

Abstract
Prediabetic NOD mice exhibit hyperglucagonemia, possibly due to an intrinsic α-cell defect. Here, we show that the expression of a potential glucagon inhibitor, the adenosine A1 receptor (Adora1), is gradually diminished in α-cells of NOD mice, autoantibody-positive (AA+) and overtly type 1 diabetic (T1D) patients during the progression of disease. We demonstrated that islet inflammation was associated with loss of Adora1 expression through the alternative splicing of Adora1. Expression of the spliced variant (Adora1-Var) was upregulated in the pancreas of 12-week-old NOD versus age-matched NOD.B10 (non–diabetes-susceptible) control mice and was detected in the pancreas of AA+ patients but not in control subjects or overtly diabetic patients, suggesting that inflammation drives the splicing of Adora1. We subsequently demonstrated that Adora1-Var expression was upregulated in the islets of NOD.B10 mice after exposure to inflammatory cytokines and in the pancreas of NOD.SCID mice after adoptive transfer of activated autologous splenocytes. Adora1-Var encodes a dominant-negative N-terminal truncated isoform of Adora1. The splicing of Adora1 and loss of Adora1 expression on α-cells may explain the hyperglucagonemia observed in prediabetic NOD mice and may contribute to the pathogenesis of human T1D and NOD disease.
Type 1 diabetes (T1D) results from the gradual autoimmune destruction of pancreatic β-cells that occurs over the course of months to years. Owing to the lack of overt symptoms, prediabetic individuals are difficult to identify, and tissues from such individuals are not readily available for study. By using the well-established non-obese diabetic (NOD) mouse model of T1D, it is possible to follow disease progression and identify defects that occur before the onset of hyperglycemia. Disease occurs spontaneously with a predictable course: Peri-insulitis occurs at 4 to 8 weeks of age, and the onset of infiltrative/destructive insulitis occurs at ∼12 weeks of age. This is followed by substantial β-cell destruction and resultant hyperglycemia. Disease does not occur in the major histocompatibility complex congenic NOD.B10 control mice.
During T1D, the control of glucagon secretion is impaired. Glucagon stimulates gluconeogenesis and glycogenolysis and thus plays a critical role in blood glucose homeostasis. Elevated fasting plasma glucagon levels (1) and reduced suppression of glucagon secretion after hyperglycemia (2,3) occurs in NOD mice and in spontaneously diabetic KK mice. Similarly, early- and late-stage T1D patients exhibit diminished suppression of glucagon secretion after the administration or ingestion of glucose (4–6). This induced hyperglucagonemia increases hepatic glucose release and exacerbates postprandial hyperglycemia (6). Hypersecretion of glucagon is primarily attributed to a deficiency of insulin or somatostatin. However, islets can regulate glucagon over a glucose concentration range that is not associated with changes in insulin or somatostatin (7), and prediabetic NOD and KK mice show elevated fasting and nonfasting plasma glucagon levels without changes in plasma insulin or blood glucose (1–3,8). In addition, increased α-cell function has been demonstrated in diabetic NOD mice (9) and enlarged glucagon-containing granules in streptozotocin-treated mice (10), suggesting that an intrinsic α-cell defect may contribute to the early stages of disease pathophysiology.
We investigated this by performing microarray analysis to identify dysregulated genes in the islets of NOD mice. Interestingly, Adora1 expression was found to be downregulated. The adenosine A1 receptor (Adora1) is a G-protein coupled receptor (GPCR) that is involved in maintaining glucose homeostasis and regulating glucagon secretion (11,12). Elevated plasma glucagon levels after a glucose challenge or ingestion of a high-fat diet and increased duration of glucagon release during hyperglycemia have been observed in Adora1 knockout (KO) mice (12–14), suggesting that loss of Adora1 expression in α-cells may contribute to the pathology of NOD disease and human T1D.
We studied the gene and protein expression of Adora1 through the progression of T1D and found that Adora1 expression was gradually diminished in α-cells of NOD mice and human autoantibody-positive (AA+) and long-term T1D patients as disease progressed. Reduced Adora1 expression was associated with increased lymphocytic infiltration and inflammation of the islets and may occur through alternative splicing of the Adora1 gene. Splicing of Adora1 in islets was induced in vitro and in vivo by inflammatory cytokines and was upregulated in the pancreas of 12-week-old NOD mice at the initiation of destructive insulitis. The dominant-negative splice variant, Adora1-Var, was also detected in the pancreas of AA+ patients but not in the noninflamed pancreata of control subjects or long-term T1D patients. These findings suggest that loss of functional ADORA1 expression may contribute to the unrestrained secretion of glucagon, and may represent an early intrinsic α-cell defect that contributes to the progression and clinical manifestation of T1D.
RESEARCH DESIGN AND METHODS
Mice.
Female NOD/LtJ (NOD), NOD.B10Sn-H2b/J (NOD.B10), NOD.CB17-Prkdcscid/J (NOD.SCID), and NOD.Cg-Tg(TcraBDC2.5)1DoiTg(TcrbBDC2.5)Doi/DoiJ (NOD.BDC2.5) mice were purchased from The Jackson Laboratory. Animals were maintained under pathogen-free conditions at the Stanford School of Medicine Animal Facility, according to institutional guidelines and under approved protocols in the Stanford Medical Center Department of Comparative Medicine. NOD mice of various ages served as a model of human T1D at different stages of disease, with 12 weeks of age being most comparable to AA+ patients. Major histocompatibility complex congenic healthy NOD.B10 mice served as controls. NOD.SCID mice were used to study the effect of insulitis on gene splicing and served as recipients for activated splenocytes of NOD.BDC.2.5 mice that migrate to the islets.
Serum glucagon measurements.
Blood glucose was measured and serum was collected from prediabetic, overnight-fasted NOD and NOD.B10 mice (10–13 weeks of age). Glucagon was measured using the Glucagon EIA kit (Sigma-Aldrich), and data analysis was performed using GraphPad Prism 6 software, according to the manufacturers’ instructions.
Human samples.
