

Background: NtdA represents a novel aminotransferase recognizing a sugar 6-phosphate.
Results: We have determined the structure of NtdA with pyridoxamine phosphate (the internal and external aldimines), identifying determinants of substrate specificity.
Conclusion: The structures suggest a canonical aminotransferase chemical mechanism, but features exclude the binding of sugar nucleotides.
Significance: A new subfamily of sugar aminotransferases is revealed, enhancing our understanding of antibiotic biosynthesis.
Keywords: Antibiotics, Carbohydrate, Cell Wall, Crystal Structure, Enzymes, X-ray Crystallography, Kanosamine, Transaminase
Abstract
NtdA from Bacillus subtilis is a sugar aminotransferase that catalyzes the pyridoxal phosphate-dependent equatorial transamination of 3-oxo-α-d-glucose 6-phosphate to form α-d-kanosamine 6-phosphate. The crystal structure of NtdA shows that NtdA shares the common aspartate aminotransferase fold (Type 1) with residues from both monomers forming the active site. The crystal structures of NtdA alone, co-crystallized with the product α-d-kanosamine 6-phosphate, and incubated with the amine donor glutamate reveal three key structures in the mechanistic pathway of NtdA. The structure of NtdA alone reveals the internal aldimine form of NtdA with the cofactor pyridoxal phosphate covalently attached to Lys-247. The addition of glutamate results in formation of pyridoxamine phosphate. Co-crystallization with kanosamine 6-phosphate results in the formation of the external aldimine. Only α-d-kanosamine 6-phosphate is observed in the active site of NtdA, not the β-anomer. A comparison of the structure and sequence of NtdA with other sugar aminotransferases enables us to propose that the VIβ family of aminotransferases should be divided into subfamilies based on the catalytic lysine motif.
Introduction
NtdA is a pyridoxal phosphate (PLP)3-dependent enzyme that we have recently shown to be a 3-oxo-d-glucose 6-phosphate:l-glutamate aminotransferase participating in the biosynthesis of kanosamine in Bacillus subtilis (1). Kanosamine is a sugar antibiotic shown to inhibit plant pathogens, growth of Saccharomyces cerevisiae, and a range of human pathogenic fungi, including Candida albicans (2–4). Kanosamine biosynthesis was first observed in Bacillus pumilus (5, 6). Various microbes have been found to produce kanosamine as a biosynthetic end product or as an intermediate of other natural products (3, 5, 7–9). Kanosamine and UDP-kanosamine are specific intermediates in the biosynthesis of 3-amino-5-hydroxybenzoate, a precursor of the mitomycin and ansamycin antibiotics, including rifamycin B (10). Besides being the biosynthetic precursor of the 3-amino-5-hydroxybenzoate biosynthetic pathway, kanosamine is also the source of nitrogen for the aminoshikimate pathway (10, 11). The best described pathway for kanosamine production involves the phosphorylation of glucose to glucose 1-phosphate, followed by pyrophosphorylation to UDP-glucose (6, 11, 12). UDP-glucose is then oxidized to UDP-3-oxo-d-glucose. Transamination of UDP-3-oxo-d-glucose yields UDP-kanosamine, which is then hydrolyzed to form kanosamine. An additional pathway for kanosamine biosynthesis has been proposed to exist in B. pumilus (6). The second pathway involves the oxidation of glucose to 3-oxo-d-glucose, followed by transamination to kanosamine. However, the authors were not able to verify this second pathway (6). More recently, we have shown that the ntd operon from Bacillus subtilis expresses three enzymes (NtdA, NtdB, and NtdC) that catalyze the biosynthesis of kanosamine from glucose 6-phosphate in a manner similar to the second pathway proposed for B. pumilus (1, 5, 6).
The ntd operon was first described by Inaoka et al. (13) as being necessary and sufficient for the production of 3,3′-neotrehalosadiamine (NTD), a disaccharide consisting of α,α,1,1′-linked kanosamine residues. NTD was originally identified as an antibiotic produced by B. pumilus (9) and Bacillus circulans (14) and shown to inhibit the growth of Staphylococcus aureus and Klebsiella pneumonia. Genome sequencing revealed that Bacillus anthracis and Bacillus licheniformis also contain orthologs of the NTD biosynthetic enzymes and its transcriptional regulator, NtdR (15). Expression of the ntdABC biosynthetic operon in Escherichia coli results in NTD production, suggesting that the ntdABC operon encodes all enzymes necessary for the production of NTD (13). Sequence comparisons suggest that the gene products of ntdA, ntdB, and ntdC are a PLP-dependent aminotransferase, a haloacid dehalogenase hydrolase superfamily member, and a NADH-dependent dehydrogenase, respectively. We have since shown that rather than producing NTD, these enzymes produce kanosamine. On the basis of our biochemical characterization of these enzymes, we have proposed a kanosamine pathway in B. subtilis involving the oxidation of glucose 6-phosphate to 3-oxoglucose 6-phosphate by NtdC, followed by transamination to kanosamine 6-phosphate (NtdA). The removal of the phosphate group by the phosphatase NtdB then yields kanosamine (Scheme 1).
SCHEME 1.

Proposed kanosamine biosynthetic pathway in B. subtilis. The kanosamine biosynthetic pathway proceeds via the oxidation of glucose 6-phosphate to 3-oxoglucose 6-phosphate (NtdC), followed by transamination to K6P (NtdA). Hydrolysis of the C6 phosphate by a K6P phosphatase (NtdB) yields kanosamine.
