

Abstract
Nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) remain the cornerstone of HIV treatment; however, they are associated with toxicities attributed in part to inhibition of mitochondrial DNA (mtDNA) polymerase γ. In this study, we compared the in vitro toxicity profiles of structurally similar NRTIs (BMS-986001 to stavudine and tenofovir to adefovir) that differ by the presence of an acetylene or methyl group, respectively. Primary cultures of human renal proximal tubule epithelium, skeletal muscle myotubes, and differentiated adipocytes were exposed to the NRTIs at the maximum concentration (Cmax) reported for the clinically approved dose (investigational dose for BMS-986001, 600 mg) and a high equimolar concentration (200 μM) for 19 days. After 19 days, BMS-986001 did not significantly decrease mtDNA or cell protein at either concentration in any cell line. In contrast, stavudine significantly decreased mtDNA in all cultures (1.5- to 2.5-fold) (except at Cmax in renal cells) and cell protein in renal cells (1.4- to 2.4-fold). By day 19, at 200 μM, tenofovir significantly reduced mtDNA in adipocytes (1.9-fold) and adefovir significantly decreased mtDNA in all cultures (3.7- to 10.2-fold); however, no significant reduction in mtDNA was observed at Cmax in any cell line. Adefovir also significantly reduced cell protein at both concentrations in renal cells (2.2- to 2.8-fold) and at 200 μM in muscle cells (2.0-fold). In conclusion, BMS-986001 and tenofovir were considerably less cytotoxic than their respective structural analogs, demonstrating that small structural differences can contribute to significant differences in toxicity.
INTRODUCTION
Nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) remain a cornerstone of combination antiretroviral therapy for HIV. Although several NRTIs are used extensively, there are ongoing concerns regarding the established and potential long-term toxicities of the class. These include cytopenias (zidovudine [AZT]), pancreatitis (didanosine [ddI]), and other issues related to mitochondrial toxicity, including peripheral neuropathy (ddI and stavudine [d4T]), lipoatrophy (d4T and AZT), and metabolic acidosis (predominantly with d4T, ddI, and AZT) (1). Major HIV type 1 (HIV-1) treatment guidelines recommend combinations of tenofovir disoproxil fumarate (TDF) or abacavir (ABC) with emtricitabine or lamivudine as part of initial HIV-1 treatment regimens (1–3). TDF is preferred due to a favorable tolerability and efficacy profile; however, concerns regarding nephrotoxicity, decreased bone mineral density, and increased risk of fractures remain (1, 4–6). Inhibition of mitochondrial DNA (mtDNA) synthesis or mitochondrial damage is commonly considered to be the cause of toxicities such as lipoatrophy, associated with d4T and AZT (7, 8), or nephrotoxicity, associated with TDF (9–11).
BMS-986001 is a thymidine NRTI that has been developed to maintain the in vitro antiviral activity demonstrated by other NRTIs without the associated toxicity concerns. BMS-986001 (previously known as 4′-Ed4T and festinavir) is a novel analog of d4T which, in preliminary in vitro experiments, demonstrated potent antiviral activity and reduced mitochondrial toxicity compared with the mitochondrial toxicity of d4T (12).
In order to further evaluate the in vitro toxicity profile of BMS-986001, we examined its effect on mtDNA levels and measures of cell viability in several fully differentiated human cell lines. The effects of BMS-986001 were compared with those of the thymidine analog d4T, from which it differs by the presence of an acetylene group on the 5-membered ring in BMS-986001 (Fig. 1). Additionally, the adenosine analogs tenofovir (TFV) and adefovir (ADV) were evaluated; these molecules are also structurally similar, with TFV having an additional methyl group compared with the structure of ADV (Fig. 1). ADV, which is not approved for use in treating HIV-1, was selected because of its structural similarity to TFV and, also, because it is associated with a significant clinical incidence of renal toxicity, the mechanism of which remains unproven but which is possibly related to mitochondrial toxicity (13, 14). Finally, the in vitro toxicity profiles of AZT, an early thymidine analog, and ABC, an NRTI commonly used in clinical practice, were evaluated.
Fig 1.
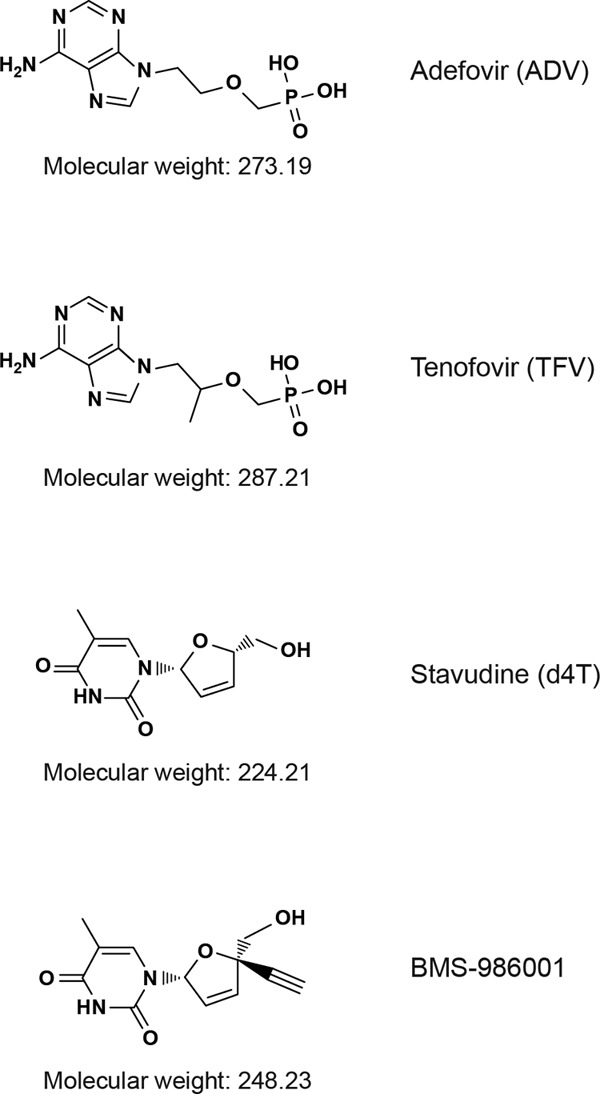
Structures of NRTIs used in this study.
