

Abstract
Maspin, a multifaceted tumor suppressor, belongs to the serine protease inhibitor superfamily, but only inhibits serine protease-like enzymes such as histone deacetylase 1 (HDAC1). Maspin is specifically expressed in epithelial cells and it is differentially regulated during tumor progression. A new emerging consensus suggests that a shift in maspin subcellular localization from the nucleus to the cytoplasm stratifies with poor cancer prognosis. In the current study, we employed a rational mutagenesis approach and showed that maspin reactive center loop (RCL) and its neighboring sequence are critical for maspin stability. Further, when expressed in multiple tumor cell lines, single point mutation of Aspartate346 (D346) to Glutamate (E346), maspinD346E, was predominantly nuclear, whereas wild type maspin (maspinWT) was both cytoplasmic and nuclear. Evidence from cellular fractionation followed by immunological and proteomic protein identification, combined with the evidence from fluorescent imaging of endogenous proteins, fluorescent protein fusion constructs, as well as bimolecular fluorescence complementation (BiFC) showed that the increased nuclear enrichment of maspinD346E was, at least in part, due to its increased affinity to HDAC1. MaspinD346E was also more potent than maspinWT as an HDAC inhibitor. Taken together, our evidence demonstrates that D346 is a critical cis-element in maspin sequence that determines the molecular context and subcellular localization of maspin. A mechanistic model derived from our evidence suggests a new window of opportunity for the development of maspin-based biologically competent HDAC inhibitors for cancer treatment.
Introduction
Tumor suppressor maspin is a member of Clade B serine protease inhibitor (serpin) superfamily, but has been shown to have deviant biological functions and molecular modes of action [1], [2]. Earlier structural and experimental evidence suggests that maspin does not inhibit an active serine protease [3]–[5]. To date, we have identified only two circumstantial serine protease targets of maspin: the zymogen form of urokinase type plasminogen activator (pro-uPA) [6]–[8] and the single-chain tissue-type plasminogen activator (sc-tPA) that is associated with fibrinogen [1]. Our laboratory also discovered that endogenous maspin binds to and inhibits the activity of histone deacetylase 1 (HDAC1) [7], [9], which is a major nuclear deacetylase of class I that is up-regulated in many types of cancers [2]. To our knowledge, maspin is the only endogenous polypeptide inhibitor of HDAC1 identified thus far. Homozygous deletion of either the maspin gene or the HDAC1 gene results in embryonic lethality in murine models, signifying the equally vital roles of maspin and HDAC1 in embryogenesis [10], [11]. As compared to pharmacological HDAC inhibitors, maspin regulates a much smaller cluster of HDAC target genes, all being implicated in controlling epithelial differentiation [12]. These findings provided a functional connection between maspin and a better differentiated phenotype as well as better prognosis of human cancer [13].
Maspin is an epithelial-specific gene shown to be differentially regulated in the progression of many types of solid tumors including prostate [14], [15], breast [16], [17] and lung [18], [19]. The differential regulation of maspin occurs not only at the level of expression, but also at the level of subcellular distribution. In accordance with the spectrum of its possible molecular targets and overall tumor suppressive activities, maspin is known to be a secreted and cell surface-associated protein [20]. In normal epithelial tissues, the intracellular maspin is predominantly nuclear. Accumulated clinical evidence in lung [19], [21], breast [22] and ovarian [23], [24] carcinoma demonstrates that nuclear retention of maspin is correlated with better overall patient survival [14]. Conversely, the shift in subcellular localization of maspin from the nucleus to the cytoplasm is associated with a gain of function during tumor progression [19].
In a recent study, the expression of bioengineered maspin targeted for nuclear exclusion failed to exert tumor suppressive effects both in vitro and in vivo [25], [26]. This evidence supports the significance of maspin translocation from the nucleus to cytoplasm in tumor progression. However, it is important to note that maspin does not have any of the currently known intrinsic “address tags” such as nuclear localization sequence (NLS), nuclear export sequence (NES) or secretory leader sequence (SLS) [27]. To date, the molecular mechanisms that control maspin trafficking and maspin nuclear localization, in particular, are unknown. As a 42 kDa protein, maspin could passively diffuse through the nuclear envelope [28]. However, considering the distinct subcellular localization of maspin at different epithelial dedifferentiation states and the evidence that maspin is not mutated in tumor progression, we speculate that maspin subcellular localization may be actively controlled by its molecular partnership that is subjected to modulations by pathological signals. In parallel, it is reasonable to hypothesize that intracellular maspin trafficking also depends on key cis-elements in maspin primary sequence. To this end, maspin contains a reactive center loop (RCL) sequence, as expected based on its overall alignment with members of the serpin superfamily. Although the most variable and deviant among serpins [4], [5], [29], the RCL of maspin is essential for its biochemical effects on the serine protease-like targets [1], [6], [7], and its biological effects on tumor cell motility and invasion [15], [20], [30]. Based on several known serpin structures including that of maspin, amino acid residues in close vicinity to RCL may play an important role in the overall affinity for the target proteins. Interestingly, it is noted that at the C-terminal end of the RCL sequence of maspin is an Aspartate 346 (D346), which is unique among all serpin homologs and maspin orthologs.
In this study, we report that a conservative substitution of maspin D346 by glutamic acid (E) resulted in its dominant nuclear distribution and increased interaction with HDAC1 in multiple cancer cell lines. Our data led to a hypothetical model that helps explain how maspin nuclear retention and its tumor suppressive competency may be bypassed in tumor progression. These novel insights may guide future development of maspin-based biologically competent HDAC inhibitors for cancer treatment.
Materials and Methods
Cell Culture, Reagents and Antibodies
Immortalized normal human epithelial cells from prostate (CRL2221), breast (MCF10A) and lung (BEAS2B), human prostate carcinoma cell lines (DU145 and PC3) and human lung carcinoma cell line H1299 were purchased from American Type Culture Collection (ATCC, Manassas, VA). All media and media supplements were purchased from Life Technologies™ (Grand Island, NY) unless stated otherwise. CRL2221 cells were grown in Keratinocyte Serum-Free Medium with supplied growth factors, MCF-10A cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/F12 with additional supplements [7] and BEAS 2B cells were cultured in LHC 8 media [19]. Carcinoma cell lines were maintained in RPMI 1640 media containing FBS from Hyclone (South Logan, UT; 5% for DU145; 10% for PC3 and H1299) [6]. All cells were cultures at 37°C with 6.5% CO2.
We used the following reagents: MG132 (#474790, Calbiochem, La Jolla, CA), Leptomycin B (#L2913-5UG, Sigma Aldrich, St. Louis, MO), goat serum (# ab7481, Abcam, Cambridge, MA), protein A/G agarose beads (# sc2003, Santa Cruz, Santa Cruz, CA), normal mouse IgG (#sc52003, Santa Cruz), normal rabbit IgG (# sc3888, Santa Cruz) and DAPI (#10236276001, Roche Applied Science, Foster City, CA). Primary antibodies used include: maspin (#554292, BD Pharmingen, San Jose, CA), HDAC1 (#06720, Millipore, Grand Island, NY), poly (ADP-ribose) polymerase (PARP, #04–575, Millipore), acetylated histone 3 at lysine 9 (Acetyl H3 K9, #07352, Millipore), Lamin B (#ab16048, Abcam), GAPDH (#9484, Abcam), β tubulin (#ab6046, Abcam), and glucose regulated protein 78 (#sc13968, GRP78, Santa Cruz). The secondary antibodies used include: anti mouse-HRP conjugated and anti-rabbit-HRP conjugated (#NXA931 and #NA934, respectively, from GE Health Care, Fairfield, CT) and fluorescent dyes, Alexa Fluor 488 and Alexa Fluor 594 (#A11029 and #A21203, respectively, from Life Technologies™).
