

Abstract
OBJECTIVE
Ketosis-prone diabetes (KPD) is characterized by diabetic ketoacidosis (DKA) in patients lacking typical features of type 1 diabetes. A validated classification scheme for KPD includes two autoantibody-negative (“A−”) phenotypic forms: “A−β−” (lean, early onset, lacking β-cell functional reserve) and “A−β+” (obese, late onset, with substantial β-cell functional reserve after the index episode of DKA). Recent longitudinal analysis of a large KPD cohort revealed that the A−β+ phenotype includes two distinct subtypes distinguished by the index DKA episode having a defined precipitant (“provoked,” with progressive β-cell function loss over time) or no precipitant (“unprovoked,” with sustained β-cell functional reserve). These three A− KPD subtypes are characterized by absence of humoral islet autoimmune markers, but a role for cellular islet autoimmunity is unknown.
RESEARCH DESIGN AND METHODS
Islet-specific T-cell responses and the percentage of proinflammatory (CD14+CD16+) blood monocytes were measured in A−β− (n = 7), provoked A−β+ (n = 15), and unprovoked A−β+ (n = 13) KPD patients. Genotyping was performed for type 1 diabetes–associated HLA class II alleles.
RESULTS
Provoked A−β+ and A−β− KPD patients manifested stronger islet-specific T-cell responses (P < 0.03) and higher percentages of proinflammatory CD14+CD16+ monocytes (P < 0.01) than unprovoked A−β+ KPD patients. A significant relationship between type 1 diabetes HLA class II protective alleles and negative T-cell responses was observed.
CONCLUSIONS
Provoked A−β+ KPD and A−β− KPD are associated with a high frequency of cellular islet autoimmunity and proinflammatory monocyte populations. In contrast, unprovoked A−β+ KPD lacks both humoral and cellular islet autoimmunity.
Ketosis-prone diabetes (KPD), characterized by presentation with diabetic ketoacidosis (DKA) in patients lacking the typical features of autoimmune type 1 diabetes, is a heterogeneous syndrome (1,2). A validated classification scheme for KPD, based on the presence or absence of β-cell autoantibodies (“A+” or “A−”) and presence or absence of β-cell functional reserve (“β+” or “β−”) (3) includes two autoantibody-negative A− phenotypic forms: “A−β−” (lean, early onset, lacking β-cell functional reserve) and “A−β+” (obese, late onset, with substantial β-cell functional reserve after the index episode of DKA). Long-term longitudinal follow-up of a large cohort of A−β+ KPD patients has revealed that this phenotype comprises two distinct subtypes distinguished by whether the index DKA episode had a defined precipitant (“provoked” A−β+) or no precipitant (“unprovoked” A−β+) (4). Provoked A−β+ KPD patients have progressive loss of β-cell function after initial recovery from the DKA episode, with relapse to insulin dependence, no sex predominance, and an increased frequency of the HLA class II alleles DQB1*0302 and DRB1*04 associated with susceptibility to autoimmune type 1 diabetes. In contrast, unprovoked A−β+ KPD patients have sustained preservation of β-cell function after recovery from the DKA episode, prolonged insulin independence, male predominance, and an increased frequency of the protective allele DQB1*0602 (4). The unique clinical features and natural histories of these two subtypes of A−β+ KPD patients suggest distinctive underlying pathophysiologic processes for each. Although an underlying “occult” autoimmune element is suggested in the provoked A−β+ subtype by progressive β-cell loss and the presence of type 1 diabetes–associated HLA susceptibility alleles, the unprovoked A−β+ subtype could represent a truly nonautoimmune syndrome of KPD.
We have previously shown that the T cells of a significant proportion of individuals with an apparent phenotype of type 2 diabetes react strongly to islet antigens, despite lacking β-cell autoantibodies, and that this reactivity is associated with low C-peptide levels, indicating underlying cellular immune damage to β-cells (5,6). This finding expands the range of diabetic phenotypes—including those labeled as having “type 2” diabetes—with a potential pathophysiologic basis in islet autoimmunity. In the current study, we extended these findings to the unique, emerging forms of A− KPD. Specifically, we hypothesized that differences in cellular immune responses might distinguish the three A− KPD subtypes (A−β−, unprovoked A−β+, and provoked A−β+) with regard to a cellular autoimmune etiology. To test this hypothesis, we measured islet-specific T-cell responses using the validated cellular immunoblotting assay and islet autoantibody responses to determine the presence of islet autoimmunity in patients carefully phenotyped for these three KPD subtypes. We further assessed the percentage of proinflammatory (CD14+CD16+) monocytes in the peripheral blood of the three KPD subtypes.
