

Abstract
We previously reported the findings from a genome-wide association study of the response of maximal oxygen uptake (V̇o2max) to an exercise program. Here we follow up on these results to generate hypotheses on genes, pathways, and systems involved in the ability to respond to exercise training. A systems biology approach can help us better establish a comprehensive physiological description of what underlies V̇o2maxtrainability. The primary material for this exploration was the individual single-nucleotide polymorphism (SNP), SNP-gene mapping, and statistical significance levels. We aimed to generate novel hypotheses through analyses that go beyond statistical association of single-locus markers. This was accomplished through three complementary approaches: 1) building de novo evidence of gene candidacy through informatics-driven literature mining; 2) aggregating evidence from statistical associations to link variant enrichment in biological pathways to V̇o2max trainability; and 3) predicting possible consequences of variants residing in the pathways of interest. We started with candidate gene prioritization followed by pathway analysis focused on overrepresentation analysis and gene set enrichment analysis. Subsequently, leads were followed using in silico analysis of predicted SNP functions. Pathways related to cellular energetics (pantothenate and CoA biosynthesis; PPAR signaling) and immune functions (complement and coagulation cascades) had the highest levels of SNP burden. In particular, long-chain fatty acid transport and fatty acid oxidation genes and sequence variants were found to influence differences in V̇o2max trainability. Together, these methods allow for the hypothesis-driven ranking and prioritization of genes and pathways for future experimental testing and validation.
Keywords: endurance training, genotypes, bioinformatics, pathway analysis, single-nucleotide polymorphisms
genome-wide association studies (GWAS) have been successful in identifying genes or loci associated with complex, quantitative traits on the basis of observational data. However, for most GWAS, the identified variants explain only a small proportion of the overall genetic variance for a given trait (32). We previously reported the findings from a GWAS of the response of cardiorespiratory fitness (as assessed by maximal oxygen uptake, or V̇o2max) to a standardized exercise training program (7). We found that 39 single nucleotide polymorphisms (SNPs) were associated with the response of V̇o2max at P < 1.5 × 10−4. Multivariate analyses found that a panel of 21 SNPs accounted for 49% of the variance in V̇o2max trainability, which corresponds to the heritability level observed in this cohort (4). The genomic predictor score created from this panel of 21 SNPs was quite successful at discriminating between high and low V̇o2max responders to exercise training. We have also produced a transcriptional map of genes regulated in a manner sensitive to the degree of V̇o2max responses to exercise training (21). Additionally, baseline transcriptomic predictors of exercise responsiveness have also been identified, and a subset of the predictor genes were found to contain sequence variants that were also associated with gains in V̇o2max (40).
The current study extends the findings from these earlier studies by utilizing the same GWAS cohort to interrogate the underlying biological mechanisms influencing V̇o2max trainability through an analysis of the collective evidence from DNA sequence variation. The core premise behind the approach exemplified in this study is that genes do not work in isolation; instead, molecular networks and cellular pathways interact in the modulation of complex phenotypes (6, 14, 18, 20, 39, 42, 44). Pathway-based approaches jointly consider multiple variants in interacting or related genes. As recently reviewed by Khatri et al. (22), pathway-based approaches complement single SNP analysis by incorporating prior biological knowledge with a focus on groups of functionally related genes. For example, analysis of type 2 diabetes risk alleles from the Wellcome Trust Case-Control Consortium cohort that focused on pathways rather than individual significance levels were consistent with risk pathways identified through RNA network analysis (13). A pathway-based approach would therefore allow us to elaborate on the connectivity among genes exhibiting sequence variants that have been shown to contribute to an ability to improve cardiorespiratory fitness with regular exercise. A systems biology approach is likely to help us establish a comprehensive physiological and metabolic model of the underpinnings of V̇o2max trainability. Furthermore, this effort will provide a foundation for the definition of new molecular and physiological hypotheses to be tested in subsequent experimental studies.
MATERIALS AND METHODS
Study Design
The sample, study design, and exercise training protocol of the HERITAGE Family Study have been extensively described elsewhere (5). Briefly, 834 subjects from 218 families were recruited to participate in an endurance exercise training study and to have measurements taken of their baseline V̇o2max. Among them, 473 adults (230 men; 243 women) from 99 families of Caucasian descent were defined as completers and were used in the current study. Parents were ≤65 years of age; offspring ranged in age from 17 to 41 years. Participants were sedentary at baseline, and normotensive or mildly hypertensive (<160/100 mmHg) without medication for hypertension, diabetes, or dyslipidemia (5). The basic characteristics of the subjects are summarized in Table 1. The study protocol had been approved by the institutional review boards at each of the five participating centers of the HERITAGE Family Study consortium. Written informed consent was obtained from each participant.
Table 1.
Descriptive data for HERITAGE Family Study subjects included in this study
| HERITAGE subjects | |
|---|---|
| Number | 473 |
| Male/Female | 230/243 |
| Age, years | 35.8 (14.5) |
| Baseline body mass index, kg/m2 | 25.8 (4.9) |
| Baseline V̇o2max, ml/min | 2458 (740) |
| Baseline V̇o2max, ml·kg−1·min−1 | 33.2 (8.9) |
| V̇o2max response, ml/min | 395 (215) |
| V̇o2max response, % | 16.9 (9.0) |
Values are mean (SD). V̇o2max response is posttraining V̇o2max minus baseline V̇o2max (positive value represents improvement in V̇o2max).
Each subject in the HERITAGE study exercised three times per wk for 20 wk on cycle ergometers. The intensity of the training was customized for each individual on the basis of heart rate (HR) and V̇o2max measurements taken at the baseline maximal exercise test. Details of the exercise training protocol can be found elsewhere (5). Briefly, subjects trained at the HR associated with 55% of baseline V̇o2max for 30 min per session for the first 2 wk. The duration and intensity were gradually increased every 2 wk, until reaching 50 min and 75% of the HR associated with baseline V̇o2max. This level was maintained for the final 6 wk of training. All training was performed on Universal Aerobicycles (Cedar Rapids, IA) and power output was controlled by direct HR monitoring using the Universal Gym Mednet (Cedar Rapids, IA) computerized system. The protocol was standardized across all clinical centers and supervised to ensure that the equipment was working properly and that participants were compliant with the protocol.
Two maximal exercise tests to measure V̇o2max were performed on 2 separate days at baseline and again on 2 separate days after training on a SensorMedics 800S (Yorba Linda, CA) cycle ergometer and a SensorMedics 2900 metabolic measurement cart (37). The tests were conducted at about the same time of day, with at least 48 h between the two tests. In the first test, subjects exercised at a power output of 50 W for 3 min, followed by increases of 25 W each 2 min until volitional exhaustion. For older, smaller, or less fit individuals, the test was started at 40 W, with increases of 10–20 W each 2 min thereafter. In the second test, subjects exercised for ∼10 min at an absolute (50 W) and at a relative power output equivalent to 60% V̇o2max. They then exercised for 3 min at a relative power output that was 80% of their V̇o2max, after which resistance was increased to the highest power output attained in the first maximal test. If the subjects were able to pedal after 2 min, power output was increased each 2 min thereafter until they reached volitional fatigue. The average V̇o2max from these two sets was taken as the V̇o2max for that subject and used in analyses if both values were within 5% of each other. If they differed by >5%, the higher V̇o2max value was used.
DNA Extraction and Genotyping
Genomic DNA was prepared from immortalized lymphoblastoid cell lines by a commercial DNA extraction kit (Gentra Systems, Minneapolis, MN). GWAS SNPs were genotyped using the Illumina Human CNV370-Quad v3.0 BeadChips on an Illumina BeadStation 500GX platform. The genotype calls were performed with Illumina GenomeStudio software and all samples were called in the same batch to eliminate batch-to-batch variation. All GenomeStudio genotype calls with a GenTrain score less than 0.885 were checked and confirmed manually. Monomorphic SNPs and SNPs with only one heterozygote, and SNPs with more than 30% missing data were filtered out with the GenomeStudio. Quality control of the GWAS SNP data confirmed all family relationships and found no evidence of DNA sample mix-ups. Only 78 SNPs (0.023%) had more than 10% missing data. Minor allele frequency was less than 1% for 1,301 SNPs (0.39%). Hardy-Weinberg equilibrium test P values were less than 10−5 and 10−6 for 55 (0.017%) and 12 (0.0037%) SNPs, respectively. Twelve samples were genotyped in duplicate with 100% reproducibility across all SNPs.
The original HERITAGE GWAS study genotyped ∼325,000 SNPs. Although this is not a small number, our current knowledge on genotype frequencies extends to tens of millions of SNPs obtained from high-density, next-generation sequencing platforms. Imputation is a statistical method that allows one to utilize this current knowledge even when all SNPs are not experimentally genotyped in a study. This occurs by inferring genotypes in an experimental sample by using the correlation between SNP markers in a reference sample drawn from the same population. These population-matched reference genotypes are provided from the very high-density SNP maps generated in a common cohort of control samples in the HapMap2 or 1000 Genomes Project (38). For the current study, we used MACH (28) to impute untyped markers for HERITAGE Caucasian subjects. To impute individuals of European ancestry, we used the Caucasian (CEU) reference panel consisting of 120 haplotypes constructed from HapMap Phase II data (release 22, build 36). From a total of 99 HERITAGE families, 200 unrelated individuals (with the lowest GWAS missing rate) were chosen to estimate the imputation model parameters. Using these estimates, imputations were performed on all 501 subjects. The final imputed data set provided information on ∼2.5 million SNPs.
Statistical and Bioinformatic Analysis
SNP-level analysis.