NtdA is the second enzyme in this pathway. On the basis of amino acid sequence analysis, NtdA is classified as a member of the sugar aminotransferase (SAT) family. All of these enzymes catalyze a PLP-dependent amination of a 3-oxo- or 4-oxo-sugar. In each case, the amino donor is either glutamate or glutamine, and the amino acceptor is a sugar nucleotide, except in the case of BtrR, which uses scyllo-inosose (16). SATs have been divided into three subgroups based on multiple sequence alignment: VIα, containing enzymes that utilize 4-oxo-sugar nucleotides; VIβ, those utilizing 3-oxo-sugar nucleotides; and VIγ, those utilizing scyllo-inosose (16, 17). NtdA could be classified as a SAT VIβ enzyme because it is proposed to catalyze the PLP-dependent amination of 3-oxoglucose 6-phosphate.
In this study, we present the structures of PLP- and pyridoxamine phosphate (PMP)-bound forms of NtdA, in addition to the external aldimine of PLP formed with the substrate α-d-kanosamine 6-phosphate (K6P). These structures are the first atomic resolution crystal structures of NtdA and provide a basis for active site discrimination between sugar phosphates and sugar nucleotides by this and other aminotransferases.
EXPERIMENTAL PROCEDURES
Overexpression and Purification
N-terminally His6-tagged NtdA was overexpressed in E. coli strain BL21-Gold(DE3) (Stratagene) containing the pET28b-NtdA overexpression construct and purified to homogeneity by Ni2+ affinity chromatography as described previously (18). NtdA was concentrated to 5 mg/ml in 25 mm Tris (pH 8.5) and 0.15 m NaCl for crystallization.
Crystallization and Data Collection
Crystals of NtdA were grown at 295 K using the microbatch method as described previously (18). Briefly, equal volumes of protein solution (1.2 μl) and precipitant solution (1.2 μl) were mixed. The crystallization drop was overlaid with paraffin oil (Hampton Research) to prevent evaporation of water from the drop. Plate-like crystals of the internal aldimine bound to NtdA (NtdA-PLP) were obtained after 1–4 days in 0.2 m ammonium acetate, 0.1 m trisodium citrate (pH 5.6), and 10–30% PEG 3350 either with or without 0.4 m NaCl. Crystals were cryoprotected in mother liquid containing 30% glycerol. Plate-like crystals of PMP bound to NtdA were obtained in 0.2 m ammonium acetate, 0.1 m trisodium citrate (pH 5.6), 20% PEG 3350, 20 mm glucose, and 55 mm glutamate. The NtdA-PMP crystals were cryoprotected in mother liquid containing 30% ethylene glycol. For co-crystallization of NtdA with K6P, the protein was first incubated for 15 min at room temperature with 10 mm K6P prior to crystallization. Pink plate-like crystals were obtained within 1 week under the same conditions as the NtdA-PLP crystals. The addition of K6P resulted in pink co-crystals, indicating the formation of the PLP external aldimine with the substrate K6P. These crystals were cryoprotected in mother liquid containing 30% ethylene glycol.
To test whether NtdA binds nucleotides, NtdA-PLP crystals were soaked for 2 h with either 20 mm TDP or 37 mm UDP in 0.2 m ammonium acetate, 0.1 m trisodium citrate (pH 5.6), and 20% PEG 3350. Co-crystallization of NtdA with UDP or TDP was done under the same solution conditions with either 20 mm TDP or 37 mm UDP in 0.2 m ammonium acetate, 0.1 m trisodium citrate (pH 5.6), and 20% PEG 3350. The crystals were cryoprotected in mother liquid with 25% glycol.
Diffraction data were collected at 100 K from single crystals in a cold nitrogen stream on beamline O8ID-1 at the Canadian Light Source (CLS), equipped with a MAR 225 CCD x-ray detector. Data were processed and scaled using HKL2000 (19) and Autoprocess (20). The data collection statistics are shown in Table 1.
TABLE 1.
Crystallographic data
Numbers in parentheses represent values for the highest resolution shell. PDB, Protein Data Bank.
| NtdA-PLP | NtdA-PMP | K6P external aldimine | |
|---|---|---|---|
| Data collection | |||
| Space group | P21 | P21 | P21 |
| Unit cell dimensions | |||
| a, b, c (Å) | 50.2, 106.5, 98.6 | 50.1, 107.0, 99.2 | 49.7, 106.4, 98.2 |
| α, β, γ | 90.0°, 96.2°, 90.0° | 90.0°, 95.8°, 90.0° | 90.0°, 96.4°, 90.0° |
| Resolution range (Å) | 30.0–2.30 (2.38–2.30) | 37.0–2.22 (2.30–2.22) | 46.7–1.71 (1.75–1.71) |
| No. of unique reflections | 45,198 (3874) | 50,732 (4827) | 108,740 (7967) |
| Completeness | 98.5 (85.1) | 99.4 (96.2) | 99.3 (98.5) |
| Redundancy | 7.9 (5.9) | 3.8 (3.1) | 4.0 (3.8) |
| Rmerge (%)a | 8.5 (29.4) | 10.4 (43.8) | 6.6 (75.5) |
| I/σI | 22.6 (4.3) | 11.5 (3.1) | 14.3 (2.2) |
| Refinement statistics | |||
| Resolution (Å) | 28.9–2.30 | 37.0–2.22 | 46.7–1.71 |
| Rwork/Rfree (%)b | 16.1/21.4 | 15.8/21.1 | 15.2/8.6 |
| B-factor (Å2) | |||
| Overall | |||
| Protein | 32.8 | 23.9 | 22.7 |
| Ligand | 24.3 | 21.3 | 22.5 |
| Water | 33.9 | 30.5 | 36.0 |
| No. of ligand or cofactor molecules | 2 LLP | 2 PMP | 2 K6P |
| r.m.s.d. | |||
| Bonds (Å) | 0.007 | 0.007 | 0.007 |
| Angles | 1.049° | 1.052° | 1.130° |
| Ramachandran (%) | |||
| Favored | 96.6 | 97.5 | 97.8 |
| Allowed | 3.4 | 2.5 | 2.2 |
| PDB codes | 4K2B | 4K2I | 4K2M |
a Rmerge = Σ|Io − 〈I〉|/ΣI, where I is the intensity of a reflection and 〈I〉 is the mean intensity of a group of equivalent reflections.
b Rwork = Σ‖Fobs| − |Fcalc‖/Σ|Fobs|. Rfree is 5% of the randomly excluded reflections from refinement.