(This work has been presented in part at the XIX International AIDS Conference, Washington, DC, 22 to 27 July 2012 [15] and the 11th International Congress on Drug Therapy in HIV Infection, Glasgow, United Kingdom, 11 to 15 November 2012 [16].)
MATERIALS AND METHODS
Materials.
Test compounds were purchased from Sigma Chemical Company (St. Louis, MO) (AZT, d4T, and ADV) or Toronto Research Chemicals (North York, Ontario, Canada) (ABC and TFV) or synthesized by Bristol-Myers Squibb (BMS-986001). Test compounds were dissolved in sterile distilled water as 20 mM stock solutions.
Cell culture.
Primary human renal proximal tubule (RPT) cells (Lonza, Allendale, NJ) were cultured using the renal epithelial cell BulletKit (Lonza), containing renal epithelial cell basal medium, according to the manufacturer's instructions. Human subcutaneous preadipocytes (Zenbio, Research Triangle Park, NC) were cultured in complete adipocyte differentiation medium (Zenbio) according to the manufacturer's instructions. Normal human skeletal muscle myoblasts, received as proliferating cells (Lonza), were grown to partial confluence in skGM-2 BulletKit medium (Lonza) and then differentiated for 5 days to form myotubes in Dulbecco's modified Eagle's medium–Ham's F-12 (1:1, vol/vol) medium (Lonza) containing 2% horse serum (Invitrogen, Carlsbad, CA). All cells were maintained in a sterile environment in 5% CO2 and 95% air at 37°C. Renal epithelial cells were passaged on days 5, 9, and 14. Mature adipocytes and myotubes do not divide and so were not passaged.
In vitro toxicity and functional assessment.
After an initial incubation period to allow cells to attach to the substrate and to fully differentiate (renal and muscle cells, 5 to 10 days, and adipocytes, 17 days), cells were exposed to test compounds or vehicle (water [1%, vol/vol]) for a total of 19 days. Fresh compound-containing medium prepared using frozen aliquots of test compounds was added to the cultures on days 5, 9, and 14. Test compounds were added at their steady-state peak plasma level maximum concentration (Cmax) in humans during HIV therapy, as reported in the product data sheets, and also at an equimolar concentration of 200 μM (Table 1). For the investigational NRTI BMS-986001, the Cmax of 40 μM used in this study corresponds to the steady-state Cmax observed after administration of a 600-mg dose daily (17). On days 9, 14, and 19, samples of culture supernatant and cells were taken for analysis. Experiments were performed in triplicate. At each time point, cell viability was assessed by total cell protein, ATP content, and lactate release, and mitochondrial toxicity was measured by mtDNA ATP8 content as described below.
Table 1.
Test agents and concentrations used
| Agenta | Mol wt | Low concn (μM)b |
|---|---|---|
| ADV | 273.2 | 1c |
| d4T | 224.2 | 7.6 |
| TFV | 287.21 | 2c |
| BMS-986001 | 248.2 | 40 |
| ABC | 286.33 | 23.0 |
| AZT | 267.24 | 14.5 |
ABC, abacavir; ADV, adefovir; AZT, zidovudine; d4T, stavudine; TFV, tenofovir.
Two concentrations were tested. The low concentration tested for each agent was the approximate reported Cmax value based on the clinically approved dose or, for BMS-986001, based on the observed Cmax at a dose of 600 mg daily (17). The high concentration tested, 200 μM for all agents, was used to provide a comparison of equimolar concentrations of the NRTIs.
Total cell protein.
Changes in total cell protein are associated with changes in cell number, with a decrease in protein concentration indicating loss of cells. Protein was measured using the bicinchoninic acid (BCA) method (BCA solution; Sigma) following lysis of the cultured cells in each well with somatic cell ATP-releasing reagent (Sigma). Absorption was measured at 562 nm, and the protein content (mg protein/ml) of each sample was determined by comparison with standard curves (protein standards; Thermo Scientific, Waltham, MA).
ATP content.
ATP is required by all cells, and consequently, a decrease in ATP levels is indicative of cell loss. Cell ATP content was measured using the CellTiter-Glo luminescent cell viability assay (Promega, Fitchburg, WI) and an EnVision plate reader (Perkin-Elmer, Waltham, MA) according to the manufacturers' instructions. Bioluminescence was measured as light intensity units (LU)/mg cell protein.
Lactate release.
Lactate production was measured as an indicator of conversion from oxidative respiration to glycolysis and also as a measure of cell viability. The amount of lactate released into the culture medium was determined by analysis of culture supernatants using lactate reagent (Roche Diagnostics, Indianapolis, IN) and a Roche/Hitachi 912 clinical chemistry analyzer according to the manufacturer's instructions.
mtDNA ATP8 content.
ATP8 is a subunit of mtDNA polymerase γ; as such, the abundance of ATP8 DNA is indicative of mitochondrial content and, thus, is a surrogate marker for mitochondrial DNA polymerase γ activity. DNA was isolated from the cells using the Qiagen AllPrep DNA/RNA minikit (Qiagen, Germantown, MD). Quantitative PCR (TaqMan; Invitrogen, Carlsbad, CA) was conducted using the ABI PRISM 7900 sequence detection system on 10 ng of DNA. The primer/probe sets were designed using ABI PRISM Primer Express software (PE Biosystems, Foster City, CA). The human gene primers and TaqMan probes are shown in Table 2 and were used at final concentrations of 600 to 900 nM for the primers and 200 nM for the probes. Cycle time values were normalized to those of the glyceraldehyde-3-phosphate dehydrogenase housekeeping gene (GAPDH) and the vehicle control (water, 1%), and the relative expression quantities were determined. The amplification efficiency of the primer/probe was above 94%.
Table 2.
Primer and probe sequences used for mitochondrial DNA analysis
| Human gene target | Sequence |
|
|---|---|---|
| Forward or probe | Reverse | |
| Primer | ||
| Nuclear GAPDH | GGTTTACATGTTCCAATATGATTCCA | ATGGGATTTCCATTGATGACAAG |
| Mitochondrial ATP8 | AATATTAAACACAAACTACCACCTACC | TGGTTCTCAGGGTTTGTTATA |
| TaqMan probe | ||
| Nuclear GAPDH | FAMATGGCACCGTCAAGGCTGAGAACGMGB | |
| Mitochondrial ATP8 | FAMCCTCACCAAAGCCCAMGB | |
GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Analysis of results.