V̇o2max training response (ΔV̇o2max) was adjusted for age, sex, and baseline V̇o2max using a stepwise regression procedure. Residuals were standardized to a zero mean and unit variance within sex-by-generation subgroups as previously described (4).
Associations between the GWAS SNPs and V̇o2max training response were analyzed using the MERLIN software package (1). The total association model of MERLIN utilizes a variance-components framework to combine a phenotypic means model and the estimates of additive genetic, residual genetic, and residual environmental variances from a variance-covariance matrix into a single likelihood model. The evidence of association is evaluated by maximizing the likelihoods under two conditions: the null hypothesis (L0) restricts the additive genetic effect of the marker locus to zero (βa = 0), whereas the alternative hypothesis does not impose any restrictions on βa. The quantity of twice the difference of the log likelihoods between the alternative and the null hypotheses {2[ln (L1) − ln (L0)]} is distributed as χ2 with 1 degree of freedom (difference in number of parameters estimated).
Gene-level analysis.
SNP-GENE MAPPING AND GENE P VALUE ASSIGNMENT.
A SNP Si was mapped to gene Gj (j = 1, … n, where n is the total number of genes represented by all SNPs) if Si was located within the primary transcript of the gene or a window of 20 kilobases on either end of the gene. Although the effects from more distant SNPs are certainly possible (e.g., SNPs in enhancer regions), having a wide window can also lead to spurious SNP-gene mappings and increase the number of false positive findings. The 20-kb window provided an optimal width for capturing potential regulatory SNPs in the 5′ and 3′ untranslated regions of a gene, while controlling for false SNP-to-gene assignments due to larger windows (26). The second most significantly associated SNP (among all SNPs mapped to a gene) was used to represent the association statistic for that gene, as recommended by Nam et al. (34). Additionally, we also performed comparative analysis using gene P values based on the best scoring SNP for the gene. If an SNP mapped to multiple genes, all such genes were included in the analysis.
CANDIDATE GENE PRIORITIZATION.
The gene prioritization tool CANDID (19) was used to generate a ranked list of candidate genes associated with changes in ΔV̇o2max. CANDID takes a list of SNPs as input and performs a weighted analysis including text mining, protein domain information, sequence conservation, gene expression, linkage, and association results to score and rank genes. In the current study, the top 10,000 ΔV̇o2max associated SNPs (sorted by their association P value) were examined with the following weighting scheme: literature (key words = exercise, aerobic, capacity, oxygen), 10%; cross-species sequence conservation, 15%; gene expression in target tissues (cardiac myocytes, skeletal muscle, lung, liver, kidney, adrenal cortex, adipocyte), 25%; ΔV̇o2max GWAS association P value (this study); and 50% (scheme 1, 10_15_25_50). The weights of other available criteria such as protein domain information and linkage analysis were set to zero. In this context, literature evidence was obtained from the PubMed database and sequence conservation scores were accessed from the National Center for Biotechnology Information Conserved Domain Database. Gene expression values refer to basal expression levels and were obtained from the GeneAtlas expression profile database (Genomics Institute of the Novartis Research Foundation). To avoid potential biases arising from our prior publications linking genomics and transcriptomics to cardiorespiratory fitness (8, 21, 40), the literature contributions were assigned a relatively small weight of 10%. The largest weight (50%) was assigned to the GWAS association score to ensure that the new results from association analysis would remain the major component of the combined CANDID score for each gene. However, to estimate the robustness of the weighting scheme described above, we also carried out analyses using alternate weights for the above four criteria and compared the overlap among the top 25 genes for each weighing scheme (scheme 2, 5_5_15_75; scheme 3, 10_15_50_25; scheme 4, 25_15_10_50).
Hypotheses linking candidate genes to relevant exercise-related phenotypes were generated and prioritized by the Biograph knowledge-mining software (29). Biograph assembles and analyzes information from heterogeneous databases via unsupervised data mining techniques and stochastic random walks to generate a map of relations linking source concepts (e.g., phenotypes, diseases) to targets (e.g., candidate genes). This network of relationships is analyzed by the Biograph algorithm to score and rank the different path linking concepts to targets, resulting in an automated formulation of one or more functional hypotheses. The relative strength of each hypothesis is computed, which allows one to assess the proximity of the association between a concept and a target. This information is used to prioritize candidate genes.
Pathway-level analysis.
Pathway analysis was conducted via two different yet complementary approaches: overrepresentation analysis (ORA) and gene-set enrichment analysis (GSEA). Both methods rely on comparison of predefined gene sets (pathways) and observed genes from an experiment (GWAS). In ORA, a subset of the most significant GWAS genes (P < 0.005 in the present study) are queried against the pathway gene sets to determine whether the GWAS genes are observed more frequently in any of the pathways than would be expected by chance alone (i.e., overrepresented). ORA was conducted in the Ingenuity Pathway Analysis (IPA) environment and the reference gene sets (pathways) were derived from the Ingenuity Knowledge Base (www.ingenuity.com). Overrepresentation of biological pathways was ascertained via Fisher's exact test and corrected for multiple testing by the Benjamini-Hochberg procedure (3). GSEA, on the other hand, utilizes all observed GWAS genes by ranking them on the basis of their statistical significance. Predetermined pathways (gene sets) are then queried against the ranked GWAS genes to test whether pathway gene sets are enriched among the most significant GWAS genes. The enrichment score calculated by GSEA algorithms reflect the degree to which a pathway is overrepresented at the extremes of the entire ranked gene list. Statistical significance of the enrichment score is ascertained by permutation testing and multiple testing is controlled by the false discovery rate. In the present study, GSEA was carried out by two separate tools: the IGSEA4GWAS (48) (IGSEA, http://gsea4gwas.psych.ac.cn/inputPage.jsp) and GSA-SNP (34) (http://gsa.muldas.org), In IGSEA, the GWAS gene list (ranked by statistical significance) was used to calculate significance-proportion-based pathway enrichment scores. The score reflects what proportion of all genes in a given pathway ranked among the most significant in the GWAS gene list. This approach guards against situations in which one (or a few) highly significant gene(s) may artificially inflate the pathway score. SNP label permutation was used to calculate statistical significance and false discovery rates for each pathway.
In GSA-SNP, a z score was calculated for each pathway on the basis of the number of genes in a given pathway, strength of the GWAS association P values for the individual genes in the pathway, and the mean and standard deviation of all GWAS gene P values. Statistical significance of each pathway was tested using the z statistic and multiple testing was controlled for by the Benjamini-Hochberg procedure (3).
For both IGSEA and GSA-SNP, biological pathway information was obtained from the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database and downloaded from the Molecular Signatures Database collection (www.broadinstitute.org/gsea/msigdb; August 2010 version). KEGG is a collection of pathway maps representing our current knowledge of the molecular interaction and reaction networks involved in biological processes such as metabolism, cellular signaling, and human diseases. A total of 186 pathways were downloaded. Pathways with <10 genes or >200 genes were excluded from further analysis to protect against spurious associations from very small pathways or the lack of sufficient specificity from very large pathways.
The SNPNEXUS tool (http://www.snp-nexus.org/) was used to characterize pathways on the basis of predicted functional consequences of the associated SNPs. For this analysis, SNPs were characterized on the basis of their location with respect to a gene and to potential regulatory elements. Possible functional consequences of an SNP was defined in terms of its location in a gene locus (intronic, exonic, splice-site junction, 5′-UTR, 3′-UTR, etc.), in regulatory elements (CpG islands, transcription factor binding sites, etc.), or in highly conserved sequence, as well as its predicted effects on gene function (structural variation such as inversions, insertions-deletions, copy number polymorphisms), or on function of the gene product [tolerated or damaging (23)]. Each pathway was scored for the number of SNPs in the different functional categories and a combined pathway score was created by summing over the individual scores in each SNP category, followed by normalization by the number of significantly associated SNPs (nominal P < 0.05) in the pathway. The corrected pathway score was an index of the predicted SNP burden for each pathway and was used to rank the pathways.
RESULTS
Gene-Level Analysis
Gene-wide association statistics.
Gene-wide association P values were obtained according to the method described by Nam et al. (34), in which the second most significant SNP P value for a given gene is used to represent the statistical significance of the gene locus. A list of the top 20 genes is shown in Table 2. For comparison purposes, the ranks of the genes on the basis of best/most significant SNP approach are also listed. Of these, SLED1, EEF1DP3 (pseudogenes) and CASC2 (long noncoding RNA) were excluded from further analyses. A comparison of the gene-ranks on the basis of best SNP and second-best SNP approach showed that ACSL1 and HCG22 scoring high in both approaches (ranked 1 and 2 in second-best SNP and 2 and 5 in best SNP approaches, respectively). However, several genes (e.g., FABP6, CAMTA1, CD44, etc.) displayed poorer ranks in the best SNP-based scoring method, whereas several genes with ranks <20 in the best SNP-based scoring were not selected in the second-best SNP approach.
Table 2.
