

Abstract
As the second dimension to the genome, the epigenome contains key information specific to every type of cells. Thousands of human epigenome maps have been produced in recent years thanks to rapid development of high throughput epigenome mapping technologies. In this review, we discuss the current epigenome mapping toolkit and utilities of epigenome maps. We focus particularly on mapping of DNA methylation, chromatin modification state and chromatin structures, and emphasize the use of epigenome maps to delineate human gene regulatory sequences and developmental programs. We also provide a perspective on the progress of the epigenomics field and challenges ahead.
Introduction
More than a decade has passed since the human genome was completely sequenced, but how genomic information directs spatial and temporal specific gene expression programs remains to be elucidated (Lander, 2011). The answer to this question is not only essential for understanding the mechanisms of human development, but also key to studying the phenotypic variations among human populations and the etiology of many human diseases. However, a major challenge remains: each of the more than 200 different cell types in the human body contains an identical copy of the genome but expresses a distinct set of genes. How does a genome guide a limited set of genes to be expressed at different levels in distinct cell types?
Overwhelming evidence now indicates that the epigenome serves to instruct the unique gene expression program in each cell type together with its genome. The word “epigenetics”, coined half a century ago by combining “epigenesis” and “genetics”, to describe the mechanisms of cell fate commitment and lineage specification during animal development (Holliday, 1990; Waddington, 1959). Today, the “epigenome” is generally used to describe the global, comprehensive view of sequence-independent processes that modulate gene expression patterns in a cell, and has been liberally applied in reference to the collection of DNA methylation state and covalent modification of histone proteins along the genome (Bernstein et al., 2007; Bonasio et al., 2010). The epigenome can differ from cell type to cell type, and in each cell it regulates gene expression in a number of ways - by organizing the nuclear architecture of the chromosomes, restricting or facilitating transcription factor access to DNA, and preserving a memory of past transcriptional activities. Thus, the epigenome represents a second dimension of the genomic sequence and is pivotal to for maintaining cell-type-specific gene expression patterns.
Not long ago, there were many points of trepidation about the value and utility of mapping epigenomes in human cells (Henikoff et al., 2008; Madhani et al., 2008). At the time, it was suggested that histone modifications simply reflect activities of transcription factors (TFs), so cataloging their patterns would offer little new information. However, some investigators believed in the value of epigenome maps and advocated for concerted efforts to produce such resources (Feinberg, 2007; Henikoff et al., 2008; Jones and Martienssen, 2005). The last five years have shown that epigenome maps can greatly facilitate the identification of potential functional sequences thereby annotating of the human genome. Now, we appreciate the utility of epigenomic maps in the delineation of thousands of lincRNA genes and hundreds of thousands of cis-regulatory elements (Consortium et al., 2012; Ernst et al., 2011; Guttman et al., 2009; Heintzman et al., 2009; Xie et al., 2013b; Zhu et al., 2013), all of which were obtained without prior knowledge of cell-type-specific master transcriptional regulators. Interestingly, bioinformatic analysis of tissue-specific cis-regulatory elements has actually uncovered novel TFs regulating specific cellular states.
Propelled by rapid technological advances, the field of epigenomics is enjoying unprecedented growth with no sign of deceleration. An expanding cadre of researchers is working to explore exciting frontiers in epigenomics. Many international consortia have been formed to tackle the fundamental problems in epigenomics by sharing resources and protocols (Table 1) (Beck et al., 2012; Bernstein et al., 2010). Consequently, the number of epigenomic datasets and publications has grown exponentially in recent years. The resulting epigenomic maps have linked genomic sequences to many nuclear processes including splicing, replication, DNA damage response, folding, chromatin packaging, and cell type specific gene expression patterns.
Table 1.
Large Scale National and International Epigenomic Consortia
| Project Name | Start Date | Affiliations | Completed and Expected Data Contributions | Selected Publication | Access Data |
|---|---|---|---|---|---|
| Encyclopedia of DNA Elements (ENCODE) | 2003 | NIH | Dnase-seq, RNA-seq, ChIP-seq, and 5C in 100s of primary human tissues and cell lines | (Consortium et al., 2012) | http://encodeproject.org/ |
| The Cancer Genome Atlas (TCGA) | 2006 | NIH | DNA methylomes in 1000s of patients samples from more than 20 cancer types | (Garraway and Lander, 2013) | http://cancergenome.nih.gov/ |
| Roadmap Epigenomics Project | 2008 | NIH | Dnase-seq, RNA-seq, ChIP-seq, and MethylC-seq in 100s of normal primary cells, hESC, and hESC derived cells | (Bernstein et al., 2010) | http://www.epigenomebrowser.org/ |
| International Cancer Genome Consortium (ICGC) | 2008 | 15 countries, includes TCGA | DNA methylation profiles in thousands of patient samples from 50 different cancers | (Consortium et al., 2010) | http://dcc.icgc.org/ |
| International Human Epigenome Consortium (IHEC) | 2010 | 7 countries, includes BLUEPRINT, Roadmap | Goal: 1,000 Epigenomes in 250 cell types | (American Association for Cancer Research Human Epigenome Task ForceEuropean Union, Network of Excellence, Scientific Advisory Board, 2008) | http://ihec-epigenomes.org |
In this review, we focus on recent progress in several areas of epigenomics. First, we describe the remarkable advances in epigenomic technologies, especially the next-generation sequencing based applications, which have fueled the growth of the field. Second, we discuss the utility of epigenomic maps, emphasizing the power of these maps in annotating transcription units, cis-regulatory elements in the context of development and disease pathogenesis. We also explore new biological insights gained through integrative analysis of epigenomic maps in mammalian cell systems, highlighting the study of pluripotency and lineage specification of embryonic stem cells. Finally, we provide a perspective on the road ahead regarding meeting the technical challenges and addressing unanswered questions in the field. As for advancements in our understanding of the mechanistic roles of sequence-specific TFs and non-coding RNAs, chromatin and DNA modifying enzymes, and chromatin binding proteins in establishing, maintaining, and removing epigenetic marks, we refer readers to recent excellent reviews (Badeaux and Shi, 2013; Calo and Wysocka, 2013; Lee and Young, 2013; Pastor et al., 2013; Rinn and Chang, 2012; Smith and Meissner, 2013).
Epigenome Mapping Technologies
The unprecedented genome-wide scope and nucleotide precision with which we can now map human epigenomes was enabled by disruptive technology advancement, namely the application of microarrays and next generation sequencing (NGS). A slew of molecular biology assays previously used to measure a single locus are now integrated with these platforms. Powerful parallel short-read sequencing technologies have proven increasingly high throughput, fast, accurate, and cost-effective at rates faster than Moore’s Law. By far, the greatest advantage of NGS is its ability to survey the entire genome in an unbiased and comprehensive manner. Due to this monumental shift in assay capacity, researchers can ask if conclusions drawn from locus-centered studies extend to other parts (even unknown parts) of the genome. In defiance of the traditional scientific method emphasizing hypothesis testing, global profiling promotes hypothesis-free exploration of new observations and correlations. Overall, the accomplishments and discoveries of the epigenomics field have hinged in large part on the iterative inventions and improvements of technologies (Figure 1). Here we discuss cutting-edge epigenome mapping techniques that integrate next generation sequencing platforms and disclose their advantages and disadvantages.
Figure 1.
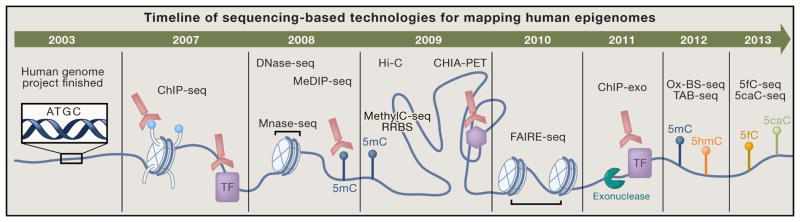
Timeline of Sequencing Based Technologies for Mapping Human Epigenomes
Mapping DNA Methylation
Of the four nucleotides composing DNA, cytosine is by far the most dynamic. Cytosine can be methylated at its 5th carbon (5mC) and in the human genome 60–80% of 28 million CpG dinucleotides are methylated (Lister et al., 2009; Ziller et al., 2013). Stressing the importance of DNA methylation during development, deletion of cytosine methyltransferases responsible for de novo (DNMT3A, DNMT3B) or maintenance (DNMT1) of methylation through cellular divisions results in embryonic and neonatal lethality in mice (Li et al., 1992; Okano et al., 1999). Detection of DNA methylation at individual loci and with promoter-focused studies established the important repressive roles of DNA methylation in imprinting, retrotransposon silencing, and X chromosome inactivation (Bird, 2002). Global DNA methylation technologies now measure DNA methylation abundance at all cytosines at base resolution in the human genome. The elucidation of complete human methylomes progressed the narrow view of 5mC as only a stable repressive mark to an epigenetic mark that is dynamically deposited and removed, can exist in non-CpG sequence contexts, and is enriched at the bodies of actively transcribed genes. (Hellman and Chess, 2007; Lister et al., 2009).
The toolkit for measuring DNA methylation includes three main molecular biology based techniques: digestion of genomic DNA with methyl-sensitive restriction enzymes, affinity-based enrichment of methylated DNA fragments, and chemical conversion methods (Bock, 2012; Laird, 2010). The choice of how to assay DNA methylation depends on the resolution and genome coverage needs, and both parameters ultimately dictate the experimental cost. Considering resolution, endonuclease digestion based assays (MRE-seq, etc.) are limited by the frequency of cut sites. It is possible to improve resolution by cutting with multiple enzymes. Affinity enrichment based assays capture methylated DNA fragments with an antibody (MeDIP-seq) or a methyl binding domains (MBD-seq) (Down et al., 2008; Serre et al., 2010). When sequencing enriched DNA fragments, at least one cytosine is certainly methylated but the exact site or combination of sites could not be directly determined. Therefore the resolution of affinity-based assays is highly dependent on the DNA fragment size, CpG density, and immunoprecipitation quality of the reagent. Lastly, the results of both restriction enzyme and affinity based sequencing methods are qualitative rather than absolute. On the other hand, because affinity and restriction enzyme based methylation assays enrich or capture methylated DNA regions the sequencing costs are restricted (Figure 2).
Figure 2.

