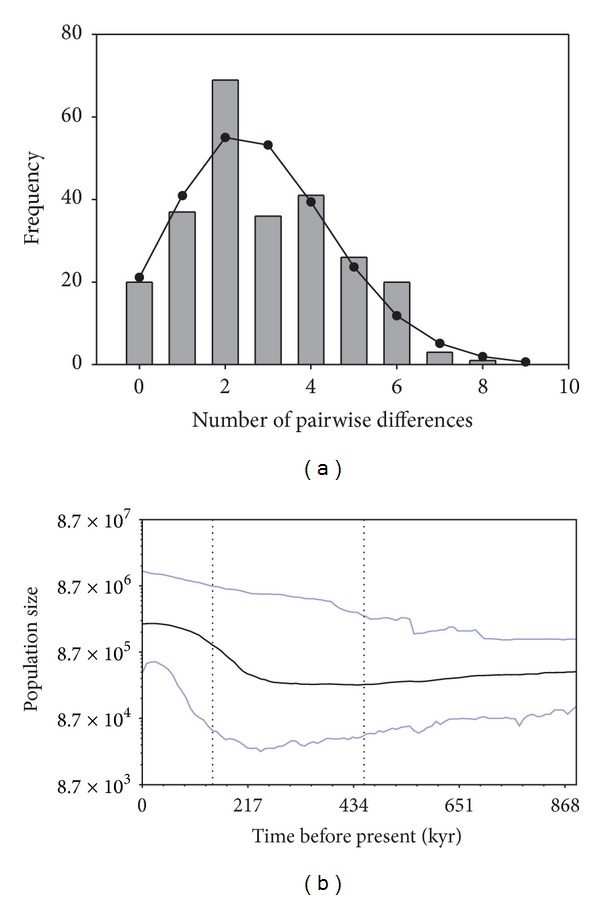
Demographic history of Culex tarsalis from the Sonoran Desert inferred from the mismatch distribution (a) and Bayesian skyline analysis (b). Vertical bars of the mismatch distribution show the observed distribution of pairwise differences among COI haplotypes, with the solid line representing the expected distribution under the sudden expansion model. The Bayesian skyline plot shows the estimated changes in effective female population size (N ef) over time given on a logarithmic scale. The solid lines represent the median estimates of population size (middle line); upper and lower lines show the 95% highest posterior density (HPD) intervals. The vertical dotted lines represent the median estimate (right) and lower 95% HPD (left) of time to the most recent common ancestor.
Bayesian skyline analysis (Figure 4) showed that Cx. tarsalis showed a clear signature of an historical population expansion, consistent with the results from FLUCTUATE and the mismatch distribution. Given the untested assumptions of a neutral mutation rate per site per generation (μ) of 1.15 × 10−8 and a single generation per year, the timing of the expansion shown in the Bayesian skyline plot is only a rough approximation. Nonetheless, the mismatch distribution and Bayesian skyline plot both suggest that the expansion began approximately 200,000 generations ago, which places it within the timeframe of the Pleistocene, unless improbable estimates of μ and generation time are assumed.
4. Discussion
4.1. Genetic Diversity
A major finding of this study was that genetic diversity in the COI gene segment of Cx. quinquefasciatus from the Sonoran Desert was much lower than that seen in Cx. tarsalis and Culex sp. 1 and sp. 2 (Table 1). One possible explanation for this difference is that Cx. quinquefasciatus has preferentially undergone repeated cycles of population fluctuations, resulting in a much lower genetic diversity, owing to vector control measures in urban areas in northwestern Mexico which are primarily aimed at controlling Ae. aegypti and the dengue virus. Subtle ecological differences in microhabitat preferences that result in less exposure to insecticides might explain why the Sonoran Desert Cx. tarsalis and Culex sp. 1 and sp. 2 maintain a relatively high genetic diversity. These three species, or putative species, show diversity indices similar to native dipterans from the Sonoran Desert region, including the cactophilic Drosophila (with the exception of D. nigrospiracula) and Odontoloxozus longicornis and O. pachycericola [38, 41–43]. Ecological studies conducted in California, USA, have shown that both Cx. quinquefasciatus and Cx. tarsalis are most abundant in riparian habitats, but that Cx. quinquefasciatus shows higher relative abundance than Cx. tarsalis in residential habitats [44]. We also noted that Cx. quinquefasciatus was typically the most abundant mosquito species in our urban collections, consistent with the observations of Reisen et al. [44].