In healthy subjects, 90–95% of classical monocytes express high levels of the cell surface marker CD14 (CD14+), without expression of CD16 (CD16−). Inflammation and infection are associated with the emergence of a distinct monocyte population characterized by coexpression of CD14 and CD16 (CD14+CD16+). CD14+CD16+ monocytes secrete high levels of proinflammatory tumor necrosis factor-α and low levels of anti-inflammatory interleukin-10, leading to their designation as proinflammatory monocytes (7).
Our results demonstrate that 1) provoked A−β+ KPD, associated with progressive loss of β-cell function, is associated with a high frequency of cellular islet autoimmunity and proinflammatory monocyte populations; 2) unprovoked A− KPD is a distinct syndrome of reversible β-cell dysfunction lacking evidence of humoral or cellular islet autoimmunity; and 3) a substantial proportion of A−β− KPD patients, who resemble patients with type 1 diabetes but lack autoantibodies, have evidence of cellular islet autoimmunity.
RESEARCH DESIGN AND METHODS
Subjects
The protocol was approved by the institutional review boards for human studies of Baylor College of Medicine, the Harris County Hospital District, and the University of Washington. All subjects enrolled in the study gave informed written consent.
All KPD patients were identified by acute presentation with an episode of DKA, defined by the presence of all of the following: anion gap ≥15, blood pH <7.30, serum bicarbonate ≤17 mmol/L, serum glucose >200 mg/dL, serum ketones ≥5.2 mmol/L, and urine ketones >13.9 mmol/L. The A− KPD patients were identified by the absence of GAD65, islet antigen-2, or ZnT8 autoantibodies, measured in sera by quantitative radioligand binding assays with recent modifications (8,9). As previously described (1), KPD patients are classified as A− if the autoantibody index for all three autoantibodies are below the ethnic-specific 98th percentile. Patients were classified as β+ or β− based on the presence or absence of β-cell functional reserve, measured by fasting serum C-peptide concentration and C-peptide response to glucagon 1–4 weeks after resolution of ketoacidosis. Cutoffs defining β-cell functional reserve were as previously established and longitudinally validated using receiver-operator characteristic curve analysis (1); briefly, β+ status is defined by fasting C-peptide >1 ng/mL and peak C-peptide after glucagon stimulation >1.5 ng/mL.
Provoked A−β+ KPD patients were defined by the presence of a clinically defined precipitating factor associated with or immediately preceding the index DKA episode (4). We have previously shown that the phenotype of provoked A−β+ KPD is characterized by progressive loss of β-cell function after recovery from the index DKA episode, no sex predominance, inability to discontinue insulin or relapsing to insulin requirement after a brief period off insulin, and an increased frequency of type 1 diabetes-associated HLA class II alleles DQB1*0302 and DRB1*04 (4). Unprovoked A−β+ KPD patients were defined by the absence of a clinically defined precipitating factor associated with or immediately preceding the index DKA episode. We have previously shown that the phenotype of unprovoked A−β+ KPD is characterized by marked improvement and sustained preservation of β-cell function after recovery from the index DKA episode, male predominance, insulin independence, and an increased frequency of the protective allele DQB1*0602 (4). A−β− KPD patients were defined exclusively by A and β status, without regard to DKA precipitant, because the latter does not affect the natural history or phenotypic characteristics of this KPD subgroup.
Blood was drawn for autoantibody, phenotyping, and T-cell responses during routine follow-up visits of the subjects to the KPD clinic at Ben Taub General Hospital. All subjects were clinically stable, without evidence of acute illness or inflammation at the time of the blood draw. Blinded whole-blood samples were transported, using a validated shipping protocol, overnight in a blinded fashion to the University of Washington, Seattle, WA, for T-cell analysis (10,11). Serum samples were shipped frozen to the University of Washington for measurement of islet autoantibodies.
Autoantibody assays
Radioligand binding assay.
GAD65, islet cell antigen (IA-2), and zinc transporter isoform 8 (ZnT8) autoantibodies were determined by radioligand binding assay, as previously described (8,9). Recombinant [35]S-antigen was produced in an in vitro coupled transcription and translation system with SP6 RNA polymerase and nuclease-treated rabbit reticulocyte lysate (Promega, Madison, WI). Sera (2.5 μL) were incubated with [35]S-antigen (25,000 of trichloroacetic acid–precipitable radioactivity). After an overnight incubation at 4°C, antibody-bound [35]S-antigen was separated from unbound antigen by precipitation with PAS (Invitrogen, Carlsbad, CA). The immunoprecipitated radioactivity was counted on a Wallac Microbeta Liquid Scintillation Counter (PerkinElmer Life and Analytical Sciences, Inc., Boston, MA). Antibody-positive control sera were included as a standard and used to express immunoglobulin-binding levels as a relative unit. Samples with a GAD65 antibody titer >65 units/mL were considered positive for GAD65-antibody. IA2-antibody positivity was established as exceeding the binding of the 98th percentile for healthy controls (30 units/mL). Autoantibody positivity for ZnT8R and ZnT8 W were set at 15 units/mL and 26 units/mL, respectively, based on the 98th percentile observed in 50 healthy human control sera. The autoantibody laboratory (C.S.H.) participated in the Diabetes Autoantibody Standardization Program (DASP) workshop (12); the GAD65 autoantibody assay showed a sensitivity of 86% and specificity of 93%, and the ICA512/IA-2 autoantibody assay showed a sensitivity of 66% and a specificity of 98%. ZnT8 autoantibodies were not included in the workshop.