Adenoviral Expression of Recombinant Maspin
Maspin recombinant proteins were generated as previously described [7]. Briefly, to generate truncation mutants maspin1–323, maspin1–340 and maspin1–348, the C-terminus of maspin after designated last amino acid residue in the pVL1393/maspin template vector [31] was fused in frame to a translation-stop codon using the following PCR primers. For maspin1–323: 5′-CTGAAGATGGTGGGGATTCCTAAGAGGTGCCAGGAGCACGG-3′, and 5′-CCGTGCTCCTGGCACCTCTTAGGAATCCCCACCATCTTCAG-3′; for maspin1–340: 5′-CCATAGAGGTGCCAGGAGCACGGTAACTGCAGCACAAGG-3′, and 5′-CCTTGTGCTGCAGTTACCGTGCTCCTGGCACCTCTGTGG-3′; for maspin1–348: 5′-CCTGCAGCACAAGGATGAATTGTAAGCTGACCATCCC-3′, and 5′-GGGATGGTCAGCTTACAATTCATCCTTGTGCTGCAGG-3′. Using the Exsite PCR-Based Site-Directed Mutagenesis kit we substituted Aspartate (D) 346 in the pVL1393/maspin template with Glutamic acid (E) to generate maspinD346E mutant. The PCR primers for mutagenesis were 5′-CGGATCCTGCAGCACAAGGAAGAATTTAATGCTGACCATCCC-3′ and 5′-GGGATGGTCAGCATTAAATTCTTCCTTGTGCTGCAGGATCCG-3′. The cDNAs encoding maspin mutants were sequence-verified and sub-cloned into the pAdenoVator-CMV5 vector (Q-Bio Gene, Carlsbad, CA) as described [7]. The resulting plasmids were purified and used to generate the recombinant adenoviral DNA and the adenovirus using an established procedure [7]. Adenoviral titer was determined using the Quick Titer™ Adenovirus Titer ELISA kit according to the manufacturer’s instructions (Cell Biolabs, Inc., San Diego, CA). For routine in vitro infections, adenovirus was added to 24 hrs-old cell culture at the multiplicity of infection (MOI) of 30 (DU145 cells) or 20 (H1299 cells). For subsequent functional and biological assays, the cells were used three days post infection, unless stated otherwise, since the expression of recombinant proteins was at its maximum.
Constructs for Fusion Proteins
The constructs for fluorescence-tagged maspin and HDAC1 were made as follows: for maspin-green fluorescent protein (GFP) chimera the coding region of human maspin cDNA was PCR amplified from pBluescript-maspin [16], [32] inserting SacI restriction sites. The amplified fragment was sub-cloned into pEGFP-N1 mammalian expression vector from Clontech (Mountain View, CA). For the construction of HDAC1-red fluorescent protein (RFP) chimera, mRFP was PCR amplified from pmRFP1-N1 vector and inserted into pcDNA3.1-HDAC1 (Addgene repository, plasmid 13820) and ligated using EcoRI. For this reason, an additional N-terminal EcoRI site had to be eliminated first by site-directed mutagenesis (QuikChange Lightning Site-Directed Mutagenesis Kit, Agilent Technologies, Santa Clara, CA). The constructs for bimolecular fluorescence complementation (BiFC) [33] were generously provided by Dr. Kerppola (University of Michigan, Ann Arbor, MI): BiFC bJunYN, BiFC bFosYC and BiFC bFosYC Δ179–193 that utilize yellow fluorescence protein (YFP) as a reporter. Mutagenesis in BiFC MaspinYC was performed using QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies) to replace bFos with maspin in BiFC bFosYC plasmid and bJun with HDAC1 in BiFC bJunYN plasmid. The resulting constructs are designated as BiFC maspinYC and BiFC HDAC1YN, respectively.
Transient Transfection
Cells grown in 6 well plates at 70% confluence were transfected or co-transfected with 1 µg of plasmid DNA for maspin-GFP and/or HDAC1-RFP, using the X-tremeGENE 9 DNA transfection reagent (Roche Applied Science, Indianapolis, IN). Fluorescence imaging of live cells expressing GFP- and/or RFP-fusion proteins was acquired 40–48 h after transient transfection using the Leica DM IRB fluorescence microscope. For transfection using the BiFC constructs, the experiments were performed as outlined by Kerppola [33], allowing the maturation of the fluorescent complexes for 24 hrs at 30°C prior to live cell imaging. In BiFC experiments, co-transfection with BiFC bJunYN and BiFC bFosYC, and the co-transfection with BiFC bJunYN and BiFC bFosYC Δ179–193 were used as positive and negative controls, respectively.
Immunofluorescence Staining, Confocal and Live Cell Imaging
Cells grown in 8-well chamber slides (#154534, Thermo Fisher Scientific, Hudson, NH) to 70% confluence were fixed with 4% paraformaldehyde (15 min at room temperature (RT)), and permeabilized with 100% ice cold methanol (10 min at −20°C). The slides were incubated with 10% normal goat serum in PBS for 1 hr, and incubated with anti-maspin (1∶100) antibody alone or in a combination with either anti-lamin B (1∶50), anti-HDAC1 (1∶50) or anti-GRP78 (1∶50) at 4°C overnight. Cells were washed and incubated for 2 hrs at room temperature (RT) with Alexa Fluor 488 (1∶500) alone or in combination with Alexa Fluor 594 (1∶500). The nuclei were counterstained with DAPI. Life cell imaging of transiently transfected cells was performed using the Leica Fluorescent microscope. The confocal imaging was assisted by the Microscopy, Imaging and Cytometry Resources Core at Karmanos Cancer Institute, Wayne State University School of Medicine.
Cellular Fractionation, Immunoprecipitation (IP) and Protein Identification by Mass Spectrophotometry (MS)
For identification and quantification of maspin in the cytosolic and nuclear compartments, cells were fractioned as previously described [34] or by using the Subcellular Protein Fractionation Kit (Thermo Fisher Scientific, Rockford, IL) according to the manufacturer’s instructions. Fractionated proteins were subjected to IP. Briefly, to block nonspecific background, protein A/G-agarose beads, at a final concentration of 5% (v/v), were incubated with 500 µg of total lysate (or fractionated cell lysates) in IP buffer as previously described [34]. The resulting mixture was centrifuged at 10,000×g for 30 s, and the harvested supernatant was incubated with 5 µg of maspin antibody or 5 µg of pre-immune mouse IgG with gentle agitation at 4°C overnight. Protein A/G beads were then added to a final concentration of 2.5% (v/v). The mixture was further incubated for 2 hrs at RT with gentle agitation and centrifuged at 10,000×g for 30 s. The harvested beads were washed four times with IP buffer and one time with PBS, re-suspended with SDS sample buffer, heat denatured, and subjected to SDS-PAGE. The proteins resolved by SDS-PAGE were either profiled by western blotting or by MS/proteomic analysis for definitive identification and quantification. For MS/proteomic analysis, SDS-PAGE gels were stained with Sypro Ruby (#S12000, Life Technologies™) and washed. Gel slices were reduced with DTT and alkylated with IAA prior to digesting proteins with trypsin as previously described [11]. Peptides extracted from the gels were identified by LC-MS/MS on a Q Exactive OrbiTrap system and quantified by spectral counting. Data analysis was performed using Proteome Discoverer 1.3, which incorporated the Mascot algorithm (Matrix Science). The UniProt human database was used and a reverse decoy protein database was run simultaneously for false discovery rate (FDR) determination. Secondary analysis was performed using Scaffold (Proteome Software). A fixed modification of +57 on cysteine (carbamidomethylation) and variable modifications of +16 on methionine (oxidation) and +42 on protein n-terminus (acetylation) were included in the search. Minimum protein identification probability was set at ≥95% with 2 unique peptides.