Top 20 GWAS associated genes on the basis of second-best SNP P values
| Gene | Second-best P value SNP | Second-best P value | Rank second-best P value | Rank best P value† |
|---|---|---|---|---|
| ACSL1 | rs6552828 | 3.80 × 10−6 | 1 | 2 |
| SLED1* | rs6552828 | 3.80 × 10−6 | 1 | 1 |
| HCG22 | rs2517512 | 3.09 × 10−5 | 2 | 5 |
| EEF1DP3* | rs2773968 | 3.67 × 10−5 | 3 | 3 |
| BIRC7 | rs6122403 | 6.24 × 10−5 | 4 | 8 |
| YTHDF1 | rs6122403 | 6.24 × 10−5 | 4 | 9 |
| ITGB8 | rs10265149 | 7.04 × 10−5 | 5 | 6 |
| TEC | rs13117386 | 7.97 × 10−5 | 6 | 22 |
| SHANK2 | rs10751308 | 8.11 × 10−5 | 7 | 7 |
| C4orf40 | rs3775758 | 1.03 × 10−4 | 8 | 18 |
| TTLL11 | rs7022103 | 1.07 × 10−4 | 9 | 25 |
| CD6 | rs175098 | 1.11 × 10−4 | 10 | 26 |
| RYR2 | rs6659362 | 1.18 × 10−4 | 11 | 27 |
| FAM19A2 | rs2168452 | 1.34 × 10−4 | 12 | 29 |
| FABP6 | rs7734683 | 1.44 × 10−4 | 13 | 36 |
| C12orf36 | rs877972 | 1.45 × 10−4 | 14 | 38 |
| CAMTA1 | rs884736 | 1.54 × 10−4 | 15 | 35 |
| LOC731789 | rs11015207 | 1.61 × 10−4 | 16 | 42 |
| CD44 | rs4756195 | 1.64 × 10−4 | 17 | 34 |
| CASC2 | rs1413184 | 1.65 × 10−4 | 18 | 43 |
Genes are sorted on the basis of their second-best SNP P values.
Pseudogenes.
Comparative gene rank on the basis of best-scoring SNP P value.
The ACSL1 genes were consistently rated at the top of the list of associated genes, regardless of the SNP P value (best or second-best) used. In this context it is noteworthy that the same SNP in ACSL1 gene (rs2556868) also showed the strongest association to V̇o2max training response in the original HERITAGE GWAS study (7). As indicated earlier, the SLED1 gene was located within an intron of the ACSL1 gene on chromosome 4 and reported to be a pseudogene for proteoglycan 3. The top ΔV̇o2max associated SNP, rs4862423 (P = 2.41 × 10−6), was also found to be located within the ACSL1 gene.
Candidate gene prioritization.
We applied the CANDID software tool to conduct a weighted and integrated analysis of GWAS-based gene association data in combination with prior literature, sequence conservation, and gene expression information, to further refine the list of candidate genes associated with V̇o2max training response. The results of the analysis are shown in Table 3. Although a few of our prior publications have investigated genomics and transcriptomics as predictors of the response to exercise training (7, 21, 40), the overall evidence from literature connecting genes to cardiorespiratory fitness trainability is relatively low, as reflected by the poor scores for the literature criterion for most of the query genes. Several of the top-ranked genes (RPTOR, SLC45A1, NLN, and PRKG1) demonstrated the maximal cross-species conservation score of 1. The expression scores were also consistently high for this group of genes (>0.9), indicating considerable expression in nearly all the tissues selected. The GWAS association P values for this set of genes ranged from 7 × 10−4 to 5 × 10−3, indicating moderate evidence of association (with respect to a significance threshold of 2.5 × 10−6, assuming 20,000 genes tested). In comparison, the gene with the strongest evidence for GWAS association, ACSL1 (P = 2.41 × 10−6), was ranked 414th overall (final score = 67.93), primarily due to lower expression in the majority of the tissues tested. When CANDID analysis was conducted by tissue-specific gene expression scores, ACSL1 was the top-scoring candidate among all adipocyte-expressed genes. The effects of the weighting scheme on gene ranks were tested by repeating the analysis in CANDID after applying varying weights to the four criteria (literature, conservation, gene expression, and GWAS association; described in Materials and Methods) and investigating the overlap among the top 25 genes for each weighting scheme. As shown in Fig. 1, a set of five genes (RPTOR, SLC45A1, PRKG1, NLN, and PINX1) were present in all weighting schemes tested, whereas the number of overlapping genes increased to 16 when schemes 1–3 were considered.
Table 3.
Top 20 candidate genes identified by CANDID
| Gene | Chromosome | Literature score (10%)* | Conservation score (15%)* | Expression score (25%)* | Association score (50%)* | Final score† |
|---|---|---|---|---|---|---|
| SLC45A1 | 1 | 0 | 1 | 0.968534 | 0.998248 | 89.12578 |
| RPTOR | 17 | 0.048193 | 1 | 0.94607 | 0.999044 | 89.0859 |
| PINX1 | 8 | 0 | 0.888889 | 1 | 0.996724 | 88.16955 |
| NLN | 5 | 0.034483 | 1 | 0.911942 | 0.998974 | 88.09209 |
| PRKG1 | 10 | 0 | 1 | 0.894647 | 0.999281 | 87.33022 |
| SNX14 | 6 | 0 | 0.777778 | 1 | 0.999779 | 86.65563 |
| MACROD2 | 20 | 0 | 0.777778 | 1 | 0.998501 | 86.59174 |
| KCNT1 | 9 | 0 | 0.777778 | 1 | 0.996373 | 86.48534 |
| ADCY5 | 3 | 0.013699 | 0.777778 | 0.979768 | 0.99703 | 86.14938 |
| BTBD9 | 6 | 0 | 0.777778 | 0.976886 | 0.999533 | 86.06546 |
| KCNQ5 | 6 | 0 | 0.777778 | 0.97928 | 0.995775 | 85.93743 |
| ACVR1C | 2 | 0 | 0.777778 | 0.975932 | 0.995639 | 85.84693 |
| CLYBL | 13 | 0 | 0.888889 | 0.889237 | 0.996524 | 85.39048 |
| NALCN | 13 | 0 | 0.888889 | 0.872696 | 0.998995 | 85.10051 |
| PARK2 | 6 | 0.040984 | 0.777778 | 0.913164 | 0.998644 | 84.83783 |
| HLCS | 21 | 0 | 1 | 0.795126 | 0.996765 | 84.71642 |
| TMEM181 | 6 | 0 | 0.666667 | 0.983658 | 0.998007 | 84.49182 |
| VPS53 | 17 | 0 | 1 | 0.778758 | 0.998109 | 84.37442 |
| LHFPL3 | 7 | 0 | 0.666667 | 0.982795 | 0.995552 | 84.3475 |
| GRIK4 | 11 | 0.019608 | 1 | 0.767733 | 0.99877 | 84.32792 |
The top 10,000 SNPs ranked by their GWAS association P values were used as input into the CANDID tool.
Numbers in parentheses represent the relative weights for the different criteria.
Genes are sorted by their final score, which is the weighted sum of individual criterion scores.
Fig. 1.
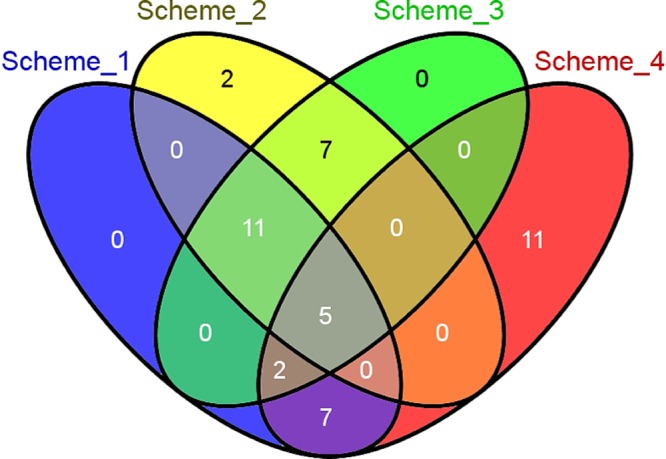
Effects of weighting schemes on candidate gene analysis using CANDID. The top 10,000 ΔV̇o2max associated genes were analyzed in CANDID using different weights for literature, sequence conservation, gene expression, and GWAS association (schemes 1–4, described in Materials and Methods). Overlap among the top 25 genes for each scheme was assessed by Venn analysis.
To explore how the top candidate genes from GWAS or CANDID could be functionally linked to the phenotype, we applied the Biograph knowledge-mining software. The top 20 genes each from GWAS and CANDID analysis were individually examined for their relation to a set of exercise-related phenotypes selected by a key word search of Biograph's knowledge repository. Although additional functional hypotheses can certainly exist between additional genes in the GWAS and CANDID outputs and the Biograph-selected phenotypes, we chose to focus on the top 20 genes from each analysis in the current study. The results of the analysis are summarized in Fig. 2. Specifically, we determined the Biograph-predicted relative proximity of each gene to each of the phenotype terms on the basis of expected strength of association of each gene to the phenotype. The proximity was scored as a relative rank for the gene compared with all genes in Biograph's knowledge base. These ranks were used to construct a two-way clustering of genes and phenotypic terms so that genes with similar proximity ranks (rows) and terms with similar relationships to the genes (columns) were clustered together. The relative ranks of genes were colored-coded (green representing stronger, white representing moderate, and red representing weaker association of a gene with a phenotype). We observed that a set of genes including PINX1, CD44, PARK2, RYR2, ADCY5, and SHANK2 displayed a strong proximity to all of the phenotypic terms tested (typically occupying the top 10th percentile of all genes). This was followed by a group of genes including KCNQ5, GRIK4, RPTOR, ACVR1C, and ACSL1 that were strongly related to some and moderately related to the other phenotype terms (typically between the 10th and 25th percentiles of all genes). Finally, there were some instances when a gene displayed a strong predicted proximity score for only a handful of phenotypes but were otherwise weakly associated. For example, the NALCN gene was strongly associated to the muscle contraction phenotype (ranked 9.85%) but otherwise weakly related to other phenotypes (next best score being 24.9%). The MACROD2 gene displayed strong to moderate associations with the majority of the phenotypes (ranks between 6–30%) but scored very poorly against cardiovascular physiological processes, musculoskeletal development, muscle contraction, and aerobic electron transport phenotypes (scores between 70–75%). Finally, genes such as HCG22 and LHFPL3 ranked poorly against all phenotypes (ranks >90% on average). Biograph did not have any information on FAM19A2, C4orf40, C12orf36,. or LOC731789 genes.