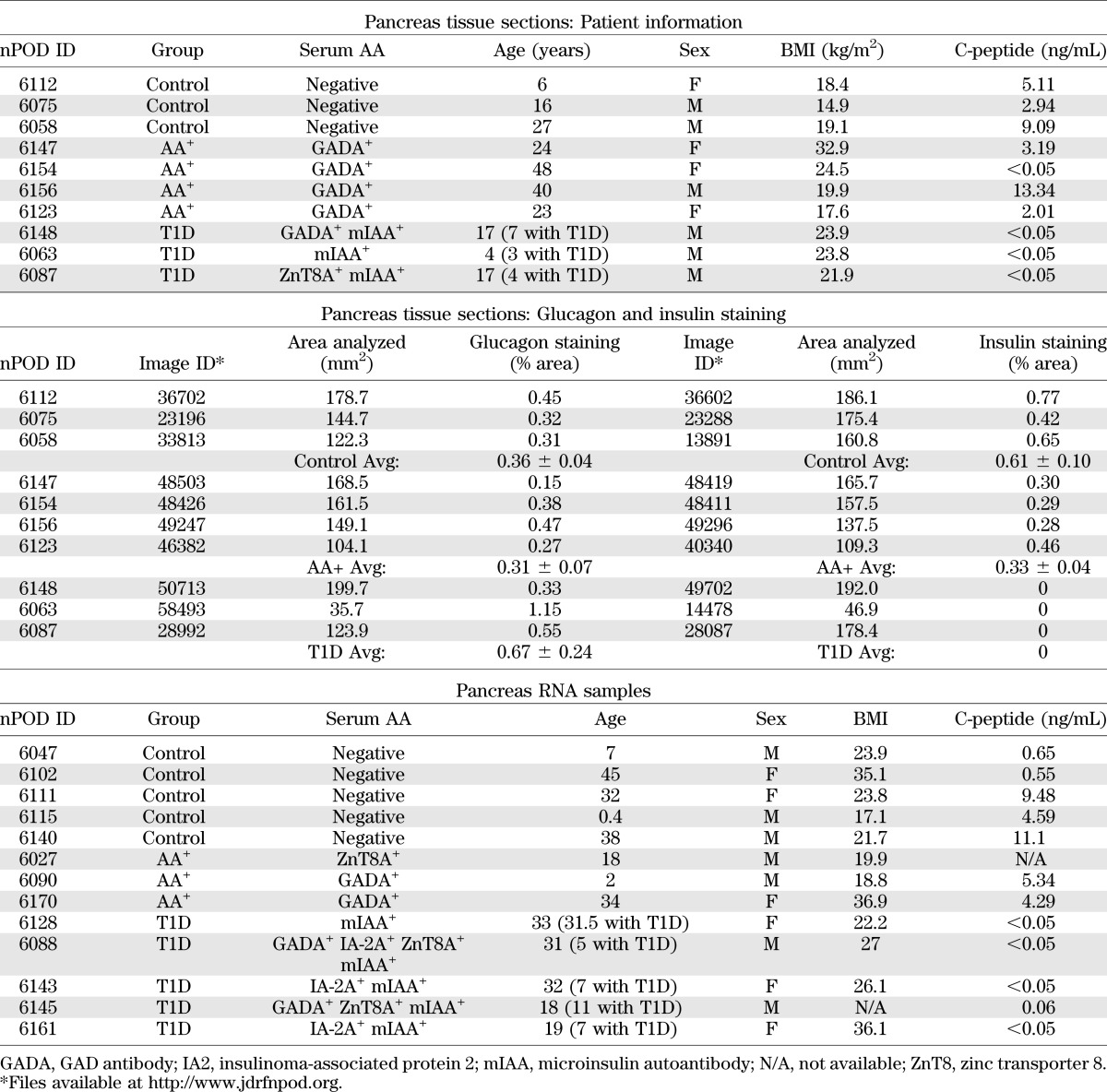
Human pancreas samples used for histology and RT-PCR were obtained through the JDRF network for Pancreatic Organ Donors with Diabetes (nPOD). High-resolution images of glucagon- and insulin-stained pancreas sections were provided by nPOD and are available at http://www.jdrfnpod.org/online-pathology.php. Glucagon and insulin staining was assessed using Adobe Photoshop CS4, and the total area analyzed was measured using Aperio WebScope (Leica). Sample information is summarized in Table 1. Additional information is available at http://www.jdrfnpod.org/online-pathology.php.
TABLE 1.
Human pancreas samples
Microarray analysis.
The pancreata of NOD and NOD.B10 mice (n = 6) were frozen, and islets were isolated by laser capture microdissection (LCM). Cryosections (8 μm) were stained with an Arcturus HistoGene Staining kit (Applied Biosystems), and LCM was performed using the Leica AS LMD system. RNA was extracted from 40 islet sections and assessed as described below. Samples were preamplified using the TrueLabeling-PicoAMP kit (SABiosciences), postlabeled with Cy5, and run against a Cy3-labeled mouse Universal RNA control (SABiosciences). Microarrays were performed using the Whole Mouse Genome Microarray Kit, 4 × 44K two-color arrays (Agilent Technologies), and data were analyzed using GeneSpring GX 11.5, as previously described (15). Samples were filtered for detected entities, and a t test was performed to identify genes that were changed by twofold or more.
Islet isolation.
Pancreatic islets were isolated from 12- and 20-week-old NOD and NOD.B10 mice, as previously described (16). To examine the effect of inflammation on Adora1 splicing, islets from three to five mice were pooled, and ∼50 individual islets were incubated for 24 h in the supernatant of activated or nonactivated 12-week-old NOD.B10 splenocytes.
Splenocyte activation.
For activation of splenocytes to obtain “inflammatory” cytokines, cells were cultured in plates previously coated with anti-CD3 and anti-CD28 (2 mg/mL) in the presence of lipopolysaccharide (1 mg/mL) and interferon-α (IFN-α) (200 units/mL) for 24 h. Nonactivated splenocytes were cultured in RPMI medium. The supernatants of these cells were collected and analyzed by Luminex arrays (Human Immune Monitoring Center, Stanford, CA).
Adoptive transfer of activated NOD.BDC2.5 splenocytes to NOD.SCID mice.
Splenocytes from 12-week-old female NOD.BDC2.5 mice were activated for 24 h as described above. Cells were washed in PBS, and 5 × 106 cells (in 200 μL PBS) were injected intraperitoneally into female 12-week-old NOD.SCID mice. Control mice were injected with an equal volume of PBS. Tissues were harvested 48 h later, and RNA was extracted as described below. Migration of NOD.BDC2.5 splenocytes into the pancreas was confirmed by PCR using BDC2.5 transgene primers available at JAX (Supplementary Fig. 1).
RNA extraction, cDNA synthesis, and quantitative PCR.
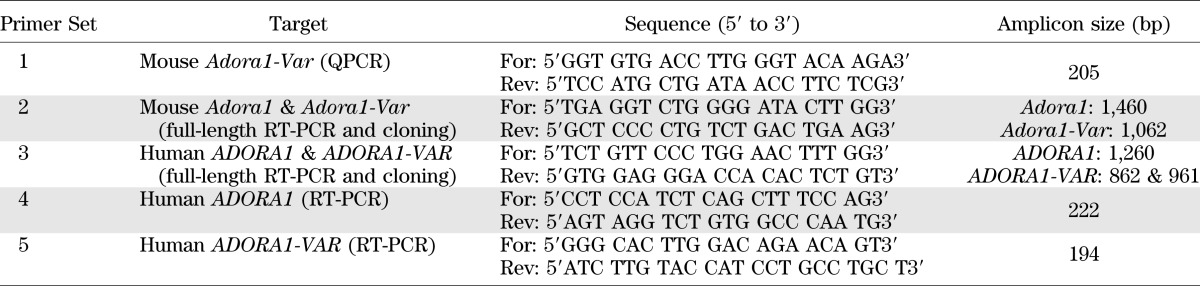
Total RNA was extracted using Trizol reagent and the Qiagen RNeasy mini or micro kit (islets), as previously described (15). Total RNA was assessed using the Agilent 2100 Bioanalyzer and the RNA 6000 Pico Reagent Kit (Agilent). First-strand cDNA was generated using SuperScript III (Invitrogen). Quantitative PCR (QPCR) was performed to measure Adora1, Adora2a, Gcg, Ifng, Gapdh, 18S rRNA, and Actb, using assays from Applied Biosystems. Adora1-Var was measured with primer set 1 (Table 2). The forward primer spans exons 1 and 3 of the mouse Adora1 gene (NM_001039510.1). This assay amplified only the product of interest and efficiently amplified up to a threshold cycle of 38 (Supplementary Fig. 3). cDNA was preamplified using the TaqMan PreAmp Master Mix (Applied Biosystems) before QPCR for measurements in islet tissues and pancreas of NOD.SCID mice. Assays were performed using the 7900HT Fast Real-Time PCR System (Applied Biosystems) and the TaqMan Gene Expression Master Mix (Applied Biosystems) or SsoFast EvaGreen Supermix (Bio-Rad). The comparative Ct method for relative quantification (ΔΔCt) was used.