“Epi-nomics” of Human DNA Methylation Technologies
Bisulfite sequencing is a chemical conversion method that directly determines the methylation state of each cytosine in a binary fashion. Treatment of genomic DNA with sodium bisulfite chemically converts unmethylated cytosines to uracil. After PCR and assuming nearly complete bisulfite conversion, all unmethylated cytosines become thymidines and remaining cytosines correspond to 5mC. Initially, individual loci were assayed from BS treated genomic DNA with locus specific PCR primers followed by Sanger sequencing (Clark et al., 1994). In a step towards increasing genomic coverage, reduced representation bisulfite sequencing (RRBS) combines restriction digestion with bisulfite sequencing for specific interrogation of high CpG density regions such as clusters of CpGs at promoters called CpG islands (Meissner, 2005). At the pinnacle of the genomic coverage spectrum, bisulfite treatment coupled with whole genome sequencing (variably referred to as MethylC-seq, BS-seq, or WGBS) features nucleotide resolution and quantitative rates of methylation for all cytosines (Cokus et al., 2008; Lister et al., 2008). Due to its global scope, highly quantitative measurement, and single base resolution, MethylC-seq is widely accepted as a gold standard for mapping DNA methylomes. However, some caveats of MethylC-seq remain, such as potential PCR biases due to unbalanced CG content of methylated and unmethylated fragments which can skew 5mC quantitation and mapping inefficiencies of bisulfite treated DNA (Laird, 2010; Lister et al., 2009).
The DNA methylation field collectively experienced an epiphany upon the recent discovery of 5-hydroxymethylcytosine (5hmC) as an intermediate of demethylation of 5mC to cytosine (Kriaucionis and Heintz, 2009; Tahiliani et al., 2009). The ten-eleven translocation (TET) family of proteins, TET1, TET2, and TET3, oxidize 5mC through 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) intermediates before being replaced by cytosine via base excision repair pathways or a yet unidentified decarboxylase (Pastor et al., 2013). As it became clear that four variants of cytosine exist, a clarification to MethylC-seq results also came to light. 5mC and 5hmC, but not 5fC and 5caC, are both resistant to bisulfite conversion and therefore cannot be distinguished from each other in MethylC-seq data (Huang et al., 2010; Jin et al., 2010). In order to understand the role of DNA demethylation, new techniques would need to be developed to accurately differentiate cytosine methylation states.
In an exciting advancement in the field, an assortment of methods was published for the detection of all cytosine methylation states. The first versions use antibodies to either directly IP 5hmC (hMeDIP-seq) or chemically modify 5hmC making it more immunogenic (anti-CMS, hMe-Seal, GLIB, JBP-1) (Ficz et al., 2011; Pastor et al., 2011; Robertson et al., 2011; Song et al., 2011; Williams et al., 2011). While these methods are an important advancement, as mentioned above, affinity-based techniques are hindered by low resolution and qualitative signal. Last year, both oxidative bisulfite sequencing (oxBS-seq) and TET assisted bisulfite sequencing (TAB-seq) were introduced as single-base resolution methods for measuring 5hmC (Booth et al., 2012; Yu et al., 2012). In oxBS-seq, 5hmC nucleotides are sensitized to bisulfite treatment after a chemical reaction to specifically oxidize all 5hmC and to 5fC. Then, DNA is successively treated with sodium bisulfite such that remaining cytosines must originally be 5mC. The sequencing results of oxBS-seq therefore accurately capture 5mC levels and subtraction of MethylC-seq data reveals true 5hmC sites. Of concern with oxBS-seq are the successive chemical treatments that can induce DNA damage and skew the results. The second approach, TAB-seq, directly assays 5hmC location and abundances. First, 5hmC is tagged with a glucose molecule using the T4 bacteriophage enzyme beta glucosyltransferase. Next, genomic DNA is treated with purified TET enzyme to oxidize 5mC to 5caC while glucosylated-5hmC is protected. Finally, after bisulfite treatment and sequencing only 5hmC is read as cytosine, while all cytosines and cytosine variants are detected as thymidine. TAB-seq’s accuracy is especially dependent on efficient oxidation and conversion by TET, such that a bottleneck is the tedious process of purifying catalytically active TET enzyme (Song et al., 2012). In a final round of progress for DNA methylation detection, methods to detect 5fC (fC-seq, fCAB-seq, 5fC-DP-Seq) and 5caC (5caC-seq) were recently described, and the only unturned stone is measuring 5caC at base resolution (Raiber et al., 2012; Shen et al., 2013; Song et al., 2013).
Methods without enrichment steps, like MethylC-seq, oxBS-seq, and TAB-seq, require an immense amount of sequencing and are expensive (Figure 2). In order to quantitatively measure the methylation rate at each cytosine, high-quality single-nucleotide resolution 5mC methylome data sets are expected to have 30X sequencing depth. A recent study by Meissner and colleagues find that only ~20% of CpGs are differentially methylated between 30 diverse human cell and tissue types tested. Therefore, up to 80% of MethylC-seq data are the result of superfluous sequencing of DNA fragments without CpGs or containing uninformative, constitutively methylated CpGs across 30 cell and tissue types examined (Ziller et al., 2013). One approach to limiting sequencing costs is to combine capture-based methods with base-resolution DNA methylation assays for targeted mapping as accomplished by bisulfite padlock probes (BSPPs) and Illumina’s Infinium BeadChIP technology (Bibikova et al., 2011; Diep et al., 2012). Theoretically, base resolution methylomes of rare cytosine variants such as 5caC should be 1000X in sequence coverage (Pastor et al., 2013). The extreme sequencing depth is necessitated by the rarity of cytosine variants; for example, in mouse embryonic stem cells (mESCs) 5hmC accounts for about 0.1% and 5caC accounts for 0.0003% of cytosines (Ito et al., 2011; Yu et al., 2012). In terms of studying DNA methylation dynamics, a technology capable of quantitatively measuring and precisely differentiating all states of cytosine methylation in one assay would the most efficient use of sequencing resources.
Unfortunately, the prohibitive cost of base resolution human methylomes has limited the number of available datasets and presumably slowed progress towards novel insights. Resolution, genomic coverage, and monetary cost serve as interdependent considerations for generating methylome datam where one parameter cannot be changed without affecting the others (Figure 2). Maximizing resolution and coverage while keeping costs low, currently an unrealistic situation, may eventually be possible with innovative sequencing platforms or a yet undiscovered alternative to bisulfite treatment. We look forward to the democratization of nucleotide resolution DNA methylation technologies and the resulting novel findings from a diverse collection of methylomes.
Mapping Chromatin Modification States
Chromosomal DNA is packaged into nucleosomes with DNA wrapped around histone octamers consisting of H2A, H2B, H3, H4 subunits and their variants. The histone tails and globular domains of histone proteins are subject to over 130 post-translational modifications (PTMs) and over 700 distinct histone isoforms have been detected in human cells (Tan et al., 2011; Tian et al., 2012). Well-studied covalent modifications on histones include methylation, acetylation, phosphorylation, and ubiquitination. State-of-the-art mass spectrometry based proteomic technologies unveiled many novel histone PTMs such as crotonylation, succinylation, malonylation and others (Tian et al., 2012). Histone modifications serve both activating and silencing roles in transcription, generally by controlling the accessibility of DNA and by serving as binding substrates that recruit or exclude protein complexes (Kouzarides, 2007). For example, H3K27ac is found at both active promoters and enhancers, H3K36me3 identifies actively transcribed gene bodies, and H3K27me3 marks heterochromatic or repressed regions (Li et al., 2007) (Table 3). Determining the genome-wide distribution of a histone mark can lead to clues about its role in transcriptional regulation and provoke follow-up mechanistic studies to further understand the PTMs deposition, removal, and role in development and disease.
Table 3.
Distinctive Chromatin Features of Genomic Elements
| Functional Annotation | Histone Marks | References |
|---|---|---|
| Promoters | H3K4me3 | (Bernstein et al., 2005; Kim et al., 2005; Pokholok et al., 2005) |
| Bivalent/Poised Promoter | H3K4me3/H3K27me3 | (Bernstein et al., 2006) |
| Transcribed Gene Body | H3K36me3 | (Barski et al., 2007) |
| Enhancer (both active and poised) | H3K4me1/H3K4me3- | (Heintzman et al., 2007) |
| Poised Developmental Enhancer | H3K4me1/H3K27me3 | (Creyghton et al., 2010; Rada-Iglesias et al., 2011) |
| Active Enhancer | H3K4me1/H3K27ac | (Creyghton et al., 2010; Heintzman et al., 2009; Rada-Iglesias et al., 2011) |
| Polycomb Repressed Regions | H3K27me3 | (Bernstein et al., 2006; Lee et al., 2006) |
| Heterochromatin | H3K9me3 | (Mikkelsen et al., 2007) |
Chromatin immunoprecipitation followed by sequencing (ChIP-seq) maps the genome wide binding pattern of chromatin associated proteins, which includes modified histones. To perform this method, DNA-protein complexes containing a specific protein of interest are immunoprecipitated from crosslinked, sonicated chromatin. DNA is purified from the enriched pool and adaptors are ligated for subsequent PCR and sequencing. The digital sequences of enriched DNA, called reads, are computationally aligned to the reference genome to define punctate peaks or broad blocks of modified histones or protein occupancy. Since its development in 2007, researchers have used ChIP-seq extensively to survey the genomic profiles of histones and their modifications, TFs, DNA and histone modifying enzymes, transcriptional machinery, and other chromatin-associated proteins (Barski et al., 2007; Johnson et al., 2007; Mikkelsen et al., 2007; Robertson et al., 2007) Furthermore, multi-dimensional datasets are now available for cell lines, primary cells, tissues, and embryos from an increasing number of species. The ENCODE and Roadmap Epigenomics Projects have contributed enormous data repositories by performing thousands of ChIP-seq experiments in hundreds of human cell types.