Data are presented as means ± standard errors in all tables and Fig. 1. The data were examined for significant changes with respect to the vehicle-treated control for the day of assay using JMP 8.0 (The SAS Institute, Cary, NC). Analysis of variance followed by the Dunnett or the Tukey-Kramer post hoc comparisons of means were performed, and statistical significance denoted by a P value of ≤0.05.
RESULTS
Control cultures.
Control cultures grown without the addition of any test compound are shown in Fig. 2. In kidney cell (RPT) cultures, there was a trend over 19 days toward loss of total cell protein, which was accompanied by an increase in ATP concentration and a reduction in lactate secretion (Table 3). This suggests that aerobic metabolism increased over time in untreated RPT cells. In control adipocyte and muscle cell cultures, there was no trend for change in ATP content, lactate secretion, or total protein content, which would be indicative of loss of viability, over 19 days (Tables 4 and 5).
Fig 2.
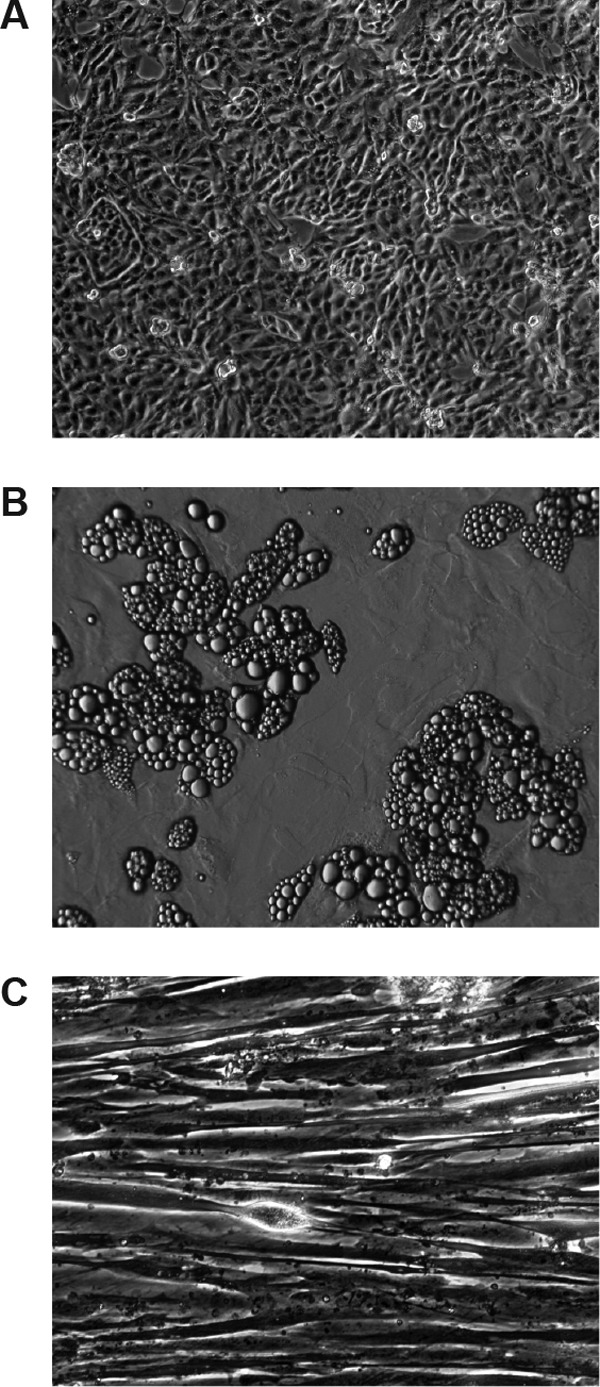
Control human primary cell cultures used in this study (magnification, ×10). (A) Renal proximal tubule cells after reaching confluence. (B) Adipocytes differentiated from subcutaneous preadipocytes. (C) Myotubes differentiated from primary human muscle satellite cells.
Table 3.
Mitochondrial DNA content and viability measures in primary cultures of human renal proximal tubule cells exposed to NRTIsa
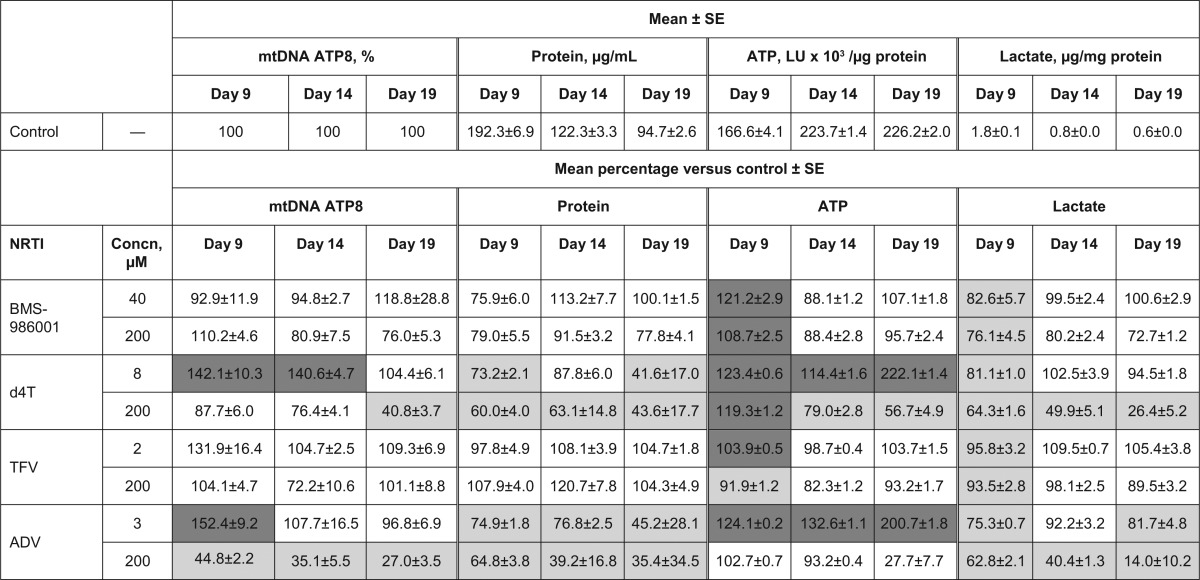
All experiments were performed in triplicate. Dark gray cells represent significant increase versus control (P < 0.05); light gray cells represent significant decrease versus control (P < 0.05). ADV, adefovir; d4T, stavudine; LU, light intensity unit; mtDNA, mitochondrial DNA; NRTI, nucleoside/nucleotide reverse transcriptase inhibitor; SE, standard error; TFV, tenofovir.