T-cell assay
Cellular immunoblotting.
Cellular immunoblotting was performed on blinded freshly isolated peripheral blood mononuclear cells (PBMCs) to test for the presence of islet-reactive T cells, as previously described (13). Briefly, normal human islet cell preparations were subjected to preparative one-dimensional 10% SDS-PAGE and electroblotted onto nitrocellulose. The nitrocellulose was cut into molecular weight regions (blots) and then solubilized to form nitrocellulose particles. The nitrocellulose particles containing islet proteins were used to stimulate PBMCs, at a concentration of 3.5 × 105 PBMCs per well (5,6,10,11,13,14). Type 1 diabetic patients have been shown to respond to 4–18 molecular weight proteins and normal control subjects (without diabetes) to 0–3 molecular weight regions (14). Human pancreatic islets were obtained from the National Institutes of Health–supported Islet Cell Resource Centers (ICR-ABCC). The specificity of the T-cell responses from diabetic patients to islet proteins has been demonstrated previously (13). Cellular immunoblotting has been validated to have excellent specificity and sensitivity for distinguishing T-cell responses to islet proteins between T1D patients and control subjects (10,11,15). The specificity and sensitivity for the cellular immunoblotting assay was similar to those for the islet autoantibody assays performed in the individual workshops and exceeded the specificity and sensitivity of other T-cell assays validated at this time.
Phenotyping
PBMCs were stained with antibodies specific to human CD14 and human CD16 (BD Pharmingen, San Jose, CA). Data acquisition was performed using BD Cellquest 5.2.1 software on a BD FACSCalibur (BD Biosciences). Data analysis was performed using FlowJo 9.3.1 software (Tree Star, Inc., Ashland, OR). The interassay coefficient of variation for the phenotyping assays was 10%.
HLA genotyping for class II type 1 diabetes susceptibility alleles
HLA genotyping for class II alleles associated with susceptibility to or protection from autoimmune type 1 diabetes was performed at Benaroya Research Institute, Seattle, WA. Briefly, HLA genotyping for DR1, DR3, DR4, DR13, DR15, DQ2, DQ6, and DQ8 was performed by real-time PCR using TaqMan specific assays, as described previously (16,17). Primers and probes specific for HLA-DRA were included as a template control. Real-time PCR was done using TaqMan GTXpress genotyping master mix (Applied Biosystems, Life Technologies, Inc.) on an ABI StepOnePlus Real-Time PCR system (Applied Biosystems), as described (17).
Statistics
ANOVAs, the Wilcoxon signed rank test, and Mann-Whitney tests were used to compare groups. Significance was established at P < 0.05. Multivariate regression analyses were performed to evaluate positivity for monocytes and islet-reactive T-cell responses to BMI, age, sex, and duration of diabetes.
RESULTS
Patient demographic, clinical, and phenotypic data are reported in Tables 1 and 2. There were no significant differences among the three groups in age at the time of T-cell testing, sex, or ethnicity (Table 1). The A−β− KPD patients had been diagnosed with diabetes at a younger age than the two A−β+ KPD groups (P < 0.01) and had a longer duration of diabetes (P < 0.01, Table 1). The initial diagnosis of diabetes coincided with the index DKA (defined as the episode of DKA that first brought the patient to the attention of the KPD research group) among unprovoked A−β+ KPD patients, but usually preceded the index DKA (by 0–7 years) among provoked A−β+ KPD patients, and was well before the index DKA among A−β− KPD patients. Fasting C-peptide and peak C-peptide levels after glucagon stimulation were significantly lower in the A−β− KPD group shortly after the index DKA (“initial” values in Table 1) (P < 0.01, Table 2). As is typical for these KPD subgroups (1), fasting and peak C-peptide values were essentially unchanged in the A−β− KPD group 6–12 months after the index DKA, whereas they increased significantly in both A−β+ KPD groups after 6–12 months. HbA1c at the time of the index DKA (“initial” HbA1c in Table 2) was similar in the three groups; however, after 6–12 months of treatment following an established and standardized protocol (18) in the same clinic, HbA1c was significantly lower in the two A−β+ KPD groups compared with the A−β− KPD group (P < 0.01), and this difference was maintained in HbA1c measured close to the time of the blood draw for T-cell testing.