As the number of ChIP-seq data sets began to grow exponentially, lab-to-lab protocol variability threatened the quality of results and downstream cross-study analysis. To stave off data inconsistencies, both the ENCODE and Roadmap consortia published optimized standard operating procedures for ChIP-seq. Of major concern was the quality of antibodies for which ChIP is undeniably dependent. Both consortia assessed histone modification antibody quality using dot blot immunoassays against histone tail peptides to ensure specific binding and minimal cross reactivity (Egelhofer et al., 2011). The ENCODE Project’s best practices include a rigorous two stage antibody validation process with a combination of immunoassays, immunofluorescence patterns, and functional assays (Landt et al., 2012). These screening methods dramatically improved the data quality and reduced costs and lost time from failed ChIP-seq experiments. However, the membrane binding conditions in immunoassays screens are not identical to immunoprecipitation in solution using magnetic beads. Lamentably, the gold standard for ChIP-grade antibody classification remains actually performing the assay itself. To this point, Bernstein and colleagues designed a method called ChIP-string to screen for effective antibodies against chromatin regulator proteins (Ram et al., 2011). In this approach, multiplexed meso-scale ChIP-seq experiments survey protein enrichment at ~500 representative loci using the nCounter probe system. High quality antibodies are distinct from IgG patterns and the occupancy distribution correlates with a logical set of chromatin states. Validating antibody reagents for enrichment specificity and robustness ensures good quality ChIP-seq data sets with high signal to noise ratios.
Given that ChIP-seq is a mature technology, the technical restrictions of the technique are well defined by its users. These restrictions include the need for large amounts of starting material, limited resolution, and the dependence on antibodies. Improvements to ChIP-seq have been developed to address these limitations and expand the possibilities of its use. Collecting enough starting material for ChIP-seq can be challenging because experiments typically require 1 million (histone modifications) to 5 million (TFs and chromatin modifiers) cells. While this is feasible when studying fast dividing cell lines, the challenge arises when studying primary cells and rare populations such as cancer stem cells or progenitor cells. ChIP-seq samples of 50,000 cells or less are possible with the ChIP-nano protocol (Adli and Bernstein, 2011). Key method modifications achieve effective chromatin fragmentation in small volumes, ensure minimal sample handling and loss by washing samples in columns, and reduce background signal. Another procedure, called ChIP-exo, improves the limited resolution from fragmentation heterogeneity after chromatin is prepared by sonication along (Rhee and Pugh, 2011). As its name suggests, sonicated and immunoprecipitated DNA is treated with a 5′-to-3′ exonuclease to digest DNA to the footprint of the crosslinked protein such that sequencing results are nucleotide resolution. This type of high resolution protein binding data is most beneficial for uncovering motifs of specific binding proteins and the effect of sequence variants on protein binding affinity. Profiling genome-wide DNA-protein interactions with ChIP-seq is technically challenging when studying novel proteins or protein isoforms, such as a histone variant, that lacks a robust or specific antibody. In this case, an obvious approach is to transiently or stably express a protein of interest (POI) with a tag or epitope that can be readily ChIP’ed. Controls are necessary to ensure the fusion protein’s localization is not altered by non-endogenous expression levels, protein instability, steric inherence, or other effects of the tag itself.
A ChIP step can be added to other genomic profiling approaches for integrated epigenomic profiling. First, two ChIP steps in a row, or Sequential-ChIP-seq, can uncover histone PTMs on the same molecule or chromatin associated proteins in the same complex. Several groups combined bisulfite sequencing with ChIP giving rise to BisChIP-seq and ChIP-BS-seq (Brinkman et al., 2012; Statham et al., 2012). Long distance DNA interactions mediated by a specific protein can be profiled using chromatin interaction analysis by paired-end-tag sequencing, or ChIA-PET (Fullwood et al., 2009). We anticipate other inventive uses of ChIP technology to continue to uncover undiscovered roles of histone modifications and histone variants in transcriptional regulation.
Mapping of Chromatin structures
Nucleosome Positioning
Moving up the organizational hierarchy of genomic organization, we now look beyond the DNA and histone modifications to the positioning of nucleosomes along the genome. Our epigenome at its most basic level is repeating units of 147 base pairs wrapped 1.7 times around each nucleosome with varying distances of linker DNA between each unit. Even this extremely simplistic model is complex because nucleosome positioning can both inhibit and promote factor binding (Bell et al., 2011). First, nucleosomes can be positioned to obstruct or reveal specific DNA sequences. Secondly, because modifications on histone tails serve as binding platforms for transcriptional regulators, nucleosome positioning regulates factor recruitment. And finally, nucleosomes are suggested to inhibit transcription by slowing progression of RNA polymerase II as it transcribes through a gene body. From a medical perspective, it will be important to determine the possible role of aberrant nucleosome positioning as caused by disease associated SNPs, insertions, deletions, and translocations.
Our understanding of the regulation of nucleosome positioning came from studies of smaller genomes, such as those in yeast and fly (Jiang and Pugh, 2009). Nucleosome positioning along DNA is influenced by favorable DNA sequence composition, the actions of ATP-dependent nucleosome remodelers, and strongly positioned nucleosomes and DNA bound proteins such as TFs and RNA Pol II are barriers to nucleosome position shifting (Mavrich et al., 2008; Narlikar et al., 2013; Yuan et al., 2005). While we understand the main determinants of nucleosome positioning, the exact contribution of each is unclear and currently under debate.
The most common method for profiling genome-wide nucleosome positioning is microcococal nuclease digestion of chromatin followed by high-throughput sequencing (MNase-seq). When native, uncrosslinked chromatin is digested with MNase, the linker DNA is cleaved while DNA wrapped around histone octamers or bound by TFs is protected. After purifying the DNA, approximately 150bp fragments corresponding to mononucleosomes are size selected on a gel and sequenced.
Given the relative size of the human genome as compared to model organisms and that most of it is nucleosomal, mapping nucleosome positioning in the human genome is no small feat. Even with only 10-fold genome coverage in CD4+ T Cells, it was clear that nucleosomes are depleted at active transcriptional start sites (TSSs) and enhancers with ordered positioning radiating outward (Schones et al., 2008). In an extraordinary sequencing effort, Gaffney et al. generated paired and single-end MNase-seq data in seven lymphoblastoid cell lines yielding 240X coverage of a single cell type and found that ~80% of the genome has non-random, albeit weakly positioned nucleosomes (Gaffney et al., 2012). MNase-seq based studies have provided great insights into the global distribution and dynamics of nucleosomes in the human genome. However, there are limitations of these datasets to consider. First, MNase digestion at the ends of nucleosomes is inconsistent such that exact position of nucleosomes can only be estimated as the center of fragments of different lengths. The results are unfortunately not single-base resolution. Moreover, MNase has AT sequence preferences and MNase protected 150bp fragments are inferred to be mononucleosomes but could in fact be created by other proteins as well (Brogaard et al., 2012).
To circumvent the weaknesses of digestion based detection of nucleosome positioning, Widom and colleagues used chemical modification of engineered histones to cleave DNA wrapped around nucleosomes (Brogaard et al., 2012). In short, DNA is precisely cleaved in a reaction with hydroxyl radicals if it interacts with a mutated residue on H4 while wound around a nucleosome. Using this method, nucleosome maps in yeast show remarkable accuracy and consistency. Interestingly, single-base pair resolution of yeast nucleosomes reveals a 10bp periodic sequence preference of flexible dinucleotides throughout the 147 bases in contact with the histone octamer. This data suggests that the role of sequence composition in the rotation positioning of the nucleosomes is stronger than previously appreciated. This method could be extended to the human genome for precise, high-resolution nucleosome positioning studies.
Another MNase-independent method for mapping nucleosome positioning uses DNA methyltransferase accessibility to footprint nucleosome positions, called nucleosome occupancy and methylome sequencing (NOMe-seq) (Kelly et al., 2012). The unique activity of the DNA methyltransferse M.CviPI, which methylates cytosine only in the GpC context, is exploited to record the DNA’s nucleosomal status because nucleosomal GpCs are protected from methylation. Next, the DNA is bisulfite treated to convert unmethylated cytosines to thymide. Following sequencing, cytosines in the CpG context were originally methylated (5mC or 5hmC) and cytosines in the GpC context were nucleosome depleted. An important advantage of NOMe-seq is the dual epigenomic information, both nucleosome position and DNA methylation abundance, comes from a single molecule rather than possibly co-occuring in a population of cells.
Chromatin Accessibility
Nucleosomes are the basic repeated structural unit of the genome and, as previously mentioned, proteins that compete for DNA binding can affect their positioning. Biochemically active regulatory elements, including promoters, enhancers, silencers, and insulators, are bound by sequence specific regulatory TFs. Open chromatin is therefore an overarching characteristic of biochemically active genomic regions. Open chromatin can be assayed genome-wide by DNase hypersensitivity followed by sequencing (DNase-seq) or formaldehyde-assisted identification of regulatory elements followed by sequencing (FAIRE-seq) (Boyle et al., 2008; Giresi et al., 2007). DNase-seq takes advantage of the protection conferred by tightly wound nucleosomes from DNaseI endonuclease digestion. Accordingly, limited digestion of native chromatin releases nucleosome-depleted fragments. Sequencing and mapping of these fragments identifies DNaseI-hypersensitive sites (DHSs) corresponding to regulatory regions. Protein and complex binding within a DHS create 6–40bp sequences of DNaseI protection, called digital DNase footprints, which can be called after deep sequencing of DNase-seq libraries (Neph et al., 2012b). Beyond being DNaseI hypersensitive, open chromatin regions are also sensitive to shearing by sonication and this concept is exploited by the FAIRE-seq assay. Initially, chromatin isolated from formaldehyde crosslinked cells is subject to sonication. Subsequently, the DNA from open chromatin regions is isolated from the aqueous phase following phenol-chloroform extraction. Similar to DNase-seq, mapping the FAIRE-seq enriched fragments genome-wide demarcate regulatory elements.
Myriad open chromatin maps are now available due to the adoption of DNase-seq by the ENCODE Project Consortium and Roadmap Epigenomics Program. In total, these consortia produced hundreds of maps encompassing 349 cell types including pluripotent stem cells, stem cell progenitors, cultured primary cells, and human fetal tissue from various types and gestational stages (Maurano et al., 2012). Meta-analysis of these datasets determined DHSs span 2.1% of the genome per cell type on average and, impressively, all ~4,000,000 sites collectively cover ~40% of the genome (Maurano et al., 2012). Mapping nucleosome-depleted chromatin is a comprehensive way to identify the global catalog of regulatory elements and factor binding sites without specific antibodies or prior knowledge of cell type specific transcriptional regulators.