Table 4.
Mitochondrial DNA content and viability measures in primary cultures of human differentiated adipocytes exposed to NRTIsa
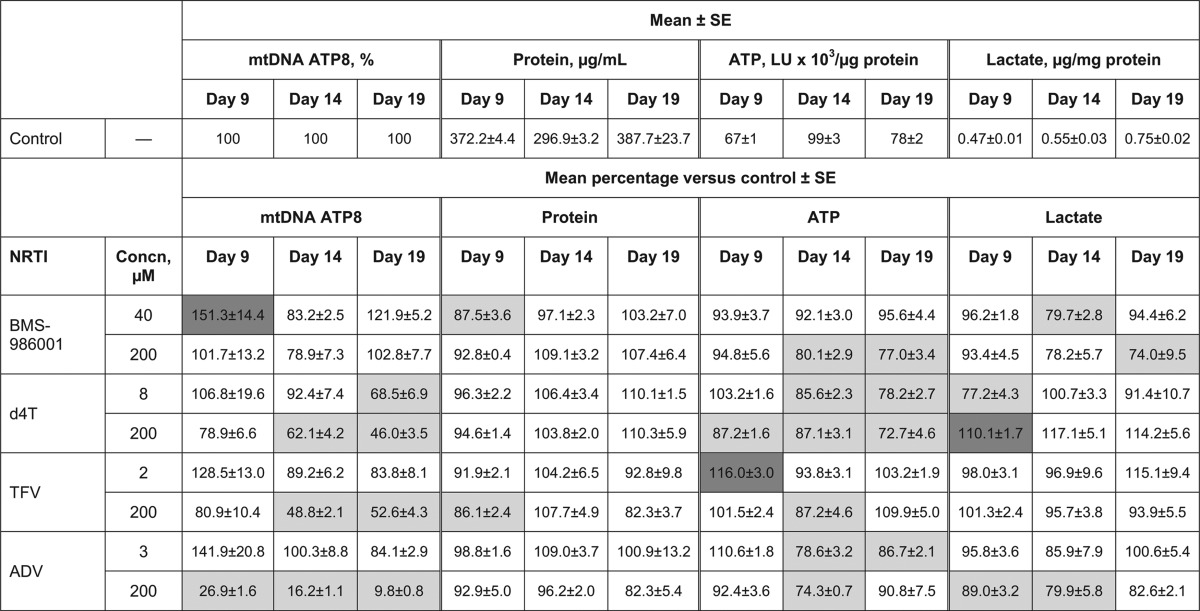
All experiments were performed in triplicate. Dark gray cells represent significant increase versus control (P < 0.05); light gray cells represent significant decrease versus control (P < 0.05). ADV, adefovir; d4T, stavudine; LU, light intensity unit; NRTI, nucleoside/nucleotide reverse transcriptase inhibitor; SE, standard error; TFV, tenofovir.
Table 5.
Mitochondrial DNA content and viability measures in primary cultures of human skeletal muscle myoblasts exposed to NRTIsa
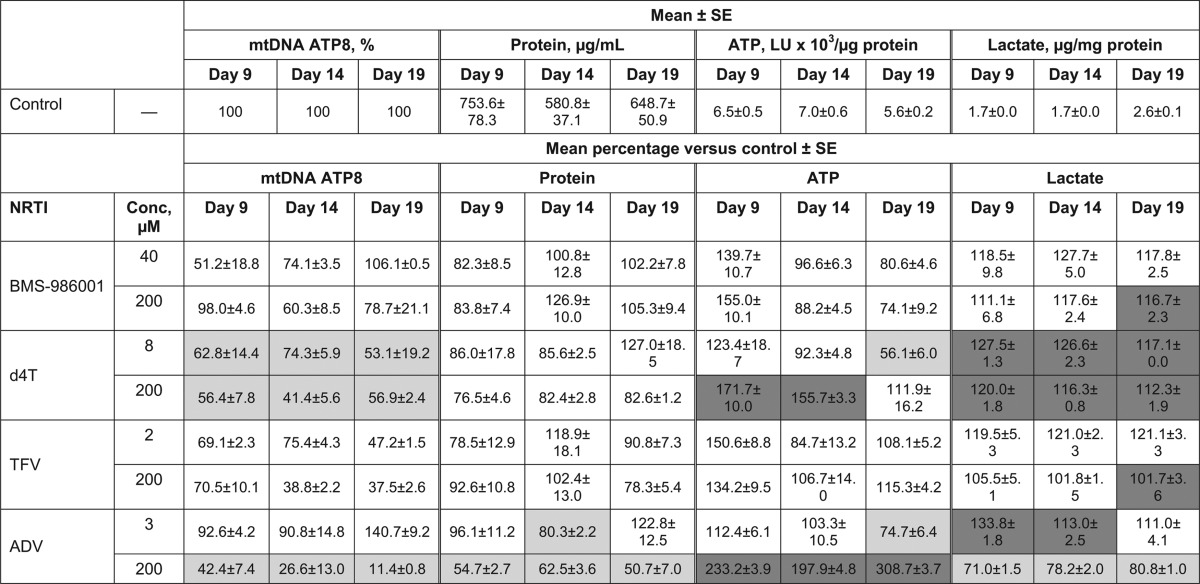
All experiments were performed in triplicate. Dark gray cells represent significant increase versus control (P < 0.05); light gray cells represent significant decrease versus control (P < 0.05). ADV, adefovir; d4T, stavudine; LU, light intensity unit; NRTI, nucleoside/nucleotide reverse transcriptase inhibitor; SE, standard error; TFV, tenofovir.
Cultures treated with BMS-986001 and d4T. (i) RPT cells.