Quantitative Real-time PCR (q-RT-PCR)
Total RNA was extracted (RNeasy Mini kit, Qiagen Valencia, CA,), and reverse-transcribed (iScript cDNA synthesis kit, Bio-Rad, Irvine, CA). The mRNA of the following genes was quantified using the following pairs of primers: AKR1C2 (5′-GTAAAGCTCTAGAGGCCGT-3′ and 5′-CTGGTCGATGGGAATTGCT-3′), Fst (5′-GTTTTCTGTCCAGGCAGCTCCAC-3′ and 5′-GCAAGATCCGGAGTGCTTTACT-3′), Glrx (5′-GGGAGCAAGAACGGTGCCTCG-3′ and 5′-ATCTGTGGTTACTGCAGAGCTCCA-3′), Kcnk1 (5′-GCACGGTGTGGCCATAACCTGT-3′ and 5′-GCCTCGGGCAACTGGAACTGG-3′), Lancl1 (5′-TGGCGCCCCTGGGGTAATCT-3′ and 5′-ACCATTCAGCAAACTTACAGGCCC-3′), Mx1 (5′-CCTATCACCAGGAGGCCAGCAAGC-3′ and 5′-TTCCGCTTGTCGCTGGTGTCG-3′), Nell2 (5′-ATGCCTGAATGGAACCATCCAGTGTG-3′ and 5′-TCGACAGCTACAAACAGCCCTGT-3′), Nmu (5′-CAGCCTCAGGCATCCAACGCA-3′ and 5′-GGAACGAGCTGCAGCAACGGA-3′), Tspan7 (5′-GGGTTGTTATGATCTGGTAACTAGTTTCATGGAGAC-3′ and 5′-GCCAGCAGCATGCCAATTAACTGG-3′), and Esrp1 (5′-CCAAGAAGAATGTACTATTACCTGAATGC-3′ and 5′-ACCTCGTGCCCTGACTACGGT-3′). RT-PCR cycle threshold of each gene was normalized using the GAPDH (5′-ATCACCATCTTCCAGGAGCGA-3′ and 5′-GCCAGTGAGCTTCCCGTTCA-3′) as an internal reference.
Miscellaneous Procedures
Protein concentration was determined using the Pierce BCA Protein Assay Kit (#23225, Thermo Scientific). Western blotting analyses were carried out as described [7]. To characterize the proliferation kinetics of adenovirus-infected cells, the cells (1×104/well) were seeded in a 6 well plate in duplicates. The next day, cells were infected with adenovirus and subsequently counted on days, 3, 6, 8, 10 and 13 using the Coulter Z1 particle counter (Beckman Coulter, Fullerton, CA) [7].
Results
Construction and Expression of Rational Maspin Mutants
To investigate whether maspin RCL sequence and its neighboring regions, including the D346 residue, play a role in dictating maspin subcellular localization and function, we used the site directed mutagenesis approach to generate maspin mutants. Considering the exposed regions near maspin RCL, as shown by the ribbon structure of maspin resolved by crystallography [29], [35] ( Figure 1A ), our scheme for rational site-directed mutagenesis included maspin1–323, maspin1–340, maspin1–348 and maspinD346E ( Figure 1B ). Previously, we successfully used an adenoviral expression system for endogenous expression of wild type maspin (maspinWT) [7] in DU145 cells that expresses a negligible level of endogenous maspin [6]. This system was used in the current study to express both wild type maspin (maspinWT) and maspin mutants. The adenovirus encoding empty vector was used as a negative control.
Figure 1. Construction and expression of rational maspin mutants.

(A) Ribbon representation of human maspin generated by UCSF Chimera (http://www.cgl.ucsf.edu/chimera/). Relative positions of reactive center loop (RCL) p′ site R340 and neighboring D346 residue are depicted as ball and stick model. (B) Schematic illustration of the wild type human maspin and four rational maspin mutants. Relative positions of RCL and D346 in a primary maspin sequence are depicted. The full-length, wild type maspin was denoted as maspinWT, the truncation mutants are maspin1–323 (C-terminal deletion starting 16 amino acids upstream of RCL), maspin1–340 (C-terminal deletion after R340) and maspin1–348 (C-terminal deletion after L348) and the point mutant is maspinD346E. (C) Western blot of maspinWT and four rational mutants re-expressed in prostate cancer cell line DU145 by adenoviral vector (multiplicity of infection, MOI = 30) in the presence or absence of proteasome inhibitor MG132 (5 µM, 6 hrs treatment). Infection of DU145 cells by adenovirus empty vector (EV) was used as a negative control. PARP was used to monitor cell viability after infection and MG132 treatment. GAPDH was used as a loading control. (D) Western blot of recombinant maspin in DU145 cells three days post-infection relative to the expression of endogenous maspin by CRL2221 cells. β tubulin was used as a loading control. (E) Growth curves of infected DU145 cells expressing either maspinWT (•), maspinD346E (○) or empty vector control (▪).
In our hands, maspinWT was expressed as early as the second day after adenoviral infection, peaked at 3–5 days post-infection and remained steady up to 15 days post-infection (data not shown). It was noted that when used at the same MOI, adenoviruses encoding different maspin mutants resulted in an unequal level of protein production ( Figure 1C ), with maspinWT expressed at the highest level, followed by the maspinD346E. To test whether maspin mutants were unstable and, thus, subjected to proteasome-mediated degradation, cells were treated with a low dose of proteasome inhibitor MG132 for 6 hrs on the third day after infection. Judging by the integrity of PARP on western blot, the cell viability was not affected by adenoviral infection or MG132 treatment. While there was no net increase in the expression of maspinWT and maspinD346E, MG132 treatment increased the level of detectable maspin1–323, maspin1–340 and maspin1–348. These data suggest that the RCL sequence and its neighboring regions are crucial for maspin stability. Regardless of treatment, maspinD346E was expressed as a stable protein suggesting that the D346 to E346 conservative substitution did not affect the backbone of the maspin structure. We subsequently focused on the comparison of maspinWT and maspinD346E. We also modified the infection MOI for each mutant to equalize the level of protein expression and made sure that the level of maspinWT or maspinD346E expressed in infected DU145 cells did not exceed the level of maspin endogenously expressed by normal immortalized prostate epithelial cells CRL2221 ( Figure 1D ). Consistent with the previous data with stably transfected DU145 cells [7], maspin expression by adenovirus did not affect cell proliferation ( Figure 1E ). Similarly, the expression of maspinD346E did not exert additional effects on the exponential growth of tumor cells. Like maspinWT, maspinD346E expressed by adenovirus did not cause cell death over a period as long as 14 days ( Figure 1E ).
Subcellular Localization of MaspinWT and MaspinD346E
DU145 cells infected with adenovirus expressing either maspinWT or maspinD346E were fixed and subjected to maspin immunofluorescence staining (green) and confocal microscope imaging. Figure 2A shows that maspinWT was predominantly localized in the cytoplasm. In contrast, maspinD346E was completely within the nuclear envelope as judged by the staining of lamin B (red). To confirm that the nuclear localization of maspinD346E was not cell line specific, lung cancer cell line H1299, which expresses no detectable level of maspin ( Figure 2B ), was infected by adenovirus. Similarly to the results with DU145 cells, maspinWT was localized in the cytoplasm, whereas maspinD346E was predominantly localized in the nuclei of H1299 cells ( Figure 2C ).
Figure 2. Subcellular localization of maspinWT and maspin D346E.

(A) Confocal immunofluorescence imaging of maspin (green) and nuclear envelope marker lamin B (red) in DU145 cells after infection. (B) Western blot of recombinant maspin in H1299 cells 3 days post-infection with adenoviral vector (MOI = 20). GAPDH was used as a loading control. (C) Confocal immunofluorescence imaging of recombinant maspin in H1299 after adenoviral infection. Nuclei were counterstained with DAPI. Scale bars = 20 µm. (D) Western blotting of maspin, histone 3, Hsp90, HDAC1 and GAPDH in fractionated lysates of infected H1299 cells. ascertain the differential subcellular distributions of maspinD346E and maspinWT, H1299 cells expressing recombinant maspinWT or maspinD346E were fractionated. As shown in Figure 2D , western blotting of HDAC1, histone 3, and Hsp90 plus GAPDH, demonstrated the purity of the nuclear soluble, nuclear chromatin-bound, and cytosolic fractions, respectively. Both maspinWT and maspin D346E were present in the cytosolic compartment. However, a higher level of maspinD346E, as compared to maspinWT, was detected in the nuclear soluble fraction. Moreover, both nuclear maspinWT and nuclear maspinD346E were detected in the chromatin-bound fractions.
To ascertain the differential subcellular distributions of maspinD346E and maspinWT, H1299 cells expressing recombinant maspinWT or maspinD346E were fractionated. As shown in Figure 2D , western blotting of HDAC1, histone 3, and Hsp90 plus GAPDH, demonstrated the purity of the nuclear soluble, nuclear chromatin-bound, and cytosolic fractions, respectively. Both maspinWT and maspin D346E were present in the cytosolic compartment. However, a higher level of maspinD346E, as compared to maspinWT, was detected in the nuclear soluble fraction. Moreover, both nuclear maspinWT and nuclear maspinD346E were detected in the chromatin-bound fractions.