Structure Determination and Refinement
The first structure of NtdA (NtdA-PLP) was solved by molecular replacement using the program MrBUMP (21) with MOLREP (22) from the CCP4 suite (23). A clear molecular replacement solution was found using Protein Data Bank code 2OGA (DesV from Streptomyces venezuelae (24)), which shows 35% sequence identity to NtdA, as the search model. All other NtdA structures were solved by molecular replacement with MOLREP (22) using the NtdA-PLP structure as a template. Prior to molecular replacement, the cofactor PLP and water molecules were removed from the template model. Refinement was carried out using CNS (25) and PHENIX (26). Initially, rigid body refinement was carried out, followed by simulated annealing using torsion angle dynamics at 5000 K to remove model bias. Iterative rebuilding of the model was done in Coot (27), followed by simulated annealing using Cartesian dynamics at 2500 K and gradually lowered to 1000 K. Clear positive density was present in the electron difference maps contoured at the 3σ level for the cofactor (PLP and PMP) and bound ligand (K6P) molecules. The cofactor and ligand molecules were built in manually in Coot. The model for K6P was generated in SKETCHER as part of the CCP4 suite. Libraries for cofactors and K6P were generated with ELBOW in PHENIX (26). The models were then further refined using restrained refinement. Non-crystallographic symmetry restraints were used throughout the refinement for all models. The refinement progress was monitored by following Rfree and inspecting the electron density maps. When Rfree dropped below 30%, water molecules were added using water update refinement in PHENIX, and their positions were manually checked using Coot. The final round of refinement was done without non-crystallographic symmetry restraints and with optimized refinement target weights for best geometry. Final refinement statistics are shown in Table 1.
Structural Analysis
The stereochemistry of all models was validated with MOLPROBITY (28) as part of PHENIX (26) and the ADIT Validation Server at the Protein Data Bank. Superpositions were calculated with DaliLite (29). Protein databases were searched with BLASTP (30) using the full-length sequence or the three domain sequences of NtdA, and multiple sequence alignments were generated with ClustalW (31). Structure-based sequence alignments were generated with PROMALS3D and SEQUOIA (32). Figures were prepared with PyMOL, ESPript (33), and LigPlus.
Protein Data Bank Accession Numbers
Coordinates have been deposited in the Protein Data Bank with accession codes 4K2B (NtdA with the PLP internal aldimine), 4K2I (NtdA with PMP), and 4K2M (NtdA with the PLP external aldimine with K6P).
RESULTS AND DISCUSSION
Structure of NtdA
The crystal structure of NtdA was solved in space group P21 to 2.3 Å resolution and refined to an overall R-factor of 16.8%. There are two NtdA monomers per asymmetric unit that form one biological homodimer. The two monomers in the homodimer are separated by a non-crystallographic 2-fold axis (Fig. 1, A and B). Both monomers superimpose well with a root mean square deviation (r.m.s.d.) of 0.9 Å for all structurally equivalent Cα atoms. The monomer-monomer interface buries an extensive, predominantly hydrophobic area of ∼2495 Å2, which corresponds to 12.7% of the total surface area of one monomer, as calculated by PISA (34).
FIGURE 1.

Crystal structure of NtdA. A and B, the overall dimer structure of NtdA shown in ribbon representation. Each monomer contains an N-terminal domain (green) and the AAT superfamily fold (red or blue). The second monomer in the homodimer is shown in a lighter color. C, the overall monomer structure of NtdA shown in ribbon representation. The N-terminal domain is colored green, the cofactor-binding domain is colored red, C-terminal domain is colored blue, and the large domain-swapping β-hairpin is colored yellow. Secondary structure elements are indicated. D, superposition of the two monomer N-terminal domains shows the large flexibility of the C termini of helices α1 and α2 and the loops following them.
Each NtdA monomer consists of three domains, as shown in Fig. 1C: an N-terminal domain (residues 1–68; green), a large cofactor-binding domain (residues 69–310; red) and a C-terminal domain (residues 311–439; blue). With the exception of the N-terminal domain, the overall fold of NtdA is characteristic of members of the aspartate aminotransferase superfamily (AAT Type 1). The large cofactor-binding domain consists of a seven-stranded mainly parallel β-sheet with a strand order of β3-β9-β8-β7-β6-β4-β5, flanked on both sides by eight α-helices (α4–α11) and two small helices (η1 and η2). Following helix α10, a long stretch of residues (positions 274–294; yellow) extends from the monomer and forms a large domain-swapping β-hairpin (β10 and β11) that interacts with the active site of the other monomer. As in the case of the AAT Type 1 superfamily, residues from both NtdA monomers form the two active sites in the homodimer. The PLP cofactor-binding site is located between the large cofactor-binding domain and the C-terminal domain (Fig. 1, A and C). The C-terminal domain consists of a four-stranded antiparallel β-sheet (β12–β15) flanked on the top by four α-helices (α12–α14 and α17) and a hairpin (residues 377–410) containing two α-helices (α15 and α16) and a small helix (η4). The C-terminal domain packs tightly on top of the large domain in a V-shaped structure.