Table 1.
Demographic data of KPD patients
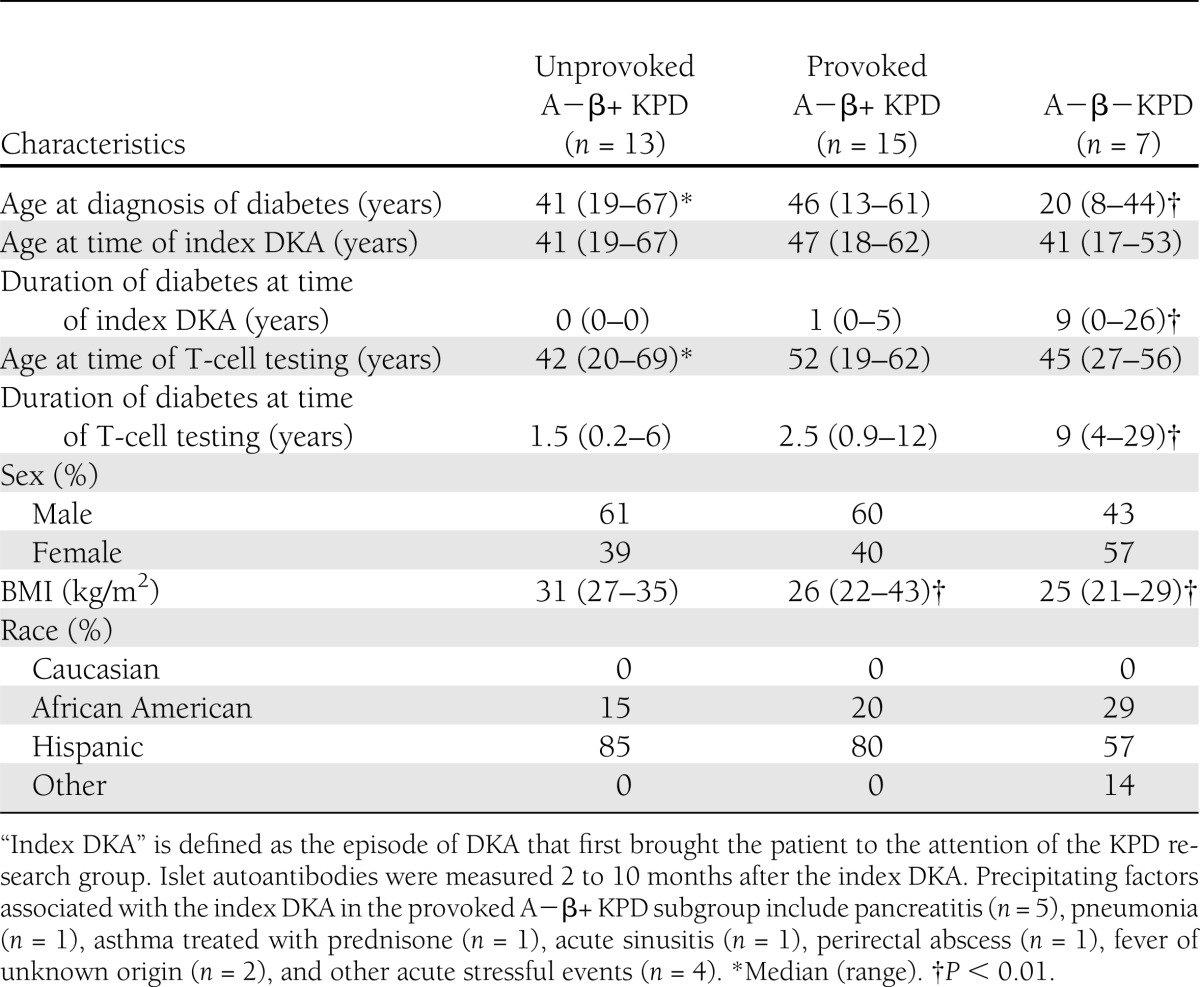
Table 2.