Fig. 2.

Application of Biograph software to formulate hypotheses on mechanisms connecting candidate genes to candidate physiological processes. Two-way hierarchical clustering of targets (query genes) and source concepts (key words). Genes are shown in rows and key words in columns. Clustering was performed on the proximity score for every gene to each source concept, as described in the text. Proximity score is represented on a green-white-red color scale with green representing greater proximity and red depicting lower proximity.
As an example of how Biograph's predictions are constructed, we have depicted the connections between the ACSL1 gene and aerobic electron transport chain phenotype in Fig. 3. ACSL1 is required for the activation and transport of long-chain fatty acids into mitochondria and has been shown to be essential for adipose and liver fatty acid oxidation in mouse models (12). ACSL1 transcript levels are also induced in the soleus muscle of B6/J mice fed a high-fat diet (24). ACSL1 is linked to the phenotype via several possible intermediates. Stronger evidence is represented by solid lines in the diagram. One possible route is through the involvement of ACSL1 in the peroxisome proliferator-activated receptor (PPAR) signaling pathway. A second route is constructed on the basis of ACSL1 reaction involving palmitic acid, which in turn, regulates UCP2, a mitochondrial protein that may play a role in free fatty acids sensing. A third possibility is via the sequential action of ACSL1 and ACSM1 on long- and medium-chain fatty acids, respectively. Finally, a fourth connection between ACSL1 and the phenotype could also be envisioned involving the diterpene quinone tanshinone.
Fig. 3.
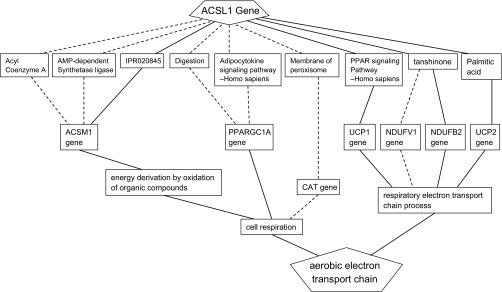
Graphical view of several functional hypotheses that link a query gene (ACSL1) to a phenotype term (aerobic electron transport chain) in Biograph. A graph is generated via the random-walk algorithm of Biograph and is based on existing information in the Biograph knowledge base. Solid and dashed lines represent greater and lesser strengths of evidence, respectively, that connect the query gene to the phenotype term.
Pathway-Level Analysis
Overrepresentation analysis.
Overrepresentation analysis by IPA led to the identification of candidate biological pathways containing a statistically significant excess of genes with association to ΔV̇o2max (P < 0.005). A comparison among the top 10 canonical pathways revealed several genes shared in common among the pathways. For example, 7 of 10 pathways shared 10 or more genes in pairwise comparisons (Fig. 4A). Of the canonical pathways, the protein kinase A (PKA) signaling pathway was a hub pathway that was connected to five other pathways (G protein-coupled receptor signaling, synaptic long-term potentiation, CREB signaling in neurons, cardiac β-adrenergic signaling, and cellular effects of sildenafil). The heat map in Fig. 4B depicts the overlapping gene contents of the top canonical pathways. For example, the PRKACG and PRKAR2B genes (encoding catalytic and regulatory subunits, respectively, of cAMP-dependent PKA) were components of all seven pathways, whereas the PLCB1 gene was a component in five pathways.
Fig. 4.

Overrepresentation analysis using IPA. A: interrelationship among the top canonical pathways associated with ΔV̇o2max. The top canonical pathways that share 10 or more genes among them are shown. The pathway name and its association P value are noted in the boxes; the number of genes shared between two pathways is noted along the line connecting them. B: heat map depicting common genes shared between the top pathways. Pathway names are in columns across the bottom, gene names are in rows. Gene membership in a pathway is indicated in red. C: top associated network derived from the Ingenuity Pathway Analysis knowledge base. The network consists of 35 genes, 31 of which are associated with ΔV̇o2max at P ≤ 0.005. Genes are colored on the basis of the strength of association (deeper shades indicate stronger association). The networks are overlaid with canonical pathway information and show the inclusion of genes from calcium signaling, nNOS signaling, and protein kinase A signaling pathways in the network. Shapes in the legend represent the types of molecules included in the network (enzymes, transporters, ion channels, etc.). The types of functional effects representing the edges are indicated by colored arrows and defined as follows: A, activation; E, expression; L, proteolysis; LO, localization; M, biochemical modification; P, phosphorylation/dephosphorylation; PD, protein-DNA binding; PP, protein-protein binding; RB, regulation of binding.
In addition to the canonical pathways, IPA also allowed for the identification of gene networks that were enriched for associated genes. These networks were dynamically created from IPA's knowledge base and represented a variety of reported functional associations among the component genes (e.g., transcriptional activation/repression, protein-protein interactions, enzyme-substrate relationships, etc.). Figure 4C depicts the top scoring network created from the list of ΔV̇o2max associated genes. For this network, 31 out of 35 genes exhibited SNPs that were associated with ΔV̇o2max (P ≤ 0.005). These associations represented various biological mechanisms including effects on gene expression, protein-DNA and protein-protein interactions, localization, phosphorylation/dephosphorylation, and others. The genes GRIN2B and APP appear as hub genes in the network with extensive connections to other genes. Additionally, when canonical pathway information was overlaid on the network, several genes belonging to the calcium signaling, nNOS signaling in neurons, and PKA signaling pathways were found to be members of this network.
Gene set enrichment analysis.
GSEA was conducted via the IGSEA and GSA-SNP software packages. To reduce false positives, we focused on the set of common ΔV̇o2max associated KEGG pathways identified by both tools. A total of 46 and 22 pathways were identified by IGSEA and GSA-SNP, respectively (false discovery rate ≤ 5%), of which 16 pathways were in common (Table 4). Additionally, we calculated the content similarity among the 16 pathways by determining the proportion of common genes shared between any two pathways (Fig. 5A). We identified large overlaps (75%) in the gene content among pathways related to immune function (type 1 diabetes mellitus, graft vs. host disease, and allograft rejection) and also between cardiomyopathy-related pathways (dilated cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy, 62% overlap). In contrast, several pathways did not show any significant overlap with others (e.g., PPAR signaling, pantothenate and CoA biosynthesis), suggesting that they make independent contributions to the association with ΔV̇o2max. An analysis of the genes found in common in three or more pathways identified ITPR1–3, PLCB1, PRKCA, and PRKCB genes shared between phosphatidylinositol signaling, calcium signaling, and vascular smooth muscle contraction pathways (Fig. 5B). The latter two pathways also shared several isoforms of adenylyl cyclase (ADCY1,3,4,7–9) along with the purine metabolism pathway. Several alpha and beta integrin isoforms (ITGA1,2,4,6,7,9 and ITGB4,6,8) were also shared between the extracellular matrix receptor interaction pathway and two pathways related to cardiomyopathies. Several genes from the major histocompatibility locus (HLA locus) were also shared among immuno-inflammation-related pathways (cell adhesion molecules, allograft rejection, type 1 diabetes mellitus, viral myocarditis, and graft vs. host disease).
Table 4.
Sixteen KEGG pathways identified in common between IGSEA4GWAS and GSA-SNP
| KEGG pathway | IGSEA4GWAS P value | GSA-SNP P value |
|---|---|---|
| KEGG_CELL_ADHESION_MOLECULES_CAMS | <0.001 | 7.41 × 10−10 |
| KEGG_ARRHYTHMOGENIC_RIGHT_VENTRICULAR_CARDIOMYOPATHY_ARVC | <0.001 | 4.97 × 10−9 |
| KEGG_CALCIUM_SIGNALING_PATHWAY | <0.001 | 1.09 × 10−8 |
| KEGG_ECM_RECEPTOR_INTERACTION | <0.001 | 3.09 × 10−7 |
| KEGG_PHOSPHATIDYLINOSITOL_SIGNALING_SYSTEM | <0.001 | 1.30 × 10−5 |
| KEGG_PANTOTHENATE_AND_COA_BIOSYNTHESIS | <0.001 | 2.73 × 10−5 |
| KEGG_VASCULAR_SMOOTH_MUSCLE_CONTRACTION | 0.003 | 3.84 × 10−5 |
| KEGG_DILATED_CARDIOMYOPATHY | <0.001 | 7.91 × 10−5 |
| KEGG_TYPE_I_DIABETES_MELLITUS | <0.001 | 8.58 × 10−5 |
| KEGG_ALLOGRAFT_REJECTION | <0.001 | 0.0004 |
| KEGG_COMPLEMENT_AND_COAGULATION_CASCADES | <0.001 | 0.0004 |
| KEGG_PPAR_SIGNALING_PATHWAY | <0.001 | 0.0004 |
| KEGG_GRAFT_VERSUS_HOST_DISEASE | <0.001 | 0.0012 |
| KEGG_VIRAL_MYOCARDITIS | <0.001 | 0.0015 |
| KEGG_ADHERENS_JUNCTION | 0.008 | 0.0043 |
| KEGG_PURINE_METABOLISM | 0.001 | 0.0046 |
All pathways have a false discovery rate of <5%.
Fig. 5.

Gene-set enrichment analysis using IGSEA4GWAS and GSA-SNP pathway analysis tools. A: overlap among 16 pathways calculated as the proportion of common genes shared by any two pathways. Each cell represents the proportion of common genes among selected pathways. The pathway names are listed on the right, and corresponding numbers are listed to the left and bottom (e.g., #1, adherens junction; #16, viral myocarditis). B: heat map depicting common genes shared between 16 pathways. Pathway names are in rows; gene names are in columns. Gene membership in a pathway is indicated in red.