NtdA possesses an additional small N-terminal domain (residues 1–68) that is not found in other aminotransferases. This domain consists of a two-stranded parallel β-sheet (β1 and β2) flanked by two α-helices (α1 and α2) and an additional helix (α3) (Fig. 1, A and C). The N-terminal domain packs tightly against the C-terminal domain with its three α-helices and forms a larger extension of the C-terminal domain. This domain is involved in numerous crystal contacts with symmetry-related molecules through its N terminus, the loop connecting helix α1 and strand β2, the loop connecting helix α2 and helix α3, and residues 55–65 of the loop after helix α3. Furthermore, the C termini of helices α1 and α2, as well as the loops following them, show a high degree of flexibility (Fig. 1D). The domain does not show any structural or functional similarity to known structures in the Protein Data Bank. An NCBI BLAST search of this N-terminal domain revealed high overall sequence identity (57–100%) to NtdA homologs from several Bacillus species (B. subtilis, Bacillus cereus, and Bacillus thuringiensis) (supplemental Fig. S1). The precise function of this domain is unknown but is likely to involve protein-protein interactions, mimicking its behavior in the crystal.
Cofactor-binding Site
The PLP cofactor is located in a deep cavity between the large domain and the C-terminal domain of each monomer, similar to other aminotransferases. This cavity is formed by the C-terminal ends of strands β7 and β8 and the N-terminal ends of strand β9 and helices α6 and α7 of one monomer and by the C terminus of helix α10 and the connecting domain-swapping hairpin from the other monomer. The active site lysine, Lys-247, is located in the loop connecting strands β8 and β9.
In the NtdA structure without substrate, i.e. the resting state of the enzyme, the cofactor PLP is covalently bound to Lys-247 via a Schiff base. During the refinement of this structure, there is a clear continuous density visible between Lys-247 Nζ and PLP C4′, indicating that the cofactor is covalently bound. Fig. 2A shows a close-up of the residues located within 3.7 Å of the cofactor. Lys-247 exists in an extended conformation. The torsion angle of the C4–C4′ bond is 43°. This directs Lys-247 Nζ out of plane of the PLP ring. However, the Nζ atom is still within hydrogen bonding distance (2.4 Å) of PLP O3. The cofactor is held in place with a number of other hydrogen bonds, as shown in Fig. 2D. In addition, Asp-222 forms a salt bridge with the protonated pyridoxal N1 atom. Asp-222 is strictly conserved in the AAT Type 1 superfamily and promotes the protonation of N1, enabling the pyridine ring to function as an effective electron sink. Asp-222 is located on the C terminus of strand β7, and its side chain is oriented by the side chain of Thr-154 and the backbone amide of Cys-224. The other monomer contributes a hydrogen bond from Asn-292′ via the long domain-swapping hairpin and a bridging water with the side chain of His-275′ of helix α10. In addition, the positive end of the α-helical dipole of helix α6 is located below the 5′-phosphoryl group, helping to stabilize the negative charge of the phosphate.
FIGURE 2.

Cofactor- and substrate-binding sites of NtdA. A, close-up of the internal aldimine bound to NtdA. The cofactor PLP is covalently bound to Lys-247 via a Schiff base. The cofactor and residues within 3.7 Å are shown in stick representation. The 2Fo − Fc electron density contoured at 1σ is shown as blue mesh. B, close-up of the cofactor-binding site. PMP is not covalently bound to Lys-247. Lys-247 is in the retracted position away from C4′ of the cofactor. PMP and residues within 3.7 Å are shown in stick representation. The 2Fo − Fc electron density contoured at 1σ is shown as blue mesh. C, left panel, close-up of the external aldimine bound to NtdA. The external aldimine and residues within 3.7 Å are shown in stick representation. The Fo − Fc electron density omit map of the external aldimine contoured at 3σ is shown as green mesh and was calculated with the external aldimine omitted. Right panel, scheme of the addition of K6P to the internal aldimine to form the external aldimine. D, schematic diagram of NtdA-K6P interactions generated by LigPlus. Hydrogen bonds are in dashed green lines. Hydrophobic contacts with the ligand are presented by red semicircles with radiating spokes. Residue labels colored in black are from the first monomer, and those colored in blue are from the second monomer.
In the first half-reaction of AATs, PLP reacts with an amino acid (amino donor) to form PMP and the 2-oxo acid product. In SATs, this amino donor is either glutamate or glutamine. Our previous study showed that glutamate is the only amino acid that functions as an amino donor for NtdA (1). In support of this, we co-crystallized NtdA in the presence of glutamate. The crystal structure was solved to 2.23 Å resolution. The electron density map shows that Lys-247 is in a retracted position away from the cofactor, with no density visible between Lys-247 and the cofactor. A UV-visible spectrum of NtdA incubated for 2 h with 55 mm glutamate shows a clear peak at ∼340 nm, indicative of PMP (data not shown), consistent with the structure representing the PMP-bound form (Fig. 2B). Comparison of the PLP- and PMP-bound structures shows that they are nearly identical, with an overall r.m.s.d. between the two structures of 0.35 Å. There are no major changes in the position of any of the cofactor-binding residues. In the PMP-bound structure, the PMP aromatic ring is ∼0.45 Å tilted over its C6–N1 bond away from Lys-247 and toward Phe-151, and PMP is not covalently bound to Lys-247, with the NH2 group of PMP now coplanar with the pyridoxal ring and forming hydrogen bonds with water molecules and O3 of the pyridoxal ring.
K6P-binding Site
The crystal structure of NtdA with K6P was solved to 1.71 Å resolution. Both active sites show a clear continuous electron density between the nitrogen atom of K6P and the C4′ atom of PLP, indicating that we have trapped the external aldimine in both active sites (Fig. 2C). The reaction mechanism depicted in Scheme 2 is completely reversible, and the presence of the external aldimine in the active site of NtdA indicates that the transamination reaction, the final step in Scheme 2, proceeded in the “reverse direction” upon incubation of K6P with NtdA. The external aldimine is buried in the active site cavity, which can be divided into two regions: the cofactor-binding site and the substrate-binding site.
SCHEME 2.