TABLE 2.
Primers for RT-PCR, cloning, and QPCR
RT-PCR and cloning
Mouse Adora1-Var.
RT-PCR was performed using the Type-it Mutation Detect PCR Kit (Qiagen) and primer set 2 (Table 2). Two amplicons, 1,460 bp and 1,062 bp, corresponding to Adora1 and Adora1-Var, respectively, were produced. The 1,062 bp product was cloned into TOPO (Invitrogen) and sequenced.
Human ADORA1-VAR.
Human pancreas RNA samples with intact 18S and 28S rRNA were used for these studies. Primers were designed to examine if a human equivalent of the mouse Adora1-Var is expressed in the pancreas (Table 2, primer set 5). The forward primer spans exon 2 and 4 and only detects a splice variant lacking exon 3 of the ADORA1 gene (NM_000674.2). Primers targeting only exon 3 were designed to detect the full-length human ADORA1 gene (Table 2, primer set 4). For cloning into TOPO, primers spanning the start and stop codon of ADORA1-VAR were used (Table 2, primer set 3). Fusion proteins were synthesized by standard subcloning techniques using the PCR primers listed in Supplementary Table 1.
Immunohistochemistry.
Frozen pancreas sections were fixed in 4% formaldehyde in PBS for 15 min, washed in PBS, and incubated in blocking buffer (5% normal goat serum containing 0.3% Triton X-100 in PBS) for 30 min and in primary antibody at 4°C for 48 h. Tissues were washed, incubated in secondary antibody overnight at 4°C, washed, and slides were overlain with cover slips. Primary and secondary antibodies were diluted in blocking buffer and are listed in Supplementary Table 2. The specificity of the Adora1 antibody (#ab82477) was confirmed by antibody neutralization using the control peptide (Abcam peptide #ab91076). Tissue sections incubated in the immunoneutralized antibody or in blocking buffer in place of the primary antibody showed no staining. Confocal images were taken using the Leica SP2 AOBS Confocal Laser Scanning Microscope and Leica Confocal 2.5 software.
Cell transfection.
Human embryonic kidney (HEK) 293 cells were transfected with various plasmids alone or in combination using Lipofectamine 2000 (Invitrogen). Adora1-tGFP and ADORA1-tGFP were purchased from OriGene. Adora1-Var-EGFP, Adora1-Var-tRFP, ADORA1-VAR-EGFP, and ADORA1-VAR-tRFP were synthesized as described above. Cells were fixed 24 h after transfection and mounted in Vectashield medium containing DAPI (Vector Laboratories). Confocal images were taken as described above.
Coimmunoprecipitation.
HEK293 cells were mock transfected or transfected with 1 μg of ADORA1-tGFP, ADORA1-VAR-tRFP, or both plasmids, as described above. After 24 h, samples were treated for 20 min with the ADORA1-selective agonist N6-cyclopentyladenosine (CPA, 100 nmol/L) and then treated with the cross-linking reagent Dithiobis(succinimidyl propionate) (2 mmol/L) for 30 min and Tris-HCl (pH 7.5, 20 mmol/L) for 15 min. Cells were lysed, and coimmunoprecipitation was performed according to manufacturer’s instructions using Protein A/G–conjugated agarose beads (Pierce) with an anti-tRFP antibody (Evrogen). Samples were eluted in 1× SDS sample buffer, and immunoblotting was performed using an anti-tGFP antibody (OriGene). The input samples were also immunoblotted for β-actin expression.
Measurement of cAMP levels in transfected HEK293 cells.
HEK293 cells (2.5 × 104) were grown to ∼80% confluency in 96-well plates and transfected with 100–200 ng of various plasmids alone or in combination using Lipofectamine 2000. Cells were treated with one or more of the following drugs for 20 min: the phosphodiesterase inhibitor R0-20-1724 (25 μmol/L), the adenylate cyclase activator forskolin (5 μmol/L), the ADORA1-selective agonist CPA (100 nmol/L), and the ADORA1-selective antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX, 100 nmol/L). Cells were lysed. Protein concentrations were determined by Bradford assay (Bio-Rad), and cAMP levels were measured using the Cyclic Nucleotide XP EIA Kit (Cell Signaling). Data analysis was performed using GraphPad Prism 6 software.
RESULTS
Microarray and QPCR analysis of the islets of NOD versus NOD.B10 mice.
For microarray analysis, islets were isolated by LCM rather than by collagenase digestion to avoid changes in gene expression that might result from a lengthy and harsh isolation procedure (17). Each sample yielded between 35 and 140 ng of RNA, with RNA integrity numbers (RIN) of ∼3, indicating degradation (Fig. 1A). Microarray analysis revealed the expression of ∼27,000 entities, with 540 and 242 entities that were up- and downregulated, respectively, by twofold or more in NOD versus NOD.B10 islets. As expected, various islet peptide genes were downregulated, whereas certain chemokine and cytokine genes were upregulated (Fig. 1B–D). The expression of Adora1 and Adora2a was examined because these receptors have been shown to regulate glucose metabolism and play a role in the development of T1D and T2D (11, 13, 18–20). Surprisingly, Adora1 was drastically reduced in the islets of NOD mice (Fig. 1E).
FIG. 1.

Gene expression in the islets of NOD vs. NOD.B10 mice. A: A bioanalyzer gel image shows RNA samples extracted from islets that were isolated by LCM. The 18S and 28S rRNA bands are indicated. Gene expression levels of pancreatic islet peptides (B), cytokines (C), chemokines (D), and adenosine receptors (E) in the islets of 12-week-old NOD vs. NOD.B10 mice (n = 6 per group), as measured by microarray analysis. F: A representative bioanalyzer gel image shows RNA samples extracted from the islets of 12- and 20-week-old NOD mice. Islets were isolated by collagenase digestion. RNA from age-matched NOD.B10 mice gave similar results (data not shown). QPCR results show Ifng (G), Gcg (H), Adora1 (I), and Adora2a (J) gene expression in the islets of 12- and 20-week-old NOD vs. NOD.B10 mice. *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001 by two-tailed unpaired Student t test of NOD vs. age-matched NOD.B10 mice (n ≥ 5 per group). Fasting serum glucagon (K) and blood glucose levels (L) in NOD and NOD.B10 mice (10–13 weeks old, n > 20 per group). Data represent mean ± SEM. wk, week.