Clinical data of KPD patients
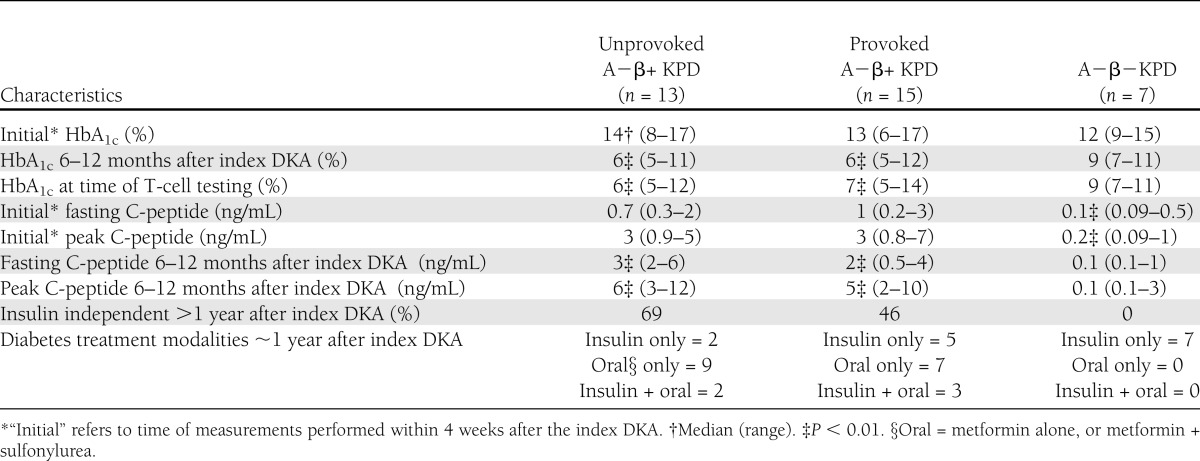
A key characteristic of provoked A−β+ KPD patients is that 3–4 years after recovery from the index DKA, their β-cell function declines and glycemic control deteriorates, whereas β-cell function remains stable and glycemic control is maintained in patients with unprovoked A−β+ KPD (4); the number of A−β+ KPD patients who participated in the current study and have been monitored long enough to compare these outcomes between provoked and unprovoked A−β+ KPD patients after 3–4 years was insufficient for analysis of this outcome. However, a substantially higher fraction of unprovoked A−β+ KPD patients (69%) than provoked A−β+ KPD patients (46%) were insulin-independent, with good glycemic control, more than 1 year after the index DKA episode.
Islet-specific T-cell reactivities among the three KPD patient groups are shown in Fig. 1. We observed that provoked A−β+ KPD patients and A−β− KPD patients had significantly (P < 0.03) higher T-cell responses to islet proteins than unprovoked A−β+ KPD patients. The T-cell responses to the islet proteins in the provoked A−β+ KPD and A−β− KPD patients were similar to what we have observed in other patient populations with autoimmune type 1 diabetes (5,6,10,11,13,14,19). There was no significant difference between the provoked A−β+ KPD and A−β− KPD patients in frequency of T-cell reactivity to islet proteins. In contrast, the patients with the distinctive syndrome of unprovoked A−β+ KPD lacked islet autoantibodies and islet-reactive T cells, suggesting that the pathophysiology of this distinctive phenotype is not associated with islet autoimmunity. Differences in the islet-specific T-cell responses among the three patient groups in Fig. 1 were not significantly affected by BMI, age, sex, or diabetes duration.
Figure 1.

Number of islet protein blot sections (cellular immunoblotting) stimulatory to T cells from unprovoked A−β+ KPD patients (n = 13), provoked A−β+ KPD patients (n = 15), and A−β− KPD patients (n = 7). The horizontal dashed line at the three blot sections represents the cutoff for T-cell positivity. Significance was taken at P < 0.05.
We next analyzed the percentages of proinflammatory monocytes (CD14+CD16+) in the peripheral blood of the three KPD subgroups and observed that the provoked A−β+ KPD patients and the A−β− KPD patients both had a significantly higher frequency of proinflammatory CD14+CD16+ monocytes than the unprovoked A−β+ KPD patients (P < 0.01) (Fig. 2). There were no differences in the frequency of proinflammatory monocytes between the provoked A−β+ KPD patients and the A−β− KPD patients. Differences in the percentages of proinflammatory monocytes observed in Fig. 2 were not significantly affected by age, sex, or diabetes duration. However, increases in proinflammatory monocytes were negatively correlated with BMI.
Figure 2.

Percentage of proinflammatory (CD14+CD16+) monocytes in the peripheral blood of unprovoked A−β+ KPD patients (n = 13), provoked A−β+ KPD patients (n = 15), and A−β− KPD patients (n = 7). Significance was taken at P < 0.01.