Ranking pathways by predicted SNP function.
We next sought to determine whether the 16 pathways could be further prioritized on the basis of functional and positional effects arising from the SNPs resident in the pathways. As a first step, we created a list of SNPs that mapped to the genes in each pathway and was nominally associated with ΔV̇o2max at P ≤ 0.05. A total of 7,774 SNPs were selected over the 16 pathways. These SNPs were then examined for functional effects through the SNPNEXUS software. Based on the predicted SNP functions, the pantothenate and CoA biosynthesis, complement and coagulation cascades, and PPAR signaling pathways were the top three pathways with the greatest SNP burden (lower ranks) (Fig. 6A), whereas lower overall SNP burdens were observed for smooth muscle contraction, adherens junction, and arrhythmogenic right ventricular cardiomyopathy pathways. We further performed two-way hierarchical clustering of the pathway ranks against the SNP categories and displayed the results as a heat map (Fig. 6B). Clustering allowed the identification of pathways that were similar to one another due to similarities in the types of SNPs contained in the pathways. We observed a clustering of the aforementioned top three pathways driven primarily by their high SNP contents in the categories of 3′UTR, 5′UTR, 3-downstream, coding, 5-upstream, transcription factor binding sites, CpG islands, and tolerated and damaging amino acid substitutions. In contrast, the SNP burden for a pathway such as vascular smooth muscle contraction was more diffuse, with high ranks observed only for intronic SNPs and SNPs that predict damage to amino acid functions.
Fig. 6.

Ranking pathways by their predicted SNP burden. A: sixteen pathways identified as being significantly associated with ΔV̇o2max by both IGSEA4GWAS and GSA-SNP were scored for functional and positional effects of the SNPs contained in their gene members. Pathways were ranked on the basis of the sum of scores (adjusted for pathway size) such that pathways with lower ranks are those with higher predicted SNP burden. B: two-way hierarchical clustering of pathways and possible SNP effects on the basis of pathway scores in each category (white, lower scores; red, higher scores). Pathways that are more similar by type of SNP burden are clustered more closely compared with pathways that have different SNP burden profiles. SNP categories are abbreviated as follows: INV, inversion; SIFT DL, amino acid substitution (damaging, low confidence); SIFT TL, amino acid substitution (tolerated, low confidence); CNP, copy number polymorphism; INDEL, insertions and deletions; SIFT DH, amino acid substitution (damaging, high confidence); NON, noncoding region; CPG, CpG island region; TFBS, transcription factor binding sites; INT, intronic; 5UP, 5′-upstream (5 kb); SIFT TH, amino acid substitution (tolerated, high confidence); COD, coding region; 5U, 5′ untranslated region (5′UTR); 3DO, 3′-downstream (5 kb); 3U, 3′ untranslated region (3′UTR).
To further understand the genetic drivers behind the pathways with high SNP burden, we investigated the pantothenate and CoA biosynthesis, and PPAR signaling pathways as exemplars. First we determined the number of statistically associated SNPs (P ≤ 0.05) in each of the genes comprising the pathway. As shown in Table 5, the pantothenate and CoA biosynthesis pathway consisted of 16 genes, of which 11 genes contained significantly associated SNPs, with the largest number of SNPs observed for the DPYD gene (62 SNPs). We further determined the predicted functional or positional effects resulting from the SNPs in these 11 genes by scoring the number of SNPs belonging in each of these categories, for each gene. No SNPs were observed in the categories of inversions or tolerated low-confidence amino acid substitutions (SIFT algorithm) (damaging low category). Among all genes, the COASY (CoA synthase) gene was found to harbor SNPs with the greatest variety of functional and positional effects, including one SNP predicting a damaging amino acid substitution (rs615942, C|A polymorphism leading to an S|Y amino acid substitution on position 55 of the protein). When a similar analysis was conducted on the PPAR signaling pathway, the ACSL1 gene was found to contain one or more SNPs belonging to the 3-downstream, 3′UTR, 5′UTR, 5-upstream, coding, intronic, and noncoding categories. SNPs in transcription factor binding sites were observed for FABP7, APOA2, and MMP1 genes, whereas damaging amino acid substitutions were predicted for MMP1, CYP4A22, and SLC27A5 genes. SNPs in the MMP1 gene were associated with the greatest variety of potential functional effects.
Table 5.
Distribution of positional and functional SNPs in Pantothenate and CoA biosynthesis and PPAR signaling pathways
| CNP | CPG | 3DO | 3UTR | 5UP | 5UTR | COD | INT | NON | INDEL | SIFT DH | SIFT TH | SIFT TL | TFBS | Total* | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pantothenate and CoA biosynthesis pathway | |||||||||||||||
| COASY | 7 | 2 | 2 | 2 | 4 | 1 | 5 | 9 | 1 | 2 | 1 | 36 | |||
| PPCS | 3 | 1 | 3 | 19 | 22 | 48 | |||||||||
| ENPP3 | 19 | 6 | 1 | 1 | 1 | 24 | 8 | 1 | 61 | ||||||
| ENPP1 | 18 | 3 | 3 | 1 | 15 | 13 | 1 | 54 | |||||||
| DPYD | 23 | 1 | 1 | 1 | 30 | 6 | 62 | ||||||||
| UPB1 | 11 | 11 | 11 | 2 | 35 | ||||||||||
| VNN2 | 7 | 7 | 9 | 4 | 27 | ||||||||||
| BCAT1 | 6 | 1 | 2 | 2 | 1 | 12 | |||||||||
| VNN1 | 1 | 2 | 4 | 7 | |||||||||||
| DPYS | 1 | 1 | 2 | ||||||||||||
| BCAT2 | 1 | 1 | |||||||||||||
| PPAR signaling pathway | |||||||||||||||
| ACSL5 | 8 | 6 | 3 | 2 | 26 | 24 | 1 | 70 | |||||||
| ACOX3 | 16 | 5 | 16 | 6 | 43 | ||||||||||
| ACSL1 | 4 | 2 | 4 | 1 | 2 | 13 | 11 | 37 | |||||||
| APOA2 | 5 | 1 | 8 | 9 | 9 | 1 | 33 | ||||||||
| MMP1 | 5 | 1 | 1 | 2 | 3 | 12 | 1 | 2 | 27 | ||||||
| CYP4A22 | 1 | 1 | 3 | 4 | 12 | 1 | 3 | 25 | |||||||
| CPT2 | 6 | 2 | 3 | 5 | 7 | 23 | |||||||||
| FABP6 | 1 | 6 | 10 | 2 | 19 | ||||||||||
| SLC27A6 | 18 | 18 | |||||||||||||
| UCP1 | 6 | 3 | 5 | 3 | 17 | ||||||||||
| CD36 | 1 | 1 | 6 | 6 | 14 | ||||||||||
| SLC27A1 | 1 | 2 | 8 | 11 | |||||||||||
| CPT2 | 10 | 10 | |||||||||||||
| FABP1 | 2 | 2 | 5 | 9 | |||||||||||
| ACOX2 | 1 | 1 | 1 | 2 | 1 | 6 | |||||||||
| ILK | 2 | 1 | 1 | 1 | 1 | 6 | |||||||||
| PPARA | 2 | 2 | 2 | 6 | |||||||||||
| CYP27A1 | 1 | 2 | 1 | 1 | 1 | 6 | |||||||||
| FABP7 | 1 | 3 | 1 | 1 | 6 | ||||||||||
| SLC27A5 | 1 | 1 | 1 | 1 | 1 | 5 | |||||||||
| SLC27A2 | 2 | 1 | 1 | 4 | |||||||||||
| PCK1 | 1 | 1 | 2 | 4 | |||||||||||
| SORBS1 | 2 | 2 | 4 | ||||||||||||
| ACOX1 | 1 | 1 | 1 | 3 | |||||||||||
| RXRA | 1 | 1 | 1 | 3 | |||||||||||
| PPARD | 3 | 3 | |||||||||||||
| APOA5 | 1 | 1 | 2 | ||||||||||||
| APOA1 | 1 | 1 | 2 | ||||||||||||
| APOC3 | 1 | 1 | 2 | ||||||||||||
| ACOX1 | 1 | 1 |
The number of nominally associated SNPs (P < 0.05) belonging in each category for each of the pathway genes are shown. Four genes in the pantothenate pathway and 39 genes in the PPAR signaling pathway did not contain any nominally associated SNP and were excluded from the table.
CNP, copy number polymorphism; CPG, CpG island region; 3DO, 3′-downstream (5 kb); 3UTR, 3′ untranslated region (3′UTR); 5UP, 5′-upstream (5 kb); 5UTR, 5′ untranslated region (5′UTR); COD, coding region; INT, intronic; NON, noncoding region; INDEL, insertions and deletions; SIFT DH, amino acid substitution (damaging, high confidence); SIFT TH, amino acid substitution (tolerated, high confidence); SIFT TL, amino acid substitution (tolerated, low confidence); TFBS, transcription factor binding sites.
Total (across rows) of functional and positional SNPs for each gene.
DISCUSSION
Previous advances in the field of exercise genomics have occurred at two broad levels. First is the discovery that intervention-based gains in maximal oxygen uptake is a heritable trait and, although genetic analysis of the HERITAGE cohort did not yield any sequence variant(s) with statistically significant association with V̇o2max training response at the genome-wide level, a relatively small set of 21 SNPs was sufficient to predict gains in V̇o2max and explain nearly the entire genetic variance of the trait (7). Second, RNA expression analysis in muscle biopsies from endurance trainees has led to the identification of training-responsive transcripts (and their candidate regulators) and a set of 29 transcripts whose preintervention expression levels were quite effective as predictors of V̇o2max training response (21, 40).