To confirm this finding, QPCR was performed in islets of 12- and 20-week-old NOD and NOD.B10 mice that were isolated by collagenase digestion. This isolation technique yielded between 500 and 2,000 ng RNA per sample, with RIN values of >7 (Fig. 1F). Similar to the microarray results, QPCR showed increased Ifng, reduced Adora1, and no significant change in Adora2a gene expression in the islets of 12-week-old NOD versus NOD.B10 mice. At 20 weeks, Ifng and Adora2a were upregulated, whereas Adora1 and Gcg were downregulated in the islets of NOD versus NOD.B10 mice (Fig. 1G–J). Because Adora1 inhibits glucagon secretion (12,13), the loss of Adora1 expression is consistent with the increased levels of fasting serum glucagon observed in prediabetic NOD mice compared with age-matched NOD.B10 mice (Fig. 1K and L). All microarray data are available at the Gene Expression Omnibus (GEO) Database (GEO series accession number: GSE45897).
Expression of Adora1/ADORA1 in the pancreas during the progression of NOD disease and T1D.
Adora1-immunoreactivity was present on all glucagon-positive α-cells and on a few insulin-positive β-cells of healthy NOD.B10 mice of various ages (Fig. 2A and Supplementary Fig. 4). This pattern was similar in 8-week-old NOD mice before the onset of destructive insulitis (Fig. 2B). At 12 weeks of age, during the onset of destructive insulitis, fewer α-cells were observed around the periphery of the islet, and some α-cells did not express Adora1. Double staining with anti-CD3 showed that loss of Adora1 expression correlated with increased CD3 expression (Fig. 2C). By 20 weeks of age, few islets were observed in the NOD pancreas, and those few islets consisted mainly of α-cells that did not express Adora1 (Fig. 2B). Surprising similarities were observed in the pattern of ADORA1 staining in human controls and NOD.B10 mice, AA+ patients and 12-week-old NOD mice, and T1D patients and 20-week-old NOD mice (Fig. 3). In particular, nondiabetic control patients who had abundant glucagon and insulin expression in their islets (Table 1 and Supplementary Fig. 2) were found to express ADORA1 on the majority of glucagon-positive α-cells and on few β-cells (Fig. 3A and D). AA+ patients, who expressed slightly lower amounts of glucagon and insulin (Table 1 and Supplementary Fig. 2), expressed ADORA1 on some but not all α-cells, and T1D patients, who expressed no insulin but abundant glucagon (Table 1 and Supplementary Fig. 2), showed little ADORA1 staining in the islets (Fig. 3B and C). Because Adora2a receptor expression has previously been reported in isolated dispersed α-cells, we also performed immunohistochemistry to examine Adora2a expression in NOD and NOD.B10 tissues. Intense Adora2a staining was observed in pancreatic ducts and cells in the insulitic lesion but was not observed in α-cells (Supplementary Figs. 5 and 6). Similar results were observed using three different antibodies against Adora2a. The expression of Adora2a in pancreatic ducts, macrophages, and various subsets of peripheral T cells have previously been demonstrated (21,22). These data suggest that the increased expression of Adora2a in isolated islets of NOD mice (Fig. 1J) may be due to Adora2a expressed in the infiltrating lymphocytes.
FIG. 2.

Immunohistochemical analysis of Adora1 expression in the islets of NOD and NOD.B10 mice. A: Confocal microscopy images show the colocalization of Adora1 (red) with glucagon (green) in all α-cells (top row) and colocalization with insulin (arrow) in a few β-cells (middle and bottom row). B: Reduced Adora1 expression (red) is observed on α-cells during the progression of NOD disease. At 8 weeks of age, before the onset of disease, Adora1 is observed on all glucagon-positive α-cells (green). At 12 weeks of age, Adora1 and glucagon expression are gradually lost around the periphery of the islets. By 20 weeks of age, the residual islets were found to express glucagon but little or no Adora1. C: Confocal microscopy data show loss of Adora1 expression (red) as islets become more infiltrated. Infiltration was assessed by CD3 staining (green). To control for variations in staining intensity between experiments, all images were taken from a single pancreas section from a 12-week-old NOD mouse. Data are representative of ≥10 similarly stained islets from at least three individual NOD and NOD.B10 mice at each age group. Scale bars represent 25 μm. Gluc, glucagon; Ins, insulin.
FIG. 3.

Localization of ADORA1 in human islets. A: Confocal microscopy images show colocalization of ADORA1 with glucagon in most of the ADORA1-positive cells of nondiabetic control patients. Some ADORA1-positive cells did not colocalize with glucagon (arrows). The islets of AA+ individuals expressed less ADORA1 than controls (B), and long-term T1D patients had little or no ADORA1 staining (C). D: ADORA1 was also found to colocalize with insulin in the islet of a control subject (arrow). nPOD patient information is reported in Table 1. Data are representative of ≥10 similarly stained islets. Scale bars represent 50 μm.
Identification of a novel alternatively spliced variant of Adora1.
RT-PCR was performed using primers that span the entire coding region of the Adora1 gene (Table 2, primer set 2). One amplicon of the expected size of 1,460 bp was produced using a pool of 12-week-old NOD or NOD.B10 pancreata cDNA (n = 5) as the template. An additional 1,062 bp amplicon was synthesized using the NOD template (Fig. 4A). This amplicon was cloned, sequenced, and found to represent an alternatively spliced isoform of Adora1 (Adora1-Var) that lacks exon 2 (GenBank Accession number: KC881107). The truncated protein corresponding to the spliced gene contains the carboxyl-terminal signaling domain, the agonist-binding site, but lacks the extracellular amino-terminal, the first three and part of the fourth transmembrane domains.
FIG. 4.

An alternatively spliced isoform of Adora1 is expressed in the pancreas of NOD and AA+ patients. A: RT-PCR was performed using primers that span the entire coding region of Adora1. Only one amplicon of the expected size was observed in pooled 12-week-old NOD.B10 pancreas samples (n = 5). An additional amplicon, corresponding to the alternatively spliced truncated isoform of Adora1 (Adora1-Var), was observed in pooled 12-week-old NOD pancreas samples (n = 5). B: RT-PCR was performed in individual 12-week-old NOD and NOD.B10 pancreas (n = 4 per group, different from those shown on panel A). Adora1-Var was expressed in one NOD.B10 mouse and in all four NOD mice examined. C: A QPCR assay was developed to specifically measure Adora1-Var (Supplementary Fig. 3). This sensitive assay demonstrated significantly higher expression of Adora1-Var in the pancreas of 12-week-old NOD vs. NOD.B10 mice. Data represent mean ± SEM. *P < 0.05, two-tailed unpaired Student t test. The expression of the full length Adora1 isoform was also measured and found not to differ. D: Bioanalyzer gel image shows RNA samples extracted from human pancreas samples. The 18S and 28S rRNA bands are indicated. E: RT-PCR was performed using primers designed to specifically detect ADORA1 or the alternatively spliced isoform of ADORA1 (ADORA1-VAR). ADORA1 was detected in all pancreas samples examined, but ADORA1-VAR was only detected in two of three AA+ patients and not in the control or T1D patients. F: An illustration of ADORA1-VAR shows the truncated transmembrane regions shaded in gray. G: An alignment of the predicted sequence of the mouse and human ADORA1-VAR isoforms.