Mapping of Higher Order Chromatin Architecture
Lower-order chromatin structures such as the 11nm fiber, also called “beads on a string”, are followed by higher-order structures like the 30nm fiber and 700nm mitotic chromosomes (Felsenfeld and Groudine, 2003). Incidentally, genome compaction brings regions that are linearly distant via short and long-range DNA-DNA interactions. Mapping the 3D structure of the nucleus is important because, like histone marks and chromatin accessibility, chromosome conformations influence mammalian gene regulation. For example, chromatin in close proximity to the lamina of the inner nuclear membrane, or lamin-associated domains (LADs), tends to be heterochromatic and transcriptionally repressed (Akhtar et al., 2013; Guelen et al., 2008). In contrast, high local concentrations of RNA polymerase II bound promoters, called transcription factories, correlate with robust gene expression (Brown et al., 2008). Historically, fluorescent microscopy imaging, though limited by resolution and throughput, has served as the gold standard for observing nuclear structure. In fact, both LADs and transcription factories were discovered in this fashion.
A paradigm shift from FISH based methods towards measuring physical DNA interactions has revolutionized our ability to broadly map nuclear architecture. The introduction of chromosome conformation capture (3C) by Dekker and colleagues provided an alternative to mapping of distances between two loci (Dekker, 2002). Succinctly, chromatin from formaldehyde crosslinked cells is digested with a restriction enzyme followed by DNA ligation under extremely dilute conditions to favor joining of ends in close proximity to each other. After reversal of crosslinks and DNA purification, the ligation frequency between two restriction fragments, measured by qPCR, indicates their interaction frequency. Such interaction frequency is generally related to the spatial distance, though this relationship could be complex, non-linear, and influenced by chromatin accessibility (Dekker et al., 2013; Williamson et al., 2012).
Differing in the number of loci tested and selection of loci, the suite of 3C-based assays include 3C, circular chromosome conformation capture (4C), chromosome conformation capture carbon copy (5C), tethered conformation capture (TCC), ChIA-PET and Hi-C (de Wit and de Laat, 2012; Sajan and Hawkins, 2012; van Steensel and Dekker, 2010). Of these, 3C and 5C are locus-centric methods, meaning the assayed regions are selected a priori. Sequence specific primers are designed for each locus of interest, for instance promoters or cis-regulatory elements. As stated previously, 3C measures the interaction frequency of one locus with another (one-to-one), between an anchor and a bait sequence, by qPCR. 5C attains higher throughput capacity by utilizing thousands of anchor and bait primers that can span an entire chromosome (many-to-many) (Dostie et al., 2006). The 4C method measures the genome-wide interaction frequency of a single anchor site. Inverse PCR of ligated and circularized interacting DNA fragments detects all interacting loci (one-to-all) (Zhao et al., 2006).
Hi-C measures the entire genome’s interaction frequency with itself as a large matrix without enrichment for specific loci (all-to-all) (Lieberman-Aiden et al., 2009). The key innovation of Hi-C is the ability to enrich for ligation junctions of interacting fragments. After digestion of crosslinked chromatin with a 6-base cutter, the overhang ends are filled in with a biotinylated base. As with all 3C-based assays, the DNA fragments are extremely dilute in the presence of ligase to promote intramolecular ligation events. The ligated sample is sonicated and a streptavidin pulldown captures all junctions of interacting DNA fragments, which are then sequenced. Hi-C interaction signal-to-noise ratio is dependent on the rate of intramolecular ligation events. TCC, a Hi-C variation, performs the ligation step on the streptavidin bead to promote these favorable ligation events (Kalhor et al., 2012). ChIA-PET is another variation of Hi-C which features an immunoprecipitation step to map DNA interactions involving a POI (Fullwood et al., 2009). This approach has been useful for understanding proteins involved in nuclear organization such as TFs, RNA polymerase, CTCF, and cohesin (Demare et al., 2013; Fullwood et al., 2009; Handoko et al., 2011; Li et al., 2012).
Due to its unbiased genome-wide scale, Hi-C promotes the discovery of novel interactions and structures. The size or level of nuclear architecture assayed with Hi-C is dependent on experimental resolution, which in turn is limited by both restriction enzyme cutting frequency and sequencing depth. At 1Mb resolution, Dekker and colleagues defined higher-order chromosome compartments that average about 5Mb in size across the genome (Lieberman-Aiden et al., 2009). Termed A and B compartments, these sequentially interchanging structures correlate with euchromatic and heterochromatic genomic regions, respectively, in a cell-type specific manner. With increase sequencing depth, we attained 40kb resolution Hi-C interaction data. In this way, our lab identified cell-type and species invariant 1 Mb regions of high local interaction frequency, called topological domains or topologically associated domains (TADs), separated by non-interacting boundary elements (Dixon et al., 2012). Boundary regions correlate with the limits of heterochromatin blocks and are enriched for CTCF binding sites, housekeeping genes, and certain transposon elements. It is may be possible to reach fragment-length resolution, or about 4kb with 6-base cutters, using Hi-C to recognize interacting regulatory elements with deeper sequencing and improved data analysis algorithms.
What has become clear using microscopy and now 3C based assays is a gradient or spectrum of DNA folding architecture (Figure 3). These interactions start as one-to-one looping of regulatory elements including enhancers, promoters, and insulators. A collection of these interactions flanked by boundary regions form local interacting neighborhoods or TADs (Dixon et al., 2012; Nora et al., 2012). Using 5C, sub-TADs were recently recognized as finer resolution segments of TADs which can be cell-type specific (Phillips-Cremins et al., 2013). Together many TADs contribute to a larger chromosome compartment and, finally, to an entire chromosome territory (Lieberman-Aiden et al., 2009). In summary, the advent of 3C technologies has revealed a novel and complex layer of genome annotation. Many questions remain regarding how nuclear architecture is regulated and how it mechanistically influences transcription, cellular identity, and possibly disease.
Figure 3.

Hierarchical Principles of Nuclear Organization
Utility of Epigenome Maps
In addition to rapid development of new epigenome mapping technologies, another important factor fueling the remarkable progress of epigenomics is the formation of many research consortia worldwide. Most notable among these consortia are the US Roadmap Epigenome Project, the ENCODE project, and the International Human Epigenome Consortium (IHEC) (Table 1). Modeling after the hugely successful Human Genome Project (Lander, 2011), these consortia standardized experimental protocols, recommended data analysis procedures, and most importantly, publicly released large amounts of data sets prior to publication. Consequently the number of epigenome maps generated has grown exponentially, from a handful in early 2007 to several thousands as of today. As detailed below, these maps have proved to be most valuable in annotation of the transcription units, cis-regulatory sequences, and other genomic features in the human genome. Integrative analysis of epigenomic maps has facilitated study of the gene regulatory programs involved in pluripotency, adipogenesis, and cardiomyocyte differentiation (Gifford et al., 2013; Hawkins et al., 2010; Mikkelsen et al., 2010; Rada-Iglesias et al., 2012; Wamstad et al., 2012; Xie et al., 2013b; Zhu et al., 2013). Further, comparison with genome-wide association studies (GWAS) revealed enrichment of disease associated sequence variants in putative cis-regulatory elements, providing insights into the pathogenesis of many common human diseases.
Annotation of cis-Regulatory Elements from Chromatin Profiles
A major challenge confronting biomedical researchers is the absence of functional annotation of the human genome, where the vast majority (98.5%) do not code for proteins yet harbor most of the disease-associated genetic variations. Several types of functional sequences are known to exist in the non-coding parts of the human genome including cis-regulatory elements. These sequences, including promoters, enhancers, and insulators, govern gene expression by recruiting sequence specific TFs that modulate local chromatin structure and assembly of transcriptional machinery. Since these functional DNAs lack consistent and recognizable sequence features, their identification and characterization had until recently been quite difficult.
Epigenome maps have proven to be a powerful tool for annotating the functional features in the human genome. This is possible because several classes of DNA elements are associated with characteristic histone modifications (Table 3). Early studies revealed that H3K4me3 predominantly associate with promoters and H3K36me3 with gene bodies. Thus, new gene units could be identified with the use of chromatin profiles. Indeed, this strategy led to the identification of several thousands of long non-coding intergenic RNA genes (lincRNA) (Guttman et al., 2009). Similarly, our lab showed that transcriptional enhancers are characterized by the presence of H3K4me1 but not H3K4me3, a combination that accurately predicted tens of thousands of new enhancers in the human genome (Heintzman and Ren, 2009; Heintzman et al., 2007). Despite initial hesitation that this signature may not universally demarcate enhancers in different cell types or in more than one species, H3K4me1 marks enhancers in all tested cell types from human to zebrafish to fly (Aday et al., 2011; Heintzman and Ren, 2009; modENCODE Consortium et al., 2010). Importantly, as enhancers have been annotated in many human cell types a persistent feature of enhancers is their cell type specificity consistent with their role in determining cellular identity. Other general properties of enhancers that are validated genome-wide, including DNaseI hypersensitivity, combinatorial TF binding, H3.3 and H2A.Z histone variant enrichment, bound RNA Pol II, and RNA production (eRNAs) (Buecker and Wysocka, 2012). Combinations of these features along with H3K4me1 allow refined and highly accurate genome-wide enhancer prediction (Rajagopal et al., 2013).
Initially, chromatin states in two human cell lines, HeLa cells and K562 cells, were mapped and used to predict 55,000 candidate enhancers in the human genome (Heintzman et al., 2009). This study demonstrated that chromatin modifications at enhancers, in particular H3K4me1 and H3K27ac, are cell type specific and correlate with cell type specific gene expression throughout the genome, suggesting a potentially critical role for enhancers in lineage-specific gene regulation. Subsequent studies confirmed this result in additional cell types. Rada-Iglesia and colleagues characterized the chromatin modification profiles in the human embryonic stem cells (hESC) and identified approximately 7,000 candidate enhancers featuring binding of TFs, presence of H3K4me1 mark and depletion of nucleosomes (Rada-Iglesias et al., 2011). Interestingly, they also classified these enhancers into “active enhancers” and “poised enhancers”, which differ mainly in the presence or absence of H3K27ac mark. Active enhancers are near genes expressed in hESCs, while poised enhancers are next to genes inactive in hESC but turned on during differentiation (Rada-Iglesias et al., 2011). Independently, Creyghton and colleagues, by examining the chromatin modification patterns in the mESCs and several differentiated mouse cell types, also found that H3K27ac can distinguish the active enhancers from poised ones (Creyghton et al., 2010). While tens of thousands of enhancers are typically marked in a cell type, a small subset of enhancers (<1%) form large domains up to 50kb marked by H3K4me1, H3K27ac, mediator, and master transcription factor binding (Whyte et al., 2013). These “super-enhancers” are posited to regulate key genes important for cell identity. For example, in mESCs super-enhancers are associated with genes necessary for pluripotency and in myotubes they are associated with skeletal muscle development. Lastly, enhancers are turned off or “decommissioned” by the removal of H3K4me1 and H3K27ac marks when they no longer pertain to the cellular state. For example, enhancers that control genes important for pluripotency must be decommissioned for proper differentiation (Whyte et al., 2012). By profiling key chromatin marks in different cell types, researchers can now annotate the location and activity state of regulatory elements across the genome to define a cell-type’s regulome.