Another possible explanation for the large differences in genetic diversity among Sonoran Desert Culex., which is not mutually exclusive of the above hypothesis, may be related to differences in population histories. Culex tarsalis is native to North America, whereas both Cx. quinquefasciatus and Cx. pipiens probably evolved in Africa [6]. Ross [45] hypothesized that Cx. quinquefasciatus was introduced from Africa via the slave trade within the last few centuries. This hypothesis, however, has been challenged [5]. Regardless of the dispersal route, if Cx. quinquefasciatus is a relatively recent arrival to the New World [8], it is possible that it would still retain the genetic signature of a founder event (i.e., reduced genetic variability) compared with the indigenous Cx. tarsalis.
A recent study reported the presence of Cx. pipiens, and hybrids between Cx. pipiens and Cx. quinquefasciatus, in Mexico City [46]. Given the close association of both species with humans, together with their potential for dispersal via commercial air traffic, clear patterns of global distributions of the two species and their hybrids [5] may become progressively obscured. Figure 2 shows that COI barcode sequences are unable to distinguish between the two species. Although a possibility thus exists that our samples are Cx. pipiens, or hybrids of Cx. pipiens and Cx. quinquefasciatus, a more thorough examination of individuals of the Cx. pipiens complex from this region with specific molecular markers that reliably separate the two cryptic species [5, 8, 10] will be required before this can be resolved.
Females of several species of Culex from North America, including Cx. restuans Theobald, Cx. nigripalpus Theobald, and Cx. salinarius Coquillett, are often indistinguishable from those of the Cx. pipiens complex and can easily be confused [5, 10]. Barcode sequences, however, have been shown to be useful for separating and identifying species of Culicidae, with interspecific K2P divergences of COI generally showing values ≥2% [11, 12, 47]. Based on the 2% cutoff, we provisionally assigned the unidentified lineages to Culex sp. 1 and sp. 2 (mean d = 2.1%), but further molecular and morphological studies will be required to confirm their identity. We have shown, however, that Culex sp. 1 and sp. 2, which were initially identified as Cx. quinquefasciatus based on morphological examination, are very closely related to Cx. nigripalpus from the Dominican Republic (Figure 2) and only more distantly related to the other Culex species shown in Figure 2. When we compared sequences of Culex sp. 1 and sp. 2 with shorter COI barcode segments (478 bp) of Culex from Brazil [48], they also did not cluster with Cx. bidens Dyar, Cx. corniger Theobald, Cx. coronator Dyar & Knab, or Cx. nigripalpus, species that have been recorded for the states of Sonora and/or Sinaloa [49].
4.2. Population Structure
Field studies suggest that both Cx. quinquefasciatus and Cx. tarsalis show relatively high dispersal capability [44]. The AMOVA results of preliminary microsatellite data on Cx. quinquefasciatus from seven widely separated Sonoran Desert localities, including localities separated by the Gulf of California (Figure 1), showed that most of the pairwise comparisons of F ST were not significant, consistent with a pattern of high gene flow. None of the pairwise comparisons of F ST between the peninsular locality at Santa Rosalía and five mainland localities were significant. The most isolated locality at Bahía Tortugas on the Pacific coast of the peninsula also showed a lack of structure between both Guaymas and Hermosillo, although the pairwise comparisons with the other three mainland localities were significant. These results are in contrast to the findings from microsatellite studies on the closely related Cx. pipiens in Colorado in which significant structure was found among populations within the state [7]. Our results, however, are similar to those obtained from microsatellite studies on Cx. pipiens populations from several states in northeastern USA in which most pairwise comparisons among populations were not significant [40]. Our findings are also consistent with results from several species of native cactophilic dipterans which show little or no structure within mainland and peninsular populations, and, in the case of Drosophila nigrospiracula and D. mettleri, no apparent structure between peninsular and mainland populations [50].
Because we were unable to distinguish Culex sp. 1 and sp. 2 from Cx. quinquefasciatus using morphological characters, we cannot rule out the possibility that individuals of these two unidentified putative species were present in our sample assigned to Cx. quinquefasciatus from Navojoa used for microsatellite analysis. None of the Navojoa DNA samples identified as Cx. quinquefasciatus and analyzed for microsatellites were sequenced for COI to confirm their identity. As mentioned earlier, DNA extracted from six individuals of Cx. quinquefasciatus from Hermosillo were analyzed for both molecular markers. The observation that no significant population structure was found between Cx. quinquefasciatus from Hermosillo and each of the other populations (Table 3), and the observation that no individuals of Culex sp. 1 and sp. 2 were found at Hermosillo, suggests that few, if any, of the individuals in our microsatellite sample from Navojoa contained Culex sp. 1 and sp. 2.