Proposed mechanism for the NtdA-catalyzed half-reaction of l-glutamate. 1, internal aldimine of PLP; 2, external aldimine with l-glutamate; 3, quinoid intermediate; 4, imine of 2-oxoglutarate and PMP; 5, PMP.
Lys-247 is located at the interface of the cofactor- and substrate-binding site and is found in the retracted position, away from C4′ of the cofactor, as observed in the PMP-bound structure. Comparison of the external aldimine with PMP-bound structures shows that they are essentially identical, with an overall r.m.s.d. between the two structures of 0.55 Å. The cofactor is bound in the same position as observed in the PMP-bound structure and makes the same interactions as described above.
The K6P-binding cavity is delineated by four loops and the long stretch of residues following helix α10 that contains the domain-swapping β-hairpin (β10 and β11). The loops are between strand β4 and the N terminus of helix α7, strands β8 and β9 (Lys-247 motif), the C terminus of strand β14 and helix α15, and helices α4 and α5 of the second monomer.
As in the case of the cofactor-binding site, the substrate-binding cavity is formed by residues from both monomers. A close-up view of the residues located within 3.7 Å of the K6P-binding cavity is presented in Fig. 2 (C and D). K6P is bound in the substrate cavity and is oriented such that its phosphate group points toward the entrance and the pyranose ring, which is in the 4C1 chair conformation, is located on top of the cofactor, and is covalently bound to it through its N3″ equatorial atom. Lys-247 is positioned such that its Nζ atom is within hydrogen bonding distance of the N3″ atom and the C4″ hydroxyl group of K6P. The C1″, C2″, and C4″ hydroxyl groups of the K6P pyranose ring all face the same direction and interact with the protein either by direct hydrogen bonds or through bridging waters. In solution, K6P exists as a mixture of α- and β-anomers in a 1:1.7 ratio. NtdA apparently recognizes only the α-anomer of K6P because the β-anomer is never observed in the crystal structure. Like the PLP cofactor, the K6P portion of the intermediate also makes a series of hydrogen bonds and hydrophobic interactions with both monomers, as shown in Fig. 2D. The C1″ axial hydroxyl group of K6P is also anchored via a bridging water molecule to His-349. The pyranose ring is stacked on one side against the phenyl ring of Phe-151 and is shielded on the other side by Tyr-246. The phosphate group is held in the cavity via hydrogen bonds to both monomers, but predominately by residues from the second monomer. Thr-98′ and Ser-99′, from the loop connecting helices α4 and α5 of the second monomer, have moved closer upon substrate binding to contribute hydrogen bonding interactions with the phosphate group of K6P.
Structural Comparison with NDP-3-oxo-SATs
The enzymes most closely related to NtdA are the SATs. Several SATs with known structure and function were identified using a Dali search (29), and the top three matches were S. venezuelae DesV (r.m.s.d. = 1.6 Å for all structurally equivalent Cα atoms, 33% sequence identity to NtdA, Protein Data Bank code 2OGA), Thermoanaerobacterium thermosaccharolyticum QdtB (r.m.s.d. = 1.6 Å, 34% identity, code 3FRK), and Pseudomonas aeruginosa WbpE (r.m.s.d. = 1.6 Å, 33% identity, codes 3NU8 and 3NUB). All of these enzymes are homodimers and catalyze a PLP-dependent equatorial transfer of the amino group to a 3-oxo-sugar nucleotide substrate and are classified in the SAT subgroup VIβ. A structure-based sequence alignment of NtdA with DesV, QdtB, and WbpE shows high conservation in the large cofactor-binding domain, including the four conserved SAT motifs I–IV (Fig. 3) (17). Of the three related structures, only the crystal structures of QdtB and WbpE have been reported with trapped external aldimines (35, 36). A superposition of the PLP external aldimines of NtdA with K6P and QdtB with TDP-3-aminoquinovose (Protein Data Bank code 3FRK) is shown in Fig. 4 (similar results were obtained using WbpE; supplemental Fig. S3). As shown in Fig. 4, the external aldimines are positioned in the same orientation within their binding cavities. The PLP cofactors make the same interactions, all with conserved active site residues (Fig. 3). All three sugar moieties are oriented the same way, with their sugar rings flanked on both sides by aromatic side chains (Tyr or Phe), sealing them off from the surrounding environment. Larkin et al. (36) proposed that this may serve to stabilize the positively charged reaction intermediate through cation-π interactions and to prevent solvent from interfering with catalysis. Furthermore, the C2″ hydroxyl group of NtdA and QdtB and the C2″ N-acetyl group of WbpE are hydrogen-bonded to a strictly conserved tyrosine (Fig. 2D). In addition, the C″ atom of each substrate is pointing into a large pocket. However, the geometry and residues that accommodate the large C6″ phosphate group of the NtdA substrate differ from the other three enzymes (Figs. 3 and 4). In NtdA, the C6″ phosphate group is tightly bound by numerous hydrogen bonding interactions compared with the much smaller C6″ methyl group of QdtB (zero H-bonds) and the C6″ carboxylate group of WbpE (two H-bonds). An additional difference between NtdA and the SAT VIβ enzymes is found in the catalytic lysine motif IV SF(Y/F)P(G/A,S/T)KxxGxxG(D/E)GG (Fig. 3, green line). The aromatic side chain (Tyr or Phe) is replaced by a smaller side chain, Gln-244 in NtdA, which creates the space required for the bulky phosphate group of K6P. In addition, the small side chain before the catalytic lysine is replaced by the aromatic side chain (Tyr-246) in NtdA, and the bulky aliphatic residues Ile and Leu from the second monomer in both WbpE and QdtB are replaced by the polar residues Thr-98′ and Ser-99′, respectively, in NtdA and have moved slightly, resulting in hydrogen bonding interactions with the phosphate group of K6P.
FIGURE 3.