To quantify maspin protein in each subcellular compartment, it is important to account for maspin in all possible molecular contexts, which may not be recapitulated by the western blot of the monomeric maspin. For this reason, fractionated cellular proteins from infected H1299 cells were subjected to IP with maspin antibody and MS. Spectral counting showed no maspin peptides in the IP control with pre-immune IgG. MaspinWT was only found in the cytosolic compartment with identification of 4 unique peptides and 5 total spectra. In parallel, maspin D346E was found both in the nuclear compartment with the identification of 11 unique peptides and 15 total spectra, and in the cytosolic fraction with the identification of 6 unique peptides and 7 total spectra. Sequence coverage of maspin is shown in Figure S1 (The differential subcellular localizations of maspinWT and maspinD346E were statistically significant (p<0.01, Fisher Exact Test). Considering that the maspin-containing multi-protein complex might be dissociated if the stringency of the cell fraction was high, an independent cell fractionation was performed at lower dissociation stringency. MS/MS detected 12 unique and 648 total histone spectra counts in the nuclear fraction; and 6 unique and 68 total histone spectra counts in the cytosolic fraction (Table S1), demonstrating the effective separation of nuclear and cytosolic fractions by this method. Consistently, maspinWT was found exclusively in the cytosolic fraction whereas maspinD346E was identified in both the cytosol (90% of spectra) and in the nucleus (10% of spectra). We speculate that the less stringent nuclear extraction may have preserved the tighter protein-protein interaction and reduced the efficiency of the IP of free maspin. Taken together, these data confirmed further enrichment of maspinD346E in the nucleus, as compared to the predominant cytosolic presence of maspinWT.
Dominant Nuclear Localization of MaspinD346E in Cells that Naturally Distribute Maspin to Cytoplasm and Nucleus
It is noted that when expressed in low grade adenocarcinoma and cancer cell lines, maspin is always distributed into the cytoplasm and the nucleus. To test whether the subcellular distribution of maspinD346E is distinct as compared to maspinWT in a cancer cell line that naturally expresses and distributes maspin, we utilized the prostate cancer cell line PC3 [7]. In an effort to differentiate between endogenous and recombinant maspin in PC3 cells, PC3 cells were transiently transfected to express maspinWT-GFP or maspinD346E-GFP. The GFP alone was used as a negative control. Figure 3A shows fluorescence imaging of live PC3 cells 48 hrs after the transfection. While the GFP alone exhibited a mixed cytosolic plus nuclear distribution as expected based on the published records [36]–[38], the maspinWT-GFP was equally distributed to cytoplasm and nucleus of each maspin-expressing cell, reminiscent of the natural distribution of endogenous maspin in PC3 cells. In contrast, maspinD346E was particularly enriched in the nucleus (75% of the cells). These data suggest that D346 may act as a dominant cis element for maspin nuclear localization.
Figure 3. Dominant nuclear localization of maspinD346E in cells that naturally distribute maspin to cytoplasm and nucleus.
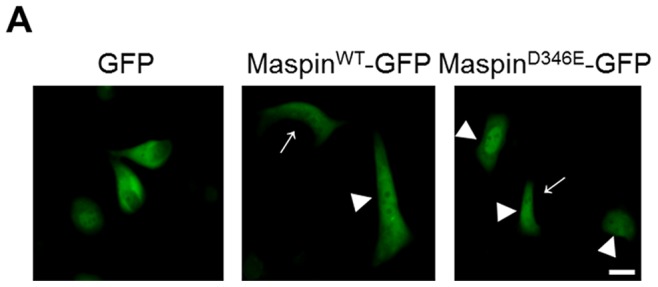
(A) Immunofluorescent imaging of live PC3 cells 48 hrs after transient transfection with pEGFP N1-maspinWT and pEGFP N1-maspinD346E → indicates cytosolic maspin whereas ▸ indicates nuclear maspin. pEGFP was used as a control. Scale bar = 20 µm.
Nuclear Localization of MaspinWT but not MaspinD346E is Inhibited by Nuclear Export Inhibitor Leptomycin B (LMB)
To date, there are no known inhibitors of nuclear import. To examine whether the increased accumulation of nuclear maspinD346E was a result of the deficiency or absence of its nuclear export, transiently transfected H1299 cells expressing either maspinWT-GFP or maspinD346E-GFP were treated with LMB, a potent inhibitor of CRM1 (chromosomal region maintenance/exportin 1), a protein required for nuclear export of proteins containing NES [39]–[41]. Under LMB treatment, molecules that depend on CRM1 export from the nucleus, such as HDAC1, are expected to be accumulated in the nuclei [42]. The essence of using live imaging was based on the need to capture LMB-induced temporal sequence of maspin trafficking. As shown in Figure 4 , to our surprise, LMB treatment of transiently transfected H1299 cells led to significant reduction of nuclear maspinWT-GFP, and had little effect on the nuclear localization of maspinD346E-GFP. It is likely that maspin trafficking was not directly controlled at the step of nuclear export. Instead, our data suggest that the nuclear retention of maspinWT, but not maspin D346E, may be competitively inhibited by a yet-to-be-identified factor (Factor X) that is exported by a CRM1-dependent mechanism and therefore sensitive to LMB treatment. An LMB-led nuclear accumulation of Factor X may consequently displace nuclear maspin. Collectively, this data demonstrates the importance of a single amino acid D346 in controlling the molecular context and nuclear retention of maspin.
Figure 4. Nuclear localization of maspinWT but not maspinD346E is inhibited by nuclear export inhibitor Leptomycin B (LMB).
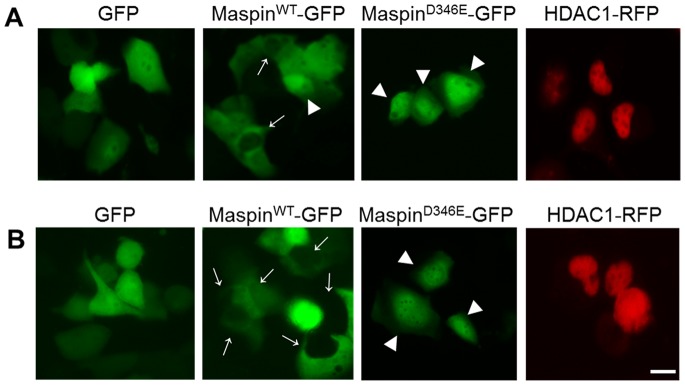
(A) Immunofluorescence imaging (x40) of live H1299 cells 40 hrs after transient transfection with pEGFP N1-maspinWT and pEGFP N1-maspinD346E. → indicates cytosolic maspin whereas ▸ indicates nuclear maspin. pEGFP N1 and was used as a control. (B) Immunofluorescence imaging of live H1299 cells 44 hrs after transient transfection with pEGFP N1-maspinWT and pEGFP N1-maspinD346E in the presence of LMB (2.5 ng/mL, 4 hrs). → indicates cytosolic maspin whereas ▸ indicates nuclear maspin. Expression of GFP alone or HDAC1-RFP was used as controls for (A) and (B). Scale bar = 20 µm.
Maspin Subcellular Localization does not Involve Endoplasmic Reticulum (ER) Retention
Upon careful study of maspin protein sequence, we have recognized that maspin D346 is within a unique intramolecular KDEL motif (K345–L348), which, if located at the very C-terminal of a protein, is also known as an endoplasmic reticulum (ER) retrieval signal [43], [44]. To test the possibility that the intramolecular KDEL motif may still play a role in maspin trafficking, we profiled maspin subcellular localization by immunofluorescence staining in three normal immortalized cell lines from prostate (CRL2221), breast (MCF10A) and lung (BEAS 2B). As shown in Figure 5A , the presence of predominantly nuclear localization of maspin (green), with some cytosolic presence, was shown in early passage CRL2221 and MCF10A cells and it was confirmed in BEAS2B cells [19]. It is worth noting that maspin subcellular distribution between the nucleus and cytoplasm became more heterogeneous as the cell culture passage number increased (data not shown). This observation was consistent with an earlier reports that maspin produced by MCF10A cells was both nucleo-cytoplasmic [45] and secreted [46] where the amount of secreted maspin depended on cell passage. Regardless, the cytosolic maspin expressed by these three cell lines did not co-localize with glucose regulated protein 78 (GRP78, red), an ER marker. Interestingly, ER function, as well as the regulation of ER-resident proteins, has been shown to be differentially regulated in tumor progression [47]–[50]. To test whether tumor cells that distribute recombinant maspinWT and maspinD346E with distinct subcellular patterns (as compared to normal cells) were affected by ER function, prostate tumor cells DU145 infected by adenovirus encoding these two proteins, respectively, were analyzed by immunofluorescence staining in parallel ( Figure 5B ). Not surprisingly, it was shown that the GRP 78 (red) immunofluorescence staining in DU145 cells was dispersed throughout the cytoplasm. MaspinWT was perinuclear and cytosolic whereas maspinD346E was predominantly nuclear. While GRP78 seemed to be an effector of ER function, it is unlikely that maspin trafficking is regulated directly by an ER-dependent mechanism.