Previous studies have examined the population genetic structure of Cx. tarsalis in western USA utilizing microsatellite markers [51, 52]. These studies have revealed a pattern of little population structure across broad areas at both the state (Colorado) and regional levels, consistent with high dispersal capability, although evidence of restriction of gene flow related to major geographic barriers (e.g., Continental Divide, Mogollon Rim, and the transition between Sonoran and Mojave Deserts) was evident. In particular, Venkatesan and Rasgon [52] found three separate population clusters of Cx. tarsalis in western USA. One of these clusters, the Sonoran cluster, occurred in southern Arizona and southeastern California, and included our sampling site at Tucson. Although we found significant structure between Guaymas and Tucson, a distance of approximately 500 km, the estimated number of migrants per generation among the two localities (N m = 2.8) suggests some gene flow. Larger sample sizes of Cx. tarsalis from the Sonoran Desert are needed, but our preliminary results based on COI are consistent with the microsatellite data in suggesting that some restrictions to gene flow may also occur in this region.
4.3. Demographic History
The large and significant negative value for Fu's F S seen in Cx. tarsalis from the Sonoran Desert (Table 1) suggested an historical population expansion. This conclusion was supported by results from FLUCTUATE, the mismatch distribution, and Bayesian skyline analysis. The mismatch distribution indicated that the population expansion began approximately 211,000 generations ago. Given the large confidence intervals surrounding the estimated number of generations since the expansion obtained from the mismatch distribution, and uncertainties in the generation time for Cx. tarsalis from the Sonoran Desert, it is impossible to arrive at a specific date for the expansion. A conservative estimate of 5–10 generations per year, however, would place the expansion at about 20,000–40,000 years ago during the late Pleistocene. This estimated timeframe is much more recent than that obtained by Venkatesan et al. [53] based on the ND4 gene in Cx. tarsalis, in which the expansion was dated to about 11,300,000 generations ago, or 375,000–560,000 years ago using their assumption of 20–30 generations per year. Although different genes, sample sizes, and assumptions were used in the two studies, a calculation error [54] is suspected in the ND4 study which probably contributed to the large discrepancy in estimated expansion dates.
Conflict of Interests
The authors declare that they have no conflict of interests.
Acknowledgments
The authors thank L. A. Hurtado, L. Matzkin, M. Polihronakis Richmond, and T. Watts for technical assistance. This work was supported by NSF Grants DEB-0075312 and OISE-0440648 to Therese A. Markow.
References
- 1.Rios-Ibarra C, Blitvich BJ, Farfan-Ale J, et al. Fatal human case of West Nile virus disease, Mexico, 2009. Emerging Infectious Diseases. 2010;16(4):741–743. doi: 10.3201/eid1604.091614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elizondo-Quiroga D, Davis CT, Fernandez-Salas I, et al. West nile virus isolation in human and mosquitoes, Mexico. Emerging Infectious Diseases. 2005;11(9):1449–1452. doi: 10.3201/eid1109.050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harbach RE. The Culicidae (Diptera): a review of taxonomy, classification and phylogeny. Zootaxa. 2007;1668:591–638. [Google Scholar]
- 4.Harbach RE. Culex classification, Mosquito taxonomic inventory. 2013. http://mosquito-taxonomic-inventory.info/
- 5.Farajollahi A, Fonseca DM, Kramer LD, Kilpatrick AM. ‘Bird biting’ mosquitoes and human disease: a review of the role of Culex pipiens complex mosquitoes in epidemiology. Infection, Genetics and Evolution. 2011;11(7):1577–1585. doi: 10.1016/j.meegid.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harbach RE. Culex pipiens: species versus species complex—taxonomic history and perspective. Journal of the American Mosquito Control Association. 2012;28(supplement 4):10–23. doi: 10.2987/8756-971X-28.4.10. [DOI] [PubMed] [Google Scholar]