At the reported Cmax and 200 μM, BMS-986001 did not induce statistically significant time-related reductions in mtDNA levels or cell viability in RPT cultures (Table 3). At both concentrations of BMS-986001, the ATP levels and lactate release were significantly different from those in the control culture on day 9; however, the differences were not significant at day 14 or day 19. As expected, treatment of RPT cells with d4T at both its Cmax and 200 μM resulted in significant changes in all measures (Table 3). Significant cell loss, as measured by reduction in total cell protein, was observed with both drug concentrations. d4T induced significant time- and concentration-related reductions in mtDNA levels, correlating with decreased cell numbers and reduced mtDNA polymerase activity. Additionally, d4T induced significant time- and concentration-related reductions in ATP, indicative of decreased cell viability, and time-related reductions in lactate release by RPT cells exposed to 200 μM d4T accompanied the loss of cells at this concentration.
(ii) Differentiated adipocytes.
There was no change over time in total cell protein following treatment with either BMS-986001 or d4T, indicating that cell numbers were not affected by either compound (Table 4). Neither concentration of BMS-986001 tested resulted in time-related reductions in mtDNA versus the amounts in the control, although there was a significant increase on day 9 in the 40 μM cultures. In contrast, mtDNA was reduced by day 19 following culture in d4T at 8 μM and 200 μM, suggesting that mtDNA polymerase γ activity was reduced in the presence of d4T. At Cmax, BMS-986001 had no significant effect on adipocyte ATP levels, but ATP concentrations were decreased by 20 to 25% on days 14 and 19 in cultures exposed to BMS-986001 at 200 μM. Treatment with both concentrations of d4T induced significant time- and concentration-related reductions in ATP levels. At the higher concentration, BMS-986001 induced a significant, 25% decrease in lactate secretion at day 19, which was not observed in the d4T-treated cultures. These observations imply that the higher concentration of BMS-986001 and both concentrations of d4T are associated with reductions in metabolic activity.
(iii) Skeletal muscle myoblasts.
At the reported Cmax, BMS-986001 did not induce statistically significant changes in any viability measure or mtDNA level in human muscle cell cultures. At the 200 μM concentration, the only significant change recorded was an increase in lactate secretion on day 19 (Table 5).
d4T had no significant effect on total cell protein at either concentration tested; however, significant increases in lactate secretion were observed at all time points for both d4T concentrations (Table 5). In addition, significant decreases in mtDNA were recorded at both concentrations and at all time points, indicating mitochondrial toxicity. ATP levels were not reduced and even increased at days 9 and 14 in muscle cells exposed to 200 μM d4T. The increased lactate production is indicative of conversion from oxidative metabolism to anaerobic metabolism and, thus, of significantly reduced mitochondrial function, due in this case to reduced mitochondrial DNA. Increases in ATP may appear to be paradoxical but are an indicator of the cell's response to respiratory stress.
Cultures treated with TFV and ADV. (i) RPT cells.
Neither concentration of TFV tested induced significant time- or concentration-dependent reductions in the viability measures of RPT cells (Table 3), with the exception of day 9, when there were significant differences versus the results for control cultures in ATP levels and lactate secretion, but these were not maintained at later time points. Furthermore, TFV did not significantly affect the mtDNA content of these cells. In contrast, ADV significantly reduced the total cell protein of RPT cultures at both test concentrations, with greater reductions recorded at the later time points (Table 3), indicating a decrease in cell numbers. At the higher, 200 μM concentration, ADV reduced both the protein and ATP concentration, indicating loss of cells due to cytotoxicity, but at the lower ADV concentration of 3 μM, the ATP concentration increased, with time-related decreases in protein, possibly indicating a reduction in cell size without loss of cell numbers. ADV significantly decreased lactate release at both concentrations, and at the 200 μM concentration, it significantly reduced the amount of mtDNA, which may either correspond to inhibition of mitochondrial DNA polymerase γ or be related to a reduction in cell size.
(ii) Differentiated adipocytes.
At Cmax, neither TFV nor ADV significantly affected cell viability measures or mtDNA, with the exception of an increase in ATP on day 9 with TFV and a decrease in ATP on days 14 and 19 with ADV (Table 4). At a concentration of 200 μM, TFV was associated with a significant decrease in mtDNA of around 50%. Total protein levels were significantly decreased on day 9 but not subsequently, and there was also a significant decrease in ATP levels on day 14. Similarly, exposure to ADV at 200 μM resulted in large decreases in mtDNA, with a level of only 10% of the level in the control culture recorded on day 19. Significant decreases in ATP (day 14) and lactate secretion (days 9 and 14) were also recorded with ADV at 200 μM. The observed reductions in mtDNA and other viability measures may be due to toxicity impacting cellular metabolic activity.
(iii) Skeletal muscle myoblasts.
As observed in RPT and adipocyte cultures, TFV did not induce significant reductions in the viability of muscle cells at its reported Cmax or at 200 μM (Table 5). The only significant difference from the control cultures was an increase in lactate excretion at day 19 in the 200 μM-treated cultures. In addition, TFV did not reduce the mtDNA content of these cells at either concentration. In contrast, at the 200 μM concentration, ADV induced significant changes in all measures, with a decrease in cell viability indicated by decreases of 40 to 50% in total cell protein and 20 to 30% in lactate release (Table 5). The changes in mtDNA levels were particularly noticeable, as they fell to 11% of the control level at day 19. The effect of ADV at its Cmax was considerably less, with only the ATP concentrations being significantly different from the concentration in the control at day 19.
Cultures treated with other NRTIs.
Toxicity data for ABC and AZT are included in Information in the supplemental material. ABC was consistently cytotoxic in all cell lines, as evidenced by reduced cell protein and lactate, especially at the higher, 200 μM concentration. The levels of cell ATP were also reduced in adipocytes but were significantly increased in skeletal muscle cells at both test concentrations and at the reported Cmax in RPT cells. ABC did not affect mtDNA consistently or in a manner that correlated with the cytotoxicity observed. AZT, an earlier thymidine analog, exhibited cytotoxicity in all cell cultures, as demonstrated by reductions in cell protein and lactate, which are indicative of reduced cell number and/or viability. In contrast, significant increases in cell ATP content were seen in RPT and skeletal muscle cultures. Significant reductions in mtDNA were only observed in RPT cells exposed to 200 μM AZT.