Interestingly, one of four NOD.B10 samples also expressed Adora1-Var when samples were run individually (Fig. 4B). Because high expression of Adora1 could out-compete Adora1-Var for reagents and prevent its detection in a competitive PCR assay, we designed primers that bind only to Adora1-Var. The assay specificity and amplification efficiency was confirmed by RT-PCR, melting curve analysis, and standard curves (Supplementary Fig. 3). Using this assay, we showed that Adora1-Var is expressed in the pancreas of 12-week-old NOD.B10 mice but at significantly lower levels than in 12-week-old NOD mice (Fig. 4C). Full-length Adora1 expression was not changed (Fig. 4C).
Next, we examined whether a similarly spliced isoform of ADORA1 is expressed in human pancreas tissues. Only RNA samples containing intact 18S and 28S rRNA were used (Fig. 4D). The human ADORA1 gene is comprised of 4 exons, with exon 3 and 4 corresponding to exon 2 and 3, respectively, of the mouse Adora1 gene. RT-PCR primers were designed against exon 3 to detect the full-length isoform of ADORA1 (Table 2, primer set 4). To detect the splice variant of ADORA1, a forward primer spanning exon 2 and 4 on the human ADORA1 gene was used (Table 2, primer set 5). All samples expressed ADORA1, but only samples from two AA+ individuals (#6027 and #6170) expressed the spliced isoform, ADORA1-VAR (Fig. 4E). The AA+ sample (#6090) that did not test positive for ADORA1-VAR belonged to a 2-year-old individual. Cloning and sequencing was performed using primers that span the predicted coding region of ADORA1-VAR (Table 2, set 3). Interestingly, two amplicons of 862 bp and 961 bp were produced. The 862 bp isoform (human ADORA1-VAR, GenBank Accession number: KC884744) was similar to the mouse isoform, containing exons 2 and 4, but lacking exon 3. The 961 bp isoform lacks 20 bp from exon 2 but includes a short 71 bp intronic insertion, 48 bp of exon 3, and exon 4 (human ADORA1-VAR2; GenBank Accession number: KC881108). Both genes encode the same truncated ADORA1 protein that lacks the N-terminal and first three transmembrane domains (Fig. 4F). The human ADORA1-VAR protein is slightly larger than the mouse isoform because exon 4 of the human ADORA1 gene contains an in-frame start codon 75 bp upstream of the mouse Adora1-Var start site. A comparison of the predicted mouse Adora1-Var and human ADORA1-VAR protein sequences is shown in Fig. 4G.
Inflammation-induced splicing of Adora1 in mouse islets.
Next, we examined whether inflammation in vitro or in vivo could drive the splicing of Adora1 in islets. Islets from 12-week-old NOD.B10 mice were incubated with the supernatant of activated or nonactivated NOD.B10 splenocytes for 24 h, and Adora1 splicing was measured. The activated splenocyte supernatant contained high levels of chemokines and cytokines (Supplementary Table 3) and induced the splicing of Adora1 in all six of the individual experiments performed (Fig. 5A). Full-length Adora1 expression was unchanged.
FIG. 5.

In vitro and in vivo inflammation of the pancreatic islets upregulates expression of Adora1-Var. A: QPCR was performed to measure Adora1-Var and Adora1 expression in 12-week-old NOD.B10 islets that were treated with the supernatant of activated NOD.B10 splenocytes or with PBS (control). Adora1-Var expression was upregulated by supernatant treatment in all six individual experiments. Ifng (B), Adora1-Var (C), and Adora1 (D) were measured in the pancreas of 12-week-old NOD.SCID mice that were treated with activated splenocytes of NOD.BDC.2.5 mice or with PBS (control). Samples with the highest Ifng expression also had the highest Adora1-Var expression. Full-length Adora1 expression was not different between splenocyte-treated and PBS-treated NOD.SCID mice.
To investigate the effect of in vivo inflammation of islets, NOD.SCID mice (n = 7) were injected intraperitoneally with 5 × 106 activated splenocytes of NOD.BDC2.5 mice. Control mice (n = 4) were treated with PBS. Expression of Ifng (Fig. 5B) and Adora1-Var was measured 48 h after injection (Fig. 5C). Splenocyte-treated NOD.SCID mice had drastically higher levels of Adora1-Var (Fig. 5C), and the change in Adora1-Var expression correlated well with Ifng expression (representing the degree of infiltration with activated splenocytes). The expression of full-length Adora1 was not changed (Fig. 5D).
ADORA1-VAR heterodimerizes with ADORA1 and inhibits ADORA1 function.
Because most truncated GPCR variants have been shown to act as dominant-negative mutants that heterodimerize with the wild-type receptor (23), we tested whether ADORA1-VAR could interact with ADORA1. HEK293 cells were transfected with fluorescently tagged ADORA1 and/or ADORA1-VAR fusion proteins (Fig. 6A–C). ADORA1 appeared to aggregate in cytoplasm when coexpressed with ADORA1-VAR (Fig. 6C), suggesting that the cell surface targeting of ADORA1, like many GPCRs, may require homodimerization of ADORA1. Results of our coimmunoprecipitation experiments suggest that ADORA1 heterodimerizes with ADORA1-VAR. Immunoprecipitation was performed using an antibody against tRFP. A band corresponding to the predicted size of ADORA1-tGFP (∼70 kDa) could be detected in cells that were transfected to express both ADORA1-tGFP and ADORA1-VAR-tRFP (Fig. 6D).
FIG. 6.

ADORA1-VAR heterodimerizes with ADORA1 and is a dominant-negative isoform of ADORA1. Confocal microscopy images show the expression of ADORA1-tGFP (A), ADORA1-VAR-EGFP (B), or ADORA1-tGFP and ADORA1-VAR-tRFP (C) in HEK293 cells. Coexpression of both ADORA1 isoforms appears to cause aggregation of the proteins in the cytoplasm. D: Coimmunoprecipitation experiments demonstrate an interaction between ADORA1-tGFP with ADORA1-VAR-tRFP. Immunoprecipitation (IP) and immunoblotting (Western blotting [WB]) was performed using an antibody against tRFP and tGFP, respectively. A band corresponding to the predicted size of ADORA1-tGFP (∼70 kDa) could be detected only in cells that express both ADORA1-tGFP and ADORA1-VAR-tRFP. As a control, cell lysates (input samples) were also immunoblotted for β-actin. cAMP accumulation was measured in HEK293 cells transfected with ADORA1-tGFP and ADORA1-VAR-EGFP alone or in combination. E: Cells transfected with ADORA1-tGFP alone inhibited cAMP accumulation induced by forskolin when treated with the ADORA1 receptor agonist CPA (100 μmol/L). This effect was abolished by coadministration with the ADORA1-receptor antagonist DPCPX (100 μmol/L). F: Untransfected cells and cells expressing an empty plasmid, ADORA1-VAR-EGFP, or ADORA1-tGFP and ADORA1-VAR-EGFP were found not to inhibit forskolin-induced cAMP accumulation after treatment with CPA. Experiments shown in panel E and F were performed in the presence of the phosphodiesterase inhibitor R0-20-1724 (25 μmol/L). Data represent mean ± SEM.