Figure 5. Maspin subcellular localization does not involve ER retention.

(A) Confocal immunofluorescence imaging of endogenous maspin (green) and GRP78 (red) in CRL2221, MCF10A and BEAS2B cells. Nuclei were counterstained with DAPI. (B) Confocal immunofluorescence imaging of recombinant maspin (green) and endogenous GRP78 (red) in DU145 cells. Nuclei were counterstained with DAPI. Scale bars = 20 µm.
Increased Nuclear Localization of Maspin Correlates with Increased HDAC1 Interaction
HDAC1 is predominantly a nuclear protein under normal physiological conditions. However, it was noted that under pathological conditions it could be translocated to the cytoplasm [42]. To date, maspin remains the only polypeptide inhibitor of HDAC1 identified. It is important to examine whether the differential trafficking of maspin is, at least in part, accompanied by a consistent trafficking pattern of HDAC1. As shown in Figure 6A , in normal immortalized epithelial cells MCF10A, maspin was primarily co-localized with HDAC1 in the nucleus, suggesting the association of maspin with HDAC1.
Figure 6. Increased nuclear localization of maspinD346E correlates with increased HDAC1 interaction.

(A) Confocal imaging of endogenous maspin (green) and HDAC1 (red) immunofluorescence staining in MCF10A cells. Scale bar = 20 µm (B). Bimolecular fluorescence complementation (BIFC) of live PC3 and DU145 cells transiently transected with pBiFC maspinWT YC or pBiFC maspinD346E YC in combination with pBiFC HDAC1 YN. The BiFC of bJunYN and BiFC bFosYC and BiFC of bJunYN and BiFC bFosYC Δ179–193 were used as positive and negative controls, respectfully, in PC3 cells. Scale bar = 10 µm (C) Immunofluorescence imaging (x40) of live PC3 cells after co-transfection with either pEGFP N1-maspinWT or pEGFP N1 maspinD346E in combination with pcDNA3.1 HDAC1-RFP. → indicates cytosolic maspin/HDAC1 interaction and ▸ indicates nuclear maspin/HDAC1 interaction. Scale bar = 20 µm. (D) Western blot of recombinant maspin and HDAC1 after immunoprecipitation (IP) with maspin antibody in DU145 cells. The mouse IgG was used as a negative control. Total levels of maspin, HDAC1, and loading control GAPDH are shown in the input panel. Numbers below represent normalized HDAC1/maspin ratio.
To test whether HDAC1 and maspin directly interact with each other, we utilized the bimolecular fluorescence complementation (BiFC) [33], [51], a method that detects the signal of protein-protein interaction only when the two proteins make physical contact. We expressed maspin-YC fusion protein and HDAC1-YN fusion protein simultaneously in PC3 and DU145 cells by double transient transfections. As shown in Figure 6B , both maspinWT and maspinD346E directly interacted with HDAC1 as evident by green fluorescence. Interestingly, while the interaction of maspinWT and HDAC1 occurred in the nucleus and the cytoplasm, a robust interaction of maspinD346E with HDAC1 occurred almost exclusively in the nucleus. The interaction of Fos/Jun and ΔFos/Jun in the nucleus was used as positive and negative controls, respectfully, for the BiFC constructs. In parallel, we also utilized the live fluorescence imaging of GFP-tagged maspin and RFP tagged HDAC1 co-expressed in PC3 cells. As shown in Figure 6C , co-localization of maspinWT-GFP and HDAC1-RFP was detected in both, the nucleus and the cytoplasm, in accordance with natural expression of maspin in PC3 cells. In contrast, increased nuclear localization of HDAC1-RFP correlated with increased nuclear interaction of maspinD346E-GFP and HDAC1-RFP. Consistent results were obtained from IP-MS proteomic analysis. As shown in Figure 6D , not only that maspinD346E was able to interact with endogenous HDAC1 but more HDAC1 co-precipitated with maspinD346E relative to the maspinWT (HDAC1 to maspin densitometry ratio 0.2 vs. 0.9 respectively). These data suggest that maspinD346E may have greater affinity towards HDAC1 as compared to maspinWT.
Maspin Nuclear Localization Correlates with Increased Histone Acetylation and Release of HDAC-repressed Gene Expression
To date, maspin remains the only known endogenous polypeptide inhibitor of HDAC1. If maspinD346E had a similar (or higher) affinity to HDAC1 as maspinWT, increased nuclear abundance of maspinD346E may lead to increased level of histone acetylation and up-regulation of HDAC1 target gene expression. Indeed, as compared to maspinWT, maspinD346E expression, via adenoviral infection, in DU145 cells led to a greater level of histone 3 (H3)-Lysine 9 (K9) acetylation ( Figure 7A ). To determine whether the correlation of maspinD346E with increased H3- K9 acetylation was consequential in the up-regulation of HDAC1 target genes, the expression level of 10 HDAC1 target genes known to be up-regulated in the presence of maspinWT was assessed by q-RT-PCR [12]. As shown in Figure 7B , 2 out of 10 HDAC1 target genes tested (Interferon-induced GTP-binding protein Mx1 and protein kinase C-binding protein NELL2) showed further increase in their mRNA expression in the presence of maspinD346E (not statistically significant). Taken together, these data suggest, that maspinD346E may function as a stronger HDAC1 inhibitor, with increased affinity for HDAC1 and increased nuclear compartmentalization.
Figure 7. Maspin nuclear localization correlates with increased histone acetylation and release of HDAC-repressed gene expression.

(A) Western blot of recombinant maspin, HDAC1, and HDAC1 target protein (acetylated Histone 3 at Lysine 9 (H3 Acetyl K9)) in DU145 cells. GAPDH was used as a loading control. (B) Q-RT-PCR of HDAC1 targeted genes differentially regulated by maspin. The threshold cycle (ct) numbers obtained from qRT-PCR were normalized by the internal GAPDH controls and presented as the fold change. MaspinWT: black bar; maspinD346E: white bar.
Discussion
In this study, using the mutagenesis approach we identified amino acid residue D346 as a unique intrinsic element of maspin that can be manipulated to redirect maspin from the cytoplasm to the nucleus, accompanied with increased inhibitory effect of maspin on HDAC1 in tumor cells. This conclusion is substantiated by our evidence derived with multiple cancer cell lines from prostate and lung, with and without natural maspin expression (DU145, PC3 and H1299), and with multiple experimental approaches to re-express intact or fusion maspin proteins (adenoviral infections, transient transfections and BiFC).
This study extends from our earlier evidence that maspin acts as an endogenous HDAC1 inhibitor. While the shift in the subcellular localization of maspin from the nucleus to the cytoplasm during cancer progression may be an early marker for tumor progression, HDAC1 is also shown to be differentially regulated during tumor progression [52]. Under pathological conditions, such as cancer, the overall expression level of HDAC1 is increased. Furthermore, HDAC1 presence and activity are noted in the cytoplasm contributing to the activity and function of certain tumor promoting molecules such as HSP90 [53]. It remains unclear whether the translocation of maspin from the nucleus to the cytoplasm in tumor progression is an adaptation in response to the adverse HDAC1 activity in the cytoplasm. Nonetheless, the ultimate loss of maspin expression in high grade carcinoma will contribute to yet an additional increase of HDAC1 activity, on top of the increased HDAC1 expression, thus promoting tumor progression.