Given this context, the present study represents a third and complementary approach encompassing a systems biology-based investigation into the biological architecture of V̇o2max training response, gleaned from the simultaneous consideration of multiple DNA sequence variants. The principal aim of this work is to generate novel hypotheses linking genotype to phenotype beyond statistical association of single-locus markers. This was accomplished through three independent but interrelated approaches: 1) building de novo evidence of gene candidacy through informatics driven literature mining; 2) aggregating evidence from statistical associations to link variant enrichment in biological pathways to the observed phenotype; and 3) predicting possible functional consequences of common variants residing in the pathways of interest. Together, these methods should allow for hypothesis-driven ranking and prioritization of genes and pathways for future experimental testing and validation.
The first step in this approach was the derivation of gene-wide ΔV̇o2max association significance levels from SNP P values. Several methods have been proposed for computing gene association statistics from SNP association measures (30, 34, 36). In the current work, we employed the method proposed by Nam et al. (34) and used the second-best SNP P value to represent a gene's association with ΔV̇o2max. Although the easiest and most widely used approach is to take the most significant P value per gene (11, 16, 41, 44, 46), this method has been reported to introduce bias in the downstream analysis, primarily due to effects from a single, highly associated potentially outlier SNP that is not consistent with any of the other SNPs mapped to the gene (17, 27). On the other hand, taking the second-most associated SNP P value significantly improves correlation among P values and FDR scores (34). To further reduce the effects of outliers, the relative ranking of the genes on the basis of their best SNP-associated P values was also compared, with the assumption that genes that ranked high in both lists were less likely to be susceptible to outlier SNPs. This criterion was satisfied for 8 of the top 10 genes (Table 3) when the genes were ranked by their second-best SNP P values. After excluding the pseudogene SLED1, the acyl CoA synthetase long-chain family member 1 (ACSL1) gene was found to have the most significant association with ΔV̇o2max (P = 3.80 × 10−6), followed by HCG22. As noted earlier, an SNP in the ACSL1 gene (rs6552828) was previously shown to display the strongest association with gains in V̇o2max in the HERITAGE GWAS study (7) and was part of the 21-SNP predictor panel, accounting by itself for ∼6% of the training response of V̇o2max. Based on the ACSL1 findings, we extended the analysis to the comparison of the genes related to the 21 predictor SNPs from HERITAGE GWAS with the top 20 most strongly associated genes in the current study, keeping in mind that the number of SNPs analyzed and the methods for SNP-to-gene mapping differed widely between the two studies. Thus whereas the HERITAGE GWAS used data from ∼325,000 genotyped SNPs and putatively assigned SNPs to their most proximal genes, the current study used imputed data on ∼2.5 million SNPs, mapped SNPs strictly on the basis of their location within, or in a 20-kb window on either side of a gene, and used the second-best SNP-association P value to represent the gene association statistic. Despite these differences, we found three other genes in common besides ACSL1 (CAMTA1, BIRC7, and CD44). Five genes did not overlap, and 12 of the 21 genes could not be compared because the genes were mapped more than 20 kb away from the SNPs.
In the next step, we extended the results obtained from GWAS analysis and performed candidate gene prioritization on the basis of a gene's likely involvement in ΔV̇o2max (33). Popular gene prioritization algorithms such as Endeavour or ToppGene (2, 10) require a training set of genes (representing genes already implicated in the phenotype of interest) to learn and then uses the learning on test data to identify new connections. However, because the a priori knowledge on the involvement of genes in ΔV̇o2max response is quite limited, we selected an unbiased prioritization tool, CANDID (19), that did not require training gene sets. We conducted a weighted analysis on the basis of sequence conservation, tissue-specific gene expression, prior literature-based evidence of relationship to phenotype, and strength of GWAS associations. The weighting scheme was designed such that the final determination of candidate genes was influenced more by the evidence from genetic association and gene expression data, although relying less on prior literature reports, thereby favoring the discovery of novel candidate genes. This approach facilitated the identification of genes that scored well in multiple categories instead of just one. Notably, it allowed for the discovery of candidate genes such as SLC45A1, RPTOR, NLN, and PRKG1 that were not top-ranked solely on the basis of GWAS P values. Conversely, the top GWAS-associated gene, ACSL1, received a considerably lower ranking in CANDID (414th) under current weighting conditions. Despite maximum sequence conservation in ACSL1, the overall low ranking was primarily driven by its relatively lower expression across the selected tissues, compared with its observed maximum (normalized expression score = 0.09). It is worth remembering that the algorithm behind CANDID rewards genes that are expressed in multiple tissues at the expense of genes with restricted tissue expression, although such expression may be highly relevant to the phenotype of interest. This appears to be the case for ACSL1, which although not highly expressed in the majority of selected tissues, has a high expression signal in adipocytes and human adipose tissue and shows a robust signal in whole blood and liver, using microarray (the BIOGPS gene annotation portal, http://www.biogps.org) or RNA sequencing-based gene expression measures (Illumina Human BodyMap 2.0 project, http://www.ensembl.info/blog/2011/05/24/human-bodymap-2–0-data-from-illumina/). Notably, when the CANDID analysis was performed one tissue at a time, ACSL1 was indeed the top-ranked candidate gene in the adipocyte-specific analysis, due to its high expression in this cell type. We further tested the robustness of weighting scheme 1 by considering other weights (schemes 2–4) that place higher emphasis on literature, gene expression, or GWAS scores. The analysis indicated significant similarity among the top-ranked genes in schemes 1–3 as assessed by the overlap of the top 25 genes in each scheme. Larger discrepancies were observed with scheme 4, which gave more weight to the literature. However, because the literature evidence is relatively scant for the phenotypes tested, the overall gene scores for scheme 4 were significantly lower compared with the other three schemes, validating our prior decision to down-weigh the literature evidence in the search for candidate genes. In brief, five genes (RPTOR, SLC45A1, PRKG1, NLN, and PINX1) were present in the top 25 genes for all schemes, suggesting their robustness to the various weighing scenarios tested. RPTOR is particularly intriguing because it is believed to be an important modulator of muscle growth signaling and we have previously proposed that a number of genes will be important mediators of adaptability to regular exercise (35, 39).
In addition to the identification and prioritization of candidate genes, we interrogated the possible biological mechanisms that could link them to the observed changes in ΔV̇o2max through use of the knowledge-mining environment implemented in Biograph. We tested a group of exercise-related concept terms (e.g., aerobic electron transport chain, musculoskeletal development, physical fitness, etc.) against the top 20 scoring genes from the GWAS and CANDID analyses and scored the genes on the basis of their proximity to these terms. The analysis allowed us to further classify the genes into four separate groups: genes with consistently high scores across all concept terms, implying broad effects on exercise-related phenotypes (ADCY5, CD44, PARK2, PINX1, RYR2); genes with moderately strong scores across all terms and higher scores against selective concepts (ACSL1, KCNQ5, GRIK4, RPTOR); genes with both high and low scores across several concepts, suggesting selective effects (NALCN, MACROD2); and genes with uniformly low scores against all concepts, suggesting weak mechanistic links (HCG22, LHFPL3). In this context it is interesting to note that although several SNPs in the HCG22 gene were also initially identified as associated with V̇o2max training response in the HERITAGE GWAS study (7), none of these SNPs were retained in the final 21-SNP genomic predictor, suggesting that their mechanistic relevance to exercise trainability may be low. Due to the high GWAS score for ACSL1 gene and its high Biograph score against the concept of aerobic electron transport chain, we examined the functional hypotheses linking ACSL1 to the concept in greater detail. The paths involved uncoupling protein UCP1, the uncoupling protein homologue UCP2, and also the compound tanshinone. Tanshinone, a diterpene quinone extracted from the root of Salvia miltiorrhiza, is a Chinese traditional herb and possesses anti-inflammatory, antioxidative, and cytotoxic properties (25). Tanshinone increased expression of both ACSL1 and NDUFB2 mRNA (47), thus providing a mechanism by which the relationship between ACSL1 and the phenotype may be pharmacologically modulated.