DISCUSSION
NRTIs have been associated with a range of different toxicities, many of which have been linked to mitochondrial toxicity (1, 7–11). Preliminary investigations found that BMS-986001 was associated with reduced mitochondrial toxicity compared with that of d4T (12). We carried out an extensive investigation of the in vitro toxicity profile of BMS-986001 and other NRTIs in order to better evaluate the toxicity profiles of these agents. These data indicate that in kidney, adipose, and muscle cells cultured for up to 19 days, both BMS-986001 and TFV have minimal adverse effects on mtDNA and other endpoints measuring cell viability and metabolism. BMS-986001 did not induce significant reductions in mtDNA in any of the primary cultures at the reported Cmax (40 μM) for the investigational dose of 600 mg once daily (17) or at the 5-fold-higher test concentration of 200 μM. Similarly, TFV did not significantly affect the mtDNA content of these cells at the reported Cmax of 2 μM or, with the exception of adipocytes, at the 100-fold-higher concentration of 200 μM. In a previous report, TFV (300 μM) did not reduce mtDNA in proliferating, undifferentiated human muscle cells (18). We have now extended this result to include differentiated, nondividing myotubes exposed to TFV.
In contrast, significant reductions in mtDNA were induced by d4T and ADV. Indeed, consistent with their known profiles, d4T and ADV were consistently more toxic than either of their close structural analogs, BMS-986001 and TFV, respectively. Toxicity at the 200 μM concentration was most often indicated by time-related, significant increases in cell death and changes in ATP or lactate concentrations. The amounts of mtDNA were consistently reduced at both the reported Cmax and the higher, 200 μM concentration of d4T and ADV; however, the magnitude of the reductions varied between cell types. For example, after 19 days, d4T reduced mtDNA at both test concentrations in both differentiated adipocytes and muscle cells but only at the higher concentration tested in kidney cells. ADV reduced mtDNA in all three cell types at the higher concentration of 200 μM but not at the reported Cmax.
One potential explanation for the observed differences in toxicity and for their apparent tissue specificity may be tissue- and NRTI-specific cellular metabolism and transport. For instance, it is well established that transporters and enzymes responsible for nucleoside analog entry and metabolism vary both qualitatively and quantitatively between tissues (19–21). ADV, which, like TDF, is associated with renal toxicity (14), is known to actively accumulate in proximal tubule epithelium in the kidney cortex through the activity of the basolateral human organic ion transporter SLC22A6 (OAT1), and mtDNA depletion has been reported in preclinical models as well as in human kidney (13, 22, 23). In our study, the high concentration of ADV (200 μM) was associated with depletion of mtDNA in all three cell types examined. However, mtDNA depletion was not seen with the concentration equivalent to the reported Cmax. It is therefore possible that ADV concentrations sufficient to deplete mtDNA are achieved in the RPT in vivo only following active accumulation and are not achieved in other tissues. TFV is also transported by OAT1, but in in vivo studies, no reduction in mtDNA was observed in the kidney (24). Furthermore, the data reported herein confirm previous observations that TFV does not reduce mtDNA in human RPT cells cultured for up to 3 weeks (18, 25, 26). Our study indicates that at a high, equimolar concentration (200 μM), TDF is significantly less cytotoxic in vitro than ADV.
mtDNA polymerase γ is the only DNA polymerase in the mitochondria, and consequently, it is intrinsic to the maintenance of mtDNA (27). Mutations in POLG (the gene encoding mtDNA polymerase γ) have been linked with a number of disorders (27), and inhibition of mtDNA polymerase γ has been implicated in some NRTI-associated toxicities (7). An explanation for the generally minimal effects of BMS-986001 and TFV on cell mtDNA content compared with the effects of their structurally similar analogs may therefore be the reported differences in their affinity for mtDNA polymerase γ. Specifically, d4T (inhibition constant [Ki], 1 μM) and ADV (Ki, 0.97 μM) are 100- and 60-fold more-potent inhibitors of this enzyme than BMS-986001 (Ki, >100 μM) and TFV (Ki, 59.5 μM), respectively (28–30).
It should be noted that the reported 40 μM Cmax of BMS-986001 used in this study was selected based on the highest dose investigated in the proof-of-concept study (Cmax of approximately 11 μg/ml, following a dose of 600 mg once daily for 10 days) (17) and is expected to be 2 to 6 times higher than the anticipated Cmax of the clinical doses under investigation in the ongoing phase IIb study (NCT01489046), which are 100 to 400 mg once daily.
ABC, which is used routinely in clinical practice, exhibited considerable cytotoxicity in the cell lines tested, as evidenced by the changes in viability measures. However, in this study, ABC did not affect mtDNA consistently or in a manner that correlated with the cytotoxicity observed. Increases in mtDNA following exposure to NRTIs have been noted both in vitro and in vivo and may be an initial response to cytotoxic stress (31, 32). ABC is a guanosine analog and has not been shown to inhibit mtDNA polymerase γ in vitro (33, 34), indicating that the cytotoxicity observed was most likely related to some other mechanism than impairment of mtDNA replication by ABC. Similarly, the general lack of effect of AZT on mtDNA is consistent with its weak inhibition of mitochondrial DNA polymerase, which is approximately equivalent to that of TFV in potency (18, 35–37). Significant reductions in mtDNA were only observed in RPT cells exposed to 200 μM AZT, and these changes appear to be in parallel with the cytotoxic effects of AZT observed in these cells and are likely to be stress related. Mechanisms other than inhibition of DNA polymerase γ have been proposed for the toxicity of AZT (38, 39).
When assessing the results of this study, several factors should be taken into account. Such long-term experiments in primary cell lines are subject to considerable variability, and in consequence, the use of a stringent post hoc statistical analysis can result in data that would appear to be biologically significant not reaching statistical significance. Furthermore, this study utilized nondividing primary human cell cultures in an effort to replicate the in vivo targets for NRTI toxicity. However, it is possible that in doing so, we have selected a system that is less sensitive than the traditionally employed dividing-cell models and that changes in cell viability measures are reduced over the time frame of these experiments. Finally, it should be noted that the cell culture media used in these studies contain significant concentrations of glucose. It is recognized that cells grown in glucose-rich media primarily use glycolysis to generate ATP and, consequently, they may have a level of protection against drugs that affect mitochondrial function (40). Despite this, as each compound was tested under the same conditions, the description herein of the effect of each compound on mtDNA can be used to assess their relative mitochondrial toxicities and allows comparison between the different NRTIs.