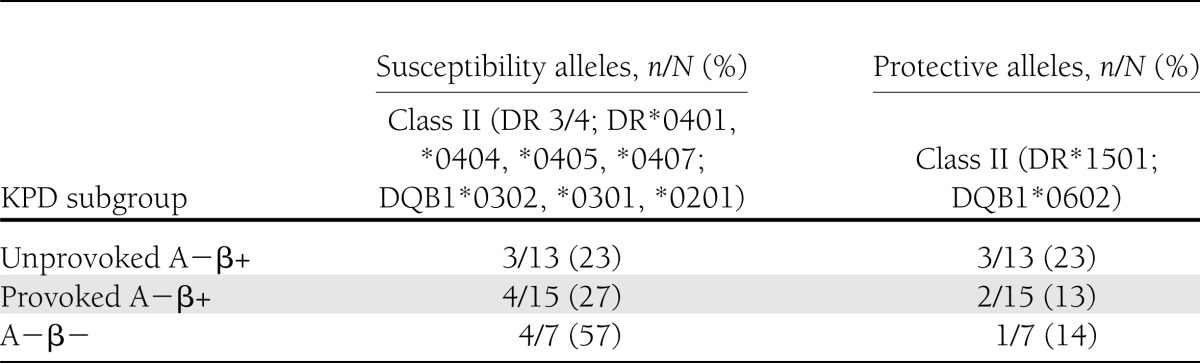
HLA genotyping was conducted to determine the frequency of the three A− subgroups of KPD patients with HLA class II alleles (in the DR and DQB loci) known to be associated with susceptibility to or protection against autoimmune type 1 diabetes. Some neutral alleles (associated with neither susceptibility nor protection) were also genotyped. Frequencies of HLA class II alleles associated with susceptibility or protection are presented in Table 3. There was no significant relationship between possessing a susceptibility allele and having a positive T-cell test. However, there was a significant relationship between possessing a protective allele and having a negative T-cell test, and no patient possessing the strong protective allele DQB1*0602 had a positive T-cell test.
Table 3.
HLA class II alleles
CONCLUSIONS
Systemic inflammation has been demonstrated to be involved in the development of type 2 diabetes. We have used the validated cellular immunoblotting assay to study islet-specific T-cell autoimmunity in autoimmune type 1 and phenotypic type 2 diabetic patients (5,6,10,11,13,14,19). These studies underscore the heterogeneity of type 2 diabetes and demonstrate an overlap between the causes of β-cell dysfunction in type 1 and type 2 diabetes because they reveal that a strikingly high percentage of patients classified as type 2 possess islet-specific autoreactive T cells. Moreover, the presence of islet-specific T cells in these type 2 diabetic patients has been linked to a more severe β-cell dysfunction (5).
The KPD syndromes include phenotypes that defy classification according to traditional definitions of type 1 and type 2 diabetes. As we have demonstrated previously (1,4,8), the “Aβ” classification of patients shortly after presentation with the index DKA is highly accurate in predicting clinical behavior (especially insulin dependence), β-cell functional reserve, and glycemic control. In particular, the characteristics of the three subgroups of KPD characterized by absence of islet autoantibodies (the A− subgroups) are distinctive, with different natural histories of β-cell dysfunction:
A−β− KPD patients have no β-cell functional reserve shortly after the episode of DKA and never recover insulin secretory capacity, indicating severe and permanent β-cell dysfunction.
Provoked A−β+ KPD patients recover β-cell function soon after the index DKA and may maintain β-cell reserve for up to 4 years, but thereafter manifest progressive loss of β-cell function and insulin dependence, suggesting an ongoing process of β-cell dysfunction that recovers only transiently upon intensive glycemic management.
Unprovoked A−β+ KPD patients recover β-cell function soon after the index DKA and maintain substantial and stable β-cell functional reserve and insulin independence for prolonged periods of time (usually >4 years).
Moreover, provoked A−β+ KPD patients have a high frequency of HLA class II alleles linked to susceptibility to autoimmune type 1 diabetes (DQB1*0302 and DRB1*04), suggesting that their pathophysiology includes a component of islet autoimmunity, whereas unprovoked A−β+ KPD patients lack the increased frequency of susceptibility alleles (4). Hence, in the current study, we used cellular immunoblotting and flow cytometry to determine whether the differences observed in the clinical characteristics and natural histories of these three A− KPD subgroups are related to the presence or absence of islet-specific T cells and/or proinflammatory monocytes (CD14+CD16+). Proinflammatory monocytes (CD14+CD16+) are increased in a variety of acute infections and severe inflammatory states such as sepsis. Therefore, all samples from subjects were obtained when they were clinically stable, without evidence of acute inflection or inflammation, to avoid the potential increases of inflammatory monocyte populations associated with inflammation or infection. We discovered an inverse relationship between BMI and the presence of proinflammatory monocytes. Because an association of obesity with increased numbers of circulating proinflammatory monocytes has been reported (20), this finding suggests that some factors unique to KPD may drive the systemic inflammatory response and override the effects of obesity per se. Further studies are warranted to investigate this aspect of KPD pathophysiology.