In a second phase, we extended the analysis from individual genes to sets of genes or pathways and applied methods to score and rank pathways for their association with ΔV̇o2max. Pathway analysis has emerged as an alternative approach to querying GWAS data in response to problems identified with traditional SNP-based data analysis (31, 45), such as the missing heritability problem (15, 43). Pathway analysis is based on the premise that biological processes are driven by several molecular pathways and a sufficient number of nonoverlapping, nonconcurrent disruptions in a sufficient fraction of those pathways (e.g., due to sequence variation) can lead to a disorder. Thus, affected individuals may share the same disrupted pathways although the associated gene variants within those pathways may be heterogeneous and the fractional contribution of each gene-variant may be low. Because the number of pathways (hundreds) is orders of magnitude lower than the number of SNPs (millions), the statistical burden of multiple testing is also significantly reduced, leading to increased power. Our pathway analysis strategy was based on increasing the scope for identifying candidate hypotheses by employing multiple analytic approaches (overrepresentation and rank-based methods) and reducing false discoveries by applying multiple tools within the same analytic approach (e.g., IGSEA and GSA-SNP for rank-based methods). By using overrepresentation analysis in the Ingenuity Pathway Analysis tool, we determined the PKA signaling mechanism to be significantly associated with ΔV̇o2max response. More specifically, we identified several genes including the catalytic and regulatory subunits of PKA (PRKACG and PRKAR2B), phospholipase B1 (PLCB1), and inositol triphosphate receptor isoforms (ITPR1 and ITPR2) that were active in a majority of the PKA-dependent pathways. The involvement of PLCB1 and ITPR genes further suggests an effect on calcium mobilization downstream of PKA function. This hypothesis was further corroborated by the results obtained from the rank-based GSEA analysis. Through GSEA, we identified 16 KEGG pathways that were significantly associated with ΔV̇o2max response in both IGSEA and GSA-SNP tools (5% FDR level). These 16 pathways included the calcium signaling and phosphatidylinositol signaling pathways, and they also shared a significant proportion of genes between them (Fig. 5C). On the basis of gene overlap estimates, we further identified the calcium signaling pathway as strongly overlapping with additional pathways related to muscle function (vascular smooth muscle contraction and dilated cardiomyopathy). More specifically, a set of core genes representing isoforms of adenylate cyclase, phospholipase B, and PKA subunits were again found to be enriched for polymorphisms, very similar to what was observed in the IPA analysis. Thus combining the results from the overrepresentation and GSEA approaches, one could postulate that sequence variations in a cluster of functionally interrelated pathways involving PKA signaling, calcium signaling, and phosphoinositide signaling were statistically associated with cardiorespiratory fitness response to exercise training and likely exerted their effects by modulating pathway functions in skeletal and cardiac muscles. In addition to the PKA-calcium signaling axis, GSEA analysis identified another cluster of pathways related to immune response that were also significantly associated to training response. The statistical significance of these pathways (allograft rejection, graft vs. host disease, and type 1 diabetes mellitus) was largely driven by the presence of several major histocompatibility antigen genes (HLA locus). Because the HLA genes are inherently polymorphic in human populations (as a mechanism for broad immune coverage) the implication of our findings in the context of exercise trainability is currently unclear. However, it is worth noting that an earlier study of RNA expression differences and exercise trainability had also identified increased expression of the HLA-A, HLA-B, HLA-C, and HLA-G genes in skeletal muscle in high responders (21).
Whereas pathways could be prioritized on the strength of statistical evidence of their association to ΔV̇o2max (via association P values or FDR) they can also be ranked on the basis of predicted effects from the SNPs contained in the pathways. Statistical association tests consider every SNP to be functionally equivalent, which in reality is almost never the case. Depending on their genomic location, SNPs can be functionally silent or exert a variety of effects that affect various aspects of gene and protein structure and function. A biological pathway containing a collection of statistically associated SNPs will be subject to these effects, which can be aggregated to estimate the relative functional liability of a pathway, compared with other pathways. We define this liability as the predicted SNP burden for the pathway. The SNPNEXUS tool (9) allowed us to classify the predicted functional and positional effects arising from the SNPs contained in the 16 common pathways and then to aggregate, quantify, and rank the pathways on the basis of their predicted SNP burdens. This analysis generated a different ranking of the pathways compared with the rankings observed by statistical associations. Two pathways that ultimately relate to lipid metabolism and cellular energetics (pantothenate and CoA biosynthesis, and PPAR signaling pathways) and one pathway related to immune function (complement and coagulation cascades) were found to have the highest levels of SNP burden. There was no overlap in the gene contents among these three pathways, suggesting that the observed SNP burden was not generated from a repository of shared SNPs.
In contrast, a pathway such as vascular smooth muscle contraction displayed lower SNP burden, although it is a part of the PKA-calcium signaling mechanism. However, one must remember that the predicted SNP burden is an aggregate of all possible SNP effects (functional and positional) within a pathway. Thus although pathways with a low SNP burden may not contain a large number of potentially consequential SNPs overall, they could still contain one or a few SNPs with significant effects on key protein functions. Indeed, the vascular smooth muscle contraction pathway was found to contain SNPs in CYP4A22 (rs2056900) and ARHGEF11 (rs868188) genes that are predicted by the SIFT algorithm to result in damaging amino acid substitutions with high confidence (23).
The implications of high SNP burden in pathways involved in coenzyme A biosynthesis and lipid metabolism are potentially significant. Common convergence points for both pathways are the metabolism of lipids and harvest of energy via the tricarboxylic acid (TCA) cycle leading to the synthesis of ATP via oxidative phosphorylation. One could postulate that variations in this process, for example due to altered substrate availability or substrate conversion, can potentially influence ATP levels during exercise training, leading to differences in ΔV̇o2max. Coenzyme A is required both for the activation of fatty acids for β-oxidation, and as a critical component of the TCA cycle (once as a component of acetyl-CoA, and again during the conversion of α-ketoglutarate to succinyl CoA). On the other hand, an analysis of the PPAR signaling pathway genes with high SNP burden implied both fatty acid transport (ACSL1, ACSL5, FABP6, and SLC27A6) and fatty acid oxidation (ACOX3, CPT2, and CYP4A22). Both of these functions eventually help produce substrates (acetyl-CoA) for the TCA cycle. It is therefore conceivable that functionally relevant SNPs within genes in either of these pathways could ultimately influence lipid metabolism processes and the TCA cycle, which in turn, would affect the availability of ATP.
Certain limitations of the current study are now discussed. First, the current analysis is restricted to common variants that were either genotyped or imputed according to HapMap Phase II reference data. Although imputation certainly increases the number of genotypes, one cannot exclude the possibility that additional, presently untyped, functionally relevant sequence variants still exist, either independently or in linkage disequilibrium with the analyzed variants. Therefore, the current estimation of SNP functional effects is likely an underestimate, especially considering that we focused on common genetic variants in this study (i.e., variants with >1% population minor allele frequency). Second, we did not have available gene expression data on the same individuals who were genotyped, resulting in an inability to identify putative expression quantitative trait loci for the associated SNPs. Finally, the choice of target tissues for gene expression analysis in CANDID reflected a relatively broad selection encompassing neural, hormonal, energy, and muscular functions that could contribute to cardiorespiratory fitness. Although inclusive, this approach will tend to reward genes with broad expression patterns and downweight genes with a more restricted tissue expression, although such genes could be equally relevant to exercise training response.
In previous studies we characterized genomic predictors associated with ΔV̇o2max response (7, 40). Identifying the genomic predictors of favorable response and poor response patterns of V̇o2max to regular exercise is clinically relevant because these predictors could be used in the design of practical prognostic clinical tools. However, before these results can be applied to the general population, the functionality of the genes identified in the present pathway analyses must be further explored. Despite its obvious limitations, the present systems biology approach has the potential to add to our knowledge of individual differences in the responsiveness of cardiorespiratory fitness-related traits to a physically active lifestyle and to our efforts toward personalized exercise prescriptions.
In conclusion, the current study exemplifies an integrative systems-biology approach that could be adopted to interrogate complex genetic data sets such as those generated from GWAS and next-generation sequencing approaches. We provided a sequential analytic model in which statistical and functional inferences are made successively at the levels of SNPs, genes, and pathways. Multiple lines of inquiry and comparison of results from a number of algorithmic approaches are used to increase confidence in the findings. Our multilevel analyses generated fairly consistent results regarding the involvement of previously established biological mechanisms related to cardiorespiratory fitness trainability (e.g., fatty acid metabolism, calcium signaling, muscle function) and the identification of novel ones (e.g., cell-cell communication and pathways central to innate immunity such as complement and coagulation cascades). These pathways constitute excellent targets for further hypothesis-driven studies to verify the individual and interactive contributions of specific genes, DNA sequence variants, and pathways to the ability to respond to regular exercise.
GRANTS
This work was supported by National Institutes of Health Grants 5R25 HL-059868–10, P20 MD-000175–09, 1R21 DK-088319–01, R01 HL-045670 (HERITAGE Family Study to T. Rankinen and C. Bouchard), and 8P20 GM-1033528 (COBRE center grant to M.A. Sarzynski); American Heart Association Grant 0SDG-4230068 to S. Ghosh; and by grants from the European Union (META-PREDICT FP7) and Medical Research Council (001873) to J.A. Timmons. C. Bouchard is partially funded by the John W. Barton Sr. Chair in Genetics and Nutrition.
DISCLOSURES
C. Bouchard is a scientific adviser to Pathway Genomics and to the NIKE SPAR program.
AUTHOR CONTRIBUTIONS
Author contributions: S.G., C.B., and T.R. conception and design of research; S.G., J.C.V., C.B., and T.R. performed experiments; S.G., J.C.V., M.A.S., Y.J.S., J.A.T., and T.R. analyzed data; S.G., J.C.V., M.A.S., Y.J.S., J.A.T., C.B., and T.R. interpreted results of experiments; S.G. and J.C.V. prepared figures; S.G. and M.A.S. drafted manuscript; S.G., J.C.V., M.A.S., Y.J.S., J.A.T., C.B., and T.R. edited and revised manuscript; S.G., C.B., and T.R. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Arthur Leon, Dr. D.C. Rao, Dr. James Skinner, Dr. Jack Wilmore, and Dr. Jacques Gagnon, and all the professional and technical staff for their contributions to the data collection of the HERITAGE Family Study.