Extending the idea that chromatin modification patterns are associated with different functional sequences, a comprehensive epigenomic study of nine human cell lines showed that the human genome could be segmented into regions carrying one of 15 different combinations of chromatin modification marks in each cell type (Ernst et al., 2011). The authors implemented a multivariate hidden markov model called ChromHMM to unbiasedly infer these chromatin states from the chromatin modification profiles. This approach found that each chromatin state corresponds to a specific category of genomic features, including active or poised promoters, enhancers, insulators and silenced domains (Table 3). Similar results have been obtained with other machine-learning algorithms (Hoffman et al., 2013; Won et al., 2013). More recently, the ENCODE consortium profiled the chromatin modification state in 46 human cell types, and found that as much as 56% of the genome is associated with specific histone modification patterns indicative of biochemical activities (Consortium et al., 2012). Ongoing studies from large epigenome mapping consortia such as the NIH Epigenome Roadmap consortium are certain to enhance human genome annotation even further (Bernstein et al., 2010).
Annotation of Long-Range Chromatin Interactions
Rather than the regulome simply being a collection of elements, it must ultimately link together enhancers, insulators, promoters, and other features in 3-dimensional space. Epigenomic maps can annotate cell-type specific long-distance interactions between regulatory elements using two approaches. First, long-range looping can be mapped by physical interaction of distant DNA loci with 3C-based assays. Considering their capabilities, 5C and ChIA-PET currently provide the best balance of resolution and reasonable coverage in the human genome for this purpose (Dekker et al., 2013; Smallwood and Ren, 2013). Several 5C and ChIA-PET datasets exist which are valuable for identifying interactions in selected human cell types, but diverse interaction maps are not yet available. The second method correlates regulatory elements and target promoters across many cells types to computationally infer pairs that regulate and likely interact with each other. These in silico predictions require the annotation of cell-type specific enhancers and active promoters with chromatin signatures, DHSs, and expression profiles (Ernst et al., 2011; Sheffield et al., 2013; Shen et al., 2012; Thurman et al., 2012). A clear advantage of the correlative approach is the necessary data in hundreds of cell types had already been produced by epigenomic consortia. For example, Thurman et al. defined almost 600,000 regulatory pairs from 79 cell types by correlating ENCODE DNase-seq and RNA-seq data (Thurman et al., 2012)., Some of the predicted interaction correlations defined by this approach were validated by both 3C and ChIA-PET but more extensive cross-validation is necessary. Furthermore, such results are limited to enhancer-promoter communication and cannot address the role of insulators or novel interacting regions.
While these two approaches use different concepts to define chromatin interactions, they independently come to similar conclusions about nuclear architecture. The main theme is consistent with the role of enhancers in the determination of cellular identity: enhancer-promoter interactions are cell-type specific (Sanyal et al., 2012; Shen et al., 2012). As previously suggested from single locus studies, enhancers target promoters at long distances and do not always target the nearest gene (Ong and Corces, 2011). This is confirmed by a broader study using 5C analysis of 1% of the human genome in three cell types which determined that ~50% of distal regulatory elements interact with the closest active gene (Sanyal et al., 2012). Surprisingly, enhancers and promoters can interact promiscuously with more than one element indicating a complicated web of transcriptional regulation (Sanyal et al., 2012; Shen et al., 2012; Thurman et al., 2012). In fact, Thurman et al. use a correlative approach to find half of TSSs regulate 10 or more distal sites and half of putative enhancers regulate more than one TSS (Thurman et al., 2012). 5C physical interaction data also confirms 49% of TSSs interact with more than one distal site but finds only 10% of enhancers interacting with more than one promoter (Sanyal et al., 2012). Another concept challenged by interactome annotations, is the classic definition of CTCF-bound insulators as having an enhancer-blocking function to limit the range of targeted enhancer activation to fine tune transcription (Phillips and Corces, 2009). Notably, enhancer-promoter interactions often span hundreds of kilobases surpassing one or more CTCF sites (Demare et al., 2013; Sanyal et al., 2012). 5C analysis predicts that almost 60% of all interactions skip a co-bound CTCF/cohesin locus (Sanyal et al., 2012). Another unexpected result is frequent insulator interactions with promoters and enhancers suggesting a possible role for CTCF in transcriptional activation (Demare et al., 2013; Handoko et al., 2011; Sanyal et al., 2012). Finally, there is an emerging role of CTCF and cohesin co-bound regulatory elements in long-range, constitutive DNA interactions in contrast to cohesin and mediator co-bound regions which facilitate short range, cell-type specific enhancer-promoter interactions (Demare et al., 2013; Phillips-Cremins et al., 2013; Sanyal et al., 2012).
On the whole, great progress has been made towards understanding how chromatin and nuclear organization jointly regulate global gene expression patterns. Still, it remains to be determined how up to 20 enhancers choose to target a particular promoter, what the functional consequence is for a promoter to be activated by more than once enhancer, and if these results are merely an artifact of averaging interactions and epigenetic maps over a population of cells. Evidence that enhancer interactions frequently bypass insulators and that CTCF can loop to transcriptional start sites suggests we must refine our understanding of CTCF-bound insulators. Certainly the conclusions from these epigenomic studies will encourage mechanistic studies to uncover the details of transcriptional regulation by long-range interactions. Continuation of this work at high resolution and in many cell types will progress the dimensions of chromatin maps from a linear chart to a 3-dimensional model of genomic annotation.
Dynamic Chromatin Landscapes During Human Development
Advances in the robustness, throughput, and accuracy of epigenomic technologies over the past decade have co-occurred with a revolution in stem cell biology and regenerative medicine. The first completed epigenomes documented the features of cultured, immortalized or cancerous cell lines. While this enabled correlation of epigenomic features with genomic elements, the resulting epigenomic landscapes were a static view. Now that lineage-specific differentiation protocols produce populations of cells with increasing purity, we can analyze dynamic epigenomic changes during development. These comprehensive studies are giving insight into how cell state transitions are influenced by chromatin states at promoters and enhancers, DNA methylation dynamics at enhancers, and the expansion of repressive domains. Finally, using chromatin signatures to define lineage specific enhancers can lead to models of TF networks that regulate cell type specification.
Unique chromatin signatures mark promoters and enhancers in pluripotent cells. Some promoters in ESCs are bivalent or co-marked with active and repressive histone marks. Sequential ChIP confirmed the presence of both H3K4me3 and H3K27me3 on the same DNA molecule or promoter allele (Bernstein et al., 2006). Due to polycomb-mediated silencing, bivalent genes are not expressed and specifically mark developmental genes that are activated in downstream cell states and are “poised” in ESCs. Consistent with this idea, differentiation of hESCs into endoderm, mesoderm, and ectoderm lineages resolves 85% of bivalent promoters into monovalent states in a lineage-specific manner (Gifford et al., 2013). H3K4me1/H3K27me3 marked poised enhancers are a special class found mostly in ESCs and tend to regulate bivalent promoters of developmental genes (Rada-Iglesias et al., 2011). Chromatin state maps can have more informative power than expression data alone, because they can identify both active genes and poised genomic elements that foreshadow a cell type’s differentiation potential.
DNA methylation can also be informative for identifying enhancers and classifying their activity. Quantitative comparisons of the first human methylomes in hESCs and fibroblasts indicated regions with dynamic DNA methylation levels. A region that is relatively hypermethylated in one cell type and hypomethylated in another is called a differentially methylated region (DMR) (Lister et al., 2009). DMRs are enriched at regulatory elements as evidenced by their overlap with DNaseI sites, TF binding sites, and enhancer chromatin marks (Hon et al., 2013; Ji et al., 2010; Ziller et al., 2013). Active enhancers correlate with the hypomethylated DMR state, also called low-methylated regions (LMRs), and the formation and maintenance of LMRs is dependent on TF binding (Stadler et al., 2011). However, whether the depletion of methylation upon TF binding occurs passively during cell cycles or through active demethylation is still unclear. Recently generated methylomes of cytosine variants find 5hmC, 5fC, and 5caC enriched at enhancers in ESCs suggest the latter is possible (Pastor et al., 2011; Shen et al., 2013; Szulwach et al., 2011; Yu et al., 2012). Even inactive enhancers can be identified by hypomethylation. We recently discovered “vestigal enhancers”, defined as regions depleted of DNA methylation and enhancer chromatin marks in adult tissues but which exhibit enhancer activity earlier in development (Hon et al., 2013). These results suggest that the methylome retains a memory of its previous cellular identities, although how and why is still unclear. Overall, dynamic DNA methylation patterns at enhancers provide yet another epigenetic feature for their annotation. In fact, unexpected regions of hypomethylation in transposable elements function helped to annotate these regions as tissues-specific enhancers (Xie et al., 2013a).
Cellular differentiation during development is characterized by gradual expansion of repressed domains. In ESCs, H3K27me3 is distributed across 8% of the genome and is most commonly observed at bivalent promoters of developmental genes (Zhu et al., 2013). First observed in IMR90 fibroblasts but since confirmed in many differentiated cell types, H3K27me3 peaks spread from bivalent promoters in ESCs to form large blocks covering up to 40% of the genome in differentiated cells (Hawkins et al., 2010; Zhu et al., 2013). These large repressed regions specifically span developmental genes, and presumably function to silence genes that do not pertain to the specified lineage. Correspondingly, chromatin accessibility at regulatory DNA is progressively lost during differentiation (Stergachis et al., 2013). The epigenome of ESCs is uniquely open and accessible, which is consistent with the role of these cells as pluripotent progenitors of all three germ layers. It is now clear that during differentiation and development, the epigenome is progressively restricted like its plasticity potential.