Frequencies of HLA class II alleles associated with susceptibility to or protection against autoimmune type 1 diabetes were measured in all of the patients in this study to assess genetic contributions to cell-mediated islet autoimmunity in the A− KPD patients. The results, in conjunction with those previously obtained in a separate, larger cohort of KPD patients, indicate that 1) in the A− KPD population, having a protective HLA class II allele is associated with absence of both humoral (4) and T-cell autoreactivity to islet antigens and that 2) compared with the other A− KPD subgroups, unprovoked A−β+ KPD, previously shown to have a lower frequency of susceptibility HLA class II alleles (4), also has a higher frequency of protective alleles. Together with the present results indicating that unprovoked A−β+ KPD has a low frequency of T-cell positivity and that having a protective allele is strongly associated with absence of T-cell positivity, these findings strengthen the notion that the cause of β-cell dysfunction in unprovoked A−β+ KPD is not related to islet autoimmunity.
In summary, our results lead to the conclusion that provoked A−β+ KPD is associated with a high frequency of islet autoimmunity and proinflammatory monocyte populations, which could contribute to the propensity to ketoacidosis and the subsequent progressive loss of β-cell function in these patients. On the other hand, unprovoked A−β+ KPD is a distinct syndrome of reversible β-cell dysfunction and sustained β-cell functional reserve comprising patients who lack evidence for humoral and cellular islet autoimmunity and whose proneness to ketosis may be explained by specific defects in oxidation of ketones and fatty acids and in the metabolism of the ketogenic amino acid leucine (21). Patients in the A−β− subgroup are demographically different from the other two A− KPD subgroups, being lean, with earlier age of onset of diabetes, and completely insulinopenic. In our earlier analyses, A−β− KPD patients were presumed to have a “nonautoimmune” etiology of severe β-cell dysfunction on the basis of absent autoantibodies and a frequency of HLA susceptibility alleles that is lower than that of A+β− KPD patients (who have classic, autoimmune type 1 diabetes) (1). The current study shows that ∼50% of these A−β− KPD patients are in fact “T-cell positive”. It appears, therefore, that A−β− KPD subsumes multiple etiologies of severe β-cell dysfunction, including cellular islet autoimmunity (“T-cell positive” patients) and monogenic causes of defective islet development or function, as we have previously reported (22). Thus, these findings demonstrate that cellular islet autoimmunity is present in some but not all A− forms of KPD and expand the autoimmune basis of β-cell dysfunction beyond the boundaries of classic, autoimmune type 1 diabetes.
Acknowledgments
This study was supported by R01-DK-083471, the Diabetes Research Center (P30-KD-017047) at the University of Washington, National Institutes of Health R21-DK-082827, and the Diabetes Research Center (P30-DK-079638) at Baylor College of Medicine.
No potential conflicts of interest relevant to this article were reported.
K.O., R.Nal., and A.B. diagnosed, managed, and phenotyped the KPD patients. D.I. performed biochemical analyses. I.C. recruited the patients and maintained the KPD database. C.S.H. performed the autoantibody analyses and analyzed data. B.M.B.-W. performed the T-cell analyses and flow cytometry and prepared figures. R.Nar. assisted with the flow cytometry. B.M.B.-W., J.P.P., and A.B. conceived the project, analyzed, and interpreted the data, and drafted the manuscript. J.P.P. guided the design of the study and analyzed and interpreted the data. All authors reviewed and edited the manuscript. B.M.B.-W., J.P.P., and A.B. are guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Mary Lou Arvizu, RN, (Ben Taub General Hospital, Houston, TX) for excellent care of the KPD patients; Jessica Reichow (Seattle Institute for Biomedical and Clinical Research, Seattle, WA) for help with the manuscript; Helena Reijonen, PhD, Karen Cerosaletti, PhD, and Vivian Gersuk, PhD, (Benaroya Research Institute, Seattle, WA) for performing HLA typing and analysis; and Dr. Edward J. Boyko (VA Puget Sound Health Care System and the University of Washington) for assistance with statistical analyses.
This study was presented as a poster presentation at the 71st Scientific Sessions of the American Diabetes Association, San Diego, California, 24–28 June 2011.