REFERENCES
- 1.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30: 97–101, 2002 [DOI] [PubMed] [Google Scholar]
- 2.Aerts S, Lambrechts D, Maity S, Van Loo P, Coessens B, De Smet F, Tranchevent LC, De Moor B, Marynen P, Hassan B, Carmeliet P, Moreau Y. Gene prioritization through genomic data fusion. Nat Biotechnol 24: 537–544, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc B 57: 289–300, 1995 [Google Scholar]
- 4.Bouchard C, An P, Rice T, Skinner JS, Wilmore JH, Gagnon J, Pérusse L, Leon AS, Rao DC. Familial aggregation of V̇o2max response to exercise training: results from the HERITAGE Family Study. J Appl Physiol 87: 1003–1008, 1999 [DOI] [PubMed] [Google Scholar]
- 5.Bouchard C, Leon AS, Rao DC, Skinner JS, Wilmore JH, Gagnon J. The HERITAGE family study. Aims, design, and measurement protocol. Med Sci Sports Exerc 27: 721–729, 1995 [PubMed] [Google Scholar]
- 6.Bouchard C, Rankinen T, Timmons JA. Genomics and genetics in the biology of adaptation to exercise. Comp Physiol 1: 1603–1648, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouchard C, Sarzynski MA, Rice TK, Kraus WE, Church TS, Sung YJ, Rao DC, Rankinen T. Genomic predictors of maximal O2 uptake response to standardized exercise training programs. J Appl Physiol 110: 1160–1170, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bray MS, Hagberg JM, Pérusse L, Rankinen T, Roth SM, Wolfarth B, Bouchard C. The human gene map for performance and health-related fitness phenotypes: the 2006–2007 update. Med Sci Sports Exerc 41: 37–72, 2009 [DOI] [PubMed] [Google Scholar]
- 9.Chelala C, Khan A, Lemoine NR. SNPnexus: a web database for functional annotation of newly discovered and public domain single nucleotide polymorphisms. Bioinformatics 25: 655–661, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 37: W305–W311, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elbers CC, van Eijk KR, Franke L, Mulder F, van der Schouw YT, Wijmenga C, Onland-Moret NC. Using genome-wide pathway analysis to unravel the etiology of complex diseases. Genet Epidemiol 33: 419–431, 2009 [DOI] [PubMed] [Google Scholar]
- 12.Ellis JM, Li LO, Wu PC, Koves TR, Ilkayeva O, Stevens RD, Watkins SM, Muoio DM, Coleman RA. Adipose acyl-CoA synthetase-1 directs fatty acids toward beta-oxidation and is required for cold thermogenesis. Cell Metab 12: 53–64, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallagher IJ, Scheele C, Keller P, Nielsen AR, Remenyi J, Fischer CP, Roder K, Babraj J, Wahlestedt C, Hutvagner G, Pedersen BK, Timmons JA. Integration of microRNA changes in vivo identifies novel molecular features of muscle insulin resistance in type 2 diabetes. Genome Med 2: 9, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh S, Dent R, Harper ME, Gorman SA, Stuart JS, McPherson R. Gene expression profiling in whole blood identifies distinct biological pathways associated with obesity. BMC Med Genomics 3: 56, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibson G. Hints of hidden heritability in GWAS. Nat Genet 42: 558–560, 2010 [DOI] [PubMed] [Google Scholar]
- 16.Holmans P, Green EK, Pahwa JS, Ferreira MA, Purcell SM, Sklar P, Owen MJ, O'Donovan MC, Craddock N. Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am J Hum Genet 85: 13–24, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hong MG, Pawitan Y, Magnusson PK, Prince JA. Strategies and issues in the detection of pathway enrichment in genome-wide association studies. Hum Genet 126: 289–301, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hutz JE, Kraja AT, McLeod HL, Province MA. CANDID: a flexible method for prioritizing candidate genes for complex human traits. Genet Epidemiol 32: 779–790, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keller P, Vollaard N, Babraj J, Ball D, Sewell DA, Timmons JA. Using systems biology to define the essential biological networks responsible for adaptation to endurance exercise training. Biochem Soc Trans 35: 1306–1309, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Keller P, Vollaard NB, Gustafsson T, Gallagher IJ, Sundberg CJ, Rankinen T, Britton SL, Bouchard C, Koch LG, Timmons JA. A transcriptional map of the impact of endurance exercise training on skeletal muscle phenotype. J Appl Physiol 110: 46–59, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khatri P, Sirota M, Butte AJ. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol 8: e1002375, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4: 1073–1081, 2009 [DOI] [PubMed] [Google Scholar]
- 24.Kus V, Prazak T, Brauner P, Hensler M, Kuda O, Flachs P, Janovska P, Medrikova D, Rossmeisl M, Jilkova Z, Stefl B, Pastalkova E, Drahota Z, Houstek J, Kopecky J. Induction of muscle thermogenesis by high-fat diet in mice: association with obesity-resistance. Am J Physiol Endocrinol Metab 295: E356–E367, 2008 [DOI] [PubMed] [Google Scholar]
- 25.Lee CY, Sher HF, Chen HW, Liu CC, Chen CH, Lin CS, Yang PC, Tsay HS, Chen JJ. Anticancer effects of tanshinone I in human non-small cell lung cancer. Mol Cancer Ther 7: 3527–3538, 2008 [DOI] [PubMed] [Google Scholar]
- 26.Lehne B, Lewis CM, Schlitt T. Exome localization of complex disease association signals. BMC Genomics 12: 92, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lehne B, Lewis CM, Schlitt T. From SNPs to genes: disease association at the gene level. PLoS One 6: e20133, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol 34: 816–834, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liekens AM, De Knijf J, Daelemans W, Goethals B, De Rijk P, Del-Favero J. BioGraph: unsupervised biomedical knowledge discovery via automated hypothesis generation. Genome Biol 12: R57, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu JZ, McRae AF, Nyholt DR, Medland SE, Wray NR, Brown KM, Hayward NK, Montgomery GW, Visscher PM, Martin NG, Macgregor S. A versatile gene-based test for genome-wide association studies. Am J Hum Genet 87: 139–145, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo L, Peng G, Zhu Y, Dong H, Amos CI, Xiong M. Genome-wide gene and pathway analysis. Eur J Hum Genet 18: 1045–1053, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature 461: 747–753, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moreau Y, Tranchevent LC. Computational tools for prioritizing candidate genes: boosting disease gene discovery. Nat Rev Genet 13: 523–536, 2012 [DOI] [PubMed] [Google Scholar]
- 34.Nam D, Kim J, Kim SY, Kim S. GSA-SNP: a general approach for gene set analysis of polymorphisms. Nucleic Acids Res 38: W749–W754, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phillips BE, Williams JP, Gustafsson T, Bouchard C, Rankinen T, Knudsen S, Smith K, Timmons JA, Atherton PJ. Molecular networks of human muscle adaptation to exercise and age. PLoS Genet 9: e1003389, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Segre AV, Groop L, Mootha VK, Daly MJ, Altshuler D. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet 6: e1001058, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skinner JS, Wilmore KM, Krasnoff JB, Jaskolski A, Jaskolska A, Gagnon J, Province MA, Leon AS, Rao DC, Wilmore JH, Bouchard C. Adaptation to a standardized training program and changes in fitness in a large, heterogeneous population: the HERITAGE Family Study. Med Sci Sports Exerc 32: 157–161, 2000 [DOI] [PubMed] [Google Scholar]
- 38.Sung YJ, Wang L, Rankinen T, Bouchard C, Rao DC. Performance of genotype imputations using data from the 1000 Genomes Project. Hum Hered 73: 18–25, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Timmons JA. Variability in training-induced skeletal muscle adaptation. J Appl Physiol 110: 846–853, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Timmons JA, Knudsen S, Rankinen T, Koch LG, Sarzynski M, Jensen T, Keller P, Scheele C, Vollaard NB, Nielsen S, Akerstrom T, MacDougald OA, Jansson E, Greenhaff PL, Tarnopolsky MA, van Loon LJ, Pedersen BK, Sundberg CJ, Wahlestedt C, Britton SL, Bouchard C. Using molecular classification to predict gains in maximal aerobic capacity following endurance exercise training in humans. J Appl Physiol 108: 1487–1496, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Torkamani A, Topol EJ, Schork NJ. Pathway analysis of seven common diseases assessed by genome-wide association. Genomics 92: 265–272, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vastrik I, D'Eustachio P, Schmidt E, Gopinath G, Croft D, de Bono B, Gillespie M, Jassal B, Lewis S, Matthews L, Wu G, Birney E, Stein L. Reactome: a knowledge base of biologic pathways and processes. Genome Biol 8: R39, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang K, Dickson SP, Stolle CA, Krantz ID, Goldstein DB, Hakonarson H. Interpretation of association signals and identification of causal variants from genome-wide association studies. Am J Hum Genet 86: 730–742, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang K, Li M, Bucan M. Pathway-based approaches for analysis of genomewide association studies. Am J Hum Genet 81: 1278–1283, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang K, Li M, Hakonarson H. Analysing biological pathways in genome-wide association studies. Nat Rev Genet 11: 843–854, 2010 [DOI] [PubMed] [Google Scholar]
- 46.Wang K, Zhang H, Kugathasan S, Annese V, Bradfield JP, Russell RK, Sleiman PM, Imielinski M, Glessner J, Hou C, Wilson DC, Walters T, Kim C, Frackelton EC, Lionetti P, Barabino A, Van Limbergen J, Guthery S, Denson L, Piccoli D, Li M, Dubinsky M, Silverberg M, Griffiths A, Grant SF, Satsangi J, Baldassano R, Hakonarson H. Diverse genome-wide association studies associate the IL12/IL23 pathway with Crohn disease. Am J Hum Genet 84: 399–405, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Wei Y, Yuan S, Liu G, Lu Y, Zhang J, Wang W. Potential anticancer activity of tanshinone IIA against human breast cancer. Int J Cancer 116: 799–807, 2005 [DOI] [PubMed] [Google Scholar]
- 48.Zhang K, Cui S, Chang S, Zhang L, Wang J. i-GSEA4GWAS: a web server for identification of pathways/gene sets associated with traits by applying an improved gene set enrichment analysis to genome-wide association study. Nucleic Acids Res 38: W90–W95, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]