In conclusion, while this study is not an extensive examination of structure-activity relationships among the NRTIs, the data do indicate that within the structural classes examined, even among thymidine analogs like BMS-986001 and d4T, small structural changes can reduce or essentially eliminate the liability for mitochondrial toxicity. In addition, the favorable in vitro toxicity profile observed for BMS-986001 compared with those of other NRTIs in this study supports its further investigation in clinical studies. The outcome of these studies will determine whether this favorable in vitro toxicity profile will also translate into the clinic.
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by Bristol-Myers Squibb. Editorial assistance was provided by Clemence Hindley from MediTech Media and was funded by Bristol-Myers Squibb.
Footnotes
Published ahead of print 30 September 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01206-13.
REFERENCES
- 1. Panel on Antiretroviral Guidelines for Adults and Adolescents 2012. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents, 2012 update. Office of AIDS Research Advisory Council, National Institutes of Health, Department of Health and Human Services, Bethesda, MD: http://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf Accessed May 2012 [Google Scholar]
- 2. Clumeck N, Pozniak A, Raffi F. 2008. European AIDS Clinical Society (EACS) guidelines for the clinical management and treatment of HIV-infected adults. HIV Med. 9:65–71 [DOI] [PubMed] [Google Scholar]
- 3. World Health Organization 2010. Antiretroviral therapy for HIV infection in adults and adolescents: recommendations for a public health approach, 2010 revision. World Health Organization. http://www.who.int/hiv/pub/arv/adult2010/en/index.html Accessed August 2012 [PubMed] [Google Scholar]
- 4. Bedimo R, Maalouf NM, Zhang S, Drechsler H, Tebas P. 2012. Osteoporotic fracture risk associated with cumulative exposure to tenofovir and other antiretroviral agents. AIDS 26:825–831 [DOI] [PubMed] [Google Scholar]
- 5. Scherzer R, Estrella M, Li Y, Choi AI, Deeks SG, Grunfeld C, Shlipak MG. 2012. Association of tenofovir exposure with kidney disease risk in HIV infection. AIDS 26:867–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reynes J, Trinh R, Pulido F, Soto-Malave R, Gathe J, Qaqish R, Tian M, Fredrick L, Podsadecki T, Norton M, Nilius A. 2013. Lopinavir/ritonavir combined with raltegravir or tenofovir/emtricitabine in antiretroviral-naive subjects: 96-week results of the PROGRESS Study. AIDS Res. Hum. Retroviruses 29:256–265 [DOI] [PubMed] [Google Scholar]
- 7. Brinkman K, Smeitink JA, Romijn JA, Reiss P. 1999. Mitochondrial toxicity induced by nucleoside-analogue reverse-transcriptase inhibitors is a key factor in the pathogenesis of antiretroviral-therapy-related lipodystrophy. Lancet 354:1112–1115 [DOI] [PubMed] [Google Scholar]
- 8. Hammond E, McKinnon E, Nolan D. 2010. Human immunodeficiency virus treatment-induced adipose tissue pathology and lipoatrophy: prevalence and metabolic consequences. Clin. Infect. Dis. 51:591–599 [DOI] [PubMed] [Google Scholar]
- 9. Herlitz LC, Mohan S, Stokes MB, Radhakrishnan J, D'Agati VD, Markowitz GS. 2010. Tenofovir nephrotoxicity: acute tubular necrosis with distinctive clinical, pathological, and mitochondrial abnormalities. Kidney Int. 78:1171–1177 [DOI] [PubMed] [Google Scholar]
- 10. Kohler JJ, Hosseini SH, Hoying-Brandt A, Green E, Johnson DM, Russ R, Tran D, Raper CM, Santoianni R, Lewis W. 2009. Tenofovir renal toxicity targets mitochondria of renal proximal tubules. Lab. Invest. 89:513–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lebrecht D, Venhoff AC, Kirschner J, Wiech T, Venhoff N, Walker UA. 2009. Mitochondrial tubulopathy in tenofovir disoproxil fumarate-treated rats. J. Acquir. Immune Defic. Syndr. 51:258–263 [DOI] [PubMed] [Google Scholar]
- 12. Dutschman GE, Grill SP, Gullen EA, Haraguchi K, Takeda S, Tanaka H, Baba M, Cheng YC. 2004. Novel 4′-substituted stavudine analog with improved anti-human immunodeficiency virus activity and decreased cytotoxicity. Antimicrob. Agents Chemother. 48:1640–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tanji N, Tanji K, Kambham N, Markowitz GS, Bell A, D'Agati VD. 2001. Adefovir nephrotoxicity: possible role of mitochondrial DNA depletion. Hum. Pathol. 32:734–740 [DOI] [PubMed] [Google Scholar]
- 14. Vigano M, Lampertico P, Colombo M. 2011. Drug safety evaluation of adefovir in HBV infection. Expert Opin. Drug Saf. 10:809–818 [DOI] [PubMed] [Google Scholar]
- 15. Flint O, Wang F 2012. XIX International AIDS Conference, Washington, DC, 22 to 27 July 2012, poster TUPE042. [Google Scholar]
- 16. Flint O, Wang F 2012. 11th International Congress on Drug Therapy in HIV Infection, Glasgow, United Kingdom, 11 to 15 November 2012, poster 051. [Google Scholar]
- 17. Cotte L, Dellamonica P, Raffi F, Yazdanpanah Y, Molina JM, Boué F, Urata Y, Chan HP, Zhu L, Chang IH, Bertz R, Hanna GJ, Grasela DM, Hwang C. 2013. Randomized, placebo-controlled study of the safety, tolerability, antiviral activity and pharmacokinetics of 10-day monotherapy with BMS-986001, a novel HIV NRTI, in treatment-experienced HIV-1-infected subjects. J. Acquir. Immune Defic. Syndr. 63:346–354 [DOI] [PubMed] [Google Scholar]
- 18. Birkus G, Hitchcock MJ, Cihlar T. 2002. Assessment of mitochondrial toxicity in human cells treated with tenofovir: comparison with other nucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 46:716–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bazzoli C, Jullien V, Le Tiec C, Rey E, Mentre F, Taburet AM. 2010. Intracellular pharmacokinetics of antiretroviral drugs in HIV-infected patients, and their correlation with drug action. Clin. Pharmacokinet. 49:17–45 [DOI] [PubMed] [Google Scholar]