Previously, we demonstrated that either purified or endogenously expressed maspin is bound to and inhibits HDAC1 [7]. Data from the current study confirmed the nuclear interaction of endogenous maspin and endogenous HDAC1 in normal epithelial cells. Considering the multi-component nature and the complex dynamics of the HDAC1 complex [54], the possibility that maspin may actually directly bind to another component in the same HDAC1 complex cannot be unambiguously excluded. Nonetheless, evidence presented in the current study is consistent with the structural consideration [55]–[57] that maspin may indeed directly interact with HDAC1. Further in line with this evidence, functional consequence of maspin/HDAC1 interaction and inhibition has been demonstrated previously by our lab, suggesting that through HDAC1 inhibition, maspin controls a small set of genes involved in epithelial differentiation [12]. Subsequently Lee et al. demonstrated that maspin inhibition of HDAC1 increased acetylation of HDAC1 target protein Ku70 resulting in an increase in apoptosis [58]. In addition to directly blocking the HDAC1-mediated gene repression and deacetylation, maspin may also play a role in dis-coupling the steps of histone modification and DNA modification, and preventing pathological DNA silencing. Our earlier study showed the effect of maspin in reversing pathological DNA methylation-silencing of glutathione S-transferase pi (GSTp), a tumor suppressor implicated in prostate cancer [59]. As compared to pharmacological HDAC inhibitors, the inhibition of HDAC1 by maspin may be more specific, as well as more coordinated in concert with other mechanisms involved in epithelial differentiation. Indeed, while maspin up-regulated HDAC1 target genes, it also led to decreased expression of a specific cluster of genes closely associated with TGFβ signaling [12], which plays a key role in epithelial cell dedifferentiation.
The subcellular localization of maspin does not appear to be regulated by the classical nuclear exclusion pathway or as a direct consequence of HDAC1 subcellular distribution. Instead, data from the current study support a novel hypothetical model as illustrated in Figure 8 . According to this model, maspin may co-exist with another HDAC1-binding factor (X) that is yet to be identified, for HDAC1. Normal epithelial tissue, benign tumor or better differentiated carcinoma with better prognosis and better overall patient survival are associated with nuclear maspin that has a stronger affinity towards HDAC1 than factor X ( Figure 8A ). This stronger affinity of nuclear maspin towards HDAC1 translates into maspin-specific HDAC1 inhibition and upregulation of a small subset of HDAC1 target genes involved in the regulation of epithelial differentiation [12]. Additionally, since the majority of maspin may be HDAC1-associated, the net efflux of maspin favors nuclear localization. Figure 8B may depict the scenario of high grade carcinoma which is characterized by less differentiated phenotype, worse patient prognosis, cytosolic maspin or, in certain cases, the complete loss of maspin. Under these circumstances, the hypothetical HDAC1- associated nuclear factor X may be replaced by X′, which displays stronger affinity towards HDAC1 and competes against maspin for nuclear retention [14].
Figure 8. A hypothetical model explaining disregulation of maspin in tumor progression.
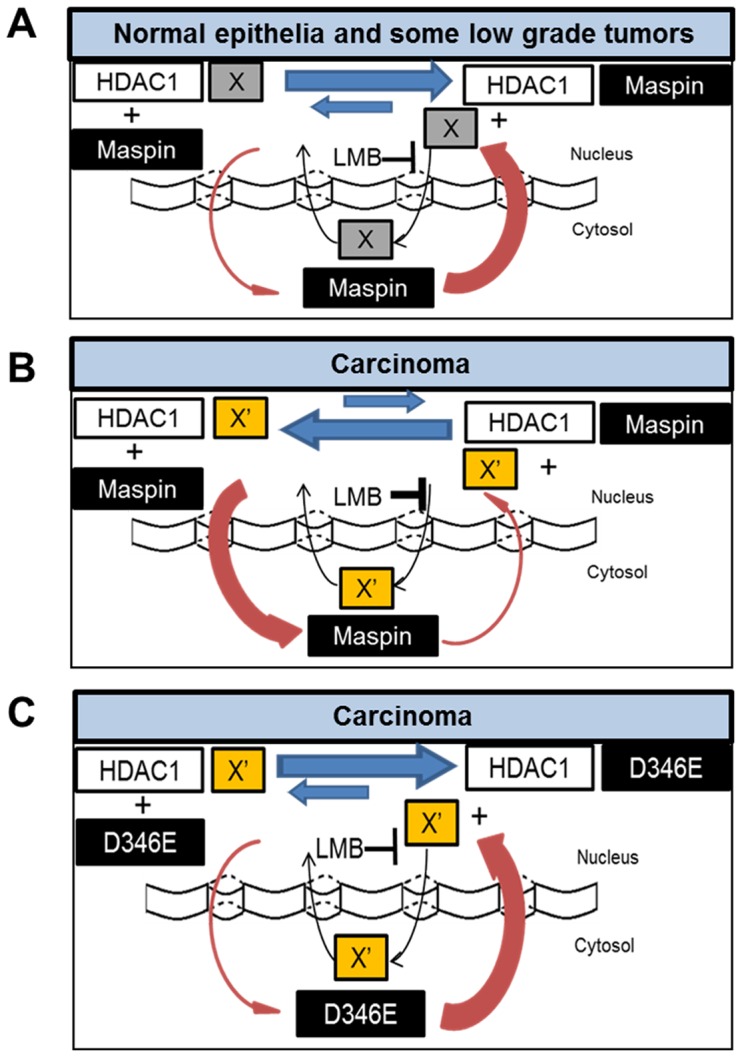
(A) Benign tumor or better differentiated cancer with better prognosis is associated with nuclear maspin. Nuclear maspin has a stronger affinity towards HDAC1 as compared to hypothetical HDAC1-associated nuclear factor X. (B) High grade carcinoma exhibiting a less differentiated phenotype and worse prognosis is associated with cytosolic maspin or loss of maspin. In cancer, hypothetical HDAC1- associated nuclear factor X is X′ and it has a stronger affinity towards HDAC1 as compared to maspin. (C) Therapeutic potential of bioengineered maspin mutant or its derivative in treating advanced disease. Maspin mutant restores maspin nuclear localization and HDAC1 inhibition in advanced disease.
Our experimental evidence suggests that where maspinWT fails to compete against X′ for HDAC1 in the nucleus of poorly differentiated tumor cells, subtle differences in maspin RCL and its neighboring regions, such as maspinD346E, may restore the nuclear presence ( Figure 8C ) as well as the nuclear activity of maspin, and ultimately restore epithelial differentiation [12]. This model helps explain why, as compared to maspinWT that was almost completely excluded from the nucleus upon LMB treatment, the nuclear localization of maspinD346E was not affected by LMB treatment. Further, based on our data and hypothetical model, the nuclear presence of maspinD346E may be dominant even in tumor cells that have already acquired the disregulated subcellular distribution of endogenous maspin. As compared to many other tumor suppressor genes such as p53 that are frequently mutated with numerous hotspots, naturally occurring maspin, with the exception of a couple of popular polymorphisms, is rarely mutated in tumor progression [60]. Thus, a maspin-mimetic drug, as exemplified by maspinD346E, may not suffer from significant interference by the background level of endogenous maspin.
It is well established that tumor cells may be sensitive to the toxicity of HDAC-targeted therapies [61]. While broad-spectrum HDAC inhibitors that target all HDACs in all subcellular compartments have a number of adverse side effects, it is intriguing to postulate that isoform-specific HDAC inhibitors that can be delivered to the correct subcellular compartment may improve the efficacy of the drugs. Lesson learned from maspin revealed the complexity and versatility of endogenous HDAC inhibitor that are not fully recapitulated by synthetic pharmacological HDAC inhibitors in clinical studies [62], [63]. Our experimental evidence with maspinD346E raised the importance of maspin molecular partnership that can be dictated by maspin sequence and may be used as the basis for better design of maspin-mimetic HDAC1-targeted anti-cancer drugs.
In summary, our findings and hypothetical model provide novel insights regarding the regulation and functional attributes of maspin translocation from the nucleus to the cytoplasm in tumor progression. The identification of a critical amino acid residue in the maspin RCL is likely to open a new window of opportunity for the development of maspin-based biologically competent HDAC inhibitor for cancer treatment.
Supporting Information
Sequence coverage of maspin in gel slices from samples isolated from nuclei. Highlighted yellow indicates a peptide containing identified amino acids. Highlighted in green indicates posttranslational modified amino acids. The modification of methionine was oxidation, +16 daltons. Five maspin spectra and corresponding peptide sequence is shown.