Structure-based sequence alignment of NtdA with NDP-3-oxo-SATs. Shown is the alignment of NtdA with the SATs QdtB (Protein Data Bank code 3FRK) from T. thermosaccharolyticum, WbpE (code 3NUB) from P. aeruginosa, and DesV (code 2OGA) from S. venezuelae. Strictly conserved residues are indicated by white letters on a red background. Conservatively substituted residues are indicated by red letters on a white background. The strictly conserved catalytic Lys and Asp residues are indicated with red asterisks. The four SAT sequence motifs are highlighted with colored bars: motif I (blue), motif II (red), the catalytic aspartate motif III (black), and the catalytic lysine motif IV (green). Residues involved in K6P binding and recognition are indicated with blue triangles.
FIGURE 4.
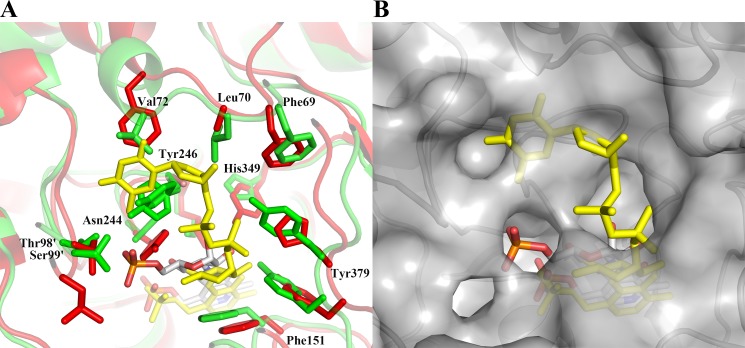
Comparison of NtdA with the external aldimines of NDP-3-oxo-SATs. A, superposition of the PLP external aldimine of NtdA with α-K6P (green ribbon) and QdtB with TDP-3-aminoquinovose (red; Protein Data Bank code 3FRK). The external aldimines are shown as stick models (Corey-Pauling-Koltun) and yellow, respectively. Residue labels refer to NtdA sequence. B, surface representation of NtdA from A showing the lack of room for the external aldimine of QdtB TDP-3-aminoquinovose (yellow sticks) in the NtdA active site.
We have recently reported that NtdA is part of a novel kanosamine biosynthetic pathway originating with glucose 6-phosphate (1). However, the resemblance of NtdA to SATs recognizing sugar nucleotides suggested that it might also recognize such substrates, which would allow the enzyme to participate in more than one pathway. To investigate the possible nucleotide binding in NtdA, we tried to both soak and co-crystallize NtdA with UDP, TDP, or TDP-kanosamine. None of these experiments resulted in nucleotide bound in the active site. A structural comparison of the nucleotide-binding region between NtdA and the SAT VIβ enzymes shows that this region is much smaller in NtdA and not conserved (Figs. 3 and 4). The loops bordering the nucleotide-binding site have similar geometry, with the exception of residues Phe-69–Glu-80 in NtdA, corresponding to where the nucleoside portion of the substrate binds in QdtB and WbpE (Figs. 3 and 4). NtdA does not contain this binding region because there is a small 310 helix (η1, Val-72–Leu-75) that, together with Leu-70 and Tyr-246, occupies this space. Helix η1 is held in position by salt bridges and hydrophobic interactions and is not likely to alter position to create the space required to bind a nucleotide. Furthermore, Tyr-378 in NtdA (which is a highly conserved His in SAT VIβ enzymes) would clash with the β-phosphate of a nucleotide moiety.
Structural Basis for Mechanism
The results of our structural studies provide excellent support for the mechanism of transamination proposed for similar enzymes (Scheme 2) (36). The PLP-bound structure represents the initial internal aldimine formed between Lys-247 and the PLP cofactor. The binding and reaction of glutamate result in the formation of the PMP-bound structure, where the amine has been transferred from the glutamate. There is no evidence of the 2-oxoglutarate, presumably due to release of this product upon formation. The K6P complex structure represents the external aldimine formed by transamination of the internal imine. Overlap of the PLP-bound structure (internal aldimine) and the K6P-bound structure (external aldimine) (Fig. 5) clearly shows the orientation of the PLP between the lysine and K6P in such a way as to facilitate the transamination reaction between the internal and external aldimines.
FIGURE 5.
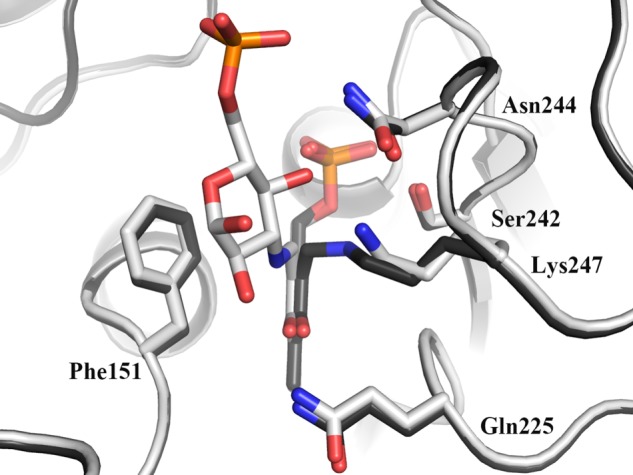
Superposition of the internal and external aldimines of NtdA. The superposition of NtdA with the internal aldimine (red) and with the external aldimine (blue) is shown. The positioning of the internal and external aldimines illustrates the orientations that will facilitate the transamination reaction.