Another major utility of epigenomic studies is the ability to identify cell state or stage specific master regulators and construct transcriptional networks. DNase-seq based methods are useful for mapping all TF binding sites genome-wide in one assay. DNase footprint analysis in 41 human cell types from ESCs to primary adult cells identified 45 million footprints and subsequent de novo motif analysis discerned over 600 unique motifs (Neph et al., 2012b). This set of motifs recovered 90% of the TRANSFAC, JASPAR, and UniPROBE motif database entries. However, nearly half of the motifs found in this study are novel and bound by an unknown protein indicating there is a substantial amount of work to be done to have a complete understanding of sequence-dependent transcriptional regulators. Similar to this approach, master transcriptional regulators can be identified by annotating putative enhancers using histone marks signatures followed by motif analysis of these regions. In this way, novel master regulators of human neural crest (NR2F1), cardiac development (Meis2), and adipogenesis (PLZF) have been successfully identified (Mikkelsen et al., 2010; Paige et al., 2012; Rada-Iglesias et al., 2012). Enhancer-driven regulatory networks can be constructed by correlating TF expression data with motif analysis at putative enhancers as predicted by histone marks or open chromatin (Ernst et al., 2011; Neph et al., 2012a). The ENCODE consortium characterized the binding patterns of 119 TFs in five cell lines by ChIP-seq which yielded ordered TF hierarchies, rules of combinatorial TF binding, and TF binding preferences for distal or proximal sites (Gerstein et al., 2012). However with over 1000 TFs in countless cell types still not assayed, the complexity of experimentally and computationally generated transcription factor regulatory networks will increase and requires further investigation.
Understanding Disease Variants with Epigenomics
The combination of data from the Human Genome Project and International HapMap Project enabled geneticists to study polygenic traits and diseases using GWAS (Frazer et al., 2009). Investigating the genetic basis of common diseases is possible by testing for DNA variants that significantly associate with cases over controls in a population. GWAS are now a common tool in genetic epidemiology, with over 1600 studies published associating 11,000+ SNPs with hundreds of distinct diseases and traits (Hindroff, 2013). In contrast to most Mendelian diseases, common disease associated risk-alleles or quantitative trait loci (QTLs) as determined by GWAS are not enriched in protein coding regions. In fact, 93% of GWAS variants are in non-coding regions, suggesting that they may influence disease phenotypes through cis-regulatory elements (Maurano et al., 2012).
A fundamental challenge for GWAS is identifying causal variants within regions containing multiple SNPs due linkage disequilibrium and, even after determining causal SNPs, understanding the function and disease-contribution of the SNP is difficult to ascertain. Assimilation of GWAS and epigenomic data can give clues about the involvement of regulatory elements and their target genes in the pathogenesis of common disease. Annotating enhancers by their chromatin state in nine cell types, Ernst el al. found disease-associated SNPs are enriched within cell-type specific enhancers from the appropriate disease cell type (Ernst et al., 2011). Maurano et al. correlated the location of over 5,000 non-coding common disease-associated SNPs with almost 4 million DHSs from hundreds of cell types (Maurano et al., 2012). DHSs containing GWAS variants tend to be active in fetal development and correlate with disease relevant cell types facilitating de novo predictions of pathogenic tissues. SNPs and indels are also associated with DNase-seq sequencing depth and these loci are called DNaseI sensitivity quantitative trait loci (dsQTLs) (Degner et al., 2012). Identification of dsQTLs in lymphoblastoid cells from 70 individuals found these genetic variants frequently fall within TF binding sites, effect local nucleosome positioning as determined by MNase-seq, and overlap with GWAS SNPs (Degner et al., 2012; Gaffney et al., 2012).
The objective of a GWAS is to identify sequence variants responsible for a disease phenotype. By generating reference epigenomes in diverse tissues, the annotation of millions of putative enhancers by two different chromatin signatures, histone mark patterns and DHSs, we can begin to study GWAS variants beyond their direct effects on protein-coding genes. Another approach, called an epigenome-wide association studies (EWAS), associates epigenetic patterns with a phenotype irrespective of genotype. Epigenomic analysis of populations is currently feasible with array-based methylation assays but could be extended to DHS or enhancer chromatin marks given a cost-effective platform (Akhtar et al., 2013; Bhandare et al., 2010; Degner et al., 2012). Since epigenetic marks are dynamic and cell type specific, a major consideration for EWAS design is choosing an appropriate cell type for investigation. Studying epigenomic determinants of cancer is especially amendable as the pathogenic tissue types are well described and samples are readily available from biopsies and invasive surgeries. Importantly, GWAS SNPs or EWAS sites enriched in regulatory elements must be functionally validated. As a first step, the effects of non-coding GWAS SNPs on enhancer function could be tested with an extension of high throughput in vivo enhancer reporter technologies (Melnikov et al., 2012; Patwardhan et al., 2012). It will be much more challenging to determine the extent to which an individual or collection of regulatory variants affect gene expression in the endogenous context.
The Road Ahead
Six decades ago, Watson and Crick put forward a model of DNA double helix structure to elucidate how genetic information is faithfully copied and propagated during cell division (Watson and Crick, 1953). Several years later, Crick famously proposed the “central dogma” to describe how information in the DNA sequence is relayed to other biomolecules such as RNA and proteins to sustain a cell’s biological activities (Crick, 1970). Now, with the human genome completely mapped, we face the daunting task to decipher the information contained in this genetic blueprint. Twelve years ago, when the human genome was first sequenced, only 1.5% of the genome could be annotated as protein coding, while the rest of the genome was thought to be mostly “junk” (Lander et al., 2001; Venter et al., 2001). Now, with the help of many epigenome maps, nearly half of the genome is predicted to carry specific biochemical activities and potential regulatory functions (Consortium et al., 2012). It is conceivable that in the near future the human genome will be completely annotated, with the catalog of transcription units and their transcriptional regulatory sequences fully mapped.
However, to reach this goal a couple of technical issues should be resolved. First, current approaches to epigenomic analysis still demand a large number of cells, limiting the cell types and developmental stages that can be examined. Robust nanoscale techniques for chromatin modification profiling, methylcytosine detection or chromatin accessibility mapping will be necessary to cover the full spectrum of cellular states. A few nanoscale ChIP-seq, whole-genome bisulfite sequencing methods have been reported, enabling epigenomic analysis of rare cell populations such as the germ cells (Adey and Shendure, 2012; Adli and Bernstein, 2011; Ng et al., 2013). We expect that the application of these methods to the rare cell types and to embryonic stages will significantly broaden our knowledge of the dynamic human epigenome, and facilitate the discovery of additional cell type specific regulatory elements and transcription units. Second, a lot of the epigenome maps currently available are from tissues consisting of heterogeneous cell populations, and do not accurately reflect the epigenomic state of a specific cell type. Today, to isolate individual cell types from a tissue in large quantity for epigenomic analysis by FACS is challenging. With the advance in nanoscale epigenomic analysis technique, it will likely be routine to obtain cell types in small quantity without affecting their cellular state and produce epigenome maps for specific cell populations in a tissue type. Eventually, the solution will likely be single cell epigenome analysis techniques. Currently, it is not yet possible to profile the chromatin state or DNA methylation status of a single cell, but this could become a reality with further development of single molecule techniques such as SCAN (Single Chromatin molecule Analysis in Nanochannels) (Murphy et al., 2013) or SMRT (single-molecule, real-time) sequencing (Clarke et al., 2009; Flusberg et al., 2010).
Ultimately, to truly understand how the genome programs human development and how certain sequence variants cause human disease, we ought to be able to predict from the sequence when and at what level a gene is expressed in different cell types. To achieve this goal, we need to overcome substantial challenges in two areas. First, we must gain a better understanding of the functional relationships between DNA methylation, chromatin modification state, or higher order chromatin structure and gene regulation. Epigenomic studies have established correlations between DNA hypomethylation, certain chromatin modification signature, and chromatin structure at cis-regulatory elements such as enhancers and promoters, but to determine whether a epigenetic state is necessary for transcription or merely coincidental will require additional mechanistic studies. To this end, new technologies are required to allow manipulation of epigenetic state of specific loci. Recent development of TALE factors and CRISPR/Cas9 as a way to target histone modification enzymes or DNA methyltransferases to specific genomic sites is a great example (Gaj et al., 2013; Ramalingam et al., 2013; Wang et al., 2013). Second, we need to better define the target genes of the distal regulatory elements such as enhancers. This is a challenge because many enhancers are located hundreds and thousands of base-pairs away from their target genes, and it is not infrequent that an enhancer and its target gene are separated by other irrelevant genes (Smallwood and Ren, 2013). Techniques such as ChIA-PET, 4C, 5C and Hi-C have been invented to identify looping interactions between enhancers and target promoters (de Wit et al., 2013; Li et al., 2012; Sanyal et al., 2012; Wei et al., 2013). However, it is currently unclear whether mere spatial proximity is sufficient for functional regulation. Thus, mapping physical interactions alone is unlikely to be adequate to resolve the target genes for enhancers. An alternative, complementary approach defines target genes for an enhancer by searching for nearby genes sharing similar chromatin state or accessibility across diverse cell types (Ernst et al., 2011; Shen et al., 2012; Thurman et al., 2012). Despite its simplicity, this approach does not work particularly well when a gene is regulated by multiple tissue-specific enhancers in different tissues. A hybrid approach combining both spatial proximity and chromatin state information will likely more accurately define the target genes for enhancers.
In summary, the last few years have witnessed an explosion of the epigenomic field. Thousands of epigenome maps from hundreds of human cell or tissue types have been produced, adding crucial insights into the second dimension of the genomic sequence. While these human epigenomes have illuminated potentially functional elements in the genome and improved our understanding of the human developmental programs, it is also clear that much more remains to be explored. Better characterization of epigenome variations in human populations and in patients will be critical for us to fully appreciate the epigenetic factors in human health and disease etiology. Indeed, projects are already underway to profile thousands more epigenomes from both healthy and diseased individuals (American Association for Cancer Research Human Epigenome Task ForceEuropean Union, Network of Excellence, Scientific Advisory Board, 2008; Bernstein et al., 2010). We anticipate an even greater revolution in our understanding of the human epigenome in the coming years.
Table 2.