- 20. Mirzaee S, Eriksson S, Albertioni F. 2010. Differences in cytosolic and mitochondrial 5′-nucleotidase and deoxynucleoside kinase activities in Sprague-Dawley rat and CD-1 mouse tissues: implication for the toxicity of nucleoside analogs in animal models. Toxicology 267:159–164 [DOI] [PubMed] [Google Scholar]
- 21. Rylova SN, Albertioni F, Flygh G, Eriksson S. 2005. Activity profiles of deoxynucleoside kinases and 5′-nucleotidases in cultured adipocytes and myoblastic cells: insights into mitochondrial toxicity of nucleoside analogs. Biochem. Pharmacol. 69:951–960 [DOI] [PubMed] [Google Scholar]
- 22. Mulato AS, Ho ES, Cihlar T. 2000. Nonsteroidal anti-inflammatory drugs efficiently reduce the transport and cytotoxicity of adefovir mediated by the human renal organic anion transporter 1. J. Pharmacol. Exp. Ther. 295:10–15 [PubMed] [Google Scholar]
- 23. Ho ES, Lin DC, Mendel DB, Cihlar T. 2000. Cytotoxicity of antiviral nucleotides adefovir and cidofovir is induced by the expression of human renal organic anion transporter 1. J. Am. Soc. Nephrol. 11:383–393 [DOI] [PubMed] [Google Scholar]
- 24. Kohler JJ, Hosseini SH, Green E, Abuin A, Ludaway T, Russ R, Santoianni R, Lewis W. 2011. Tenofovir renal proximal tubular toxicity is regulated by OAT1 and MRP4 transporters. Lab. Invest. 91:852–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cihlar T, Birkus G, Greenwalt DE, Hitchcock MJ. 2002. Tenofovir exhibits low cytotoxicity in various human cell types: comparison with other nucleoside reverse transcriptase inhibitors. Antiviral Res. 54:37–45 [DOI] [PubMed] [Google Scholar]
- 26. Vidal F, Domingo JC, Guallar J, Saumoy M, Cordobilla B, Sanchez de la Rosa R, Giralt M, Alvarez ML, Lopez-Dupla M, Torres F, Villarroya F, Cihlar T, Domingo P. 2006. In vitro cytotoxicity and mitochondrial toxicity of tenofovir alone and in combination with other antiretrovirals in human renal proximal tubule cells. Antimicrob. Agents Chemother. 50:3824–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hudson G, Chinnery PF. 2006. Mitochondrial DNA polymerase-gamma and human disease. Hum. Mol. Genet. 15(Spec No 2):R244–R252 [DOI] [PubMed] [Google Scholar]
- 28. Kramata P, Votruba I, Otova B, Holy A. 1996. Different inhibitory potencies of acyclic phosphonomethoxyalkyl nucleotide analogs toward DNA polymerases alpha, delta and epsilon. Mol. Pharmacol. 49:1005–1011 [PubMed] [Google Scholar]
- 29. Yang G, Dutschman GE, Wang CJ, Tanaka H, Baba M, Anderson KS, Cheng YC. 2007. Highly selective action of triphosphate metabolite of 4′-ethynyl D4T: a novel anti-HIV compound against HIV-1 RT. Antiviral Res. 73:185–191 [DOI] [PubMed] [Google Scholar]
- 30. Cherrington JM, Allen SJ, Bischofberger N, Chen MS. 1995. Kinetic interaction of the diphosphates of 9-(2-phosphonylmethoxyethyl)adenine and other anti-HIV active purine congeners with HIV reverse transcriptase and human DNA polymerases a, b and g. Antivir. Chem. Chemother. 6:217–221 [Google Scholar]
- 31. Papp E, Gadawski I, Cote HC. 2008. Longitudinal effects of thymidine analogues on mtDNA, mtRNA and multidrug resistance (MDR-1) induction in cultured cells. J. Antimicrob. Chemother. 61:1048–1052 [DOI] [PubMed] [Google Scholar]
- 32. Kunz A, Wurmb-Schwark N, Sewangi J, Ziske J, Lau I, Mbezi P, Theuring S, Hauser A, Dugange F, Katerna A, Harms G. 2012. Zidovudine exposure in HIV-1 infected Tanzanian women increases mitochondrial DNA levels in placenta and umbilical cords. PLoS One 7:e41637. 10.1371/journal.pone.0041637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maagaard A, Kvale D. 2009. Long term adverse effects related to nucleoside reverse transcriptase inhibitors: clinical impact of mitochondrial toxicity. Scand. J. Infect. Dis. 41:808–817 [DOI] [PubMed] [Google Scholar]
- 34. Martin JL, Brown CE, Matthews-Davis N, Reardon JE. 1994. Effects of antiviral nucleoside analogs on human DNA polymerases and mitochondrial DNA synthesis. Antimicrob. Agents Chemother. 38:2743–2749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Anderson PL, Kakuda TN, Lichtenstein KA. 2004. The cellular pharmacology of nucleoside- and nucleotide-analogue reverse-transcriptase inhibitors and its relationship to clinical toxicities. Clin. Infect. Dis. 38:743–753 [DOI] [PubMed] [Google Scholar]
- 36. Johnson AA, Ray AS, Hanes J, Suo Z, Colacino JM, Anderson KS, Johnson KA. 2001. Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J. Biol. Chem. 276:40847–40857 [DOI] [PubMed] [Google Scholar]
- 37. Lee H, Hanes J, Johnson KA. 2003. Toxicity of nucleoside analogues used to treat AIDS and the selectivity of the mitochondrial DNA polymerase. Biochemistry 42:14711–14719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lund KC, Wallace KB. 2004. Direct effects of nucleoside reverse transcriptase inhibitors on rat cardiac mitochondrial bioenergetics. Mitochondrion 4:193–202 [DOI] [PubMed] [Google Scholar]
- 39. Lynx MD, Bentley AT, McKee EE. 2006. 3′-Azido-3′-deoxythymidine (AZT) inhibits thymidine phosphorylation in isolated rat liver mitochondria: a possible mechanism of AZT hepatotoxicity. Biochem. Pharmacol. 71:1342–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y. 2007. Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol. Sci. 97:539–547 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.