NtdA Is a Member of a Novel Subfamily of SATs
Although NtdA is structurally and functionally closely related to the SAT VIβ family, it shows structural differences to accommodate the large C6″ phosphate group, and it lacks a nucleotide moiety. Also, the sequence in NtdA containing the active site lysine (S242FNPYK247NFGVCGKAGA257) is different from the reported sugar nucleotide aminotransferases in that an aromatic side chain (Tyr-246) is positioned next to the catalytic Lys-247. On the basis of these differences, we suggest dividing the SAT VIβ family into two subfamilies: VIβ-1, made up of enzymes utilizing 3-oxo-sugar nucleotide substrates; and VIβ-2, containing those utilizing 3-oxo-sugar substrates (i.e. the 3-oxo-α-d-glucose 6-phosphate subfamily). We expect that members of this latter subfamily all have the same geometry of the binding pocket and that substrate-binding residues are conserved. An NCBI BLAST search with the catalytic lysine motif SFNPYKNFGVCGKAGA identified proteins with high overall sequence identity (53–100%) to NtdA annotated as either NtdA homologs or as yet uncharacterized proteins from several Bacillus species (B. subtilis, Bacillus mojavensis, Bacillus vallismortis, B. pumilus, B. anthracis, B. licheniformis, B. cereus, and B. thuringiensis) (supplemental Fig. S2). Additionally, proteins were identified with lower overall sequence identity to NtdA from Mycobacterium abscessus, Mycobacterium massiliense (49% sequence identity), E. coli (41% sequence identity), and Erwinia species (40% sequence identity) (supplemental Fig. S3). These proteins have been annotated as UDP-4-amino-4-deoxy-l-arabinose:oxoglutarate aminotransferases (ArnB), putative NtdA homologs, or as yet uncharacterized aminotransferases. ArnB is classified in the SAT VIα family and catalyzes the axial conversion of UDP-4-oxoarabinose to UDP-4-amino-4-deoxy-l-arabinose. The putative ArnB enzymes found by searching with the NtdA catalytic lysine motif are different from the known ArnB subfamily. According to the ArnB subfamily in the UniProt list (EC 2.6.1.87), members of this family contain the catalytic lysine motif SF(F/Y)HAIKNxxxAEGG and do not have an additional N-terminal domain. As shown in supplemental Figs. S2 and S3, the proteins identified in our search contain an additional N-terminal domain, and the residues that would line the putative nucleotide-binding site are highly conserved as NtdA-like, indicating that these proteins most likely do not possess a nucleotide-binding site. Furthermore, all of the residues involved in PLP binding (black triangles) and K6P binding (blue triangles) are highly conserved (supplemental Fig. S2 and S3). Thus, our observations suggest that these enzymes are PLP-dependent sugar-6-phosphate aminotransferases.
Conclusion
The structural studies of NtdA reported in this work support the role of this enzyme in the conversion of 3-oxoglucose 6-phosphate to K6P through a PLP-dependent aminotransferase-type mechanism with glutamate as the amino donor. Our structures show the internal and external aldimine forms of NtdA and the activated PMP form. A comparison of NtdA with other similar SATs has revealed a novel motif based on the active site lysine region that we propose is predictive for identifying SATs that recognize sugar 6-phosphate substrates.
Acknowledgment
We thank Dr. David Jakeman (Dalhousie University) for the gift of TDP-kanosamine.
This work was supported by a Saskatchewan Health Research Foundation Team grant to the Molecular Design Research Group and by the University of Saskatchewan.

This article contains supplemental Figs. S1–S3.
The atomic coordinates and structure factors (codes 4K2B, 4K2I, and 4K2M) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PLP
- pyridoxal phosphate
- NTD
- 3,3′-neotrehalosadiamine
- SAT
- sugar aminotransferase
- PMP
- pyridoxamine phosphate
- K6P
- kanosamine 6-phosphate
- r.m.s.d.
- root mean square deviation
- AAT
- aspartate aminotransferase.
REFERENCES
- 1. Vetter N. D., Langill D. M., Anjum S., Boisvert-Martel J., Jagdhane R. C., Omene E., Zheng H., van Straaten K. E., Asiamah I., Krol E. S., Sanders D. A. R., Palmer D. R. J. (2013) A previously unrecognized kanosamine biosynthesis pathway in Bacillus subtilis. J. Am. Chem. Soc. 135, 5970–5973 [DOI] [PubMed] [Google Scholar]
- 2. Janiak A. M., Milewski S. (2001) Mechanism of antifungal action of kanosamine. Med. Mycol. 39, 401–408 [DOI] [PubMed] [Google Scholar]
- 3. Milner J. L., Silo-Suh L., Lee J. C., He H., Clardy J., Handelsman J. (1996) Production of kanosamine by Bacillus cereus UW85. Appl. Environ. Microbiol. 62, 3061–3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shang H., Chen J., Handelsman J., Goodman R. M. (1999) Behavior of Pythium torulosum zoospores during their interaction with tobacco roots and Bacillus cereus. Curr. Microbiol. 38, 199–204 [DOI] [PubMed] [Google Scholar]
- 5. Umezawa S., Umino K., Shibahara S., Omoto S. (1967) Studies of aminosugars. XVII. Production of 3-amino-3-deoxy-d-glucose by Bacillus species. Bull. Chem. Soc. Jpn. 40, 2419–2421 [DOI] [PubMed] [Google Scholar]
- 6. Umezawa S., Shibahara S., Omoto S., Takeuchi T., Umezawa H. (1968) Studies on the biosynthesis of 3-amino-3-deoxy-d-glucose. J. Antibiot. 21, 485–491 [DOI] [PubMed] [Google Scholar]
- 7. Dolak L. A., Castle T. M., Dietz A., Laborde A. L. (1980) 3-Amino-3-deoxyglucose produced by a Streptomyces sp. J. Antibiot. 33, 900–901 [DOI] [PubMed] [Google Scholar]