(TIF)
Quantification of histones in the nuclear and cytosolic preparations based on total spectral counts.
(PPTX)
Acknowledgments
The authors thank to Mr. Benjamin Jakupovic and Jason Liu for their critical proofreading of the article.
Funding Statement
This work was supported by NIH grants R01CA127735, R01CA84176, R21CA154319 (to Dr. Shijie Sheng). The Microscopy, Imaging and Cytometry Resources Core is supported, in part, by NIH Center grant P30CA22453 to The Karmanos Cancer Institute, Wayne State University and the Perinatology Research Branch of the National Institutes of Child Health and Development, Wayne State University. The Wayne State University Proteomics Core Facility is supported by the NIH Center Grants P30 ES06639, P30 CA22453 and P60 DK020572. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Sheng SJ, Truong B, Fredrickson D, Wu RL, Pardee AB, et al. (1998) Tissue-type plasminogen activator is a target of the tumor suppressor gene maspin. Proceedings of the National Academy of Sciences of the United States of America 95: 499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yoshida M, Furumai R, Nishiyama M, Komatsu Y, Nishino N, et al. (2001) Histone deacetylase as a new target for cancer chemotherapy. Cancer Chemotherapy and Pharmacology 48: S20–S26. [DOI] [PubMed] [Google Scholar]
- 3. Pemberton PA, Wong DT, Gibson HL, Kiefer MC, Fitzpatrick PA, et al. (1995) The Tumor-Suppressor Maspin Does Not Undergo the Stressed to Relaxed Transition or Inhibit Trypsin-Like Serine Proteases - Evidence That Maspin Is Not a Protease Inhibitory Serpin. Journal of Biological Chemistry 270: 15832–15837. [DOI] [PubMed] [Google Scholar]
- 4. Al-Ayyoubi M, Gettins PGW, Volz K (2004) Crystal structure of human maspin, a serpin with antitumor properties - Reactive center loop of maspin is exposed but constrained. Journal of Biological Chemistry 279: 55540–55544. [DOI] [PubMed] [Google Scholar]
- 5. Silverman GA, Bird PI, Carrell RW, Church FC, Coughlin PB, et al. (2001) The serpins are an expanding superfamily of structurally similar but functionally diverse proteins - Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. Journal of Biological Chemistry 276: 33293–33296. [DOI] [PubMed] [Google Scholar]
- 6. Biliran H Jr, Sheng S (2001) Pleiotrophic inhibition of pericellular urokinase-type plasminogen activator system by endogenous tumor suppressive maspin. Cancer Res 61: 8676–8682. [PubMed] [Google Scholar]
- 7. Li X, Yin S, Meng Y, Sakr W, Sheng S (2006) Endogenous inhibition of histone deacetylase 1 by tumor-suppressive maspin. Cancer Res 66: 9323–9329. [DOI] [PubMed] [Google Scholar]
- 8. McGowen R, Biliran H Jr, Sager R, Sheng S (2000) The surface of prostate carcinoma DU145 cells mediates the inhibition of urokinase-type plasminogen activator by maspin. Cancer Res 60: 4771–4778. [PubMed] [Google Scholar]
- 9. Lonardo F, Li X, Kaplun A, Soubani A, Sethi S, et al. (2010) The natural tumor suppressor protein maspin and potential application in non small cell lung cancer. Curr Pharm Des 16: 1877–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gao F, Shi HY, Daughty C, Cella N, Zhang M (2004) Maspin plays an essential role in early embryonic development. Development 131: 1479–1489. [DOI] [PubMed] [Google Scholar]
- 11. Lagger G, O’Carroll D, Rembold M, Khier H, Tischler J, et al. (2002) Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J 21: 2672–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bernardo MM, Meng Y, Lockett J, Dyson G, Dombkowski A, et al. (2011) Maspin reprograms the gene expression profile of prostate carcinoma cells for differentiation. Genes Cancer 2: 1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kaplun A, Dzinic S, Bernardo MM, Sheng SJ (2012) Tumor Suppressor Maspin as a Rheostat in HDAC Regulation to Achieve the Fine-Tuning of Epithelial Homeostasis. Critical Reviews in Eukaryotic Gene Expression 22: 249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pierson CR, McGowen R, Grignon D, Sakr W, Dey J, et al. (2002) Maspin is up-regulated in premalignant prostate epithelia. Prostate 53: 255–262. [DOI] [PubMed] [Google Scholar]
- 15. Cher ML, Biliran HR, Bhagat S, Meng YH, Che MX, et al. (2003) Maspin expression inhibits osteolysis, tumor growth, and angiogenesis in a model of prostate cancer bone metastasis. Proceedings of the National Academy of Sciences of the United States of America 100: 7847–7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang M, Magit D, Sager R (1997) Expression of maspin in prostate cells is regulated by a positive Ets element and a negative hormonal responsive element site recognized by androgen receptor. Proc Natl Acad Sci U S A 94: 5673–5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zou Z, Anisowicz A, Hendrix MJ, Thor A, Neveu M, et al. (1994) Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science 263: 526–529. [DOI] [PubMed] [Google Scholar]
- 18. Xie L, Ying WT, Li BY, Zhang KT, Qian XH, et al. (2003) [Proteomics-based identification of Maspin differential expression in bronchial epithelial immortalized cells and malignant transformation cells]. Ai Zheng 22: 463–466. [PubMed] [Google Scholar]
- 19. Lonardo F, Li XH, Siddiq F, Singh R, At-Abbadi M, et al. (2006) Maspin nuclear localization is linked to favorable morphological features in pulmonary adenocarcinoma. Lung Cancer 51: 31–39. [DOI] [PubMed] [Google Scholar]
- 20. Lockett J, Yin S, Li X, Meng Y, Sheng S (2006) Tumor suppressive maspin and epithelial homeostasis. J Cell Biochem 97: 651–660. [DOI] [PubMed] [Google Scholar]
- 21. Frey A, Soubani AO, Adam AK, Sheng S, Pass HI, et al. (2009) Nuclear, compared with combined nuclear and cytoplasmic expression of maspin, is linked in lung adenocarcinoma to reduced VEGF-A levels and in Stage I, improved survival. Histopathology 54: 590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mohsin SK, Zhang M, Clark GM, Craig Allred D (2003) Maspin expression in invasive breast cancer: association with other prognostic factors. J Pathol 199: 432–435. [DOI] [PubMed] [Google Scholar]
- 23. Solomon LA, Munkarah AR, Schimp VL, Arabi MH, Morris RT, et al. (2006) Maspin expression and localization impact on angiogenesis and prognosis in ovarian cancer. Gynecol Oncol 101: 385–389. [DOI] [PubMed] [Google Scholar]
- 24. Sood AK, Fletcher MS, Gruman LM, Coffin JE, Jabbari S, et al. (2002) The paradoxical expression of maspin in ovarian carcinoma. Clin Cancer Res 8: 2924–2932. [PubMed] [Google Scholar]
- 25.Goulet B, Chan G, Chambers AF, Lewis JD (2011) An emerging role for the nuclear localization of maspin in the suppression of tumor progression and metastasis (1) (1) This article is part of Special Issue entitled Asilomar Chromatin and has undergone the Journal’s usual peer review process. Biochem Cell Biol. [DOI] [PubMed]
- 26. Goulet B, Kennette W, Ablack A, Postenka CO, Hague N, et al. (2012) Nuclear localization of maspin is essential for its inhibition of tumor growth and metastasis. Biochemistry and Cell Biology-Biochimie Et Biologie Cellulaire 90: 109–109. [Google Scholar]
- 27. Bodenstine TM, Seftor REB, Khalkhali-Ellis Z, Seftor EA, Pemberton PA, et al. (2012) Maspin: molecular mechanisms and therapeutic implications. Cancer and Metastasis Reviews 31: 529–551. [DOI] [PubMed] [Google Scholar]
- 28. Feldherr CM, Akin D (1994) Role of Nuclear Trafficking in Regulating Cellular-Activity. International Review of Cytology - a Survey of Cell Biology, Vol 151 151: 183–228. [DOI] [PubMed] [Google Scholar]
- 29. Law RH, Irving JA, Buckle AM, Ruzyla K, Buzza M, et al. (2005) The high resolution crystal structure of the human tumor suppressor maspin reveals a novel conformational switch in the G-helix. Journal of Biological Chemistry 280: 22356–22364. [DOI] [PubMed] [Google Scholar]