- 7.Kothera L, Godsey MS, Jr., Doyle MS, Savage HM. Characterization of Culex pipiens complex (Diptera: Culicidae) populations in Colorado, USA using microsatellites. PLoS ONE. 2012;7(10) doi: 10.1371/journal.pone.0047602.e47602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang S, Molaei G, Andreadis TG. Reexamination of Culex pipiens hybridization zone in the eastern United States by ribosomal DNA-based single nucleotide polymorphism markers. American Journal of Tropical Medicine and Hygiene. 2011;85(3):434–441. doi: 10.4269/ajtmh.2011.10-0679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ratnasingham S, Hebert PDN. BOLD: the Barcode of life data system. Molecular Ecology Notes. 2007;7(3):355–364. doi: 10.1111/j.1471-8286.2007.01678.x. http://www.barcodinglife.org/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanogo YO, Kim C, Lampman R, Novak RJ. A real-time TaqMan polymerase chain reaction for the identification of Culex vectors of West Nile and Saint Louis encephalitis viruses in North America. American Journal of Tropical Medicine and Hygiene. 2007;77(1):58–66. [PubMed] [Google Scholar]
- 11.Cywinska A, Hunter FF, Hebert PDN. Identifying Canadian mosquito species through DNA barcodes. Medical and Veterinary Entomology. 2006;20(4):413–424. doi: 10.1111/j.1365-2915.2006.00653.x. [DOI] [PubMed] [Google Scholar]
- 12.Wang G, Li C, Guo X, et al. Identifying the main mosquito species in China based on DNA barcoding. PLoS ONE. 2012;7(10) doi: 10.1371/journal.pone.0047051.e47051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology. 1994;3(5):294–299. [PubMed] [Google Scholar]
- 14.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research. 1997;25(24):4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fonseca DM, Atkinson CT, Fleischer RC. Microsatellite primers for Culex pipiens quinquefasciatus, the vector of avian malaria in Hawaii. Molecular Ecology. 1998;7(11):1617–1619. [PubMed] [Google Scholar]
- 16.Dieringer D, Schlötterer C. Microsatellite analyser (MSA): a platform independent analysis tool for large microsatellite data sets. Molecular Ecology Notes. 2003;3(1):167–169. [Google Scholar]
- 17.Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources. 2010;10(3):564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 18.Guo S, Thompson EA. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics. 1992;48(2):361–372. [PubMed] [Google Scholar]
- 19.Pfeiler E, Erez T, Hurtado LA, Markow TA. Genetic differentiation and demographic history in Drosophila pachea from the Sonoran Desert. Hereditas. 2007;144(2):63–74. doi: 10.1111/j.2007.0018-0661.01997.x. [DOI] [PubMed] [Google Scholar]
- 20.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution. 1980;16(2):111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 21.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 23.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu Y. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147(2):915–925. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramos-Onsins SE, Rozas J. Statistical properties of new neutrality tests against population growth. Molecular Biology and Evolution. 2002;19(12):2092–2100. doi: 10.1093/oxfordjournals.molbev.a004034. [DOI] [PubMed] [Google Scholar]
- 26.Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Molecular Ecology. 2000;9(10):1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- 27.Reidenbach KR, Cook S, Bertone MA, Harbach RE, Wiegmann BM, Besansky NJ. Phylogenetic analysis and temporal diversification of mosquitoes (Diptera: Culicidae) based on nuclear genes and morphology. BMC Evolutionary Biology. 2009;9(1, article 298) doi: 10.1186/1471-2148-9-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 29.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17(8):754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 30.Posada D. jModelTest: phylogenetic model averaging. Molecular Biology and Evolution. 2008;25(7):1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- 31.Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131(2):479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rice WR. Analyzing tables of statistical tests. Evolution. 1989;43:223–225. doi: 10.1111/j.1558-5646.1989.tb04220.x. [DOI] [PubMed] [Google Scholar]
- 33.Brower AVZ. Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(14):6491–6495. doi: 10.1073/pnas.91.14.6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rogers AR, Harpending H. Population growth makes waves in the distribution of pairwise genetic differences. Molecular Biology and Evolution. 1992;9(3):552–569. doi: 10.1093/oxfordjournals.molbev.a040727. [DOI] [PubMed] [Google Scholar]
- 35.Harpending HC. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Human Biology. 1994;66(4):591–600. [PubMed] [Google Scholar]