- 8. Fusetani N., Ejima D., Matsunaga S., Hashimoto K., Itagaki K., Akagi Y., Taga N., Suzuki K. (1987) 3-Amino-3-deoxy-d-glucose: an antibiotic produced by a deep-sea bacterium. Experientia 43, 464–465 [DOI] [PubMed] [Google Scholar]
- 9. Tsuno T., Ikeda C., Numata K., Tomita K., Konishi M., Kawaguchi H. (1986) 3,3′-Neotrehalosadiamine (BMY-28251), a new aminosugar antibiotic. J. Antibiot. 39, 1001–1003 [DOI] [PubMed] [Google Scholar]
- 10. Floss H. G., Yu T.-W., Arakawa K. (2011) The biosynthesis of 3-amino-5-hydroxybenzoic acid (AHBA), the precursor of mC7N units in ansamycin and mitomycin antibiotics: a review. J. Antiobiot. 64, 35–44 [DOI] [PubMed] [Google Scholar]
- 11. Guo J., Frost J. W. (2002) Kanosamine biosynthesis: a likely source of the aminoshikimate pathway's nitrogen atom. J. Am. Chem. Soc. 124, 10642–10643 [DOI] [PubMed] [Google Scholar]
- 12. Arakawa K., Müller R., Mahmud T., Yu T.-W., Floss H. G. (2002) Characterization of the early stage aminoshikimate pathway in the formation of 3-amino-5-hydroxybenzoic acid: the RifN protein specifically converts kanosamine into kanosamine 6-phosphate. J. Am. Chem. Soc. 124, 10644–10645 [DOI] [PubMed] [Google Scholar]
- 13. Inaoka T., Takahashi K., Yada H., Yoshida M., Ochi K. (2004) RNA polymerase mutation activates the production of a dormant antibiotic 3,3′-neotrehalosadiamine via an autoinduction mechanism in Bacillus subtilis. J. Biol. Chem. 279, 3885–3892 [DOI] [PubMed] [Google Scholar]
- 14. Numata K., Satoh F., Hatori M., Miyaki T., Kawaguchi H. (1986) Isolation of 3,3′-neotrehalosadiamine (BMY-28251) from a butirosin-producing organism. J. Antiobiot. 39, 1346–1348 [DOI] [PubMed] [Google Scholar]
- 15. Inaoka T., Ochi K. (2007) Glucose uptake pathway-specific regulation of synthesis of neotrehalosadiamine, a novel autoinducer produced in Bacillus subtilis. J. Bacteriol. 189, 65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hwang B.-Y., Cho B.-K., Yun H., Koteshwar K., Kim B.-G. (2005) Revisit of aminotransferase in the genomic era and its application to biocatalysis. J. Mol. Catal. B Enzym. 37, 47–55 [Google Scholar]
- 17. Hwang B.-Y., Lee H. J., Yang Y. H., Joo H. S., Kim B.-G. (2004) Characterization and investigation of substrate specificity of the sugar aminotransferase WecE from E. coli K12. Chem. Biol. 11, 915–925 [DOI] [PubMed] [Google Scholar]
- 18. van Straaten K. E., Langill D. M., Palmer D. R. J., Sanders D. A. R. (2009) Purification, crystallization and preliminary X-ray analysis of NtdA, a putative pyridoxal phosphate-dependent aminotransferase from Bacillus subtilis. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 65, 426–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 20. Fodje M. N., Berg R., Black G., Grochulski P., Janzen K. (2010) Automation of the macromolecular crystallography beamlines at the Canadian light source. in Proceedings of PCaPAC-2010, Saskatoon, Saskatchewan, pp. 130–132, International Workshop on Personal Computers and Particle Accelerator Controls [Google Scholar]
- 21. Keegan R. M., Winn M. D. (2007) Automated search-model discovery and preparation for structure solution by molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 63, 447–457 [DOI] [PubMed] [Google Scholar]
- 22. Vagin A., Teplyakov A. (1997) MOLREP: an automated program for molecular replacement, J. Appl. Cryst. 30, 1022–1025 [Google Scholar]
- 23. Collaborative Computational Project Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 24. Burgie E. S., Thoden J. B., Holden H. M. (2007) Molecular architecture of DesV from Streptomyces venezuelae: a PLP-dependent transaminase involved in the biosynthesis of the unusual sugar desosamine. Protein Sci. 16, 887–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 26. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 28. Davis I. W., Murray L. W., Richardson J. S., Richardson D. C. (2004) MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 32, W615–W619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Holm L., Sander C. (1995) Dali: a network tool for protein structure comparison. Trends Biochem. Sci. 20, 478–480 [DOI] [PubMed] [Google Scholar]
- 30. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thompson J. D., Higgins D. G., Gibson T. J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bruns C. M., Hubatsch I., Ridderström M., Mannervik B., Tainer J. A. (1999) Human glutathione transferase A4-4 crystal structures and mutagenesis reveal the basis of high catalytic efficiency with toxic lipid peroxidation products. J. Mol. Biol. 288, 427–439 [DOI] [PubMed] [Google Scholar]
- 33. Gouet P., Courcelle E., Stuart D. I., Métoz F. (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 [DOI] [PubMed] [Google Scholar]
- 34. Krissinel E., Henrick K. (2005) Multiple alignment of protein structures in three dimensions. in Computational Life Sciences (Berthold M. R., Glen R., Diederichs K., Kohlbacher O., Fischer I., eds) pp 67–78, Springer-Verlag, Berlin [Google Scholar]
- 35. Thoden J. B., Schäffer C., Messner P., Holden H. M. (2009) Structural analysis of QdtB, an aminotransferase required for the biosynthesis of dTDP-3-acetamido-3,6-dideoxy-α-d-glucose. Biochemistry 48, 1553–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Larkin A., Olivier N. B., Imperiali B. (2010) Structural analysis of WbpE from Pseudomonas aeruginosa PAO1: a nucleotide sugar aminotransferase involved in O-antigen assembly. Biochemistry 49, 7227–7237 [DOI] [PMC free article] [PubMed] [Google Scholar]