Next, we tested whether ADORA1-VAR may be a dominant-negative isoform of ADORA1. Activation of ADORA1 inhibits adenylate cyclase activity and reduces cAMP levels. Inhibition of forskolin-induced cAMP accumulation was assessed in transfected HEK293 cells after treatment with 100 nmol/L CPA, an ADORA1-selective agonist. Cells transfected to express ADORA1-tGFP alone could inhibit forskolin-induced accumulation of cAMP after treatment with 100 nmol/L CPA (Fig. 6E). This was abolished by coadministration of 100 nmol/L DPCPX (ADORA1-selective antagonist). Untransfected cells and cells transfected to express an empty plasmid, ADORA1-VAR-EGFP alone, or ADORA1-VAR-EGFP with ADORA1-tGFP, were not able to inhibit accumulation of cAMP after treatment with CPA (Fig. 6F), indicating that ADORA1-VAR is not functional and acts as a dominant-negative isoform of ADORA1. Surprisingly, CPA had a small stimulatory effect on cAMP accumulation in untransfected cells and in cells transfected with the empty plasmid or ADORA1-VAR-tRFP. This may be due to nonselective binding of CPA to ADORA2A and ADORA2B receptors that are abundantly expressed on HEK293 cells (24).
DISCUSSION
The results of our study reveal an early intrinsic α-cell defect that may explain the hyperglucagonemia that is observed during the progression of diabetes. We showed that Adora1/ADORA1 expression was reduced in α-cells of prediabetic and diabetic NOD mice and in AA+ and long-term T1D patients. α-Cells respond to hypoglycemia by closing ATP-sensitive potassium channels. This leads to membrane depolarization and opening of voltage-gated Ca2+ channels, including N-, P/Q-, and/or L-type Ca2+ channels (25). The influx of Ca2+ triggers the exocytotic release of glucagon. Activation of ADORA1 has been shown to mediate the opening of ATP-sensitive potassium channels (26) to inhibit N-type (27), L-type, and P/Q-type Ca2+ currents (27). Thus, ADORA1 can inhibit exocytosis of glucagon-containing granules in α-cells, and loss or inhibition of ADORA1 may lead to the unrestrained secretion of glucagon. This may explain why NOD mice, T1D patients, and Adora1-KO mice show diminished suppression of glucagon after a glucose challenge (3–6,12,13).
The Adora1 receptor is activated by adenosine. In mouse islets, adenosine is released by exocytosis (28) and can act on Adora1 or Adora2a receptors to inhibit or stimulate glucagon secretion, respectively (12–14,29,30). Our immunohistochemistry results suggest that Adora2a receptors are not localized directly on α-cells (Supplementary Figs. 5 and 6). However, Tudurí et al. (29) clearly showed Adora2a expression on isolated dispersed α-cells. It is possible that the fairly harsh isolation procedure used to extract these cells resulted in the induction or upregulation of Adora2a expression. Studies have shown that isolation of islets by collagenase digestion leads to upregulation of >4,500 genes (17). Because Adora1 and Adora2a mediate opposing actions on glucagon secretion, the specific response will depend on the local concentration of extracellular adenosine.
Adora1 has higher affinity for adenosine than Adora2a and is activated by relatively low adenosine concentrations (31). In mouse islets, adenosine levels decrease as extracellular glucose concentrations increase from 3 mmol/L (hypoglycemic) to 16.7 mmol/L (hyperglycemic) (28), suggesting that hypoglycemia stimulates glucagon secretion via activation of Adora2a receptors while hyperglycemia inhibits glucagon secretion via activation of Adora1 receptors. The opposing actions of Adora1 and Adora2a thus allow reciprocal control and fine-tuning of glucagon release in response to changes in the glycemic status. Altered adenosine receptor expression can occur in response to various stimuli and in different pathophysiological states (32). During the pathophysiological progression of T1D, the balance of adenosine receptors may be disrupted by the loss of Adora1 receptors in α-cells (Figs. 1–3), leading to excessive activation of Adora2a and increased glucagon secretion.
Loss of Adora1 function may result from alternative splicing and expression of a dominant-negative isoform of Adora1. Human islets have been shown to express ∼20,000 genes and most of the known splicing factors (33), suggesting that islets are poised to respond quickly to various environmental stimuli. After exposure to interleukin-1β and IFN-γ, 20 and 35% of expressed genes undergo changes in expression and alternative splicing, respectively (33,34). Interestingly, the expression of Adora1 was not altered in these studies (34). This is consistent with our studies showing a lack of Adora1 splicing in NOD.B10 islets treated with interleukin-1β and IFN-γ (data not shown). Adora1 splicing may instead be modulated by other inflammatory signals, among those reported in Supplementary Table 3. Several of these are significantly upregulated in the islets of 12-week-old NOD versus NOD.B10 mice (Fig. 1C and D). Because inflammation may be required to maintain splicing, the absence of Adora1-Var in the pancreas of long-term T1D patients is not surprising (Fig. 4E).
Two transcripts of the ADORA1 gene (variants 1 and 2) that differ in the 5′ untranslated region have been identified in humans (35). Both encode the same protein. Variant 1 is detected in all tissues expressing ADORA1, whereas variant 2 is detected only in tissues that express high levels of ADORA1 (35,36). Here, we identified a novel isoform of ADORA1 that encodes a truncated protein that heterodimerizes with and acts as a dominant-negative isoform of the full length ADORA1 isoform.
The islets of newly diabetic NOD mice have previously been shown to exhibit distorted endocrine cell populations, with a significantly increased ratio of α-cells to β-cells but a reduced overall abundance of α-cells (37). We showed a loss of α-cells in the islets of 12-week-old NOD mice and found that the remaining α-cells do not all express Adora1 and are localized throughout the islet instead of around the periphery (Fig. 2B). By 20 weeks of age, after diabetes is well established, islets consist mainly of α-cells that are devoid of Adora1 expression (38). Because glucagon stimulates insulin secretion, hyperglucagonemia may lead to paracrine stimulation of β-cells. Elevated glucagon secretion has been shown to precede β-cell hyperactivity in diabetic mice (39,40), and hyperinsulinemia is observed before the onset of diabetes in mice (41) and in high-risk HLA-identical siblings of diabetic patients (42). Increased sensitivity to inflammatory cytokines (43) and higher expression of autoantigens (44,45) on hyperactive β-cells suggest that these cells are more prone to autoimmune attack (46). Consistent with this idea, prevention of β-cell hyperactivity was shown to delay the onset of diabetes (47).