Major Conceptual Advances Convey the Utility of Epigenome Maps
| Before Next-Gen Sequencing | The Next-Gen Sequencing Era | Future | ||
|---|---|---|---|---|
| CConcepts | DNA Methylation |
|
|
|
| Histone Modification |
|
|
||
| Chromatin Structure |
|
|
||
| Nuclear Architecture |
|
|
Acknowledgments
We apologize to those authors whose works are not covered here owing to space limitations. We thank Peter Jones, Bradley Bernstein, Fred Tyson, John Satterlee and members of the Ren Lab for their comments on earlier versions of this manuscript. This work is supported by funds from the Ludwig Institute for Cancer Research, NIH (grants U01ES017166 and U54 HG006997), and the California Institute for Regenerative Medicine (RN2-00905) to B.R C.M.R. was supported in part by the UCSD Genetics Training Program through an institutional training grant from the National Institute of General Medical Sciences, T32 GM008666.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aday AW, Zhu LJ, Lakshmanan A, Wang J, Lawson ND. Identification of cis regulatory features in the embryonic zebrafish genome through large-scale profiling of H3K4me1 and H3K4me3 binding sites. Dev Biol. 2011;357:450–462. doi: 10.1016/j.ydbio.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adey A, Shendure J. Ultra-low-input, tagmentation-based whole-genome bisulfite sequencing. Genome Res. 2012;22:1139–1143. doi: 10.1101/gr.136242.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adli M, Bernstein BE. Whole-genome chromatin profiling from limitednumbers of cells using nano-ChIP-seq. Nature Protocols. 2011;6:1656–1668. doi: 10.1038/nprot.2011.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar W, de Jong J, Pindyurin AV, Pagie L, Meuleman W, de Ridder J, Berns A, Wessels LFA, Van Lohuizen M, van Steensel B. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell. 2013;154:914–927. doi: 10.1016/j.cell.2013.07.018. [DOI] [PubMed] [Google Scholar]
- American Association for Cancer Research Human Epigenome Task Force, European Union, Network of Excellence, Scientific Advisory Board. Moving AHEAD with an international human epigenome project. Nature. 2008;454:711–715. doi: 10.1038/454711a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badeaux AI, Shi Y. Emerging roles for chromatin as a signal integration and storage platform. Nat Rev Mol Cell Biol. 2013;14:211–224. doi: 10.1038/nrm3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Beck S, Bernstein BE, Campbell RM, Costello JF, Dhanak D, Ecker JR, Greally JM, Issa JP, Laird PW, Polyak K, et al. A Blueprint for an International Cancer Epigenome Consortium. A Report from the AACR Cancer Epigenome Task Force. Cancer Research. 2012;72:6319–6324. doi: 10.1158/0008-5472.CAN-12-3658. [DOI] [PubMed] [Google Scholar]
- Bell O, Tiwari VK, Thomä NH, Schübeler D. Determinants and dynamics of genome accessibility. Nat Rev Genet. 2011;12:554–564. doi: 10.1038/nrg3017. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, Gingeras TR, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. The NIH Roadmap Epigenomics Mapping Consortium. Nature Biotechnology. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandare R, Schug J, Le Lay J, Fox A, Smirnova O, Liu C, Naji A, Kaestner KH. Genome-wide analysis of histone modifications in human pancreatic islets. Genome Res. 2010;20:428–433. doi: 10.1101/gr.102038.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes & Development. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Bock C. Analysing and interpreting DNA methylation data. Nat Rev Genet. 2012;13:705–719. doi: 10.1038/nrg3273. [DOI] [PubMed] [Google Scholar]
- Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330:612–616. doi: 10.1126/science.1191078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, Balasubramanian S. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–937. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. High-Resolution Mapping and Characterization of Open Chromatin across the Genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman AB, Gu H, Bartels SJJ, Zhang Y, Matarese F, Simmer F, Marks H, Bock C, Gnirke A, Meissner A, et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res. 2012;22:1128–1138. doi: 10.1101/gr.133728.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogaard K, Xi L, Wang J-P, Widom J. A map of nucleosome positions in yeast at base-pair resolution. Nature. 2012 doi: 10.1038/nature11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Green J, Neves das RP, Wallace HAC, Smith AJH, Hughes J, Gray N, Taylor S, Wood WG, Higgs DR, et al. Association between active genes occurs at nuclear speckles and is modulated by chromatin environment. J Cell Biol. 2008;182:1083–1097. doi: 10.1083/jcb.200803174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buecker C, Wysocka J. Enhancers as information integration hubs in development: lessons from genomics. Trends Genet. 2012;28:276–284. doi: 10.1016/j.tig.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Wysocka J. Modification of Enhancer Chromatin: What, How, and Why? Mol. Cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22:2990–2997. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J, Wu H-C, Jayasinghe L, Patel A, Reid S, Bayley H. Continuous base identification for single-molecule nanopore DNA sequencing. Nature Nanotech. 2009;4:265–270. doi: 10.1038/nnano.2009.12. [DOI] [PubMed] [Google Scholar]
- Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium, T.E.P., Consortium, T.E.P., data analysis coordination, O.C., data production, D.P.L., data analysis, L.A., group, W., scientific management, N.P.M., steering committee, P.I., Boise State University and University of North Carolina at Chapel Hill Proteomics groups data production and analysis , Broad Institute Group (data production and analysis), et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;488:57–74. [Google Scholar]
- Consortium, T.I.C.G., committee, E., committee, E.A.P., group, T.A.C.A.W., group, T.W., group, B.A.W., group, D.C.A.M.W., Data release, D.T.A.P.W.G., centre, D.C., committee, I.D.A., et al. PERSPECTIVES. Nature. 2010;464:993–998. [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. Central dogma of molecular biology. Nature. 1970;227:561–563. doi: 10.1038/227561a0. [DOI] [PubMed] [Google Scholar]
- de Wit E, de Laat W. A decade of 3C technologies: insights into nuclear organization. Genes & Development. 2012;26:11–24. doi: 10.1101/gad.179804.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E, Bouwman BAM, Zhu Y, Klous P, Splinter E, Verstegen MJAM, Krijger PHL, Festuccia N, Nora EP, Welling M, et al. The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature. 2013 doi: 10.1038/nature12420. [DOI] [PubMed] [Google Scholar]
- Degner JF, Pai AA, Pique-Regi R, Veyrieras JB, Gaffney DJ, Pickrell JK, De Leon S, Michelini K, Lewellen N, Crawford GE, et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012;482:390–394. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J. Capturing Chromosome Conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- Dekker J, Marti-Renom MA, Mirny LA. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet. 2013;14:390–403. doi: 10.1038/nrg3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demare LE, Leng J, Cotney J, Reilly SK, Yin J, Sarro R, Noonan JP. The genomic landscape of cohesin-associated chromatin interactions. Genome Res. 2013;23:1224–1234. doi: 10.1101/gr.156570.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diep D, Plongthongkum N, Gore A, Fung HL, Shoemaker R, Zhang K. Library-free methylation sequencing with bisulfite padlock probes. Nat Meth. 2012;9:270–272. doi: 10.1038/nmeth.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, et al. Chromosome Conformation Capture Carbon Copy (5C): A massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299–1309. doi: 10.1101/gr.5571506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, Gräf S, Johnson N, Herrero J, Tomazou EM, et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nature Biotechnology. 2008;26:779–785. doi: 10.1038/nbt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelhofer TA, Minoda A, Klugman S, Lee K, Kolasinska-Zwierz P, Alekseyenko AA, Cheung MS, Day DS, Gadel S, Gorchakov AA, et al. An assessment of histone-modification antibody quality. Nat Struct Mol Biol. 2011;18:91–93. doi: 10.1038/nsmb.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421:448–453. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, Marques CJ, Andrews S, Reik W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Meth. 2010;7:461–465. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet. 2009;10:241–251. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]
- Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney DJ, McVicker G, Pai AA, Fondufe-Mittendorf YN, Lewellen N, Michelini K, Widom J, Gilad Y, Pritchard JK. Controls of nucleosome positioning in the human genome. PLoS Genet. 2012;8:e1003036. doi: 10.1371/journal.pgen.1003036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T, Gersbach CA, Barbas CF., III ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends in Biotechnology. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford CA, Ziller MJ, Gu H, Trapnell C, Donaghey J, Tsankov A, Shalek AK, Kelley DR, Shishkin AA, Issner R, et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell. 2013;153:1149–1163. doi: 10.1016/j.cell.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007;17:877–885. doi: 10.1101/gr.5533506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CWH, Ye C, Ping JLH, Mulawadi F, et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nature Publishing Group. 2011;43:630–638. doi: 10.1038/ng.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell. 2010;6:479–491. doi: 10.1016/j.stem.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Ren B. Finding distal regulatory elements in the human genome. Curr Opin Genet Dev. 2009;19:541–549. doi: 10.1016/j.gde.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–1143. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Strahl BD, Warburton PE. Epigenomics: a roadmap to chromatin. Science. 2008;322:853. doi: 10.1126/science.322.5903.853a. [DOI] [PubMed] [Google Scholar]
- Hoffman MM, Ernst J, Wilder SP, Kundaje A, Harris RS, Libbrecht M, Giardine B, Ellenbogen PM, Bilmes JA, Birney E, et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013;41:827–841. doi: 10.1093/nar/gks1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday R. Mechanisms for the control of gene activity during development. Biol Rev Camb Philos Soc. 1990;65:431–471. doi: 10.1111/j.1469-185x.1990.tb01233.x. [DOI] [PubMed] [Google Scholar]
- Hon GC, Rajagopal N, Shen Y, McCleary DF, Yue F, Dang MD, Ren B. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nature Publishing Group; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Pastor WA, Shen Y, Tahiliani M, Liu DR, Rao A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS ONE. 2010;5:e8888. doi: 10.1371/journal.pone.0008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Ehrlich LIR, Seita J, Murakami P, Doi A, Lindau P, Lee H, Aryee MJ, Irizarry RA, Kim K, et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010;467:338–342. doi: 10.1038/nature09367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet. 2009;10:161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S-G, Kadam S, Pfeifer GP. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res. 2010;38:e125. doi: 10.1093/nar/gkq223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Jones PA, Martienssen R. A blueprint for a Human Epigenome Project: the AACR Human Epigenome Workshop. 2005:11241–11246. doi: 10.1158/0008-5472.CAN-05-3865. [DOI] [PubMed] [Google Scholar]