- 36.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Molecular Biology and Evolution. 2005;22(5):1185–1192. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- 37.Kuhner MK, Yamato J, Felsenstein J. Maximum likelihood estimation of population growth rates based on the coalescent. Genetics. 1998;149(1):429–434. doi: 10.1093/genetics/149.1.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pfeiler E, Bitler BG, Castrezana S, Matzkin LM, Markow TA. Genetic diversification and demographic history of the cactophilic pseudoscorpion Dinocheirus arizonensis from the Sonoran Desert. Molecular Phylogenetics and Evolution. 2009;52(1):133–141. doi: 10.1016/j.ympev.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song H, Buhay JE, Whiting MF, Crandall KA. Many species in one: DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(36):13486–13491. doi: 10.1073/pnas.0803076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang S, Molaei G, Andreadis TG. Genetic insights into the population structure of Culex pipiens (Diptera: Culicidae) in the northeastern United States by using microsatellite analysis. American Journal of Tropical Medicine and Hygiene. 2008;79(4):518–527. [PubMed] [Google Scholar]
- 41.Hurtado LA, Erez T, Castrezana S, Markow TA. Contrasting population genetic patterns and evolutionary histories among sympatric Sonoran Desert cactophilic Drosophila . Molecular Ecology. 2004;13(6):1365–1375. doi: 10.1111/j.1365-294X.2004.02169.x. [DOI] [PubMed] [Google Scholar]
- 42.Reed LK, Nyboer M, Markow TA. Evolutionary relationships of Drosophila mojavensis geographic host races and their sister species Drosophila arizonae . Molecular Ecology. 2007;16(5):1007–1022. doi: 10.1111/j.1365-294X.2006.02941.x. [DOI] [PubMed] [Google Scholar]
- 43.Pfeiler E, Richmond MP, Riesgo-Escovar JR, Tellez-Garcia AA, Johnson S, Markow TA. Genetic differentiation, speciation and phylogeography of cactus flies (Diptera: Neriidae: Odontoloxozus) from Mexico and southwestern USA. Biological Journal of the Linnean Society. 2013;110(2):245–256. [Google Scholar]
- 44.Reisen WK, Milby MM, Meyer RP. Population dynamics of adult Culex mosquitoes (Diptera: Culicidae) along the Kern River, Kern County, California, in 1990. Journal of Medical Entomology. 1992;29(3):531–543. doi: 10.1093/jmedent/29.3.531. [DOI] [PubMed] [Google Scholar]
- 45.Ross HH. The colonization of temperate North America by mosquitoes and man. Mosquito News. 1964;24:103–118. [Google Scholar]
- 46.Diaz-Badillo A, Bolling BG, Perez-Ramirez G, et al. The distribution of potential West Nile virus vectors, Culex pipiens pipiens and Culex pipiens quinquefasciatus (Diptera: Culicidae), in Mexico City. Parasites and Vectors. 2011;4(1, article 70) doi: 10.1186/1756-3305-4-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ruiz-Lopez F, Wilkerson RC, Conn JE, et al. DNA barcoding reveals both known and novel taxa in the Albitarsis Group (Anopheles: Nyssorhynchus) of Neotropical malaria vectors. Parasites & Vectors. 2012;5, article 44 doi: 10.1186/1756-3305-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Demari-Silva B, Vesgueiro FT, Sallum MAM, Marrelli MT. Taxonomic and Phylogenetic relationships between species of the genus Culex (Diptera: Culicidae) from Brazil inferred from the cytochrome c oxidase I mitochondrial gene. Journal of Medical Entomology. 2011;48(2):272–279. doi: 10.1603/me09293. [DOI] [PubMed] [Google Scholar]
- 49.Vargas L. Especies y distribución de mosquitos mexicanos no anofelinos (Insecta Diptera) Revista del Instituto de Salubridad y Enfermedades Tropicales. 1956;16(1):19–36. [PubMed] [Google Scholar]
- 50.Pfeiler E, Markow TA. Phylogeography of the cactophilic Drosophila and other arthropods associated with cactus necroses in the Sonoran Desert. Insects. 2011;2:218–231. doi: 10.3390/insects2020218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barker CM, Bolling BG, Black WC, IV, Moore CG, Eisen L. Mosquitoes and West Nile virus along a river corridor from prairie to montane habitats in Eastern Colorado. Journal of Vector Ecology. 2009;34(2):276–293. doi: 10.1111/j.1948-7134.2009.00036.x. [DOI] [PubMed] [Google Scholar]
- 52.Venkatesan M, Rasgon JL. Population genetic data suggest a role for mosquito-mediated dispersal of West Nile virus across the western United States. Molecular Ecology. 2010;19(8):1573–1584. doi: 10.1111/j.1365-294X.2010.04577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Venkatesan M, Westbrook CJ, Hauer MC, Rasgon JL. Evidence for a population expansion in the West Nile virus vector Culex tarsalis . Molecular Biology and Evolution. 2007;24(5):1208–1218. doi: 10.1093/molbev/msm040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schenekar T, Weiss S. High rate of calculation errors in mismatch distribution analysis results in numerous false inferences of biological importance. Heredity. 2011;107(6):511–512. doi: 10.1038/hdy.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]