The clinical manifestations of T1D can be treated by inhibition of glucagon secretion or function (48–50). Streptozotocin-induced β-cell destruction does not lead to hyperglycemia in glucagon-receptor KO mice (48) or in rats treated with a glucagon receptor antagonist (50). Immunoneutralization of glucagon also prevents hyperglycemia in alloxan-treated rabbits (49). Suppression of glucagon may also be beneficial early in the progression of T1D, when loss of ADORA1 expression and hyperglucagonemia are observed but before substantial β-cell loss has occurred (3). Alternatively, targeting the cytokines and/or chemokines that drive the splicing of ADORA1 may help reestablish the balance of ADORA1 to ADORA2A expression. Future experiments focusing on the identification and inhibition of such inflammatory mediators may be useful in the treatment of hyperglucagonemia and the early prevention of NOD disease and T1D.
Supplementary Material
ACKNOWLEDGMENTS
C.G.F. provided most of the funding (National Institutes of Health grants DK-078123 and AI-083628). L.Y. was supported by a JDRF Advanced Postdoctoral Fellowship. This research was performed with the support of the nPOD, a collaborative T1D research project sponsored by the JDRF. Organ Procurement Organizations partnering with nPOD to provide research resources are listed at www.jdrfnpod.org/our-partners.php.
No potential conflicts of interest relevant to this article were reported.
L.Y. performed the microarray, histology, ELISA assays, RT-PCR, and QPCR experiments; identified and cloned the human ADORA1-VAR and mouse Adora1-Var isoforms; synthesized the human and mouse ADORA1-VAR fluorescently-tagged fusion proteins; performed the cAMP assays and coimmunoprecipitation assays; prepared the manuscript; and composed the figures. C.T. performed the LCM experiments, collected serum, and isolated pancreatic islets by collagenase digestion. C.C.W. activated and injected splenocytes from NOD.BDC.2.5 mice into NOD.SCID mice and collected tissues from these mice. L.Y. and C.G.F. were involved in the planning and direction of this work. All authors reviewed and edited the manuscript. L.Y. and C.G.F. are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Irina Kusmartseva and Tiffany Heiple (nPOD) for their assistance in obtaining high-resolution image files of glucagon and insulin staining of human pancreata sections, Remi J. Creusot (Columbia University) for his assistance and advice on the activation of splenocytes, and Rahima Atker (Stanford University) and Rebecca Fuhlbrigge (Stanford University) for their technical assistance.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-0614/-/DC1.
REFERENCES
- 1.Ohneda A, Kobayashi T, Nihei J, Tochino Y, Kanaya H, Makino S. Insulin and glucagon in spontaneously diabetic non-obese mice. Diabetologia 1984;27:460–463 [DOI] [PubMed] [Google Scholar]
- 2.Ohneda A, Kobayashi T, Nihei J, Nishikawa K. Glucagon in spontaneously diabetic KK mice. Horm Metab Res 1981;13:207–211 [DOI] [PubMed] [Google Scholar]
- 3.Ohneda A, Kobayashi T, Nihei J, Tochino Y, Kanaya H, Makino S. Secretion of glucagon in spontaneously diabetic NOD mice. J Jap Diabet Soc 1981;24:202. [DOI] [PubMed] [Google Scholar]
- 4.Greenbaum CJ, Prigeon RL, D’Alessio DA. Impaired beta-cell function, incretin effect, and glucagon suppression in patients with type 1 diabetes who have normal fasting glucose. Diabetes 2002;51:951–957 [DOI] [PubMed] [Google Scholar]
- 5.Unger RH, Aguilar-Parada E, Müller WA, Eisentraut AM. Studies of pancreatic alpha cell function in normal and diabetic subjects. J Clin Invest 1970;49:837–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dinneen S, Alzaid A, Turk D, Rizza R. Failure of glucagon suppression contributes to postprandial hyperglycaemia in IDDM. Diabetologia 1995;38:337–343 [DOI] [PubMed] [Google Scholar]
- 7.Walker JN, Ramracheya R, Zhang Q, Johnson PR, Braun M, Rorsman P. Regulation of glucagon secretion by glucose: paracrine, intrinsic or both? Diabetes Obes Metab 2011;13(Suppl 1):95–105 [DOI] [PubMed] [Google Scholar]
- 8.Pelegri C, Rosmalen JG, Durant S, et al. Islet endocrine-cell behavior from birth onward in mice with the nonobese diabetic genetic background. Mol Med 2001;7:311–319 [PMC free article] [PubMed] [Google Scholar]
- 9.Fujita T, Yui R, Kusumoto Y, Serizawa Y, Makino S, Tochino Y. Lymphocytic insulitis in a 'non-obese diabetic (NOD)' strain of mice: an immunohistochemical and electron microscope investivation. Biomed Res 1982;3:429–443 [Google Scholar]
- 10.Huang YC, Rupnik MS, Karimian N, et al. In situ electrophysiological examination of pancreatic α cells in the streptozotocin-induced diabetes model, revealing the cellular basis of glucagon hypersecretion. Diabetes 2013;62:519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng JT, Chi TC, Liu IM. Activation of adenosine A1 receptors by drugs to lower plasma glucose in streptozotocin-induced diabetic rats. Auton Neurosci 2000;83:127–133 [DOI] [PubMed] [Google Scholar]
- 12.Johansson SM, Salehi A, Sandström ME, et al. A1 receptor deficiency causes increased insulin and glucagon secretion in mice. Biochem Pharmacol 2007;74:1628–1635 [DOI] [PubMed] [Google Scholar]
- 13.Yang GK, Fredholm BB, Kieffer TJ, Kwok YN. Improved blood glucose disposal and altered insulin secretion patterns in adenosine A(1) receptor knockout mice. Am J Physiol Endocrinol Metab 2012;303:E180–E190 [DOI] [PubMed] [Google Scholar]
- 14.Salehi A, Parandeh F, Fredholm BB, Grapengiesser E, Hellman B. Absence of adenosine A1 receptors unmasks pulses of insulin release and prolongs those of glucagon and somatostatin. Life Sci 2009;85:470–476 [DOI] [PubMed] [Google Scholar]
- 15.Yip L, Su L, Sheng D, et al. Deaf1 isoforms control the expression of genes encoding peripheral tissue antigens in the pancreatic lymph nodes during type 1 diabetes. Nat Immunol 2009;10:1026–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shizuru JA, Gregory AK, Chao CT, Fathman CG. Islet allograft survival after a single course of treatment of recipient with antibody to L3T4. Science 1987;237:278–280 [DOI] [PubMed] [Google Scholar]
- 17.Marselli L, Thorne J, Ahn YB, et al. Gene expression of purified beta-cell tissue obtained from human pancreas with laser capture microdissection. J Clin Endocrinol Metab 2008;93:1046–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burnstock G, Novak I. Purinergic signalling in the pancreas in health and disease. J Endocrinol 2012;213:123–141 [DOI] [PubMed] [Google Scholar]