- Kalhor R, Tjong H, Jayathilaka N, Alber F, Chen L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nature Biotechnology. 2012;30:90–98. doi: 10.1038/nbt.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly TK, Liu Y, Lay FD, Liang G, Berman BP, Jones PA. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012;22:2497–2506. doi: 10.1101/gr.143008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–197. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152:1237–1251. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono KI, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. Highly Integrated Single-Base Resolution Maps of the Epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhani HD, Francis NJ, Kingston RE, Kornberg RD, Moazed D, Narlikar GJ, Panning B, Struhl K. Epigenomics: a roadmap, but to where? Science. 2008;322:43–44. doi: 10.1126/science.322.5898.43b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, Schuster SC, Albert I, Pugh BF. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 2008;18:1073–1083. doi: 10.1101/gr.078261.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–5877. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, Rogov P, Feizi S, Gnirke A, Callan CG, Kinney JB, et al. systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nature Biotechnology. 2012:1–9. doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Xu Z, Zhang X, Wang L, Gimble JM, Lander ES, Rosen ED. Comparative Epigenomic Analysis of Murine and Human Adipogenesis. Cell. 2010;143:156–169. doi: 10.1016/j.cell.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Ernst J, Kharchenko PV, Kheradpour P, Nègre N, Eaton ML, Landolin JM, Bristow CA, Ma L, et al. modENCODE Consortium. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science. 2010;330:1787–1797. doi: 10.1126/science.1198374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy PJ, Cipriany BR, Wallin CB, Ju CY, Szeto K, Hagarman JA, Benitez JJ, Craighead HG, Soloway PD. Single-molecule analysis of combinatorial epigenomic states in normal and tumor cells. Proc Natl Acad Sci USA. 2013;110:7772–7777. doi: 10.1073/pnas.1218495110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar GJ, Sundaramoorthy R, Owen-Hughes T. Mechanisms and Functions of ATP-Dependent Chromatin-Remodeling Enzymes. Cell. 2013;154:490–503. doi: 10.1016/j.cell.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Stergachis AB, Reynolds A, Sandstrom R, Borenstein E, Stamatoyannopoulos JA. Circuitry and dynamics of human transcription factor regulatory networks. Cell. 2012a;150:1274–1286. doi: 10.1016/j.cell.2012.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, John S, Sandstrom R, Johnson AK, et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012b;489:83–90. doi: 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng JH, Kumar V, Muratani M, Kraus P, Yeo JC, Yaw LP, Xue K, Lufkin T, Prabhakar S, Ng HH. In vivo epigenomic profiling of germ cells reveals germ cell molecular signatures. Developmental Cell. 2013;24:324–333. doi: 10.1016/j.devcel.2012.12.011. [DOI] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Ong CT, Corces VG. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet. 2011;12:283–293. doi: 10.1038/nrg2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige SL, Thomas S, Stoick-Cooper CL, Wang H, Maves L, Sandstrom R, Pabon L, Reinecke H, Pratt G, Keller G, et al. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell. 2012;151:221–232. doi: 10.1016/j.cell.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14:341–356. doi: 10.1038/nrm3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Pape UJ, Huang Y, Henderson HR, Lister R, Ko M, McLoughlin EM, Brudno Y, Mahapatra S, Kapranov P, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–397. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan RP, Hiatt JB, Witten DM, Kim MJ, Smith RP, May D, Lee C, Andrie JM, Lee SI, Cooper GM, et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nature Biotechnology. 2012;30:265–270. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Cremins JE, Sauria MEG, Sanyal A, Gerasimova TI, Lajoie BR, Bell JSK, Ong CT, Hookway TA, Guo C, Sun Y, et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–1295. doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005;122:517–527. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Prescott S, Brugmann SA, Swigut T, Wysocka J. Epigenomic Annotation of Enhancers Predicts Transcriptional Regulators of Human Neural Crest. Stem Cell. 2012;11:633–648. doi: 10.1016/j.stem.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiber E-A, Beraldi D, Ficz G, Burgess HE, Branco MR, Murat P, Oxley D, Booth MJ, Reik W, Balasubramanian S. Genome-wide distribution of 5-formylcytosine in embryonic stem cells is associated with transcription and depends on thymine DNA glycosylase. Genome Biol. 2012;13:R69. doi: 10.1186/gb-2012-13-8-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal N, Xie W, Li Y, Wagner U, Wang W, Stamatoyannopoulos J, Ernst J, Kellis M, Ren B. RFECS: a random-forest based algorithm for enhancer identification from chromatin state. PLoS Comput Biol. 2013;9:e1002968. doi: 10.1371/journal.pcbi.1002968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram O, Goren A, Amit I, Shoresh N, Yosef N, Ernst J, Kellis M, Gymrek M, Issner R, Coyne M, et al. Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell. 2011;147:1628–1639. doi: 10.1016/j.cell.2011.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalingam S, Annaluru N, Chandrasegaran S. A CRISPR way to engineer the human genome. Genome Biol. 2013;14:107. doi: 10.1186/gb-2013-14-2-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF. Comprehensive Genome-wide Protein-DNA Interactions Detected at Single-Nucleotide Resolution. Cell. 2011;147:1408–1419. doi: 10.1016/j.cell.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson AB, Dahl JA, Vågbø CB, Tripathi P, Krokan HE, Klungland A. A novel method for the efficient and selective identification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2011;39:e55. doi: 10.1093/nar/gkr051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Meth. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- Sajan SA, Hawkins RD. Methods for Identifying Higher-Order Chromatin Structure. Annu Rev Genom Human Genet. 2012;13:59–82. doi: 10.1146/annurev-genom-090711-163818. [DOI] [PubMed] [Google Scholar]
- Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre D, Lee BH, Ting AH. MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2010;38:391–399. doi: 10.1093/nar/gkp992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield NC, Thurman RE, Song L, Safi A, Stamatoyannopoulos JA, Lenhard B, Crawford GE, Furey TS. Patterns of regulatory activity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions. Genome Res. 2013;23:777–788. doi: 10.1101/gr.152140.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Wu H, Diep D, Yamaguchi S, D’Alessio AC, Fung HL, Zhang K, Zhang Y. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell. 2013;153:692–706. doi: 10.1016/j.cell.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood A, Ren B. Genome organization and long-range regulation of gene expression by enhancers. Curr Opin Cell Biol. 2013;25:387–394. doi: 10.1016/j.ceb.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- Song CX, Szulwach KE, Dai Q, Fu Y, Mao SQ, Lin L, Street C, Li Y, Poidevin M, Wu H, et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 2013;153:678–691. doi: 10.1016/j.cell.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, Li Y, Chen CH, Zhang W, Jian X, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nature Biotechnology. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CX, Yi C, He C. Mapping recently identified nucleotide variants in the genome and transcriptome. Nature Biotechnology. 2012;30:1107–1116. doi: 10.1038/nbt.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–495. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- Statham AL, Robinson MD, Song JZ, Coolen MW, Stirzaker C, Clark SJ. Bisulfite sequencing of chromatin immunoprecipitated DNA (BisChIP-seq) directly informs methylation status of histone-modified DNA. Genome Res. 2012;22:1120–1127. doi: 10.1101/gr.132076.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergachis AB, Neph S, Reynolds A, Humbert R, Miller B, Paige SL, Vernot B, Cheng JB, Thurman RE, Sandstrom R, et al. Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell. 2013;154:888–903. doi: 10.1016/j.cell.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulwach KE, Li X, Li Y, Song C-X, Han JW, Kim S, Namburi S, Hermetz K, Kim JJ, Rudd MK, et al. Integrating 5-hydroxymethylcytosine into the epigenomic landscape of human embryonic stem cells. PLoS Genet. 2011;7:e1002154. doi: 10.1371/journal.pgen.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, et al. Identification of 67 Histone Marksand Histone Lysine Crotonylationas a New Type of Histone Modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Z, Tolić N, Zhao R, Moore RJ, Hengel SM, Robinson EW, Stenoien DL, Wu S, Smith RD, Paša-Tolić L. Enhanced top-down characterization of histone post-translational modifications. Genome Biol. 2012;13:R86. doi: 10.1186/gb-2012-13-10-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, Dekker J. Genomics tools for unraveling chromosome architecture. Nature Biotechnology. 2010;28:1089–1095. doi: 10.1038/nbt.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Waddington CH. Canalization of development and genetic assimilation of acquired characters. Nature. 1959;183:1654–1655. doi: 10.1038/1831654a0. [DOI] [PubMed] [Google Scholar]
- Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–220. doi: 10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JD, Crick FH. Genetical implications of the structure of deoxyribonucleic acid. Nature. 1953;171:964–967. doi: 10.1038/171964b0. [DOI] [PubMed] [Google Scholar]
- Wei Z, Gao F, Kim S, Yang H, Lyu J, An W, Wang K, Lu W. Klf4 organizes long-range chromosomal interactions with the oct4 locus in reprogramming and pluripotency. Cell Stem Cell. 2013;13:36–47. doi: 10.1016/j.stem.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, Foster CT, Cowley SM, Young RA. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature. 2012;482:221–225. doi: 10.1038/nature10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PAC, Rappsilber J, Helin K. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson I, Eskeland R, Lettice LA, Hill AE, Boyle S, Grimes GR, Hill RE, Bickmore WA. Anterior-posterior differences in HoxD chromatin topology in limb development. Development. 2012;139:3157–3167. doi: 10.1242/dev.081174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won K-J, Zhang X, Wang T, Ding B, Raha D, Snyder M, Ren B, Wang W. Comparative annotation of functional regions in the human genome using epigenomic data. Nucleic Acids Res. 2013;41:4423–4432. doi: 10.1093/nar/gkt143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Hong C, Zhang B, Lowdon RF, Xing X, Li D, Zhou X, Lee HJ, Maire CL, Ligon KL, et al. DNA hypomethylation within specific transposable element families associates with tissue-specific enhancer landscape. Nature Publishing Group. 2013a;45:836–841. doi: 10.1038/ng.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, Whitaker JW, Tian S, Hawkins RD, Leung D, et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell. 2013b;153:1134–1148. doi: 10.1016/j.cell.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, Li X, Dai Q, Shen Y, Park B, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368–1380. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan GC, Liu YJ, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005;309:626–630. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Tavoosidana G, Sjölinder M, Göndör A, Mariano P, Wang S, Kanduri C, Lezcano M, Singh Sandhu K, Singh U, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38:1341–1347. doi: 10.1038/ng1891. [DOI] [PubMed] [Google Scholar]
- Zhu J, Adli M, Zou JY, Verstappen G, Coyne M, Zhang X, Durham T, Miri M, Deshpande V, De Jager PL, et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;152:642–654. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller MJ, Gu H, Müller F, Donaghey J, Tsai LTY, Kohlbacher O, De Jager PL, Rosen ED, Bennett DA, Bernstein BE, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013:1–5. doi: 10.1038/nature12433. [DOI] [PMC free article] [PubMed] [Google Scholar]