- 30. Ravenhill L, Wagstaff L, Edwards DR, Ellis V, Bass R (2010) G-helix of Maspin Mediates Effects on Cell Migration and Adhesion. Journal of Biological Chemistry 285: 36285–36292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sheng SJ, Pemberton PA, Sager R (1994) Production, Purification, and Characterization of Recombinant Maspin Proteins. Journal of Biological Chemistry 269: 30988–30993. [PubMed] [Google Scholar]
- 32. Zou ZQ, Anisowicz A, Hendrix MJC, Thor A, Neveu M, et al. (1994) Maspin, a Serpin with Tumor-Suppressing Activity in Human Mammary Epithelial-Cells. Science 263: 526–529. [DOI] [PubMed] [Google Scholar]
- 33. Kerppola TK (2006) Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nature Protocols 1: 1278–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yin SP, Li XH, Meng YH, Finley RL, Sakr W, et al. (2005) Tumor-suppressive maspin regulates cell response to oxidative stress by direct interaction with glutathione S-transferase. Journal of Biological Chemistry 280: 34985–34996. [DOI] [PubMed] [Google Scholar]
- 35. Al-Ayyoubi M, Gettins PG, Volz K (2004) Crystal structure of human maspin, a serpin with antitumor properties: reactive center loop of maspin is exposed but constrained. J Biol Chem 279: 55540–55544. [DOI] [PubMed] [Google Scholar]
- 36. Grebenok RJ, Pierson E, Lambert GM, Gong FC, Afonso CL, et al. (1997) Green-fluorescent protein fusions for efficient characterization of nuclear targeting. Plant J 11: 573–586. [DOI] [PubMed] [Google Scholar]
- 37. Ogawa H, Inouye S, Tsuji FI, Yasuda K, Umesono K (1995) Localization, trafficking, and temperature-dependence of the Aequorea green fluorescent protein in cultured vertebrate cells. Proc Natl Acad Sci U S A 92: 11899–11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Morin X, Daneman R, Zavortink M, Chia W (2001) A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. Proceedings of the National Academy of Sciences of the United States of America 98: 15050–15055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wolff B, Sanglier JJ, Wang Y (1997) Leptomycin B is an inhibitor of nuclear export: Inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chemistry & Biology 4: 139–147. [DOI] [PubMed] [Google Scholar]
- 40. OssarehNazari B, Bachelerie F, Dargemont C (1997) Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science 278: 141–144. [DOI] [PubMed] [Google Scholar]
- 41. Nishi K, Yoshida M, Fujiwara D, Nishikawa M, Horinouchi S, et al. (1994) Leptomycin-B Targets a Regulatory Cascade of Crm1, a Fission Yeast Nuclear-Protein, Involved in Control of Higher-Order Chromosome Structure and Gene-Expression. Journal of Biological Chemistry 269: 6320–6324. [PubMed] [Google Scholar]
- 42. Kim JY, Shen S, Dietz K, He Y, Howell O, et al. (2010) HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nature Neuroscience 13: 180–U163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pelham HRB (1988) Evidence That Luminal Er Proteins Are Sorted from Secreted Proteins in a Post-Er Compartment. Embo Journal 7: 913–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Munro S, Pelham HRB (1987) A C-Terminal Signal Prevents Secretion of Luminal Er Proteins. Cell 48: 899–907. [DOI] [PubMed] [Google Scholar]
- 45. Teoh SSY, Whisstock JC, Bird PI (2010) Maspin (SERPINB5) Is an Obligate Intracellular Serpin. Journal of Biological Chemistry 285: 10862–10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Endsley MP, Hu Y, Deng Y, He X, Warejcka DJ, et al. (2011) Maspin, the molecular bridge between the plasminogen activator system and beta1 integrin that facilitates cell adhesion. J Biol Chem 286: 24599–24607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shin BK, Wang H, Yim AM, Le Naour F, Brichory F, et al. (2003) Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. Journal of Biological Chemistry 278: 7607–7616. [DOI] [PubMed] [Google Scholar]
- 48. Delpino A, Piselli P, Vismara D, Vendetti S, Colizzi V (1998) Cell surface localization of the 78 kD glucose regulated protein (GRP 78) induced by thapsigargin. Molecular Membrane Biology 15: 21–26. [DOI] [PubMed] [Google Scholar]
- 49.Ni M, Zhou H, Wey SA, Baumeister P, Lee AS (2009) Regulation of PERK Signaling and Leukemic Cell Survival by a Novel Cytosolic Isoform of the UPR Regulator GRP78/BiP. Plos One 4. [DOI] [PMC free article] [PubMed]
- 50. Belfi CA, Chatterjee S, Gosky DM, Berger SJ, Berger NA (1999) Increased sensitivity of human colon cancer cells to DNA cross-linking agents after GRP78 up-regulation. Biochemical and Biophysical Research Communications 257: 361–368. [DOI] [PubMed] [Google Scholar]
- 51. Hu CD, Chinenov Y, Kerppola TK (2002) Visualization of interactions among bZip and Rel family proteins in living cells using bimolecular fluorescence complementation. Molecular Cell 9: 789–798. [DOI] [PubMed] [Google Scholar]
- 52. Song Y, Shiota M, Tamiya S, Kuroiwa K, Naito S, et al. (2011) The significance of strong histone deacetylase 1 expression in the progression of prostate cancer. Histopathology 58: 773–780. [DOI] [PubMed] [Google Scholar]
- 53. Nishioka C, Ikezoe T, Yang J, Takeuchi S, Koeffler HP, et al. (2008) MS-275, a novel histone deacetylase inhibitor with selectivity against HDAC1, induces degradation of FLT3 via inhibition of chaperone function of heat shock protein 90 in AML cells. Leukemia Research 32: 1382–1392. [DOI] [PubMed] [Google Scholar]
- 54. Kato T, Shimono Y, Hasegawa M, Jijiwa M, Enomoto A, et al. (2009) Characterization of the HDAC1 Complex That Regulates the Sensitivity of Cancer Cells to Oxidative Stress. Cancer Research 69: 3597–3604. [DOI] [PubMed] [Google Scholar]
- 55. Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, et al. (1999) Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401: 188–193. [DOI] [PubMed] [Google Scholar]
- 56. Min J, Landry J, Sternglanz R, Xu RM (2001) Crystal structure of a SIR2 homolog-NAD complex. Cell 105: 269–279. [DOI] [PubMed] [Google Scholar]
- 57. Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, et al. (2004) Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 12: 1325–1334. [DOI] [PubMed] [Google Scholar]
- 58. Lee SJ, Jang H, Park C (2012) Maspin increases Ku70 acetylation and Bax-mediated cell death in cancer. Int J Mol Med 29: 225–230. [DOI] [PubMed] [Google Scholar]
- 59. Li X, Kaplun A, Lonardo F, Heath E, Sarkar FH, et al. (2011) HDAC1 inhibition by maspin abrogates epigenetic silencing of glutathione S-transferase pi in prostate carcinoma cells. Mol Cancer Res 9: 733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jang HL, Nam E, Lee KH, Yeom S, Son HJ, et al. (2008) Maspin polymorphism associated with apoptosis susceptibility and in vivo tumorigenesis. International Journal of Molecular Medicine 22: 333–338. [PubMed] [Google Scholar]
- 61. Spiegel S, Milstien S, Grant S (2012) Endogenous modulators and pharmacological inhibitors of histone deacetylases in cancer therapy. Oncogene 31: 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Balasubramanian S, Verner E, Buggy JJ (2009) Isoform-specific histone deacetylase inhibitors: the next step? Cancer Lett 280: 211–221. [DOI] [PubMed] [Google Scholar]
- 63. Delcuve GP, Khan DH, Davie JR (2013) Targeting class I histone deacetylases in cancer therapy. Expert Opin Ther Targets 17: 29–41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequence coverage of maspin in gel slices from samples isolated from nuclei. Highlighted yellow indicates a peptide containing identified amino acids. Highlighted in green indicates posttranslational modified amino acids. The modification of methionine was oxidation, +16 daltons. Five maspin spectra and corresponding peptide sequence is shown.
(TIF)
Quantification of histones in the nuclear and cytosolic preparations based on total spectral counts.
(PPTX)