- 19.Németh ZH, Bleich D, Csóka B, et al. Adenosine receptor activation ameliorates type 1 diabetes. FASEB J 2007;21:2379–2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dhalla AK, Chisholm JW, Reaven GM, Belardinelli L. A1 adenosine receptor: role in diabetes and obesity. Handb Exp Pharmacol 2009:271–295 [DOI] [PubMed] [Google Scholar]
- 21.Koshiba M, Rosin DL, Hayashi N, Linden J, Sitkovsky MV. Patterns of A2A extracellular adenosine receptor expression in different functional subsets of human peripheral T cells. Flow cytometry studies with anti-A2A receptor monoclonal antibodies. Mol Pharmacol 1999;55:614–624 [PubMed] [Google Scholar]
- 22.Novak I. Purinergic receptors in the endocrine and exocrine pancreas. Purinergic Signal 2008;4:237–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wise H. The roles played by highly truncated splice variants of G protein-coupled receptors. J Mol Signal 2012;7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sumi Y, Woehrle T, Chen Y, Yao Y, Li A, Junger WG. Adrenergic receptor activation involves ATP release and feedback through purinergic receptors. Am J Physiol Cell Physiol 2010;299:C1118–C1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rorsman P, Braun M, Zhang Q. Regulation of calcium in pancreatic α- and β-cells in health and disease. Cell Calcium 2012;51:300–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li DP, Chen SR, Pan HL. Adenosine inhibits paraventricular pre-sympathetic neurons through ATP-dependent potassium channels. J Neurochem 2010;113:530–542 [DOI] [PubMed] [Google Scholar]
- 27.Ambrósio AF, Malva JO, Carvalho AP, Carvalho CM. Inhibition of N-,P/Q- and other types of Ca2+ channels in rat hippocampal nerve terminals by the adenosine A1 receptor. Eur J Pharmacol 1997;340:301–310 [DOI] [PubMed] [Google Scholar]
- 28.Yang GK, Squires PE, Tian F, Kieffer TJ, Kwok YN, Dale N. Glucose decreases extracellular adenosine levels in isolated mouse and rat pancreatic islets. Islets 2012;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tudurí E, Filiputti E, Carneiro EM, Quesada I. Inhibition of Ca2+ signaling and glucagon secretion in mouse pancreatic alpha-cells by extracellular ATP and purinergic receptors. Am J Physiol Endocrinol Metab 2008;294:E952–E960 [DOI] [PubMed] [Google Scholar]
- 30.Chapal J, Loubatières-Mariani MM, Petit P, Roye M. Evidence for an A2-subtype adenosine receptor on pancreatic glucagon secreting cells. Br J Pharmacol 1985;86:565–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daly JW, Padgett WL. Agonist activity of 2- and 5′-substituted adenosine analogs and their N6-cycloalkyl derivatives at A1- and A2-adenosine receptors coupled to adenylate cyclase. Biochem Pharmacol 1992;43:1089–1093 [DOI] [PubMed] [Google Scholar]
- 32.St Hilaire C, Carroll SH, Chen H, Ravid K. Mechanisms of induction of adenosine receptor genes and its functional significance. J Cell Physiol 2009;218:35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eizirik DL, Sammeth M, Bouckenooghe T, et al. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet 2012;8:e1002552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ortis F, Naamane N, Flamez D, et al. Cytokines interleukin-1beta and tumor necrosis factor-alpha regulate different transcriptional and alternative splicing networks in primary beta-cells. Diabetes 2010;59:358–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren H, Stiles GL. Characterization of the human A1 adenosine receptor gene. Evidence for alternative splicing. J Biol Chem 1994;269:3104–3110 [PubMed] [Google Scholar]
- 36.Ren H, Stiles GL. Posttranscriptional mRNA processing as a mechanism for regulation of human A1 adenosine receptor expression. Proc Natl Acad Sci USA 1994;91:4864–4866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pechhold K, Zhu X, Harrison VS, et al. Dynamic changes in pancreatic endocrine cell abundance, distribution, and function in antigen-induced and spontaneous autoimmune diabetes. Diabetes 2009;58:1175–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gómez Dumm CL, Cónsole GM, Luna GC, Dardenne M, Goya RG. Quantitative immunohistochemical changes in the endocrine pancreas of nonobese diabetic (NOD) mice. Pancreas 1995;11:396–401 [DOI] [PubMed] [Google Scholar]
- 39.Leiter EH, Coleman DL, Eppig JJ. Endocrine pancreatic cells of postnatal “diabetes” (db) mice in cell culture. In Vitro 1979;15:507–521 [DOI] [PubMed] [Google Scholar]
- 40.Coulaud J, Durant S, Homo-Delarche F. Glucose homeostasis in pre-diabetic NOD and lymphocyte-deficient NOD/SCID mice during gestation. Rev Diabet Stud 2010;7:36–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amrani A, Durant S, Throsby M, Coulaud J, Dardenne M, Homo-Delarche F. Glucose homeostasis in the nonobese diabetic mouse at the prediabetic stage. Endocrinology 1998;139:1115–1124 [DOI] [PubMed] [Google Scholar]
- 42.Hollander PH, Asplin CM, Kniaz D, Hansen JA, Palmer JP. Beta-cell dysfunction in nondiabetic HLA identical siblings of insulin-dependent diabetics. Diabetes 1982;31:149–153 [DOI] [PubMed] [Google Scholar]
- 43.Palmer JP, Helqvist S, Spinas GA, et al. Interaction of beta-cell activity and IL-1 concentration and exposure time in isolated rat islets of Langerhans. Diabetes 1989;38:1211–1216 [DOI] [PubMed] [Google Scholar]
- 44.Aaen K, Rygaard J, Josefsen K, et al. Dependence of antigen expression on functional state of beta-cells. Diabetes 1990;39:697–701 [DOI] [PubMed] [Google Scholar]
- 45.Buschard K, Brogren CH, Röpke C, Rygaard J. Antigen expression of the pancreatic beta-cells is dependent on their functional state, as shown by a specific, BB rat monoclonal autoantibody IC2. APMIS 1988;96:342–346 [DOI] [PubMed] [Google Scholar]
- 46.Buschard K. The functional state of the beta cells in the pathogenesis of insulin-dependent diabetes mellitus. Autoimmunity 1991;10:65–69 [DOI] [PubMed] [Google Scholar]
- 47.Ziegler AG, Bachmann W, Rabl W. Prophylactic insulin treatment in relatives at high risk for type 1 diabetes. Diabetes Metab Rev 1993;9:289–293 [DOI] [PubMed] [Google Scholar]
- 48.Lee Y, Wang MY, Du XQ, Charron MJ, Unger RH. Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes 2011;60:391–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brand CL, Jørgensen PN, Svendsen I, Holst JJ. Evidence for a major role for glucagon in regulation of plasma glucose in conscious, nondiabetic, and alloxan-induced diabetic rabbits. Diabetes 1996;45:1076–1083 [DOI] [PubMed] [Google Scholar]
- 50.Johnson DG, Goebel CU, Hruby VJ, Bregman MD, Trivedi D. Hyperglycemia of diabetic rats decreased by a glucagon receptor antagonist. Science 1982;215:1115–1116 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.