References
- 1.Maldonado M, Hampe CS, Gaur LK, et al. Ketosis-prone diabetes: dissection of a heterogeneous syndrome using an immunogenetic and beta-cell functional classification, prospective analysis, and clinical outcomes. J Clin Endocrinol Metab 2003;88:5090–5098 [DOI] [PubMed] [Google Scholar]
- 2.Nalini R, Gaur LK, Maldonado MR, et al. HLA class II alleles specify phenotypes of ketosis-prone diabetes. Diabetes Care 2008;31:1195–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balasubramanyam A, Garza G, Rodriguez L, et al. Accuracy and predictive value of classification schemes for ketosis-prone diabetes. Diabetes Care 2006;29:2575–2579 [DOI] [PubMed] [Google Scholar]
- 4.Nalini R, Ozer K, Maldonado M, et al. Presence or absence of a known diabetic ketoacidosis precipitant defines distinct syndromes of “A-β+” ketosis-prone diabetes based on long-term β-cell function, human leukocyte antigen class II alleles, and sex predilection. Metabolism 2010;59:1448–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goel A, Chiu H, Felton J, Palmer JP, Brooks-Worrell B. T-cell responses to islet antigens improves detection of autoimmune diabetes and identifies patients with more severe β-cell lesions in phenotypic type 2 diabetes. Diabetes 2007;56:2110–2115 [DOI] [PubMed] [Google Scholar]
- 6.Brooks-Worrell BM, Reichow JL, Goel A, Ismail H, Palmer JP. Identification of autoantibody-negative autoimmune type 2 diabetic patients. Diabetes Care 2011;34:168–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ziegler-Heitbrock L. The CD14+ CD16+ blood monocytes: their role in infection and inflammation. J Leukoc Biol 2007;81:584–592 [DOI] [PubMed] [Google Scholar]
- 8.Hampe CS, Nalini R, Maldonado MR, et al. Association of amino-terminal-specific antiglutamate decarboxylase (GAD65) autoantibodies with beta-cell functional reserve and a milder clinical phenotype in patients with GAD65 antibodies and ketosis-prone diabetes mellitus. J Clin Endocrinol Metab 2007;92:462–467 [DOI] [PubMed] [Google Scholar]
- 9.Vaziri-Sani F, Oak S, Radtke J, et al. ZnT8 autoantibody titers in type 1 diabetes patients decline rapidly after clinical onset. Autoimmunity 2010;43:598–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seyfert-Margolis V, Gisler TD, Asare AL, et al. Analysis of T-cell assays to measure autoimmune responses in subjects with type 1 diabetes: results of a blinded controlled study. Diabetes 2006;55:2588–2594 [DOI] [PubMed] [Google Scholar]
- 11.Herold KC, Brooks-Worrell B, Palmer J, et al. Type 1 Diabetes TrialNet Research Group Validity and reproducibility of measurement of islet autoreactivity by T-cell assays in subjects with early type 1 diabetes. Diabetes 2009;58:2588–2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bingley PJ, Bonifacio E, Mueller PW. Diabetes Antibody Standardization Program: first assay proficiency evaluation. Diabetes 2003;52:1128–1136 [DOI] [PubMed] [Google Scholar]
- 13.Brooks-Worrell BM, Starkebaum GA, Greenbaum C, Palmer JP. Peripheral blood mononuclear cells of insulin-dependent diabetic patients respond to multiple islet cell proteins. J Immunol 1996;157:5668–5674 [PubMed] [Google Scholar]
- 14.Brooks-Worrell B, Gersuk VH, Greenbaum C, Palmer JP. Intermolecular antigen spreading occurs during the preclinical period of human type 1 diabetes. J Immunol 2001;166:5265–5270 [DOI] [PubMed] [Google Scholar]
- 15.Brooks-Worrell B, Warsen A, Palmer JP. Improved T cell assay for identification of type 1 diabetes patients. J Immunol Methods 2009;344:79–83 [DOI] [PubMed] [Google Scholar]
- 16.Gersuk VH, Nepom GT. A real-time PCR approach for rapid high resolution subtyping of HLA-DRB1*04. J Immunol Methods 2006;317:64–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gersuk VH, Nepom GT. A real-time polymerase chain reaction assay for the rapid identification of the autoimmune disease-associated allele HLA-DQB1*0602. Tissue Antigens 2009;73:335–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balasubramanyam A, Nalini R, Hampe CS, Maldonado M. Syndromes of ketosis-prone diabetes mellitus. Endocr Rev 2008;29:292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brooks-Worrell BM, Juneja R, Minokadeh A, Greenbaum CJ, Palmer JP. Cellular immune responses to human islet proteins in antibody-positive type 2 diabetic patients. Diabetes 1999;48:983–988 [DOI] [PubMed] [Google Scholar]
- 20.Ghanim H, Aljada A, Hofmeyer D, Syed T, Mohanty P, Dandona P. Circulating mononuclear cells in the obese are in a proinflammatory state. Circulation 2004;110:1564–1571 [DOI] [PubMed] [Google Scholar]
- 21.Patel SG, Hsu JW, Jahoor F, et al. Pathogenesis of A⁻β⁺ ketosis-prone diabetes. Diabetes 2013;62:912–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haaland WC, Scaduto DI, Maldonado MR, et al. A-β-subtype of ketosis-prone diabetes is not predominantly a monogenic diabetic syndrome. Diabetes Care 2009;32:873–877 [DOI] [PMC free article] [PubMed] [Google Scholar]